
14. Utilizing Genetically Engineered Organisms
In Chapter 8, “How Cellular Information Is Altered,” we discussed how cells could be genetically engineered. The techniques of genetic engineering are fairly straightforward; the design of the best production system is not. The choice of host cell, the details of the construction of the vector, and the choice of promoter must all fit into a processing strategy. That strategy includes plans not only for efficient production but also for how a product is to be recovered and purified. The development of processes for making products from genetically engineered organisms requires that many choices should be evaluated. Any choice at the molecular level imposes processing constraints. As indicated in Figure 14.1, an interdisciplinary team approach to process development is necessary to make sure that a well-designed process results.
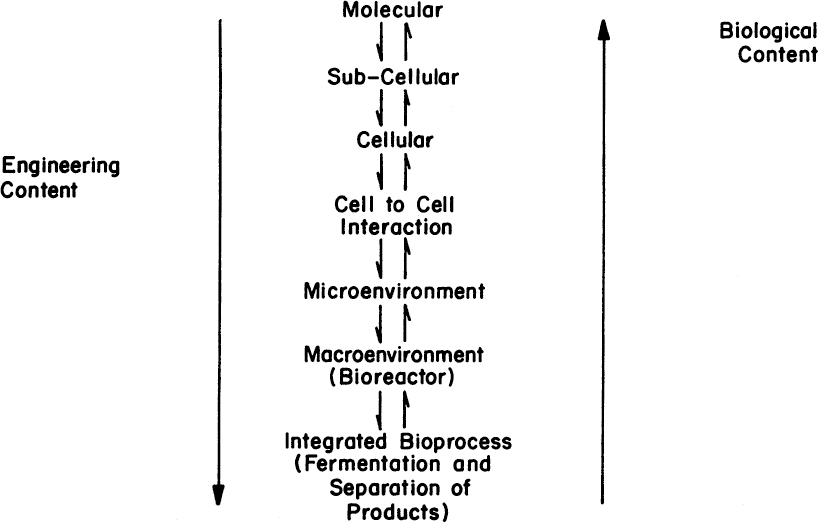
Figure 14.1. Biocatalyst and bioreactor design are strongly interconnected. Choices made at the molecular level can have profound effects on bioreactor design. A multidisciplinary approach is necessary to ensure that a well-designed process is developed.
In this chapter, we discuss some of the questions that must be considered in building processes using genetically engineered cells.
14.1. How the Product Influences Process Decisions
Genetically engineered cells can be used to make two major classes of products: proteins and nonproteins. Nonprotein products can be made by metabolically engineering cells, inserting DNA-encoding enzymes that generate new pathways or pathways with an enhanced capacity to process the precursors to a desired metabolite. Nonprotein products can also include nucleic acid- and polysaccharide-based products that are derived using recombinant DNA technology. However, most current industrial emphasis has been on proteins. Table 14.1 lists some examples. The majority of these proteins are human therapeutics, but proteins that can be used in animal husbandry, in food processing, or as industrial catalysts are also of interest. The global market for recombinant protein therapeutics, or biopharmaceuticals, which currently includes approximately 250 approved products, is forecasted to exceed $200 billion by 2020.
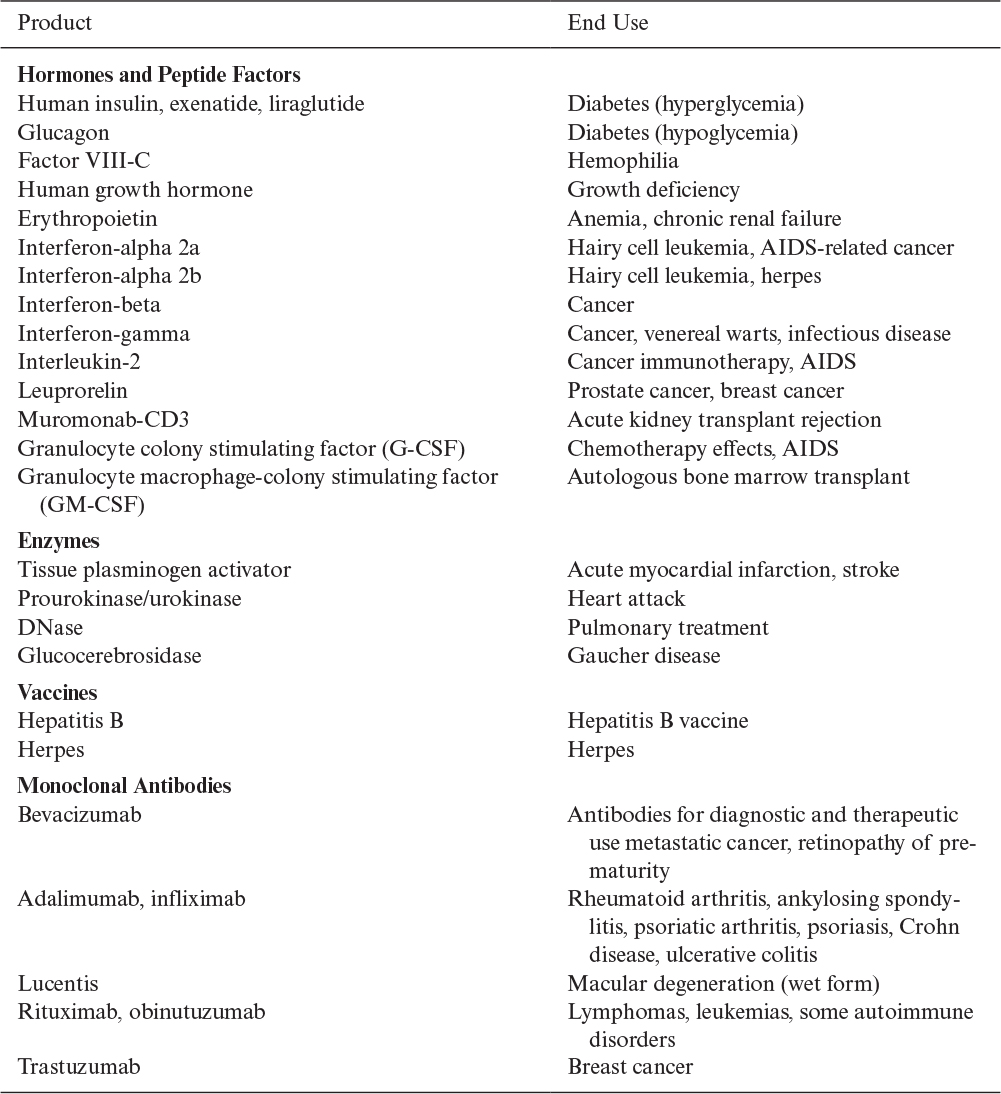
With therapeutic proteins that are injectable, the prime concern is the clinical efficiency of the product. Such products must be highly pure, since strong immunogenic reactions by patients or other side effects can be disastrous. The authenticity of the product is often critical. Correct or near-correct posttranslational processing of the protein (e.g., glycosylation or phosphorylation) is sometimes essential to its therapeutic action. Any variant forms of the protein (e.g., the modification of side groups on amino acids) are highly undesirable and present very difficult purification problems.
The processing challenges in making therapeutic proteins are to ensure product quality and safety. Process efficiency to reduce manufacturing cost, although important, is of less concern because these products are required in relatively small amounts, because they can command high prices, and because the selling price is mainly determined by the costs of process development and regulatory approvals, particularly clinical trials. Thus, for these products, the choice of biological system and processing equipment is dictated by the need to produce highly purified material in an absolutely consistent manner.
Other protein products are purchased strictly on an economic basis, and manufacturing costs play a much more critical role in the viability of a proposed process. In this case, regulatory demands are of less importance than in the production of therapeutic proteins. For animal vaccines or animal hormones, the products must be very pure and render favorable cost ratios. For example, the use of bovine somatropin to increase milk production requires that the increased value of the milk produced be substantially greater than the cost of the hormone and any increase in feed costs due to increased milk production.
For food use, product safety is important, but purity requirements are less stringent than for an injectable. The volume of the required product is often substantial (several metric tons per year), and price is critical because alternative products from natural sources may be available. Proteins used as specialty chemicals (e.g., adhesives and enzymes for industrial processes) usually can tolerate the presence of contaminating proteins and compounds. The manufacturing costs for such proteins will greatly influence the market penetration.
For nonprotein products based on metabolically engineered cells, processing costs compared to costs of other routes of manufacture (usually nonbiological) will determine the success of such a process.
Note that the constraints on production can vary widely from one product class to another. These constraints determine which host cells, vectors, genetic constructions, processing equipment, and processing strategies are chosen.
14.2. Guidelines for Choosing Host–Vector Systems
The success or failure of a process often depends on the initial choice of host organism and expression system. Such choices must be made in the context of processing strategy.
Table 14.2 summarizes many of the salient features of common host systems. The most important initial judgment must be whether posttranslational modifications of the product are necessary. If they are, then an animal cell host system is typically chosen, although the emergence of engineered yeast or bacteria that perform humanlike glycosylation reactions may also be an option. If some simple posttranslational processing is required (e.g., modification with paucimannose glycans), yeast, fungi, or insect cells may be acceptable. Whether posttranslational modifications are necessary for proper activity of a therapeutic protein cannot always be predicted with certainty, and clinical trials may be necessary.
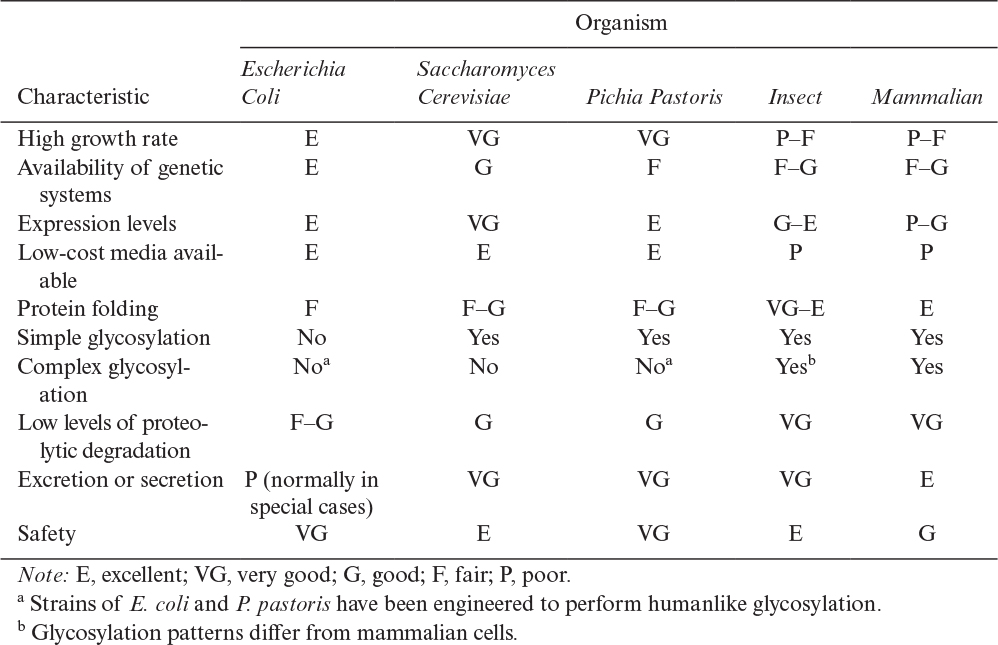
Another important consideration is whether the product will be used in foods. For example, some yeasts (e.g., Saccharomyces cerevisiae) are on the FDA GRAS (generally regarded as safe) list, which would greatly simplify obtaining regulatory approval for a given product. In some cases, edible portions of transgenic plants can be used to deliver vaccines or therapeutic proteins.
14.2.1. Escherichia Coli
If posttranslational modifications are unnecessary, E. coli is usually chosen as the initial host. The main reason for the popularity of E. coli is the broad knowledge base surrounding this heavily studied organism. E. coli physiology and its genetics are probably far better understood than any other living organism. A wide range of host backgrounds (i.e., combinations of specific mutations) is available, as well as vectors and promoters. This large knowledge base greatly facilitates sophisticated genetic manipulations. The well-defined vectors and promoters greatly speed the development of an appropriate biological catalyst.
The relatively high growth rates for E. coli coupled with the ability to grow E. coli to high cell concentrations (>50 gDW/l) and with the high expression levels possible from specific vector-promoter combinations (about 25% to 50% or more of total protein) can lead to extremely high volumetric productivities. Also, E. coli will grow on simple and inexpensive media. These factors give E. coli many economic advantages.
An important engineering contribution was the development of strategies to grow cultures of E. coli to high cell densities. The buildup of acetate and other metabolic by-products can significantly inhibit growth. Controlled feeding of glucose to prevent the accumulation of large amounts of glucose in the medium prevents overflow metabolism and the formation of acetate. Glucose feeding can be coupled to consumption rate if the consumption rate can be estimated on-line.
However, E. coli is not a perfect host. The major problems arise because E. coli does not normally secrete proteins. When proteins are retained intracellularly and produced at high levels, the amount of soluble active protein present is usually limited due to either proteolytic degradation or insolubilization into inclusion bodies.
The production of large amounts of foreign protein may trigger a heat-shock response. One response of the heat-shock regulon is increased proteolytic activity. In some cases, intracellular proteolytic activity results in product degradation at a rate nearly equal to the rate of production.
More often, the target protein forms an inclusion body. Although the heterologous protein predominates in an inclusion body, other cellular material is also often included. The protein in the inclusion body is misfolded. The misfolded protein has no biological activity and is worthless. If the inclusion bodies are recovered from the culture, they can be resolubilized and their activity (and value) can be restored. The difficulty of resolubilization can vary tremendously from one protein to another. When resolubilization is straightforward and recoveries are high, the formation of inclusion bodies can be advantageous, as it simplifies the initial steps of recovery and purification. It is important that during resolubilization the protein be checked by several analytical methods to ensure that no chemical modifications have occurred. Even slight changes in a side group can alter the effectiveness of the product.
Other consequences of cytoplasmic protein production can be important. The intracellular environment in E. coli might not allow the formation of disulfide bridges. Also, the protein will usually start with a methionine, whereas that methionine would have been removed in normal posttranslational processing in the natural host cell. If the product is retained intracellularly, then the cell must be lysed (broken) during recovery. Lysis usually results in the release of endotoxins (or pyrogens) from E. coli. Endotoxins are lipopolysaccharides (found in the outer membrane) and can result in undesirable side effects (e.g., high fevers) and death. Thus, purification is an important consideration.
Many of the limitations of E. coli can be circumvented with protein secretion and excretion. Secretion is defined here as the translocation of a protein across the inner membrane of E. coli. Excretion is defined as release of the protein into the extracellular compartment.
About 20% of all protein in E. coli is translocated across the inner membrane into the periplasmic space or incorporated into the outer membrane. Recall from Chapter 4, “How Cells Work,” that secreted proteins are made with a signal or leader peptide. The presence of a signal peptide is a necessary (but not sufficient) condition for secretion. The signal peptide is a sequence of amino acids attached to the mature protein, and the signal sequence is cleaved during secretion.
Many benefits are possible if a protein is secreted. Secretion eliminates an undesired methionine from the beginning of the protein. Secretion also often offers some protection from proteolysis. Periplasmic proteases exist in E. coli, but usually at a low level. They are most active at alkaline pH values, and pH control can be used to reduce target protein degradation. The environment in the periplasmic space promotes the correct protein folding in some cases (including the formation of disulfide bridges). Proteins in the periplasmic space can be released by gentle osmotic shock so that fewer contaminating proteins are present than if the whole cell were lysed.
Even more attractive would be the extracellular release of target proteins. Normally, E. coli excretes very few of its own proteins (colicin, hemolysin, OsmY, and YebF are a few exceptions), but a variety of schemes to obtain excretion in E. coli are being developed. Strategies usually involve either trying to disrupt the structure of the outer membrane or attempting to use the colicin, hemolysin, OsmY, or YebF excretion systems by constructing a fusion of the target protein with components of these excretion systems. More sophisticated strategies include genetically modifying E. coli with multicomponent excretion machineries (e.g., type II, type III systems) that are derived from excretion-competent bacteria. Excretion without cell lysis can simplify recovery and purification even more than secretion alone while achieving the same advantages as secretion with respect to protein processing. Excretion also facilitates the potential use of continuous immobilized cell systems. The lack of established excretion systems in E. coli has led to interest in alternative expression systems. Also, in some cases, patent considerations may require the use of alternative hosts.
Many of the aforementioned limitations can also be circumvented using genetically engineered host strains. For example, proteolytic degradation of intracellular proteins can typically be reduced or even eliminated by using a protease-deficient host strain of E. coli in which one or more of the major intracellular proteases (e.g., Lon protease) are deleted from the genome. The need for disulfide bonds in cytoplasmic proteins has prompted the development of redox-engineered host strains in which cytoplasmic enzymes that keep cysteines in their reduced form (e.g., thioredoxin reductase TrxB, glutathione reductase Gor) are deleted from the genome. These strains have been used to produce a wide range of valuable human protein products, including tissue plasminogen activator and monoclonal antibodies that require multiple disulfide bonds for proper folding. Likewise, the need for protein glycosylation has prompted the development of glyco-engineered E. coli in which a synthetic pathway is introduced for the production of eucaryotic N-glycans and the transfer of these N-glycans to target proteins. To circumvent concerns associated with endotoxin contamination, endotoxin-free strains of E. coli have been engineered by deleting the genes involved in producing the offending endotoxin signal that is normally part of the lipopolysaccharide while still retaining competency and protein expression capability.
14.2.2. Gram-Positive Bacteria
The gram-positive bacterium Bacillus subtilis is the best studied bacterial alternative to E. coli. Since it is gram positive, it has no outer membrane, and it is a very effective excreter of proteins. Many of these proteins, amylases, and proteases are produced commercially using B. subtilis. If heterologous proteins could be excreted as efficiently from B. subtilis as from amylases and proteases, then B. subtilis would be a very attractive production system.
However, B. subtilis has a number of problems that hinder its commercial adoption. A primary concern is that B. subtilis produces a large amount and variety of proteases. These proteases can degrade the product very rapidly. Mutants with greatly reduced protease activity have become available, but even these mutants may have sufficient amounts of minor proteases to be troublesome. B. subtilis is also much more difficult than E. coli to manipulate genetically because of a limited range of vectors and promoters. Also, the genetic instability of plasmids is more of a problem in B. subtilis than in E. coli. Finally, the high levels of excretion that have been observed with native B. subtilis proteins are not yet obtainable with heterologous protein (i.e., foreign proteins produced from recombinant DNA).
Other gram-positive bacteria that have been considered as hosts include Lactococcus lactis subsp. cremoris and Streptomyces sp. These other systems are less well characterized than B. subtilis but have also shown promise. For example, L. lactis has been evaluated up to phase II clinical trials as an oral anti-inflammatory protein delivery vector in situ in the human gut as treatment for inflammatory bowel disease.
Both gram-negative and gram-positive bacteria have limitations on protein processing that can be circumvented with eucaryotic cells.
14.2.3. Lower Eucaryotic Cells
The yeast Saccharomyces cerevisiae has been used extensively in food and industrial fermentations and is among the first organisms harnessed by humans. It can grow to high cell densities and at a reasonable rate (about 25% of the maximum growth rate of E. coli in similar medium). Yeasts are larger than most bacteria and can be recovered more easily from a fermentation broth.
Further advantages include the capacity to do simple glycosylation of proteins and to secrete proteins. However, S. cerevisiae tends to hyperglycosylate proteins, adding large numbers of mannose units. In some cases, the hyperglycosylated protein may be inactive. These organisms are also on the GRAS list, which simplifies regulatory approval and makes yeast particularly well suited to production of food-related proteins.
Generally, the limitations on S. cerevisiae are the difficulties of achieving high protein-expression levels, hyperglycosylation, and good excretion. Although the genetics of S. cerevisiae are better known than the genetics of any other eucaryotic cell, the range of genetic systems is limited, and stable high protein-expression levels are more difficult to achieve compared to E. coli. Also, the normal capacity of the secretion pathways in S. cerevisiae is limited and can provide a bottleneck on excreted protein production even when high expression levels are achieved.
The methylotrophic yeasts Pichia pastoris and Hansenula polymorpha are very attractive hosts for some proteins. These yeasts can grow on methanol as a carbon–energy source; methanol is also an inducer for the AOX 1 promoter, which is typically used to control expression of the target protein. Very high cell densities (e.g., up to 100 g/l) can be obtained. Due to high densities and, for some proteins, high expression levels, the volumetric productivities of these cultures can be higher than with E. coli. Protein folding and secretion often are also better than in E. coli. These yeasts do simple glycosylation and are less likely than S. cerevisiae to hyperglycosylate. Like many host systems, their effectiveness is often a function of the target protein. The disadvantages of the methylotrophic yeast are due to the high cell density and rate of metabolism, which create high levels of metabolic heat that must be removed and high oxygen demand. Effectively inducing expression while maintaining cell activity requires very good process control because of methanol’s dual rate as growth substrate and inducer. Further, high levels of methanol are inhibitory (i.e., substrate inhibition), which also demands good process control. Scale-up to large reactors often is very challenging, since heat removal, oxygen supply, and process control are typically more difficult in large reactors with longer mixing times (see Chapter 10, “Selection, Scale-Up, Operation, and Control of Bioreactors”). Also, methanol is flammable, and handling large volumes of methanol is a safety concern. Nonetheless, these methylotrophic yeasts are of increasing importance.
Fungi, such as Aspergillus nidulans and Trichoderma reesei, are also potentially important hosts. They generally have greater intrinsic capacity for protein secretion than S. cerevisiae. Their filamentous growth makes large-scale cultivation somewhat difficult. However, commercial enzyme production from these fungi is well established, and the scale-up problems have been addressed. The major limitation has been the construction of expression and secretion systems that can produce extracellular heterologous proteins in amounts as large as some of the native proteins. A better understanding of the secretion pathway and its interaction with protein structure will be critical for this system to reach its potential.
All these lower eucaryotic systems are inappropriate when complex glycosylation and posttranslational modifications are necessary. Historically, in such cases, animal cell tissue culture has been employed. However, researchers have expanded the utility of the yeast P. pastoris by engineering it to secrete human glycoproteins with fully complex, terminally sialylated N-linked glycans. This achievement involved genetic knockout of four genes to eliminate yeast-specific glycosylation, followed by introduction of 14 heterologous genes, allowing replication of the sequential steps of human glycosylation. The reported cell lines were able to produce functional recombinant erythropoietin. Recent efforts have focused on engineering humanlike O-linked protein glycosylation pathways to operate alongside the previously engineered N-linked pathways.
14.2.4. Mammalian Cells
Mammalian cell culture is chosen when the virtual authenticity of the product protein must be complete. Authenticity implies not only the correct arrangement of all amino acids but also that all posttranslational processing is identical to that in the whole animal. In some cases, the cells in culture may not do the posttranslation modifications identically to those done by the same cell while in the body. However, for bioreactor processes, mammalian cell tissue culture will provide the product closest to its natural counterpart. Another advantage is that most proteins of commercial interest are readily excreted.
Slow growth, expensive media, and low protein-expression levels all make mammalian cell culture expensive. As discussed in Chapter 12, “Bioprocess Considerations in Using Animal Cell Cultures,” a wide variety of reactor systems are being used with animal cell cultures. Although many of these can improve efficiency significantly, processes based on mammalian cells remain expensive.
Several cell lines have been used as hosts for the production of proteins using recombinant DNA. The most popular hosts are probably lines of CHO (Chinese hamster ovary) cells.
In addition to the cost of production, mammalian cells face other severe constraints. Normal cells from animals are capable of dividing only a few times; these cell lines are mortal. Some cells are immortal or continuous and can divide continuously, just as a bacterium can. Continuous cell lines are transformed cells. Cancer cells are transformed also (i.e., have lost the inhibition of cell replication). The theoretical possibility that a cancer-promoting substance could be injected along with the desired product necessitates extreme care in the purification process. It is particularly important to exclude nucleic acids from the product. The use of transformed cells also requires precautions to ensure worker safety.
Additionally, the vectors commonly used with mammalian cell cultures have been derived from primate viruses. Again, there is concern about the reversion of such vectors back to a form that could be pathogenic in humans.
Most of these vectors cannot give high expression levels of the target protein in common host cells (usually <5% of total protein). However, higher levels of expression can be obtained (e.g., >1 g/l of secreted, active protein) through amplification of the number of gene copies. It may take 6 months with a CHO cell line to achieve stable, high-level expression. It is often easier to obtain high titers (or product concentration) when producing monoclonal antibodies from hybridoma cultures. However, the technology for producing proteins, in general, is improving rapidly and high-level production can be achieved for many proteins from animal cells.
The quality of the protein product, especially as determined by producing proteins with consistent and authentic posttranslational modifications, may change upon scale-up. Since animal cultures are chosen primarily as the best hosts to produce humanlike glycosylated proteins, it is critical to understand how process conditions affect protein quality. Since culture conditions (shear, glucose, amino sugars, dissolved oxygen [DO], etc.) can change upon scale-up, the efficiency of cellular protein processing can change, altering the level of posttranslational processing. Further, it has been shown that protein quality may change with harvest time in batch cultures. This change may be due to alterations in intracellular machinery, but often it is due to release of proteases and sialidase (an enzyme that removes the sialic acid cap from glycosylated proteins) from dead cells. Also, excessive levels of protein production may saturate the intracellular protein-processing organelles (i.e., endoplasmic reticulum [ER] and Golgi apparatus), leading to incompletely processed proteins. These problems can significantly impact process strategy. One well-known company has been forced to harvest 24 hours early to maintain the sialic acid/protein ratio specified for the product. Early harvest resulted in a 30% loss in protein concentration (as compared with delayed harvest).
Strategies to reduce such problems include selection of cell lines or genetic manipulation of cell lines with reduced levels of sialidase production or enhanced protein-processing capacity. Historically, this task has been difficult, but cell line engineering has become greatly simplified through targeted genome editing. The bacterial clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system enables rapid, easy, and efficient engineering of mammalian genomes. It has a wide range of applications from modification of individual genes to genome-wide screening or regulation of genes. The facile genome editing afforded by CRISPR/Cas9 along with the recent publication of CHO genome sequences should open the door to engineering high-level production of proteins and product quality attributes of interest in this important industrial cell line. Redesign of medium can be beneficial; chemicals that inhibit undesirable extracellular enzyme activity can be added or precursors added (e.g., amino sugars) to improve processing. Cell lysis can be reduced by adding genes (e.g., bcl-2) to the host cell that reduce apoptosis. An engineering solution is to remove the product from the medium as it is formed. For example, perfusion systems with an integrated product capture step can be used.
14.2.5. Insect Cell–Baculovirus System
