
Chapter 16
Electrochemical Affinity and Nucleic Acid Sensors
This chapter introduces affinity and nucleic acid sensors based on dynamic electrochemistry transduction methods. Most of the methods reviewed here belong to the amperometric/voltammetric class, but applications of reciprocal derivative chronopotentiometry are also emphasized as this method provides excellent sensitivity in certain transduction processes based on irreversible electrochemical reactions.
In both classes of sensor presented in this chapter, the recognition occurs by formation of molecular complexes involving multiple noncovalent bonds. However, nucleic acid sensors present the particular feature of recognition by hydrogen bonding that involves specific nucleobase pairs.
Another common feature of sensors in both above classes is the extensive use of transduction labels that can be either enzymes or electrochemically active small molecules.
16.1 Amperometric Affinity Sensors
A great deal of research work on amperometric transduction in immunosensors has led to important advances in this field [1–4]. As compounds involved in immunosensing are usually not electrochemically active, amperometric immunosensors rely on labeling with either electrochemically-active compounds or redox enzymes.
Application of synthetic receptors (such as molecularly imprinted polymers) provides opportunities for substituting expensive and unstable antibodies with stable materials for the determination of small molecules. In this area, much progress has been made in the development of sensors for the determination of electrochemically active compounds, although inactive species can also be determined by means of redox probes or by competitive assay.
16.1.1 Redox Labels in Amperometric Immunosensors
In principle, one can resort to either the sandwich or competitive format using immunoreagents tagged with redox labels in order to assess amperometrically the extent of the recognition reaction. However, the sensitivity of this method is often not sufficient for the analysis of real samples. Signal amplification by electrochemical recycling of the redox label can be applied in such instances. Recycling can also be performed by means of an enzyme if the redox probe can act as its substrate. Ferrocenes are among the most common redox labels in such applications.
16.1.2 Enzyme-Linked Amperometric Immunosensors
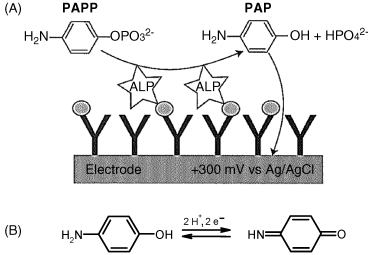
Enzyme labels allow very sensitive immunosensors to be designed since one single label produces a large amount of detectable product. Both hydrolases (alkaline phosphatase and ![]() -galactosidase) and oxidases (peroxidase, glucose oxidase and laccase) are suitable for such applications. A common example is alkaline phosphatase that catalyzes the dephosphorylation of p-aminophenyl phosphate (PAPP) leading to p-aminophenol (PAP) that can be detected by its electrochemical reaction (Figure 16.1). The limit of detection can be improved to a great extent by electrochemical recycling PAP.
-galactosidase) and oxidases (peroxidase, glucose oxidase and laccase) are suitable for such applications. A common example is alkaline phosphatase that catalyzes the dephosphorylation of p-aminophenyl phosphate (PAPP) leading to p-aminophenol (PAP) that can be detected by its electrochemical reaction (Figure 16.1). The limit of detection can be improved to a great extent by electrochemical recycling PAP.
Figure 16.1 An amperometric immunosensor based on alkaline phosphatase (ALP) as label. (A) Sensor mechanism; (B) electrochemical reaction of PAP. Adapted with permission from [4]. Copyright John Wiley & Sons, Ltd.
Redox enzyme labels provide various opportunities for developing amperometric immunosensors. For example, Figure 16.2A outlines the principles of an enzyme-linked competitive format immunosensor. As is typical in this format, an enzyme-labeled analyte is added to the sample containing the target analyte. The enzyme is brought to near the surface of the electrode by the immunoreaction and catalyzes the conversion of the substrate into an electrochemically active product that undergoes an electrochemical reaction to generates the response current.
Figure 16.2 Amperometric immunoassay formats using a redox enzyme label (Ez). (A) antigen detection by a competitive immunoassay format. (B) Antibody detection by the sandwich immunoassay format. Reproduced with permission from [5]. Open access.