A popular alternative for protein production at small (<100 l) or laboratory scale is the insect cell–baculovirus system. This system is particularly attractive for rapidly obtaining biologically active protein for characterization studies. Typical host cell lines come from the fall armyworm (Spodoptera frugiperda) and the cabbage looper (Trichoplusia ni). The baculovirus Autographa californica nuclear polyhedrosis virus (AcNPV) is used as a vector for insertion of recombinant DNA into the host cell.
This virus has an unusual biphasic replication cycle in nature. An insect ingests the occluded form in which multiple virus particles are embedded in a protein matrix. The protein matrix protects the virus when it is on a leaf from environmental stresses (e.g., UV radiation). This protein matrix is from the polyhedrin protein. In the midgut of the insect, the matrix dissolves, allowing the virus to attack the cells lining the insect’s gut; this is the primary infection. These infected cells release a second type of virus; it is nonoccluded (no polyhedrin matrix) and buds through the cell envelope. The nonoccluded virus (NOV) infects other cells throughout the insect (secondary infection).
In insect cell culture, only NOVs are infectious, and the polyhedrin gene is unnecessary. The polyhedrin promoter is the strongest known animal promoter and is expressed late in the infection cycle. Replacing the polyhedrin structural gene with the gene for a target protein allows high-level target protein production (up to 50% of cellular protein). Proteins that are secreted and glycosylated are often made at much lower levels than nonsecreted proteins.
In addition to high expression levels, the insect–baculovirus system offers safety advantages over mammal–retrovirus systems. The insect cell lines derived from ovaries or embryos are continuous but not transformed. The baculovirus is not pathogenic toward either plants or mammals. Thus, the insect–baculovirus system offers potential safety advantages. Another important advantage is that the molecular biology and high-level expression of correctly folded proteins can be achieved in less than a month.
This system also has the cellular machinery to do almost all the complex posttranslational modifications that mammalian cells do. However, even when the machinery is present, at least some of the proteins produced in the insect cell–baculovirus system are not processed identically to the native protein. In some cases, their slight variations may be beneficial (e.g., increased antigenic response in the development of an AIDS vaccine), while in others they may be undesirable. While complex glycoforms (including sialic acid) have been made, it is more common to observe only simple glycoforms. Production of complex forms requires special host cell lines and is sensitive to culture conditions.
The insect cell–baculovirus system is a good system to illustrate a holistic perspective on heterologous protein production. Any bioprocess for protein production is complex, consisting of the nonlinear interaction of many subcomponents. Thus, the optimal process is not simply the sum of individually optimized steps. Figure 14.2 presents a holistic view for the insect cell–baculovirus system. Because of the viral component, this system is even more complex than most other bioprocesses, as the infection process and resulting protein expression kinetics must be considered. One factor is the ratio of infectious particles to cells (e.g., multiplicity of infection, or MOI), which alters the synchrony of infection and the resulting protein expression kinetics. Another is the genetic design of the virus (which shares many of the general features of vector design). Also, the quality of the virus stock is important; if the virus stock is maintained incorrectly, mutant virus can form. One example is the formation of defective interfering particles (DIPs) that reduce protein expression in the culture by 90% when high MOIs are used.
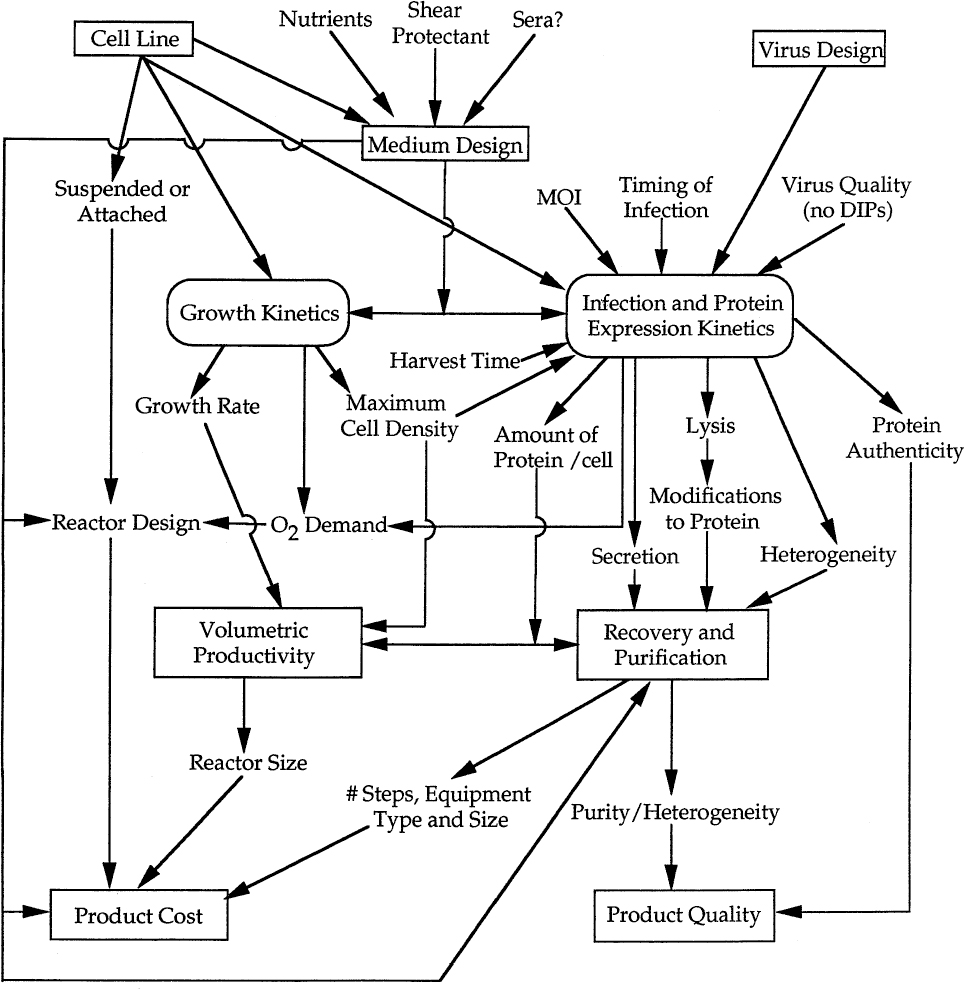
Figure 14.2. Holistic perspective on heterologous protein production with the baculovirus expression system.
Working from the bottom of Figure 14.2 toward the top should reveal the desired product quality and the product worth. Knowing these factors guides selection of bioprocess strategies to achieve the cost and quality desired. To develop that strategy requires an understanding of the basic kinetics and capabilities of the biological system. Understanding these requirements guides selection of the specific host cell line, the medium, and the molecular design of the virus. For example, the addition of serum to the medium of some Ti ni cell lines results in production of proteins with complex N-linked glycosylation, including a sialic acid cap, which may be a requirement for product quality. However, the use of serum alters growth kinetics, expression levels (often less), and the difficulty of purification, which may alter cost. Such trade-offs need to be considered with respect to alternative approaches (e.g., development of a genetically engineered host that could perform the same reactions but in serum-free medium).
14.2.6. Transgenic Animals
In some cases, proteins with necessary biological activity cannot be made in animal cell culture. Although posttranslational protein processes, such as N-linked glycosylation, can be done in cell culture, other more subtle forms of posttranslational processing may not be done satisfactorily. An alternative to cell culture is the use of transgenic animals. Animals are engineered to express the protein and release it into specific fluids, such as milk or urine. For example, transgenic goats have been engineered to produce the protein components of spider silk in milk. High concentrations of complex proteins can be achieved, and such approaches can be cost effective for complex proteins. In these cases, the role for the bioprocess engineer is in protein recovery and purification, although significant issues exist for agricultural engineers and animal scientists in devising appropriate systems to obtain the protein-containing fluid (e.g., pig milking stations). While transgenic animals can be developed from many mammals, sheep, goats, and pigs are the primary species used commercially.
There are significant limitations on this technology. In some cases, the protein of interest will cause adverse health problems in the producing animal. The use of animals raises safety concerns with respect to virus or prion transmission. The process to generate and screen for high-producing animals is inefficient and costly (e.g., $100,000 for a goat and $500,000 for a cow). Perhaps surprisingly, not all of the complex posttranslational processing steps necessary to achieve the desired product occur when the protein is expressed in milk, urine, or blood. Nonetheless, transgenic animals will be critical for production of some proteins.
14.2.7. Transgenic Plants and Plant Cell Culture
Proteins, including many complex protein assemblies, such as antibodies and viruslike particles (as vaccines), can be made inexpensively in plants. Transgenic plants offer many potential advantages in addition to cost. Since plant viruses are not infective for humans, there are no safety concerns with respect to endogenous viruses or prions. Scale-up is readily accomplished by planting more acreage. The protein can be targeted for sterile, edible compartments, either reducing the need for rigorous purification or making it an ideal vehicle for oral delivery of a therapeutic protein. Indeed, development of edible vaccines for use in developing countries is being actively pursued.
The disadvantages of transgenic plants are that expression levels are often low (1% of total soluble protein is considered good), N-linked glycosylation is incomplete, and some other mammalian posttranslational processing is missing. While inexpensive, with easy scale-up, it takes 30 months to test and produce sufficient seed for unlimited commercial use. Such long lead times are undesirable. Further, environmental control on field-grown crops is difficult, so the amount (and possibly quality) of the product can vary from time to time and place to place.
While many crops could be used, much of the commercial interest centers on transgenic corn. Some corn products are used in medicinals, so there exist some FDA guidelines (e.g., contamination with herbicides and mycotoxins), and there is considerable processing experience. Production costs vary with degree of desired purity. For high-purity material (95% pure) a cost of about $4 to $8/g can be estimated. For higher-purity material (99%) with full quality assurance and control, a cost of about $20 to $30/g is reasonable. At least two enzymes are being produced commercially from transgenic plants. Large-scale production of monoclonal antibodies (e.g., 500 kg/yr) for topical uses is being considered.
The use of plant cell cultures is also being explored. The primary advantage of such cultures over transgenic plants is the much higher level of control that can be exercised over the process. Plant cell cultures, compared to animal cell cultures, grow to very high cell density, use defined media, and are intrinsically safer. See Chapter 13, “Bioprocess Considerations in Using Plant Cell Cultures,” for a detailed discussion.
14.2.8. Cell-Free Protein Synthesis
Cell-free protein synthesis (CFPS) is the production of proteins in vitro without using intact, living cells. The typical components of a cell-free reaction include a cell extract, an energy source, a supply of amino acids, cofactors such as magnesium, and the DNA (linear template or circular plasmid) encoding the desired genes. Cell extracts are obtained by lysing the cell of interest and centrifuging out the cell walls, DNA genome, and other debris. The remains are the necessary cell machinery, including ribosomes, aminoacyl-tRNA synthetases, translation initiation and elongation factors, nucleases, and so on. Common cell extracts in use today are made from E. coli, rabbit reticulocytes, wheat germ, and insect cells.
CFPS systems have played an essential role in the discovery of the genetic code and, more recently, are used to meet the increasing demands for simple and efficient protein synthesis. Protein yields can now reach g protein/l reaction volume, batch reactions last for multiple hours, and reaction scale has reached the 100 l milestone. In one notable example, CFPS of 700 mg/l human granulocyte-macrophage colony-stimulating factor (GM-CSF) was achieved at the 100 l manufacturing scale.
There are many advantages gained when using cell-free systems. First, the open nature of the reaction allows the user to directly influence the biochemical systems of interest (e.g., protein synthesis, metabolism). As a result, new components (natural and nonnatural) can be added or synthesized, and these can be maintained at precise concentrations. Second, the chemical environment can be controlled, actively monitored, and rapidly sampled. Third, cell-free systems separate catalyst synthesis (cell growth) from catalyst utilization (protein production), circumventing a major challenge afflicting cell-based engineering efforts. Finally, there are no cell viability constraints. As a result, CFPS has found a wide range of applications from the production of pharmaceutical proteins to protein evolution. Additionally, CFPS systems can be used to test and model our understanding of biological systems.
14.2.9. Comparison of Strategies
The choice of host-vector system is complicated. The characteristics desired in the protein product and the cost are the critical factors in the choice. The dominant systems for commercial production are E. coli and CHO cell cultures. An interesting study of the process economics of these two systems for production of tissue plasminogen activator, tPA, has been published by Datar, Cartwright, and Rosen.† Although this study is dated and current CHO systems show much higher productivities and advances in making soluble protein from E. coli have been made, this analysis provides insight into the factors that need to be considered in comparing production strategies. Their analysis was for plants making 11 kg/yr of product. The CHO cell process was assumed to produce 33.5 mg/l of product, while E. coli made 460 mg/l. The CHO cell product was correctly folded, biologically active, and released into the medium. The E. coli product was primarily in the form of inclusion bodies, and thus biologically inactive, misfolded, and insoluble. The process to resolubilize and refold the E. coli product into active material requires extra steps. The recovery process for the CHO cell material requires five steps, whereas 16 steps are required for the E. coli process. The larger the number of steps, the greater the possibility of yield loss. Total recovery of 47% with the CHO–produced material was possible compared to only 2.8% for the E. coli–produced material.
† R. V. Darter, T. Cartwright, and C. G. Rosen, “Process Economics of Animal Cell and Bacterial Fermentations,” Bio/Technology 11: 349, 1993.
The extra steps in the E. coli process are for cell recovery, cell breakage, recovery of inclusion bodies, resolubilization of inclusion bodies, concentration, sulfonation, refolding, and concentration of the renatured protein. The difficulty of these processes depends on the nature of the protein; tPA is particularly difficult. The concentration of tPA must be maintained at 2.5 mg/l or less, and refolding is slow, requiring 48 hours. A 20% efficiency for renaturation was achieved. Many proteins can be refolded at higher concentrations (up to 1 g/l) and much more quickly. For tPA, the result is unacceptably large tanks and very high chemical usage. In this case, 5 tons of urea and 26 tons of guanidine would be necessary to produce only 11 kg of active tPA.
For tPA, the required bioreactor volumes were 14,000 l for the CHO process and 17,300 l for the E. coli process. The capital costs were $11.1 million for the CHO process and $70.9 million for the E. coli process, with 75% of that capital cost being required for the refolding tanks. Under these conditions, the unit production costs are $10,660/g for the CHO process versus $22,000/g for the E. coli process. The rate of return on investment (ROI) for the CHO process was 130% versus only 8% for the E. coli process. However, if the refolding step yield were 90% instead of 20%, the overall yield would improve to 15.4%, and the unit production cost would fall to $7,530 with an ROI of 85% for the improved E. coli process at production of 11 kg/yr. If the E. coli plant remained the original size (17,300 l fermenter) so as to produce 61.3 kg/yr, the unit production cost would drop to $4,400/g. The cost of tPA from the CHO process is very sensitive to cost of serum in the medium. If the price of media dropped from $10.5/l to $2/l (e.g., 10% to 2% serum in the medium), the cost of the CHO cell product would drop to $6,500/g. (Modern CHO processes are serum free not necessarily to save media costs but to simplify product recovery.)
A primary lesson from this exercise is the difficulty of making choices of host–vector systems without a complete analysis. The price will depend on the protein, its characteristics, and intended use. Changes in process technology (e.g., low serum medium for CHO cells or protein secretion systems in E. coli) can have dramatic effects on manufacturing costs and choice of the host-vector system.
14.3. Process Constraints: Genetic Instability
There is a tension between the goal of maximal target-protein production and the maintenance of a vigorous culture. The formation of large amounts of foreign protein is always detrimental to the host cell, often lethal. Cells that lose the capacity to make the target protein often grow much more quickly and can displace the original, more productive strain, leading to genetic instability (see Figure 14.3).
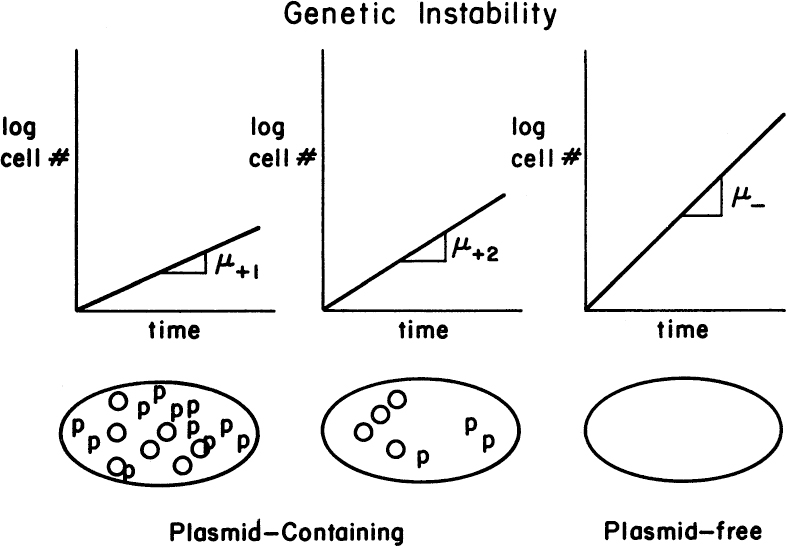
Figure 14.3. Cells that contain plasmids (O) actively making protein (p) must direct many of their cellular resources away from growth and hence grow more slowly than plasmid-free cells (that is, μ+1 < μ+2 < μ–).
Genetic instability can occur due to segregational loss, structural instability, host cell regulatory mutations, and the growth-rate ratio of plasmid-free or altered cells to plasmid-containing unaltered cells. Genetic instability can occur in any expression system. We illustrate this problem by considering gene expression from plasmids in bacteria.
14.3.1. Segregational Loss
Segregational loss occurs when a cell divides such that one of the daughter cells receives no plasmids (see Figure 14.4). Plasmids can be described as high-copy-number plasmids (>20 copies per cell) and low-copy-number plasmids (sometimes as low as one or two copies per cell). Low-copy-number plasmids usually have specific mechanisms to ensure their equal distribution among daughter cells. High-copy-number plasmids are usually distributed randomly among daughter cells following a binomial distribution. For high copy numbers, almost all the daughter cells receive some plasmids, but even if the possibility of forming a plasmid-free cell is low (one per million cell divisions), a large reactor contains so many cells that some plasmid-free cells will be present (e.g., 1000 l with 109 cells/ml yields 1015 cells and 109 plasmid-free cells being formed every cell generation).
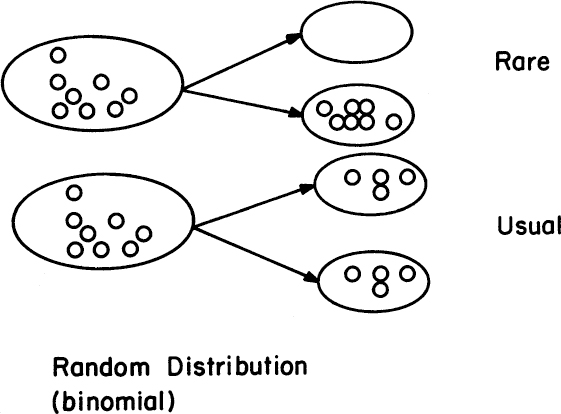
Figure 14.4. Segregational instability results when a dividing cell donates all its plasmids to one progeny and none to the other. (With permission, from G. Georgiou, Optimizing the production of recombinant proteins in microorganisms, AIChE J. 34(8):1233 [1988].)
The segregational loss of plasmid can be influenced by many environmental factors, such as dissolved oxygen, temperature, medium composition, and dilution rate in a chemostat. Many plasmids will also form multimers, which are multiple copies of the same plasmid attached to each other to form a single unit. The process of multimerization involves using host cell recombination systems. A dimer is a replicative unit in which two separate plasmids have been joined, and a tetramer is a single unit consisting of four separate monomers fused together.
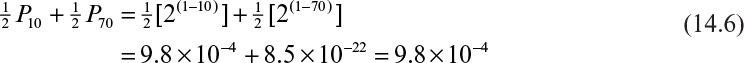
14.3.2. Plasmid Structural Instability
In addition to the problems of segregational instability, some cells retain plasmids but alter the plasmid to reduce its harmful effects on the cell (structural instability). For example, the plasmid may encode both for antibiotic resistance and for a foreign protein. The foreign protein drains cellular resources away from growth toward an end-product of no benefit to the cell. However, if the investigator has added antibiotics to the medium, the cell will benefit from retaining the gene encoding the antibiotic resistance. Normal mutations will result in some altered plasmids that retain the capacity to encode for desirable functions (e.g., antibiotic resistance) while no longer making the foreign protein. In other cases, cellular recombination systems will integrate the gene for antibiotic resistance into the chromosome. Cells containing structurally altered plasmids can normally grow much more quickly than cells with the original plasmids. A culture having undergone a change in which the population is dominated by cells with an altered plasmid has undergone structural instability.
14.3.3. Host Cell Mutations
Mutations in host cells can also occur that make them far less useful as production systems for a given product. These mutations often alter cellular regulation and result in reduced target-protein synthesis. For example, if the promoter controlling expression of the foreign protein utilizes a host cell factor (e.g., a repressor), then modification of the host cell factor may greatly modulate the level of production of the desired plasmid-encoded protein. The lac promoter (see our discussion in Chapter 4 of the lac operon) can be induced by adding chemicals; for a lac promoter lactose or a chemical analog of lactose (e.g., isopropyl-β-D-thiogalactoside [IPTG]) can be used. Such promoters are often used in plasmid construction to control the synthesis of a plasmid-encoded protein. If induction of plasmid-encoded protein synthesis from this promoter reduces cellular growth rates (as in Figure 14.2), then a mutation that inactivates lac permease would prevent protein induction in that mutant cell. The lac permease protein is necessary for the rapid uptake of the inducer. Thus, the mutant cell would grow faster than the desired strain. Alternatively, a host cell mutation in the repressor, so that it would not recognize the inducer, would make induction impossible.
The key feature of this category of genetic instability is that a host cell mutation imparts a growth advantage to the mutant so that it will eventually dominate the culture. In this case, the mutant cell will contain unaltered plasmids but will make very little of the target plasmid-encoded protein.
14.3.4. Growth-Rate-Dominated Instability
The importance of all three of these factors (segregational loss of plasmid, structural alterations of the plasmid, and host cell mutations) depends on the growth-rate differential of the changed cell-plasmid system to the original host–vector system. If the altered host–vector system has a distinct growth advantage over the original host–vector system, the altered system will eventually dominate (i.e., genetic instability will occur).
The terms used to describe the cause of genetic instability are based on fairly subtle distinctions. For example, if genetic instability is due to segregational instability, we would infer that the rate of formation of plasmid-free cells is high. In this case, the number of plasmid-free cells would be high irrespective of whether the plasmid-free cells had a growth-rate advantage. If, on the other hand, we claimed that the genetic instability is growth-rate dependent, we would imply that the rate of formation of plasmid-free cells is low, but the plasmid-free cells have such a large growth-rate advantage that they outgrow the original host–vector system. In most cases, growth-rate-dependent instability and one of the other factors (segregational loss, structural changes in the plasmid, or host cell mutations) are important.
The growth-rate ratio can be manipulated to some extent by the choice of medium (e.g., the use of selective pressure such as antibiotic supplementation to kill plasmid-free cells) and the use of production systems that do not allow significant target-protein production during most of the culture period. For example, an inducible promoter can be turned on only at the end of a batch growth cycle when only one or two more cell doublings may normally occur. Before induction, the metabolic burden imposed by the formation of the target protein is nil, and the growth ratio of the altered to the original host–vector system is close to 1 (or less if selective pressure is also applied). This two-phase fermentation can be done as a modified batch system, or a multistage chemostat could be used. In a two-stage system, the first stage is optimized to produce viable plasmid-containing cells, and production formation is induced in the second stage. The continual resupply of fresh cells to the second stage ensures that many unaltered cells will be present.
The problem of genetic instability is more significant in commercial operations than in laboratory-scale experiments. The primary reason is that the culture must go through many more generations to reach a density of 1010 cells/ml in a 10,000 l tank than in a shake flask with 25 ml. Also, the use of antibiotics as selective agents may not be desirable in the large-scale system owing either to cost or to regulatory constraints on product quality.
In the next section, we discuss some implications of these process constraints on plasmid design. In the section following, we discuss how simple mathematical models of genetic instability can be constructed.
14.4. Avoiding Process Problems in Plasmid Design
When we design vectors for genetic engineering, we are concerned with elements that control plasmid copy number, the level of target-gene expression, and the nature of the gene product, and we must also allow for the application of selective pressure (e.g., antibiotic resistance). The vector must also be designed to be compatible with the host cell.
Different origins of replication exist for various plasmids. The origin often contains transcripts that regulate copy number. Different mutations in these regulatory transcripts will yield greatly different copy numbers. In some cases, these transcripts have temperature-sensitive mutations, and temperature shifts can lead to runaway replication in which plasmid copy number increases until cell death occurs.
Total protein production depends on both the number of gene copies (e.g., the number of plasmids) and the strength of the promoter used to control transcription from these promoters. Increasing copy number while maintaining a fixed promoter strength increases protein production in a saturable manner. Typically, doubling copy numbers from 25 to 50 will increase protein production twofold, but an increase from 50 to 100 will increase protein production less than twofold. If the number of replicating units is above 50, pure segregational plasmid loss is very low. Most useful cloning vectors in E. coli have stable copy numbers from 25 to 250.
Many promoters exist. Some of the important ones for use in E. coli are listed in Table 14.3. An ideal promoter would be both very strong and tightly regulated. A zero basal level of protein production is desirable, particularly if the target protein is toxic to the host cell. A rapid response to induction is desirable, and the inducer should be cheap and safe. Although temperature induction is often used on a small scale, thermal lags in a large fermentation vessel can be problematic. Increased temperatures may also activate a heat-shock response and increased levels of proteolytic enzymes. Many chemical inducers are expensive or might cause health concerns if not removed from the product. Some promoters respond to starvation for a nutrient (e.g., phosphate, oxygen, and energy), but the control of induction with such promoters can be difficult to do precisely. The recent isolation of a promoter induced by oxygen depletion may prove useful because oxygen levels can be controlled relatively easily in fermenters.

Anytime a strong promoter is used, a strong transcriptional terminator should be used in the construction. Recall from Chapter 4 that a terminator facilitates the release of RNA polymerase after a gene or operon is read. Without a strong terminator, the RNA polymerase may not disengage. If the RNA polymerase reads through, it may transcribe undesirable genes or may disturb the elements controlling plasmid copy numbers. In extreme cases, this might cause runaway replication and cell death.
The nature of the protein and its localization are important considerations in achieving a good process. To prevent proteolytic destruction of the target protein, a hybrid gene for the production of a fusion protein can be made. Typically, a small part of a protein native to the host cell is fused to the sequence for the target protein with a linkage that can be easily cleaved during downstream processing. Also, fusion proteins may be constructed to facilitate downstream recovery by providing a “handle” or “tail” that adheres easily to a particular chromatographic medium.
Another approach to preventing intracellular proteolysis is to develop a secretion vector in which a signal sequence is coupled to the target protein. If the protein is secreted in one host, it will usually be excreted in another, at least if the right signal sequence is used. Replacement of the protein’s natural signal sequence (e.g., from an eucaryotic protein) with a host-specific signal sequence can often improve secretion.
The secretion process is complicated, and the fusion of a signal sequence with a normally nonsecreted protein (e.g., cytoplasmic) does not ensure secretion, although several cases of secretion of normally cytoplasmic proteins have been reported. Apparently, the mature form of the protein contains the “information” necessary in the secretion process, but no general rules are available to specify when coupling a signal sequence to a normally cytoplasmic protein will lead to secretion.
To ensure the genetic stability of any construct and to aid in the selection of the desired host–vector combination, the vector should be developed to survive under selective growth conditions. The most common strategy is to include genes for antibiotic resistance. The common cloning plasmid pBR322 contains both ampicillin and tetracycline resistance. Multiple resistance genes are an aid in selecting for human-designed modifications of the plasmid.
Another strategy for selection is to place on the vector the genes necessary to make an essential metabolite (e.g., an amino acid). If the vector is placed in a host that is auxotrophic for that amino acid, then the vector complements the host.† In a medium without that amino acid, only plasmid-containing cells should be able to grow. Because the genes for the synthesis of the auxotrophic factors can be integrated into the chromosome or because of reversions on the parental chromosome, double auxotrophs are often used to reduce the probability of nonplasmid-containing cells outgrowing the desired construction.
† Recall that an auxotrophic mutant would be unable to synthesize an essential compound on its own.
One weakness in both of these strategies is that, even when the cell loses the plasmid, the plasmid-free cell will retain for several divisions enough gene product to provide antibiotic resistance or the production of an auxotrophic factor (see Figure 14.5). Thus, cells that will not form viable colonies on selective plates (about 25 generations are required to form a colony) can still be present and dividing in a large-scale system. These plasmid-free cells consume resources without making product.
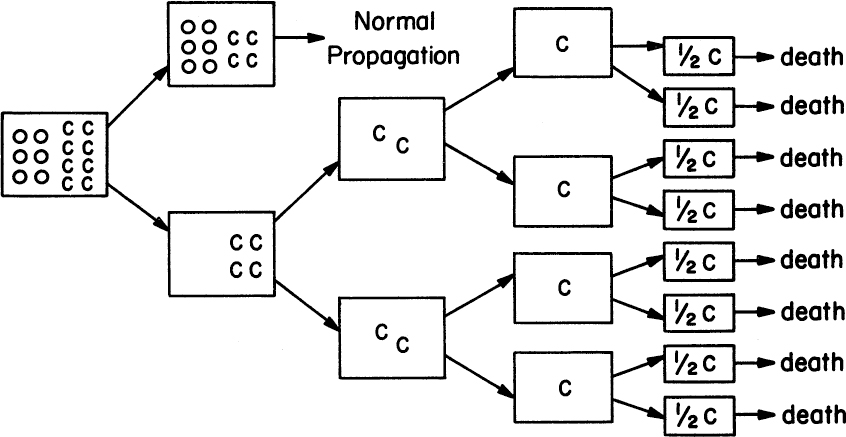
Figure 14.5. Newly born plasmid-free cells usually contain sufficient complementing factor (C) to withstand killing by a selective agent or starvation from the lack of a growth factor. In this case, the plasmid-free cell undergoes three divisions before the complementing factor is reduced to an ineffective level.
Another related problem, particularly in large-scale systems, is that plasmid-containing cells may protect plasmid-free cells from the selective agent. For example, auxotrophic cells with a plasmid may leak sufficient levels of the auxotrophic factor that plasmid-free cells can grow. With an antibiotic, the enzyme responsible for antibiotic degradation may leak into the medium. Also, the enzyme may be so effective, even when retained intracellularly, that all the antibiotic is destroyed quickly in a high-density culture, reducing the extracellular concentration to zero. Although genes allowing the placement of selective pressure on a culture are essential in vector development, the engineer should be aware of the limitations of selective pressure in commercial-scale systems.
The other useful addition to plasmid construction is the addition of elements that improve plasmid segregation. Examples are the so-called par and cer loci. These elements act positively to ensure more even distribution of plasmids. The mechanisms behind these elements are incompletely understood, although they may involve promoting plasmid–membrane complexes (the par locus) or decreasing the net level of multimerization (the cer locus). Recall from Example 14.1 that multimerization decreases the number of independent, inheritable units, thus increasing the probability of forming a plasmid-free cell.
Any choice of vector construction must consider host cell characteristics. Proteolytic degradation may not be critical if the host cell has been mutated to inactivate all known proteases. Multimerization can be reduced by choosing a host with a defective recombination system. However, host cells with a defective recombination system tend to grow poorly. Many other possible host cell modifications enter into considerations of how to best construct a vector for a commercial operation.
These qualitative ideas allow us to anticipate to some extent what problems may arise in the maintenance of genetic stability and net protein expression. However, a good deal of research has been done on predicting genetic instability.
14.5. Predicting Host–Vector Interactions and Genetic Instability
Many of the structured mathematical models we discussed previously can be extended to include component models for plasmid replication. Such models can then predict how plasmid-encoded functions interact with the host cell. The quantitative prediction of the growth-rate ratio and the development of plasmid-free cells due to segregational losses can be readily made. The most sophisticated models will predict the distribution of plasmids within a population and even the effects of multimerization on genetic stability. These models are too complex to warrant discussion in an introductory course.
We consider some simple models that mimic many of the characteristics we discussed with models of mixed cultures. A number of simple models for plasmid-bearing cells have been proposed. The key parameters in such models are the relative growth rates of plasmid-free and plasmid-containing cells and the rate of generation of plasmid-free cells (i.e., segregational loss). These parameters can be determined experimentally or even predicted for more sophisticated models of host–vector interactions.
Consider how a simple model may be constructed and how the parameters of interest may be determined experimentally.†
† This analysis is adapted from the paper of N. S. Cooper, M. E. Brown, and C. A. Cauleott, Journal of General Microbiology 133: 1871, 1987.
The simplest model considers only two cell types: plasmid free (n–) and plasmid containing (n+), where n– and n+ are the number of concentrations of plasmid-free and plasmid-containing cells, respectively. The model assumes that all plasmid-containing cells are identical in growth rate and in the probability of plasmid loss. This assumption is the same as assuming that all cells have exactly the same copy number. As we showed in Example 14.1, the actual distribution of copy numbers can make a significant difference on plasmid loss. Also, plasmid-encoded protein production is not a linear function of copy number, so assuming that all cells have the same copy number may lead to incorrect estimates of the growth rate of plasmid-bearing cells. The assumption of a single type of plasmid-bearing cell is a weak assumption, but other assumptions result in a level of complexity inconsistent with this book’s purpose. However, models that recognize the segregated nature of the plasmid population are available in the research literature.
Let us further restrict our initial considerations to a single-stage chemostat:
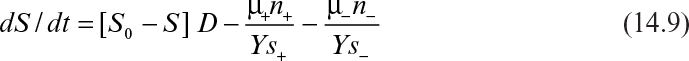
where R is the rate of generation of plasmid-free cells from plasmid-containing cells and Ys+ and Ys– are cell-number yield coefficients (i.e., the number of cells formed per unit mass of limiting nutrient consumed). R can be represented by
where P = probability of forming a plasmid-free cell. P can be estimated by equation 14.1 if the copy number is known or can be predicted with a more sophisticated structured–segregated model. A value for P could be estimated from an experimentally determined copy-number distribution as in Example 14.1(c), which would be more realistic than assuming a monocopy number. As we will soon see, R can be determined experimentally without a knowledge of copy number.
Equations 14.7 through 14.9 assume only simple competition between plasmid-containing and plasmid-free cells. No selective agents are present, and the production of complementing factors from the plasmid is neglected. Following is the simplest assumption for cellular kinetics:


The situation can be further simplified if we assume that after a few generations in a chemostat with constant operating conditions the total number of cells (Nt) is constant. This approximation will be acceptable in many cases as long as the metabolic burden imposed by plasmid-encoded functions is not too drastic and D is less than 80% of either μ+max or μ–max. For allowable dilution rates, these assumptions allow us to decouple the substrate balance (equation 14.9) from immediate consideration. Then, addition of equations 14.7 and 14.8 yields
Nt is constant, and
Therefore, at quasi-steady state, equation 14.12 becomes
or
where f+ is the fraction of the total population that is plasmid-containing cells and f– is the fraction of plasmid-free cells. Since f+ + f– = 1,
Substituting equation 14.15 into equation 14.7 after equation 14.7 is divided by Nt yields
And, after rearrangement,
Equation 14.17 is a Bernoulli’s equation. It can be solved in terms of a dummy variable v = 1/f+, assuming a constant ∆μ and R. A constant ∆μ and R would be achieved only if the copy number distribution of plasmid-containing cells remained constant during the experiment. Also, strictly speaking, the assumption of constant ∆μ is inconsistent with constant Nt. This analysis is best applied to situations where ∆μ is not extremely large. The solution is
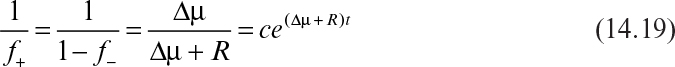
where c is the constant of integration. The initial condition is f– = f–0 at t = 0. Then c must be
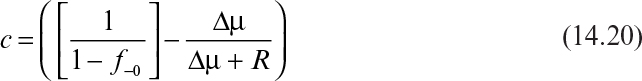
Once c is evaluated, equation 14.19 can be rearranged to yield