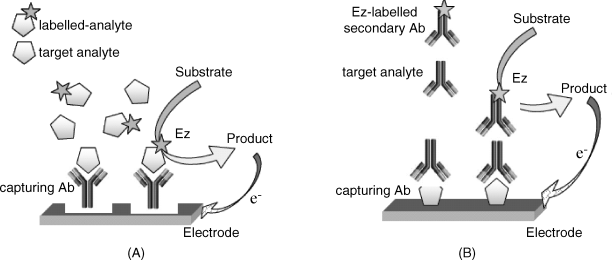
The principle of the sandwich format applied to amperometric immuno sensors is illustrated in Figure 16.2B. In this format, the analyte interacts with the immobilized receptor at the electrode surface to form a binary complex. Then, an enzyme-labeled secondary antibody binds to the binary complex and, in the presence of the enzyme substrate, an electrochemically active product forms. The electrochemical reaction of this product provides the sensor response in the form of an electric current.
In the case of an oxidase label, the product can be hydrogen peroxide that form by the reduction of dissolved oxygen. In order to obtain an oxygen-free immunosensor, one can resort to an electron-transfer mediator such as a ferrocene derivative or hydroquinone. After incorporating the enzyme label in the sensing part, the immunosensor acts as an amperometric enzyme sensor composed of an enzyme monolayer. Therefore, it functions under the condition of external diffusion control (see Section 15.1). As the enzyme loading is rather low, the device has the advantage that it functions under a zero-order kinetic regime and generates a response proportional to the amount of enzyme present at the interface. As the amount of enzyme label is determined by the analyte concentration, the response current is itself a function of this concentration.
Screen-printing technology that allows facile fabrication of cheap, disposable sensors, has proved to be very convenient for manufacturing amperometric immunosensors [6]. For example, the device presented in Figure 16.3A has been designed as a sensor for determining the 2,4-dichlorophenoxyacetic acid pesticide. It is built up on the screen-printed transducer strip (I) that includes the working electrode and an Ag/AgCl reference electrode connected to the measuring instrument via silver paste paths coated with an insulator. The capture antibody can be immobilized on the working electrode surface. Alternatively, it can be bound to a nylon mesh disk (II) which is stored and assembled with the transducer just prior to the assay. In order to deal with very small volume samples, a miniature cell (III) is attached over the working area. As this sensor has been designed to work in the competitive format, horseradish peroxidase has been conjugated with analyte molecules and added to the sample along with hydroquinone and hydrogen peroxide. Hydroquinone is catalytically oxidized to p-benzoquinone that is then reduced at the electrode back to hydroquinone. The reduction current represents the response signal. As expected for a competitive assay, the response current decreases with increasing analyte concentration as shown in Figure 16.3B.
Figure 16.3 A screen-printed amperometric immunosensor. (A) sensor configuration. (I) The screen-printed strip. (1) working electrode; (2) reference electrode; (3) insulator layer; (4) ceramic support; (5) electric contacts. (II) Nylon mesh disk with immobilized antibody. (III) A piece of plastic tube forming a minicell. (IV) Assembly of the sensor. (B) Calibration graph for the determination of the 2,4-dichlorophenoxyacetic acid pesticide with the above sensor. (1) Maximum current recorded after adding the sample; (2) steady-state current. Adapted with permission from [6]. Copyright 1995 Elsevier.
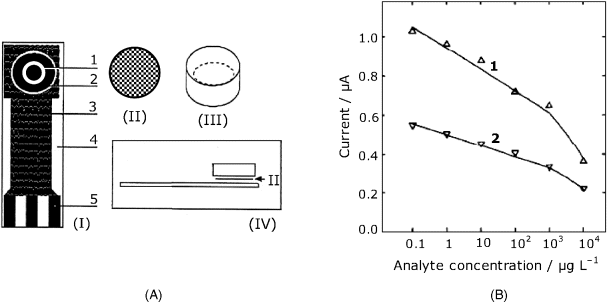
A similar manufacturing technology has been used to produce a 5-channel multiple sensor on a single strip, which affords simultaneous assay of different samples [7].
16.1.3 Separationless Amperometric Immunosensors
Clearly, the approaches outlined above require the labeled compound to be washed out prior to signal evaluation. This is not a major problem when the assay is run in an automated flow-analysis system, but avoiding separation simplifies the protocol and shortens the analysis time.
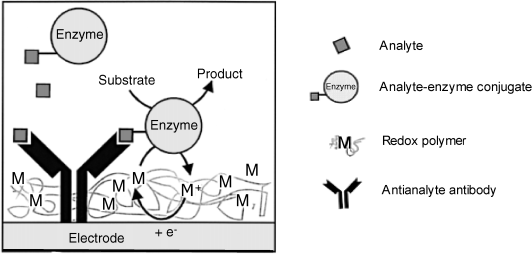
The separation step can be eliminated by a proper design of the immunosensor if a redox enzyme is used as label. This is based on the following principle. Redox enzymes perform electron transfer from substrate to the electrode via a mediator that can be immobilized within a redox polymer in order to allow for enzyme wiring (see Section). Hence, the immunosensor sensing layer can be obtained by assembling the capture receptor along with a redox polymer at the electrode surface. Binding of the labeled reagent into the immunocomplex brings the enzyme into the sensing layer where electron transfer is feasible and gives rise to a current proportional to the surface enzyme concentration. The bulk solution enzyme can hardly assume a favorable orientation at the surface and is hence not operative. Consequently, the current can be monitored with no prior separation of the labeled reagent. A redox polymer sensor therefore elicits spatial resolution as it can distinguish between the dissolved labeled species and the same species included in the immunocomplex.
Figure 16.4 presents a competitive format immunosensor based on this principle. The antianalyte antibody is incorporated within a redox polymer while the enzyme-labeled analyte is added to the sample and competes with the analyte itself in the antibody-binding process. Therefore, the amount of enzyme brought to the surface increases with decreasing analyte concentration. Ideally, in this arrangement, only the enzyme included in the immunocomplex is able to perform substrate conversion by electron transfer to the electrode via the redox polymer.
Figure 16.4 An amperometric competitive format immunosensor based on wired enzyme transduction. Adapted with permission from [3]. Copyright 2000 Springer Science + Business Media B.V.
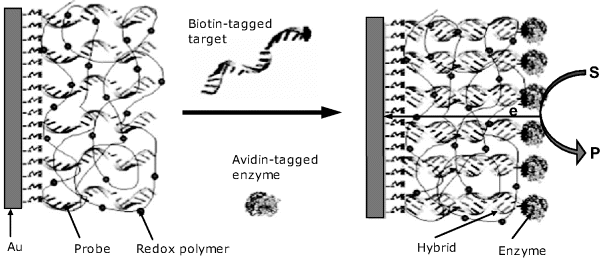
A separationless sandwich assay amperometric immunosensor is shown in Figure 16.5A. Here, a receptor antibody is immobilized within a redox polymer matrix at the electrode surface by avidin–biotin linking. After analyte binding to the receptor, the peroxidase-labeled signaling antibody is added to form the sandwich complex. The enzyme included in this complex catalyzes the reduction of hydrogen peroxide coupled with electron transport via the redox polymer. The contribution of the bulk enzyme to this process is negligible. In this particular example, choline oxidase is integrated with the sensing layer in order to produce hydrogen peroxide needed in the peroxidase reaction.
Figure 16.5 An amperometric sandwich immunosensor based on wired enzyme transduction. (A) Sensor configuration; (B) Response function. Broken lines show the 95% confidence interval limits. Adapted with permission from [8]. Copyright 1999 Springer-Verlag.