Since f–0 is usually ≪1, equation 14.21 becomes

Further simplification is possible by considering three limiting cases:
Case 1: ∆μ ≫ R (growth-rate-dependent instability dominant)
Case 2: ∆μ ≤ R (segregational instability dominant)
Case 3: ∆μ < 0 and |∆μ| ≫ R (effective selective pressure)
These limiting cases have the following yields:
Case 1: Where t is sufficiently small so that ∆μ ≫ (f–0∆μ + R)e(∆μt):
Case 2: Where f–0 ∆μ will be ≪ R and using a binomial expansion:
Case 3: The denominator is ≈∆μ, since |∆μ| ≫ R and f–0 ≪ 1, and we denote ∆μ = –|∆μ|:

For each of these cases, a straight-line portion of the data will allow estimates of ∆μ and R if the basic assumptions for each case are met.
Figure 14.6 shows the behavior that would be expected for each case in a chemostat. The appropriate plots to estimate the parameters ∆μ and R are also given.
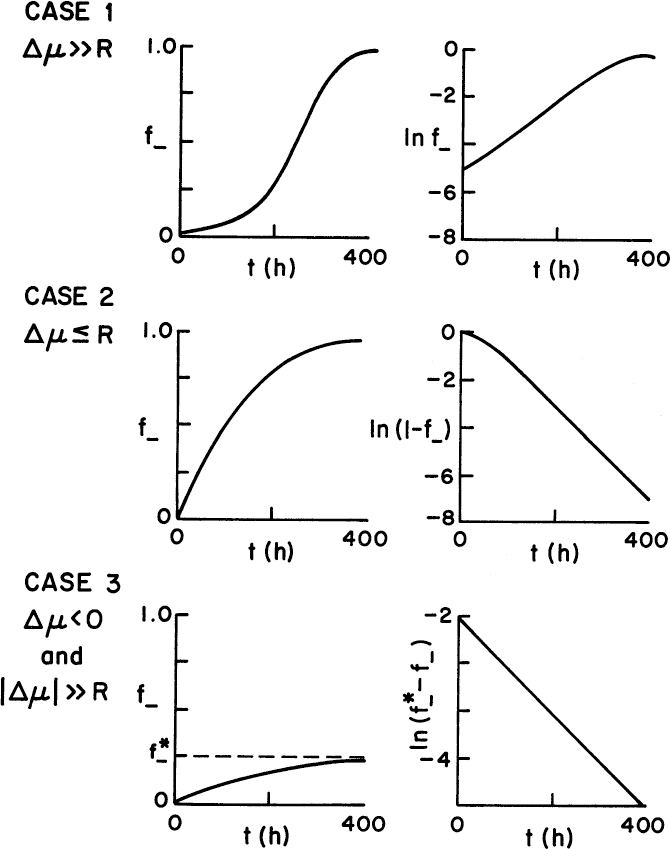
Figure 14.6. The shape of a plot of f– versus t is diagnostic for limiting cases of equation 14.21. For each case, a plot with a substantial linear region can be constructed and used to evaluate ∆μ and R.
In case 1 for ∆μt > 1, we have the following:
Thus, the slope, m, gives ∆μ, and R is given by the intercept:
where f–i is the value of f– at the intercept.
Equation 14.27 has interesting implications in helping to quantify the conditions for which equation 14.26 is valid. That is, t must be sufficiently small when ∆μ ≫ R:
Δμ >> (f–0Δμ + R)eΔμt
Thus,
or
or
or

The analysis also assumes that ∆μt > 1; thus, the linear region suitable for use in case 1 is

In case 2, for t >/(∆μ + R),
The intercept is used to evaluate f+i and the slope m = –(∆μ + R):
In case 3, we note that f– assumes a constant value between 0 and 1, which we denote as ![]() . Equation 14.25 yields
. Equation 14.25 yields ![]() by allowing t → ∞:
by allowing t → ∞:
A plot of ![]() versus time will give a slope, m:
versus time will give a slope, m:
Equations 14.36 and 14.37 can be combined to give


This analysis provides a method to estimate experimentally the parameters of importance in predicting genetic instability. Once these parameters are known, equation 14.21 can be used to predict chemostat performance. However, recall the large number of assumptions that went into these expressions. Case 1 is the one relevant to most commercial systems, where high expression levels are coupled with high-copy-number plasmids. Case 2 will occur only for low-copy-number plasmids that do not have a par locus or other stabilizing features. The results for case 3 must be applied very cautiously. For μ– to be less than μ+, selective pressure usually must be applied (e.g., antibiotic resistance or auxotrophic hosts). Equations 14.7 and 14.8 do not recognize the possibility of leakage of nutrients or enzymes, which would degrade the antibiotics. If there were no leakage and the antibiotic-to-cell ratio were high enough to leave an effective residual level of antibiotic, then the analysis for case 3 could be applied if μ– were interpreted correctly. For a newly born plasmid-free cell, there will be a carryover of the complementing factor, which will be gradually reduced by turnover and dilution (recall Figure 14.5). Thus, in the first generation after plasmid loss, μ– may even be greater than μ+, but μ– will decrease from generation to generation until it becomes zero. The analysis from case 3 will give an effective average value of μ–.
Removal of many of these assumptions to yield a more realistic analysis increases complexity and makes the development of simple analytical expressions difficult. Let us consider how this simple analysis may be used and how it can be extended.