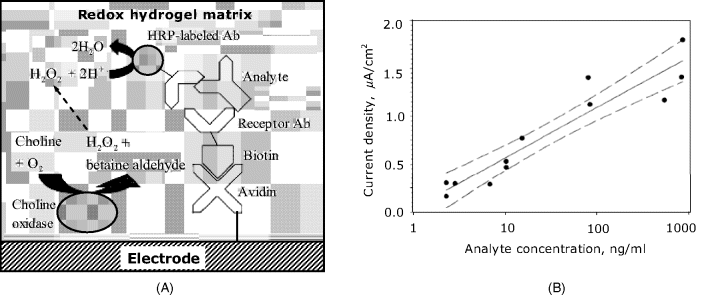
An electrode modified with avidin, redox polymer and choline oxidase forms a generic platform to which a suitable biotinylated antibody can be added in order to determine a particular analyte. A typical response function for rabbit IgG determination is shown in Figure 16.4B that demonstrates a linear trend on a semilogarithmic plot extending over almost three orders of magnitude.
It should be noted, however, that the contribution of the solution phase enzyme to the total current cannot be totally eliminated. Random interactions between the labeled reagent in the solution and the redox polymer are possible to some extent and give rise to current fluctuations. That is why the points on the graph in Figure 16.5B show a relatively large spread around the average trend. In order to minimize this effect, the labeled antibody to antigen concentration ratio should not exceed 4 : 1.
16.1.4 Nanomaterials Applications in Amperometric Immunosensors
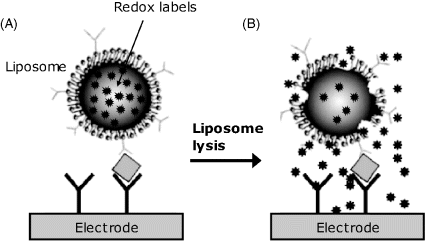
Application of nanomaterials in amperometric immunosensors brought about new opportunities for sensor design and signal amplification [9]. As an example, Figure 16.6 presents a method of signal amplification in a sensor based on a redox probe. In this case, the secondary antibody used in the sandwich format is tagged with liposomes that encapsulate a redox probe solution. After forming the tertiary complex (Figure 16.6A), liposomes are broken down by a surfactant and this releases the redox probe. Released redox probe molecules diffuses to the electrode and undergoes electrochemical conversion. This approach secures a large amount of redox compound which induce a high sensitivity particularly when a sensitive detection methods (such as differential pulse voltammetry) is applied.
Figure 16.6 Amperometric immunoassay based on a redox probe loaded within liposome labels. Adapted with permission from [9]. Copyright 2007 Elsevier.
Gold nanoparticles used as labels can function as catalysts for certain chemical reactions and, combined with electrochemical recycling, allows achieving detection limits as low as 1 fg/μL with the dynamic range stretching over nine orders of magnitude [10].
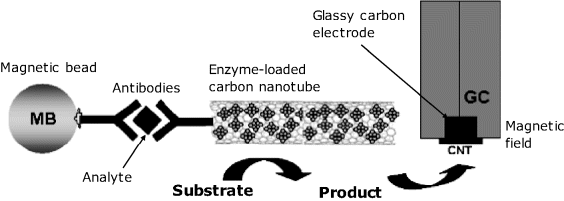
Carbon nanotubes have been applied as immobilization supports for functional molecules involved with sensor functioning while magnetic beads tags are useful for magnetic accumulation at the electrode surface. Both these principles are outlined in Figure 16.7. In this example, the immunoreaction proceeds in the homogeneous sandwich format, the analyte being coupled to two distinct antibodies. One of them is labelled with an enzyme-loaded carbon nanotube while the second one is tagged with magnetic beads. The ternary complex formed in the solution phase is then collected at the electrode surface by applying a magnetic field. The high enzyme loading achieved by means of the nanotube imparts this sensor with a 100-times enhanced sensitivity with respect to the version based on a single enzyme labeled antibody. On the other hand, magnetic beads integrated with the recognition system allows the immunocomplex to be easily detected without pretreatment steps such as preconcentration or purification, which are implicitly required in standard methods [11].
Figure 16.7 An application of carbon nanotubes and magnetic beads in amperometric immunosensors. Adapted with permission from [9] (Copyright 2007 Elsevier B.V.) and [12] (Copyright 2004 American Chemical Society).
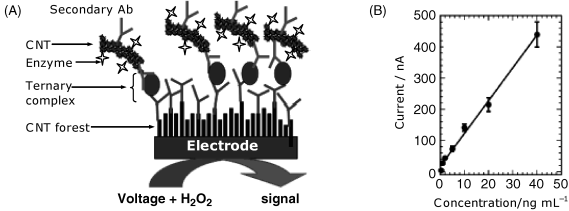
The enzyme labeling principle illustrated above has also been applied to develop an amperometric immunosensor for prostate specific antigen (PSA), which is a prostate cancer biomarker [13]. As above, the secondary antibody has been attached to carbon nanotubes along with the peroxidase label (Figure 16.8A). The capture antibody has been immobilized to a carbon nanotube forest appended to the underlying electrode. The current signal is generated by the peroxidase-catalyzed reduction of hydrogen peroxide, the electron transfer from the enzyme to the electrode being achieved directly, via the carbon nanotubes. This sensor elicits a linear response within the concentration range of clinical interest (Figure 16.7B). The error bars demonstrate a good reproducibility. Excellent accuracy has been demonstrated by the good agreement with results obtained by the standard ELISA test. If amplification is necessary, it can be obtained by adding hydroquinone as mediator.
Figure 16.8 Carbon nanotubes as support for antibody immobilization at the electrode surface and support for the enzyme label. (A) Sensor configuration; (B) calibration graph. Adapted with permission from [13]. Copyright 2006 American Chemical Society.
16.1.5 Imprinted Polymers in Amperometric Affinity Sensors
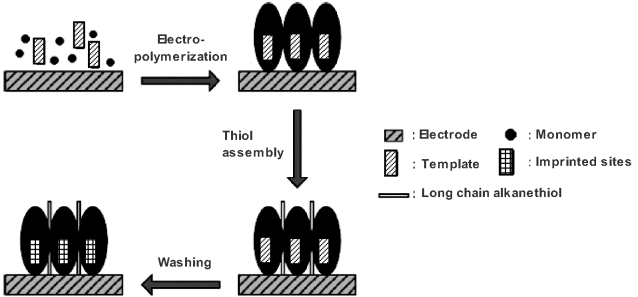
Recognition by molecularly imprinted polymers has been used to develop amperometric sensors in which the polymer serves to selectively accumulate the analyte at the electrode surface [14, 15]. A facile method for the in situ synthesis of imprinted polymers involves electrochemical polymerization at the sensor electrode surface, as demonstrated in Figure 16.9. The polymerization is conducted in a solution containing both the monomer and the template yielding a polymer layer which includes template molecules. In order to block the exposed electrode surface, a long-chain alkanethiol is chemisorbed onto the surface afterwards. Finally, embedded template molecules are washed out, leaving imprinted sites within the polymer layer.
Figure 16.9 In situ preparation of imprinted polymers by electrochemical polymerization at a gold electrode. Adapted with permission from [15]. Copyright 2010 Wiley-VCH Verlag GmbH & Co.
Electrochemically active analytes can be detected by their own electrochemical reaction. In order to achieve a reasonable sensitivity, highly sensitive voltammetric techniques (such as differential pulse voltammetry or square-wave voltammetry) should be applied.
Preformed imprinted polymers can be immobilized as particles embedded in a suitable matrix. Alternatively, they can be cast over the electrode surface to form thin films or can be grown in situ by electrochemical polymerization in the presence of the template. Overoxidized polypyrrole proved to be a convenient material for this type of application.