Figure 14.7. Determination of and R using data from Example 14.2.

† Physically, these changes in μ+ and μ– could occur due to small changes in residual substrate concentration. This observation implies that the assumption of constant and R used in integrating equation 14.17 would fail if were very large.

A change in the probability of segregational losses as a function of dilution rate corresponds to the copy number, which also changes with dilution rate. The pDW17 plasmid has an average copy number of about 40 to 50 in exponential growth in minimal medium.
Consider equation 14.1, repeated here, which predicts the probability of plasmid loss by random segregation:
where Z = 40 and P = 1.8 × 10–12, which is certainly less than the maximum value calculated in these experiments. Note from Example 14.1, however, that if the average copy number were distributed so that half the cells had 10 plasmids and half had 70, then the probability of plasmid loss would be 9.8 × 10–4, which is very close to the maximum probabilities allowed by these experimental data.


Our discussion and examples so far have been for continuous reactors. For most industrial applications, batch or fed-batch operations will be used. While continuous reactors are particularly sensitive to genetic instability, genetic instability can be a significant limitation for batch systems.
For a batch reactor, the cell-number balances are
where
Solving equation 14.44 with these initial conditions results in equation 14.46:
And equation 14.45, using equation 14.46, yields
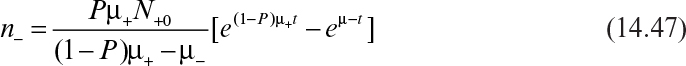
Since ![]() ,
,

Equation 14.48 is usually recast in terms of number of generations of plasmid-containing cells (ng), a dimensionless growth-rate ratio (a), and the probability of forming a plasmid-free cell upon division of a plasmid-containing cell (P). The mathematical definitions of ng and a are


With these definitions, equation 14.48 becomes

The use of this approach is illustrated in the following example.
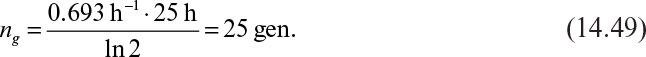
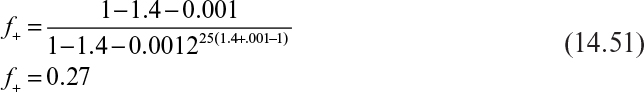
14.6. Regulatory Constraints on Genetic Processes
When genetic engineering was first introduced, there was a great deal of concern over whether the release of genetically modified cells could have undesirable ecological consequences. Reports in the popular press led to fears of “genetic monsters” growing in our sewers, on our farmlands, or elsewhere. Consequently, the use of genetic engineering technology is strictly regulated.
The degree of regulatory constraint varies with the nature of the host, vector, and target protein. For example, consider a scenario where serious harm might arise. The gene for a highly toxic protein is cloned into E. coli to obtain enough protein to study that protein’s biochemistry. Assume that a plasmid that is promiscuous (i.e., the plasmid will shuttle across species lines) is used. Also assume that laboratory hygiene is not adequate and a small flying insect enters the laboratory and comes into contact with a colony on a plate awaiting destruction. If that insect leaves the laboratory and returns to its natural habitat, then the target gene is accidentally released into the environment. Laboratory strains of E. coli are fragile and usually will not survive long in a natural environment. However, a very small probability exists that the plasmid could cross over species lines and become incorporated into a more hardy soil bacterium (e.g., Pseudomonas sp.). The plasmid would most certainly contain antibiotic-resistance factors as well. The newly transformed soil bacterium could replicate. Many soil bacteria are opportunistic pathogens. If they enter the body through a wound, they can multiply and cause an infection. If, in addition, the bacterium makes a toxic protein, the person or animal that was infected could die from the toxic protein before the infection was controlled. If the plasmid also confers antibiotic resistance, the infection would not respond to treatment by the corresponding antibiotic, further complicating control of the spread of the gene for the toxin.
This scenario requires that several highly improbable events occur. No case of significant harm to humans or the environment due to the release of genetically modified cells has been documented. However, the potential for harm is real.
Regulations controlling genetic engineering concentrate on preventing the accidental release of genetically engineered organisms. The deliberate release of genetically engineered cells is possible, but an elaborate procedure must be followed to obtain permission for such experiments.
The degree of containment required depends on the ability for the host to survive if released, the ability for the vector to cross species lines or for a cell to be transformed by a piece of naked DNA and then have it incorporated into the chromosome via recombination, and the nature of the genes and gene products being engineered. The National Institutes of Health (NIH) have issued guidelines that regulate the use of recombinant DNA technology. Special regulations apply to large-scale systems (defined as >10 l). There are three levels of containment: BL1-LS, BL2-LS, and BL3-LS. BL1-LS (biosafety level 1, large scale) is the least stringent. Experiments involving overproduction of E. coli proteins in E. coli using plasmids derived from wild populations are readily approved and do not require elaborate containment procedures (see Table 14.5). Experiments that would move the capacity to produce a toxin from a higher organism into bacteria or yeast would be subjected to a much more thorough evaluation, and more elaborate control facilities and procedures would be required.

Table 14.6 compares the requirements for the three containment levels.