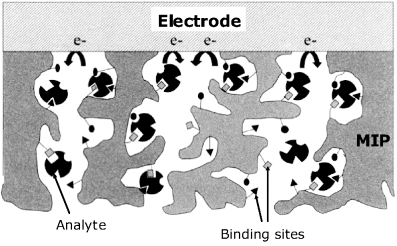
The assay with an imprinted polymer sensor proceeds via three main steps. First, analyte accumulation is performed in a nonaqueous polar solvent such as acetonitrile. The solvent swells the polymer and facilitates access of the analyte to the binding sites within the polymer pores (Figure 16.10). The accumulation time is selected by preliminary trials so as to obtain a sufficiently large response. Next, the sensor is carefully washed with the same solvent in order to remove the adsorbed analyte molecules. Finally, quantitation is carried out in an aqueous solution with the pH sufficiently low to promote the dissociation of the analyte-receptor hydrogen bonds. The analyte is thus released and diffuses within the pores to the electrode surface where the electrochemical reaction takes place. If sensor regeneration is to be attempted, this should be done by means of a solvent that dissolves the reaction products. Often, regeneration of the sensor is hardly achievable. As such sensors are inexpensive and easily manufactured it is suitable therefore to design them as disposable, single-use sensors.
Figure 16.10 Response mechanism of an amperometric sensor based on imprinted polymer recognition. Adapted with permission from [16]. Copyright 2002 Elsevier.
Molecularly imprinted polymers should be prepared so that diffusion of both reactant and product within the polymer pores is possible. Otherwise, the polymer pores become clogged with the product.
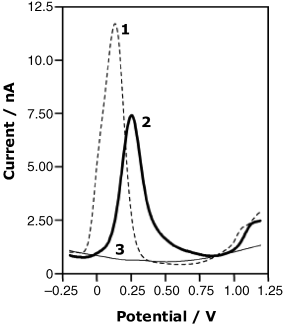
An application to the determination the macrocyclic antibiotic rifamycin SV is demonstrated in Figure 16.11. This compound undergoes an anodic reaction at the bare electrode (curve 1) but such a determination can be impaired by the interference from other electroactive compounds. The signal generated after accumulation at an imprinted polymer electrode (curve 2) is somewhat lower and shifted towards more positive potentials, but the risk of interference is greatly reduced. Nonspecific accumulation is negligible, as is demonstrated by curve 3 that represents the voltammogram recorded with an electrode coated with the same polymer in the nonimprinted form.
Figure 16.11 Differential pulse voltammograms for the determination of rifamycin SV at a bare carbon electrode (1), at an electrode coated with an imprinted polymer (2) and at a nonimprinted polymer modified electrode (3). 2.5 mM rifamycin in a pH 7.5. phosphate buffer. Reference electrode: Ag|AgCl|KCl(sat.). Adapted with permission from [17]. Copyright 2004 Springer Verlag.
If the analyte itself is not electrochemically active, the response current can be produced by a redox probe whose permeation through the polymer film is modulated by the recognition event. The competitive detection scheme is also applicable with imprinted polymer sensors. In this version, the analyte is first bound to the receptor layer and the sensor is then incubated with an inactive analyte analog. The analyte thus released is finally detected by its electrochemical reaction.
The quality of imprinted polymer sensors can be assessed by means of the response current enhancement factor CE, defined as follows [15]:
(16.1) ![]()
Here, ![]() is the response of an imprinted polymer coated electrode and
is the response of an imprinted polymer coated electrode and ![]() is the response of a control electrode coated with the same polymer in the nonimprinted form. The selectivity of the sensor can be estimated by comparing CE values for the analyte itself and for possible interferents.
is the response of a control electrode coated with the same polymer in the nonimprinted form. The selectivity of the sensor can be estimated by comparing CE values for the analyte itself and for possible interferents.
The response time is determined by the rate of analyte diffusion within the pores to the electrode surface. That is why the response time with sensors using MIP particles are longer than that with thin films. Imprinted polymers in the form of nanoparticles can elicit even shorter response time.
Long-term stability is determined by the stability of the polymer itself. Best stability has been reported for highly crosslinked polymers such as acrylic or vinyl polymers.
Although much progress has been made in the application of imprinted polymers, it is still important to expand their application to real samples. Also, elimination of nonaqueous solvents in the assay protocol is sought.
16.1.6 Outlook
The design of amperometric immunosensors relies on strategies based on either redox labels or on enzyme labels. In both cases, the capture receptor is immobilized at the surface of an electrode and the label is brought to the electrode surface by suitable immunoreactions, either in the competitive or the sandwich format.
Redox label immunosensors provide satisfactory sensitivity only when combined with a recycling method. On the other hand, use of enzyme labels leads to very sensitive sensors owing to the fact that one single enzyme molecule catalyzes the conversion of a large number of substrate molecules per unit time. High sensitivity is provided by enzymes with a large turnover number. The enzyme-substrate couple is selected so as to produce an electrochemically active compound whose electrochemical reaction generates the response current. Hydrolase or oxidase enzymes are suited as labels in amperometric immunosensors.
Oxidase enzymes offer the possibility of developing separationless immunosensors. In this configuration, the capture receptor is immobilized at the electrode surface within a redox polymer matrix, which provides mediated electron transfer from the enzyme active site to the electrode. Therefore, the enzyme tagged to an immunoreagent present in the solution phase does not contribute to the sensor response and need not be washed out.
Application of nanomaterials is advantageous in that large numbers of labels can be attached to an immunoreagent. Thus, liposomes allow the incorporation of a large number of small-molecule redox probes, whilst carbon nanotubes used as supports for enzyme immobilization are useful for preparing multienzyme molecule tags. Tagging with magnetic beads eliminates the necessity for immobilization of the capture receptor as the immunocomplex formed in the bulk of the solution can be accumulated at the electrode surface by means of a magnetic field.
Affinity amperometric sensors for various compounds can be produced by means of molecularly imprinted polymers. Imprinted polymers for such applications can be readily synthesized by electrochemical polymerization at the sensor electrode surface. If the analyte is electrochemically active, the imprinted polymer imparts selectivity by preferential accumulation of the analyte. In the case of nonelectrochemically inactive analytes, one can resort to a competitive assay, using an electrochemically active analyte analog.
16.2 Electrochemical Nucleic Acid-Based Sensors
Amperometric transduction in nucleic acid sensors can be achieved in several ways [18, 19]. First, certain nucleobases are themselves electrochemically active even when included in a nucleic acid sequence. Therefore, nucleic acids containing such nucleobases are self-indicating and need, in principle, no redox labels for amperometric transduction. However, much better sensitivity is achieved when mediated electron transfer to the active nucleobase is used instead. In this case, carefully structured probes are needed in order to prevent the mediator contributing directly to the overall current. This trouble can be avoided by using redox labels that are either intercalated in the duplex species formed by hybridization or are covalently attached to a species involved with this process. Considerable amplification of the response signal is achieved by using redox enzyme or nonbiotic catalysts as labels. IUPAC recommendations concerning concepts, terms and methodology in electrochemical nucleic acid-based sensors are available in ref. [20].
16.2.1 Electrochemical Reactions of Nucleobases
The most straightforward transduction method is based on the electrochemical reactions of nucleobases residues that are a components of the nucleic acid molecule. The electrochemical activity of nucleic acids was first noted by Pale![]() ek using a mercury electrode [21]. Further studies showed that the electrochemical response is due to the reduction of adenine and cytosine residues. The electrochemical activity is sensitive to changes in the nucleic acid structure and conformation. Therefore, these electrochemical reactions form the basis of a series of methods for assessing DNA hybridization, damage and denaturation [22–25].
ek using a mercury electrode [21]. Further studies showed that the electrochemical response is due to the reduction of adenine and cytosine residues. The electrochemical activity is sensitive to changes in the nucleic acid structure and conformation. Therefore, these electrochemical reactions form the basis of a series of methods for assessing DNA hybridization, damage and denaturation [22–25].
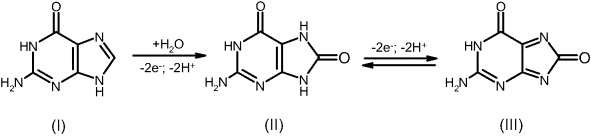
Research work based on mercury electrodes laid the fundament of the electrochemistry of nucleic acids and revealed its potential analytical applications. Nevertheless, a liquid mercury electrode is not suitable for developing biosensors and solid electrodes represent the practical alternative for electrochemical transduction. Solid electrodes are best suited for transduction by anodic reactions of nucleobases, particularly guanine and adenine. These compounds, as well as their nucleosides and nucleotides yield well-developed voltammetric signals at graphite electrodes over a broad pH range [26, 27]. The anodic reaction of guanine involves two successive two-electron transfer processes, according to the scheme in Figure 16.12. However, the product (III) is not stable and subsequently decays to other final products.