TABLE 14.6. Physical Containment Requirements for Large-Scale Fermentations Using Organisms Containing Recombinant DNA Molecules
In all cases, no viable organisms can be purposely released. Gas vented from the fermenter must be filtered and sterilized. All cells in the liquid effluent must be killed. The latter can present some operating issues, since the inactivation of the host cell must be done in such a way as not to harm what are often fragile products. Emergency plans and devices must be on hand to handle any accidental spill or loss of fluid in the fermentation area. These extra precautions increase manufacturing costs.
The issue of the regulation of cells and recombinant DNA will undoubtedly undergo further refinement with time. Both laboratory and manufacturing personnel need to keep abreast of any such changes.
14.7. Metabolic Engineering
Metabolic engineering involves controlling and redirecting metabolic pathways to improve product formation rates and yields (and to suppress formation of undesired products) by using metabolic, enzymatic, and/or genetic manipulations of a host organism. Often, the tools of genetic engineering are used to generate a totally new metabolic pathway (series of enzymes), amplify an existing pathway, disable an undesired pathway, or alter the regulation of a pathway. The principle motivations for metabolic engineering are the production of specialty chemicals (e.g., secondary metabolites, amino acids, biofuels, flavorings, fragrances, polymer precursors), utilization of alternative substrates (e.g., pentose sugars from hemicellulose), or bioremediation of hazardous wastes (e.g., benzoates or trichloroethylene; see Figure 14.8 for an example). Metabolic production of chemicals is desired over chemical synthesis routes due to the high specificity of biological catalysts, mainly enzymes, to produce desired enantiomers as well as to their ability to catalyze reactions for which a chemical synthesis route is unknown or too lengthy. Additionally, metabolic synthesis can occur at more labile reaction conditions than other chemical routes and is a sustainable way of producing chemicals. Three values that are often optimized for metabolic production are final titer (g/l), yield (g/g), and specific productivity rate (g/l hr).
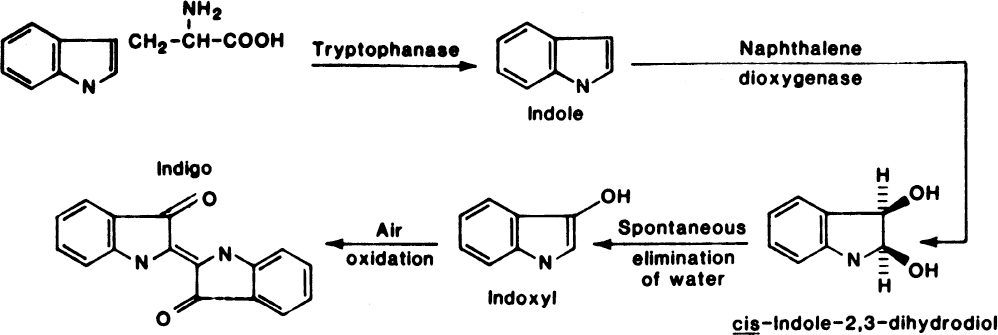
Figure 14.8. Proposed pathway for indigo biosynthesis in a recombinant strain of E. coli. Indole is formed from tryptophan by tryptophanase, a natural enzyme in E. coli. Naphthalene dioxygenase formed by the expression of the cloned Pseudomonas DNA oxidizes indole to indigo; cis-2,3-dihydroxy-2,3-dihydroindole and indoxyl have not been isolated. Their inclusion is based on the known activities of aromatic hydrocarbon dioxygenases and established mechanisms for the chemical synthesis of indigo. (With permission, from B. D. Ensley et al., Expression of naphthalene oxidation genes in Escherichia coli results in the biosynthesis of indigo, Science 222:167, 1983, and American Association for the Advancement of Science.)
One of the first choices a metabolic engineer must make is whether to produce a desired compound in a native host that naturally expresses the metabolic pathway or to port the pathway into a chassis organism for heterologous expression. Due to recent advances in synthetic biology and systems biology, engineering nonmodel organisms has become easier and more widespread; however, there are still many advantages to heterologous hosts. Following are some guidelines for using each.
Heterologous hosts:
• The desired metabolic pathway can be put under the control of a regulated promoter. The investigator can turn on the pathway in situations where the pathway might normally be suppressed (e.g., degradation of a hazardous compound to a concentration lower than necessary to induce the pathway in the natural isolate) as well as at desired times to not interfere with cell growth (e.g., late exponential phase to ensure enough biomass is built up and to not have the pathway interfere with cell growth). Often, the environmental signals that cause expression of pathways in native hosts are poorly understood.
• High levels of enzymes in desired pathways can be obtained with strong promoters; only low activity levels may be present in the natural isolate.
• Genetic manipulation is easier in well-understood heterologous hosts (e.g., deleting competing pathways, addition of genes to the chromosome, or production of enzymes from plasmids). Additionally, most chassis organisms are fully sequenced permitting the generation of metabolic models and prediction of knockouts.
• Transformation protocols are well established, permitting the addition of many pathway variants to the organism to select optimal performers. “Omics” (transcriptomics, proteomics, and metabolomics) techniques and data are established.
• Several pathways can be combined in a single organism by selecting enzymes from more than one organism.
• A product is toxic if it is produced in high quantities in the native host. Similarly, feedback pathway regulation may be poorly understood in the native organism.
• Pathways moved from lower eucaryotes to bacteria can be controlled by a single promoter; in lower eucaryotes, each protein has a separate promoter.
• Model organisms typically grow quickly using inexpensive media. Bioreactors are available for scale-up.
• There are safety concerns for culturing large amounts of pathogenic native hosts.
• The genetically engineered cell can be proprietary property.
Native host:
• Any of the enzymes required for the biological conversion are unknown, eliminating the ability to transfer specific genes to a heterologous host.
• The desired product is toxic to heterologous host.
• The native host has unique growth properties (e.g., utilization of unique carbon sources, necessity of specific substrate uptake to form product of interest, growth in high-temperature environments). Additionally, many organisms have noncanonical pathways, including novel reduction pathways to make them advantageous to produce specific compounds.
• The host has the ability to grow in a microbial consortia to produce the desired compound.
Equally important to the selection of host is the identification of pathway enzymes. Advances in genomic sequencing and annotation have rapidly allowed the construction of pathways never before found in nature. Increased DNA synthesis capability has permitted the use of genes from organisms that cannot be easily cultured or are pathogenic. Inexpensive gene synthesis has also enabled researchers to test many homologous enzymes for a single pathway step to find ones with optimal conversion, specificity, and solubility. Detailed metabolic maps are available through the Kyoto Encyclopedia of Genes and Genomes (KEGG), allowing for easy determination of the presence of pathways to make precursor compounds as well as competing pathways. DNA sequencing and synthesis combined with omics data has been used to elucidate novel enzyme functions for previously nonannotated enzymes, permitting their use in heterologous hosts. While addition of new reactions may be accomplished by the addition of plasmids encoding enzymes of interest, the modification or elimination of pathways in a cell requires editing of its genomic DNA. Currently, genome editing can be accomplished in many bacteria using phage-induced homologous recombination. In eucaryotes, natural homologous recombination can be induced by targeted creation of double-stranded breaks in the chromosomal DNA. This targeting can be accomplished by fusions of nucleases to DNA-binding proteins such as zinc fingers and transcription activator-like effectors (TALEs). More recently, CRISPR-Cas systems, which comprise RNA-guided nucleases, have shown remarkable versatility in targeting genomes of bacteria, yeast, plant, insect, zebrafish, and even human cell lines.
Engineering pathways require good quantitative information on the flow of metabolites in a cell. Structured models of cells can be used in conjunction with experiments to optimize the design of new pathways. Metabolic engineering has been a fertile area for engineering contributions.
Since the first mention of metabolic engineering in the early 1990s, the field has greatly progressed due to advancements in genomic sequencing, enzyme characterization and pathway implementation. Metabolic engineers have been able to create a vast array of chemicals from those to be used as second-generation biofuels to secondary metabolites such as terpenoids, polyketides and nonribosomal peptides. One of the largest success stories of metabolic products, due to the scale of its production, is 1,3-propanediol. Biofuels such as isobutanol and n-butanol can now be made at large scales and high concentrations (>50 g/l for isobutanol). Engineered microbes also contribute a key step to industrial vitamin C production, which is over 110 kilotons annually. Additionally, high-value therapeutic precursors are being created, such as artemisinic acid that can be converted into antimalarial drugs and opioids such as hydrocodone. Biosynthetic routes for polymeric precursors such as glucaric acid and adipic acid have been developed. High-value fragrances and flavors such as acetoin, vanillin, and methyl ketones are possible through modification to central carbon metabolism, amino acid synthesis, and fatty acid metabolism.
14.8. Synthetic and Systems Biology
Successful implementation of genetically engineered cells for an industrial process requires exquisite control of the complex cellular environment. The discipline of synthetic biology arose out of the application of control principles from electrical engineering to biological systems. A primary doctrine of synthetic biology is that biological systems are composites of “parts” that can be replaced or modified to accomplish specific goals. Examples of these parts include promoters, ribosome binding sites (RBSs), terminators, and plasmid origins of replication, as well as genes of interest. For example, expression of a gene of interest might be increased by replacement of a weak promoter with a promoter of high or intermediate strength.
Another thrust in synthetic biology has been the creation of cellular circuits, whereby engineered cells can sense changes in the environment (input) and respond (e.g., by production of a reporter protein) using Boolean logic (output). Inducible promoters factor heavily into the creation of cellular circuits and repressor–promoter pairs have been assembled into multi-input NOT, AND, XOR, and other logic gates. Additionally, repressors can be layered to create feedback loops that lead to oscillatory or toggle switchlike behavior. Finally, multilayered kill-switch circuits have been developed to prevent the growth of engineered microbes outside of a designated environment.
Many efforts in synthetic biology research are focused on standardization—that is, characterization of the performance of individual parts under different conditions to reliably predict outcomes when parts are combined. These efforts seek to make construction of large, complex assemblies of parts easy for researchers to construct and implement.
Computational modeling efforts to understand the complexity of biological/metabolic/synthetic networks fall into the discipline of systems biology. Due to the highly nonlinear, dynamic nature of a cell and its metabolism, uninformed changes intended to improve production of a specific compound often fail to give the desired result. The challenge of metabolic engineering and synthetic biology is to express appropriate genes in the right amount. Too much of a key enzyme may result in little change, if it is not a rate-influencing step, as there is insufficient reactant to increase the flow of substrate (or flux) into the reaction product. Additionally, overexpression of non-rate-limiting enzymes leads to increased metabolic burden, diverting cellular resources away from product formation to unnecessary protein expression. If it is rate influencing, the increased reaction of the substrate will alter fluxes of precursors in other pathways; sometimes these changes compromise the cell’s ability to grow or to provide coreactants when needed. Metabolic engineering and synthetic biology require a delicate balance of activities and are a problem of quantitative optimization rather than simple maximization of expression of a gene. Using various computational techniques, systems biologists model reactions that contribute to particular reaction networks such as cell-signaling or blood clotting. Such networks can comprise hundreds to thousands of reactions, even up to the genome scale in microbes.
Metabolic control theory and the closely allied activity of metabolic flux analysis are mathematical tools that can be applied to such problems. These tools are used to predict the flux through a metabolic network, allowing determination of the right amount of enzyme expression and whether competing pathways need to be removed. A flux balance equation consists of the product of a stoichiometric matrix and an intracellular flux vector to yield an overall rate vector. Using whole genome sequencing and knowledge of pathway organization, it is possible to construct a metabolic model and the stoichiometric matrix. Typically, equations in such a model are highly underdetermined due to the fluxes being unknown and require assumptions (e.g., on energy stoichiometry) that may not be justified for a metabolically engineered cell. However, improved experimental techniques to measure the intracellular flux vector (e.g., gas chromatography or liquid chromatography combined with advanced mass spectrometry, nuclear magnetic resonance [NMR], carbon isotope labeling and tracking, high-throughput transcriptomics via RNA-Seq) coupled with genomic/proteomic information may provide the data to allow complete solution of the flux balance equations.
By assumption of steady state, the dot product of the stoichiometric matrix and flux vector is set to zero, creating a linear set of equations to be solved. A numeric solution to the underdetermined fluxes is possible when given a function to be maximized, often cell growth. Heterologous pathway enzymes and reactions can be added to the model to see how fluxes are changed upon their addition. Such models have been useful in predicting pathway knockouts to eliminate enzymes from diverting flux away from desired products as well as determining optimal expression amounts for heterologous enzymes. Additionally, these models provide detailed information about generation of endogenous precursors necessary for product formation and redox factor regeneration, which may be overlooked upon first implementation of the heterologous pathway. One caution is that analysis of pathways in isolation can be misleading. An assumption of such analysis is that the products of the pathway do not influence inputs into that pathway. Given the complexity of a cell, it is difficult to assure that such an assumption is satisfied. Due to improved experimental techniques, it is now possible to measure fluxes for hosts expressing heterologous pathways, eliminating previous concerns of its effect on endogenous metabolism. Increased computational speed, transcriptomics, proteomics, and metabolomics have enabled a new set of kinetic-based models that do not assume steady state to provide more quantitative prediction of nonlinear metabolism.
Metabolic control theory is based on a sensitivity analysis to calculate the response of a pathway to changes in the individual steps in a pathway. This approach allows calculations of flux control coefficients, which are defined as the fractional change of flux expected for a fractional change in the amount of each enzyme. This process involves a linearization, so that flux coefficients can vary significantly if growth conditions and the cell’s physiological step change. An important result of the application of this theory to many cases is that it is rare for a single enzyme step to be rate-controlling. Typically, several enzymatic steps influence rate. Maximization of flux through a particular pathway would require several enzyme activities to be altered simultaneously. The potential of such analysis to contribute to rational design of microbes is clear but not yet routinely applicable.
Quantitative modeling of metabolism with heterologous pathways has shaped the design and implementation of such pathways. Balancing enzyme expression level with flux and metabolic burden is often the first concern. Enzyme production can be varied using different transcriptional promoters, plasmid copy numbers, ribosome binding sites, transcript stability, genomic integration copies, compartmentalization, and posttranscriptional stability to achieve balance. However, these strategies provide static control of enzyme production. Dynamic production strategies make use of biosensors that provide feedback control of pathway expression to permit an even more balanced expression profile. Searching through the immense expression space is not possible for many pathways due to low-throughput titer measurements; modular metabolic engineering groups enzymes to facilitate quicker optimization of expression. Lastly, but importantly, overall pathway reaction must be considered, especially to achieve redox balance. Balancing of metabolism and expression is important to obtaining high titer product but also aids in pathway stability.
14.9. Protein Engineering
Not only can cells be engineered to make high levels of naturally occurring proteins or to introduce new pathways, but we can also make novel proteins. It is possible to make synthetic genes and design proteins in silico to form totally new proteins with folds and chemistries not observed in nature. Through protein crystallography, NMR, and computational modeling, we are beginning to understand the rules by which a protein’s primary structure is converted into its three-dimensional form to relate a protein’s shape to its functional properties, stability, and catalytic activity.
The goal of protein engineering is to endow a protein with superior characteristics from those observed in nature to facilitate their use in biotechnology. Some properties that are commonly altered include the enhancement of catalytic rate, improvement in stability (thermostability, resistance to proteases, intracellular solubility), modification in binding specificity, alteration of cofactor preference, detoxification of the protein for the desired host compatibility, modification of protein–protein interactions, alteration of posttranslational modification/regulation, and differentiation of protein localization. Protein engineering has typically followed two paradigms: rational design and directed evolution. Rational design utilizes knowledge of how protein structure affects functional characteristics (i.e., structure–function relationships) in order to make specific changes to the primary sequence of the protein in a manner that alters the function in a desired way. Directed evolution involves introducing mutations randomly to the protein sequence, generating a library of protein variants that is subjected to a screen or selection to identify beneficial mutations.
In general, for rational design, the protein structure must be known from X-ray crystallography and/or NMR. Key amino acids in the structure are selected for alteration based on computer modeling and previously developed structure–function relationships. Residues in and around the active site are often mutated to change interactions of the protein with substrates to enhance catalytic rates and alter specificity. Stability improvement can occur through mutation of surface residues or introducing additional bonding pairs (e.g., noncovalent hydrogen bonds, covalent disulfide bonds). The technique used to generate genes encoding the desired changes in protein structure is called site-directed mutagenesis. Using this approach, any desired amino acid can be inserted precisely into the desired position. If a protein structure is not known, often related proteins are crystalized and a homology model can be generated to permit rational design.
It might not be clear why site-directed mutagenesis would be preferred to simple random mutation-selection procedures. One reason may be that mutation followed by selection for particular properties may be difficult when the alterations in protein properties are subtle and confer no advantages or disadvantages on the mutant cell. It may also prove difficult to interrogate all or most of the members in a directed evolution library (often containing 106 unique variants or greater), especially if the assay for characterizing the protein variants is low throughput.
A second reason is that site-directed mutagenesis can be used to generate the insertion of an amino acid in a particular location, while a random mutation giving the same result would occur so infrequently as to be unobtainable.
To make this point more evident, consider the degenerate nature of the genetic code. Each codon consists of three letters. The odds for mutation in one of these three letters is about 10–8 per generation. The odds that two letters would simultaneously be altered is much lower (order of 10–16). The codon UAC (for tyrosine) could be altered by single-letter substitutions to give AAC (asparagine), GAC (aspartate), CAC (histidine), UCC (serine), UUC (phenylalanine), UGC (cysteine), UAA (stop signal), UAG (stop signal), and UAU (tyrosine). Random mutants in this case are very unlikely to carry substitutions for 13 of the 20 amino acids. Thus, most of the potential insertions can be generated reliably only by using site-directed mutagenesis.
It is often the case that structure–function information is not available, making rational strategies for protein engineering difficult or even impossible to implement. Under these circumstances, an alternative approach is that of directed evolution. This process is based on random mutagenesis of a gene and the subsequent isolation of proteins with desired properties. Large libraries of mutant genes must be made so that the rare beneficial variants are present. One technique to generate a library of mutants is error-prone PCR, where the fidelity of a DNA polymerase is decreased in a manner that permits the user to control the mutation rate and thereby generate genetic diversity. Another technique for generating gene libraries is DNA shuffling, whereby homologous or nonhomologous genes are randomly fragmented and reassembled in vitro to create a library of chimeras. Once a library of mutants is generated, a high-throughput isolation step in the form of a screen or selection must be applied to identify library members that exhibit the desired characteristic(s). Screens analyze the entire protein library by coupling the mutation that leads to a desired protein property with a high-throughput assay such as flow-assisted cell sorting (FACS), cell-surface display/phage display, or colorimetric readouts. Selections impose a hard cutoff for library members by coupling the desired characteristic(s) directly to survival or death of the host cell, similar to antibiotic selection for plasmid-containing cells. Critical to the success of a directed-evolution strategy is the coupling of genotype (DNA sequence of specific variants) to phenotype (output of a screen/selection) to determine the identity of the mutations that gave rise to the desired property change and to permit an iterative approach where the improved variant is used as the template for another directed evolution cycle.
Directed-evolution and rational design are often not performed in a vacuum and are most effective when combined through what is called semirational design. Due to incomplete protein function–structure knowledge, often several residues must be altered to find protein variants with the desired properties. By mutating only specific residues, semirational design eliminates the need for a high-throughput screen/selection by focusing the library on variants that have a high likelihood of generating desired functional changes. Saturation mutagenesis (site directed mutagenesis where the codons for all possible amino acids are substituted), and iterative cycles of mutation and characterization are utilized to find beneficial mutations. Additionally, these techniques combined with improved biochemical methods are leading to a better understanding of the complex relationship between protein structure and function by connecting amino acid changes with altered protein characteristics.
14.10. Summary
The application of recombinant DNA technology at the commercial level requires a judicious choice of the proper host–vector system. E. coli greatly facilitates sophisticated genetic manipulations, but process or product considerations may suggest alternative hosts. S. cerevisiae is easy to culture and is already on the GRAS list, simplifying regulatory approval, although productivities are low with some proteins and hyperglycosylation is a problem. P. pastoris can produce very high concentrations of proteins, but the use of methanol presents challenges in reactor control and safety. Bacillus and the lower fungi may have well-developed secretion systems that would be attractive if they can be harnessed. Animal cell culture is required when posttranslational modifications are essential. Mammalian cells offer the highest degree of fidelity to the authentic natural product. Insect cell systems potentially offer high expression levels and greater safety than mammalian cells at the cost of a potential decrease in the fidelity of posttranslational processing. Transgenic animals are good alternatives for complex proteins requiring extensive posttranslational processing. Transgenic plants and plant cell cultures are emerging as production systems for high-volume protein products. Plants are well suited to produce proteins for oral or topical delivery. The growing number of available genome sequences combined with new tools for facile genome editing (e.g., gene knockout, gene knock-in) makes it theoretically possible to improve the utility of virtually any of these hosts (or many others) for a given application.
A host–vector system is useful only if it can persist long enough to make commercially important product quantities. Genetic instability is the loss of the genetic information to make the target protein. Segregational instability arises when a plasmid-free cell is formed during cell division. Structured instability results when the cell loses the capacity to make the target protein in significant quantities due to changes in plasmid structure. Host-cell-derived instability occurs when chromosomal mutations decrease effective plasmid-encoded protein production while the cell retains the plasmid. Growth-rate-dependent instability is a function of the growth-rate differential between plasmid-free and plasmid-containing cells; it is usually a determining factor in deciding how long a fermentation can be usefully maintained. Simple models to evaluate genetic instability are available.
The vector must be designed to optimize a desired process. Factors to be considered include vector copy number, promoter strength and regulation, the use of fusion proteins, signal sequences and secretion, genes providing selective pressure, and elements enhancing the accuracy of vector partitioning.
The engineer must be aware of the regulatory constraints on the release of cells with recombinant DNA. These are particularly relevant in plant design, where guidelines for physical containment must be met. Deliberate release of genetically modified cells is possible, but extensive documentation will be required.
Two increasingly important applications of genetic engineering are metabolic or pathway engineering for the production or destruction of nonproteins (along with the related fields of synthetic and systems biology) and protein engineering for the production of novel or specifically modified proteins.
Suggestions for Further Reading
Genetic Engineering for Protein Production
BANEYX, F., AND M. MUJACIC, Recombinant Protein Folding and Misfolding in Escherichia coli, Nat. Biotechnol. 22: 1399–1408, 2004.
DATAR, R. V., T. CARTWRIGHT, AND C-G. ROSEN, Process Economics of Animal Cell and Bacterial Fermentations: A Case Study Analysis of Tissue Plasminogen Activator, Nat. Biotechnol. 11: 349, 1993.
FERNANDEZ, J. M., AND J. P. HOEFFLER, Gene Expression Systems, Academic Press, New York, 1999.
GEORGIOU, G., Optimizing the Production of Recombinant Proteins in Microorganisms, AIChE J. 34: 1233, 1988.
GLICK, B. R., J. J. PASTERNAK, AND C. L. PATTEN, Molecular Biotechnology: Principles and Applications of Recombinant DNA, ASM Press, Washington, DC, 2010.
GOOCHEE, C. F., AND T. MONICA, Environmental Effects on Protein Glycosylation, Nat. Biotechnol. 8: 421, 1990.
HUANG, C. J., H. LIN, AND X. YANG, Industrial Production of Recombinant Therapeutics in Escherichia coli and Its Recent Advancements, J. Ind. Microbiol. Biotechnol. 39: 383–99, 2012.
KUNKEL, J. P., D. C. JAN, M. BUTLER, AND J. C. JAMIESON, Comparisons of the Glycosylation of a Monoclonal Antibody Produced under Nominally Identical Cell Culture Conditions in Two Different Bioreactors, Biotechnol. Progr. 16: 462–470, 2000.
MA, J. K., P. DRAKE, AND P. M. CHRISTOU, Genetic Modification: The Production of Recombinant Pharmaceutical Proteins in Plants, Nat. Rev. 4: 794–805, 2003.
SHULER, M. L., H. A. WOOD, R. R. GRANADOS, AND D. A. HAMMER, Baculovirus Expression Systems and Biopesticides, Wiley, New York, 1995.
VELANDER, W. H., H. LUBON, AND W. N. DROHAN, Transgenic Livestock as Drug Factories, Sci. Am. 276: 70–74, 1997.
Aspects of Metabolic Engineering
BAILEY, J. E., Towards a Science of Metabolic Engineering, Science 252: 1668–1675, 1991.
KEASLING, J. D. Manufacturing Molecules through Metabolic Engineering, Science 330: 1355–1358, 2010.
LEE, S. Y., AND E. T. PAPOUTSAKIS, Metabolic Engineering, Marcel Dekker, New York, 1999.
MORENO-SANCHEZ, R., E. SAAVEDRA, S. RODRIGUEZ-ENRIQUEZ, AND V. OLIN-SANDOVAL, Metabolic Control Analysis: A Tool for Designing Strategies to Manipulate Metabolic Pathways, J. Biomed. Biotechnol. 2008: 597913, 2008.
NIELSEN, J., AND J. D. KEASLING, Engineering Cellular Metabolism, Cell, 164: 1185–1197, 2016.
STEPHANOPOULOS, G., J. NIELSEN, AND A. ARISTIDOU, Metabolic Engineering: Principles and Methodologies, Academic Press, San Diego, CA, 1998.
SUN, J., AND H. S. ALPER, Metabolic Engineering of Strains: From Industrial-Scale to Lab-Scale Chemical Production, J. Ind. Microbiol. Biotechnol. 42: 423–436, 2015.
Synthetic and Systems Biology
BARRETT, C. L., T. Y. KIM, H. U. KIM, B. Ø. PALSSON, AND S. Y. LEE, Systems Biology as a Foundation for Genome-Scale Synthetic Biology, Curr. Opin. Biotechnol. 17: 488–492, 2006.
CHENG, A. A., AND T. K. LU, Synthetic Biology: An Emerging Engineering Discipline, Annu. Rev. Biomed. Eng. 14: 155–178, 2012.
IDEKER, T., T. GALITSKI, AND L. HOOD, A New Approach to Decoding Life: Systems Biology, Annu. Rev. Genomics Hum. Genet. 2: 343–372, 2001.
SMANSKI, M. J., H. ZHOU, J. CLAESEN, B. SHEN, M. A. FISCHBACH, AND C. A. VOIGT, Synthetic Biology to Access and Expand Nature’s Chemical Diversity, Nat. Rev. Microbiol. 14: 135–149, 2016.
STEPHANOPOULOS, G., Synthetic Biology and Metabolic Engineering, ACS Synthet. Biol. 1: 514–525, 2012.
Protein Engineering
Biocatalysis: Synthesis Methods that Exploit Enzymatic Activities, Nature Insight 409, No. 6817, 2001. (This issue contains excellent review articles on protein engineering and the industrial use of such enzymes.)
BLOOM, J. D., AND F. H. ARNOLD, In the Light of Directed Evolution: Pathways of Adaptive Protein Evolution, PNAS 106: 9995–10000, 2009.
BLUNDELL, T. L., Problems and Solutions in Protein Engineering—Towards Rational Design, Trends Biotechnol. 12: 145–148, 1994.
LUTZ, S., Beyond Directed Evolution: Semi-Rational Protein Engineering and Design, Curr. Opin. Biotechnol. 21: 734–743, 2010.
KUCHNER, O., AND F. H. ARNOLD, Directed Evolution of Enzyme Catalysts, Trends Biotechnol. 15: 523–530, 1997.
WATTERS, A. L., AND D. BAKER, Searching for Folded Proteins In Vitro and In Silico, Eur. J. Biochem. 271: 1615–1622, 2004.
Regulatory Issues
GIORGIOU, R. J., AND J. J. WU, Design of Large Scale Containment Facilities for Recombinant DNA Fermentation, Trends Biotechnol. 4: 60, 1986.
NATIONAL ACADEMY OF SCIENCES, Introduction of Recombinant DNA-Engineered Organisms into the Environment: Key Issue, National Academies Press, Washington, DC, 1987.
NATIONAL INSTITUTES OF HEALTH, Guidelines for Research Involving Recombinant DNA Molecules, Federal Register 51 (May 7) 16958, 1987.
Modeling Genetic Instability
AIBA, S., AND T. IMANAKA, A Perspective on the Application of Genetic Engineering: Stability of Recombinant Plasmid, Ann. N.Y. Acad. Sci. 369: 1, 1981.
COOPER, N. S., M. E. BROWN, AND C. A. CAULCOTT, A Mathematical Method for Analyzing Plasmid Stability in Microorganisms, J. Gen. Microbiol. 133: 1871, 1987.
Problems
14.1. Calculate the probability of forming a plasmid-free cell due to random segregation for a cell with 50 plasmid monomer equivalents. Forty percent of the total plasmid DNA is in dimers and 16% in tetramers. Redo for the case where there is a population with the distribution of copy numbers per cell as follows, assuming monomers only:

14.2. Assume that all plasmid-containing cells have eight plasmids; that an antibiotic is present in the medium, and the plasmid-containing cells are totally resistant; and that a newly born, plasmid-free cell has sufficient enzyme to protect a cell and its progeny for three generations. Estimate the fraction of plasmid-containing cells in the population in a batch reactor starting with only plasmid-containing cells after five generations.
14.3. Consider an industrial-scale batch fermentation. A 10,000 l fermenter with 5 × 1010 cells/ml is the desired scale-up operation. Inoculum for the large tank is brought through a series of seed tanks and flasks, beginning with a single pure colony growing on an agar slant. Assume that a colony (106 plasmid-containing cells) is picked and placed in a test tube with 1 ml of medium. Calculate how many generations will be required to achieve the required cell density in the 10,000 l fermenter. What fraction of the total population will be plasmid-free cells if μ+ = 1.0 h–1, μ– = 1.2 h–1, and P = 0.0005?
14.4. Assume that you have been assigned to a team to produce human epidermal growth factor (hEGF). A small peptide, hEGF, speeds wound healing and may be useful in treating ulcers. A market size of 50 to 500 kg/yr has been estimated. Posttranslational processing is not essential to the value of the product. It is a secreted product in the natural host cell. Discuss what recommendations you would make to the molecular-biology team leader for the choice of host cell and the design of a reactor. Make your recommendations from the perspective of what is desirable to make an effective process. You should point out any potential problems with the solution you have proposed as well as defend why your approach should be advantageous.
14.5. Develop a model to describe the stability of a chemostat culture for a plasmid-containing culture. For some cultures, plasmids make a protein product (e.g., colicin in E. coli) that kills plasmid-free cells but does not act on plasmid-containing cells. Assume that the rate of killing by colicin is kTCn–, where kT is the rate constant for the killing and C is the colicin concentration. Assume that the colicin production is first order with respect to n+.
14.6. Consider the following data for E. coli B/r-pDW17 grown in a minimal medium supplemented with amino acids. Estimate Δμ and R. Compare the stability of this system to one with a glucose-minimal medium (Example 14.2).
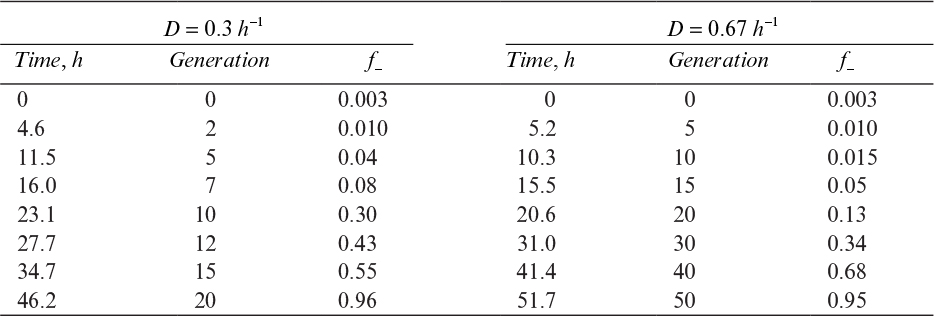
14.7. It has been claimed that gel immobilization stabilizes a plasmid-containing population. A factor suggested to be responsible for the stabilization is compartmentalization of the population into very small pockets. For example, the pocket may start with an individual cell and grow to a level of 200 cells per cavity. Develop a mathematical formula to compare the number of plasmid-free cells in a gel to that in a large, well-mixed tank.
14.8. You must design an operating strategy to allow an E. coli fermentation to achieve a high cell density (> 50 g / l) in a fed-batch system. You have access to an off-gas analyzer that will measure the pCO2 in the exit gas. The glucose concentration must be less than 100 mg/l to avoid the formation of acetate and other inhibitory products. Develop an approach to control the glucose feed rate so as to maintain the glucose level at 100 ± 20 mg/l. What equations would you use, and what assumptions would you make?
14.9. Develop a simple model for a population in which plasmids are present at division with copy numbers 2, 4, 6, 8, or 10. The model should be developed in analogy to equations 14.7 through 14.9. You can assume that dividing cells either segregate plasmids perfectly or generate a plasmid-free cell.
14.10. Assume you have an inoculum with 95% plasmid-containing cells and 5% plasmid-free cells in a 2 l reactor with a total cell population of 2 × 1010 cells/ml. You use this inoculum for a 1000 l reactor and achieve a final population of 4 × 1010cells/ml. Assuming μ+ = 0.69 h–1, μ– = 1.0 h–1, and P = 0.0002, predict the fraction of plasmid-containing cells.
14.11. Assume you scale up from 1 1 of 1 × 1010 cells/ml of 100% plasmid-containing cells to 20,000 l of 5 × 109 cell/ml, at which point overproduction of the target protein is induced. You harvest 6 hours after induction. The value of P is 0.0005. Before induction, μ+ = 0.95 h-1 and μ– = 1.0 h–1. After induction, μ+ is 0.15 h-1. What is the fraction of plasmid-containing cells at induction? What is the fraction of plasmid-containing cells at harvest?
14.12. A metabolic pathway involves enzymes A, B, and C. Given that enzyme B is the rate-limiting enzyme, in this pathway, would you expect this enzyme to be tightly regulated? Why or why not? Name two or three strategies that could be taken to increase flux through this pathway.
14.13. You desire to change the specificity of an antibody fragment using semirational design. You have discovered a 3-amino acid stretch in complimentary determining region 3 (CDR3) that you hypothesize directs the specificity of the antibody. Your goal is to study this region by saturation mutagenesis (substituting all possible amino acids in each position).
a. How large of a protein library would you need to screen if you wish to have threefold library coverage (screening three times more members than the theoretical library size that contains all possible variants)?
b. After your first attempt, you decide to screen a similar 3-amino acid region in CDR1 and CDR2. How large of a library will you need to ensure threefold library coverage in all three CDRs simultaneously?
c. Why is it necessary to have threefold library coverage?
d. How many library members in the CDR3 library do you expect to be truncated due to the inclusion of a stop codon? Can you think of a way of biasing the library not to include stop codons in these positions?
14.14. You are a fledgling protein engineer and wish to study the diversity of every possible 40-amino acid peptide. As a first test, you want to synthesize all possible amino acid combinations for the peptide using all 20 amino acids. Ignoring the DNA needed to make each peptide, what is the mass of your peptide library if you synthesized only one molecule of each peptide variant? Are you surprised by this mass?