Figure 16.12 Electrochemical oxidation of guanine.
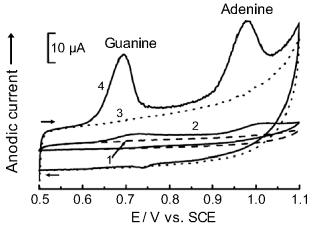
Anodic reactions of guanine and adenine are demonstrated in Figure 16.13 that displays cyclic voltammograms of calf tymus DNA at a bare glassy carbon electrode (curve 1), the same electrode coated with a nonionic surfactant (curve 2) and at same electrode modified with multiwalled carbon nanotubes (MWCNTs) with no prior accumulation (curve 3), and after prior adsorptive accumulation of DNA at the MWCNT-modified surface (curve 4). Curve 4 displays distinct anodic peaks due to the oxidation of guanine and adenine. Figure 16.13 also demonstrates the large background current, which is a common feature of the anodic reaction of nucleobases included in nucleic acid. This characteristic severely impairs the limit of detection [28].
Figure 16.13 Cyclic voltammograms of a 50 μg L−1 calf tymus DNA solution at a bare and modified glassy carbon electrode. ![]() ; 2H+ (1) Bare glassy carbon electrode; (2) DHP-coated electrode; (3) MWNT-DHP coated electrode; (4) Same electrode as in (3), after 2 min DNA accumulation at the surface. 0.1 M phosphate buffer (pH 7); potential scan rate, 0.1 V s−1. DHP denotes the nonionic surfactant dihexadecyl hydrogen phosphate. Adapted with permission from [30]. Copyright 2003 Springer.
; 2H+ (1) Bare glassy carbon electrode; (2) DHP-coated electrode; (3) MWNT-DHP coated electrode; (4) Same electrode as in (3), after 2 min DNA accumulation at the surface. 0.1 M phosphate buffer (pH 7); potential scan rate, 0.1 V s−1. DHP denotes the nonionic surfactant dihexadecyl hydrogen phosphate. Adapted with permission from [30]. Copyright 2003 Springer.
A solution to the above-mentioned background interference was advanced by Wang et al. [29] who employed a carbon paste electrode and monitored the electrochemical reaction by reciprocal derivative chronopotentiometry instead of linear scan voltammetry. This method is characterized by an excellent signal/background ratio and allows the determination of DNA at concentrations as low as 0.5 mg L−1.
16.2.2 Amperometric Nucleic Acid Sensors Based on Self-Indicating Hybridization
Direct amperometric transduction is based on the electrochemical oxidation of guanine residues in the target after hybridization with the electrode-immobilized nucleic acid probe. However, guanine, if present in the probe, also reacts giving rise to a disturbing background current. This problem can be avoided if the probe is synthesized such as to contain inosine instead of guanine. Inosine is not electrochemically active and the background current in this case is very low. Based on these principles, a method for microfabrication of thick-film hybridization nucleic acid sensors has been developed [31]. The best quantitation method is reciprocal derivative chronopotentiometry (Section 13.8.8). Such single-use sensors proved suitable for decentralized trace quantitation of DNA in order to detect pathogenic microorganisms.
Nevertheless, when dealing with real samples, various concomitants (such as mismatched and noncomplementary oligomers, chromosomal DNA, RNA and proteins) can undergo electrochemical oxidation along with guanine in the target and lead to a poor sensitivity owing to the enhanced background current. This drawback has been circumvented by linking the probe molecules to magnetic beads and effecting the hybridization in the solution phase. Bead-tagged hybrids are drawn to the electrode surface by a magnet and by this means are separated from the concomitants [32].
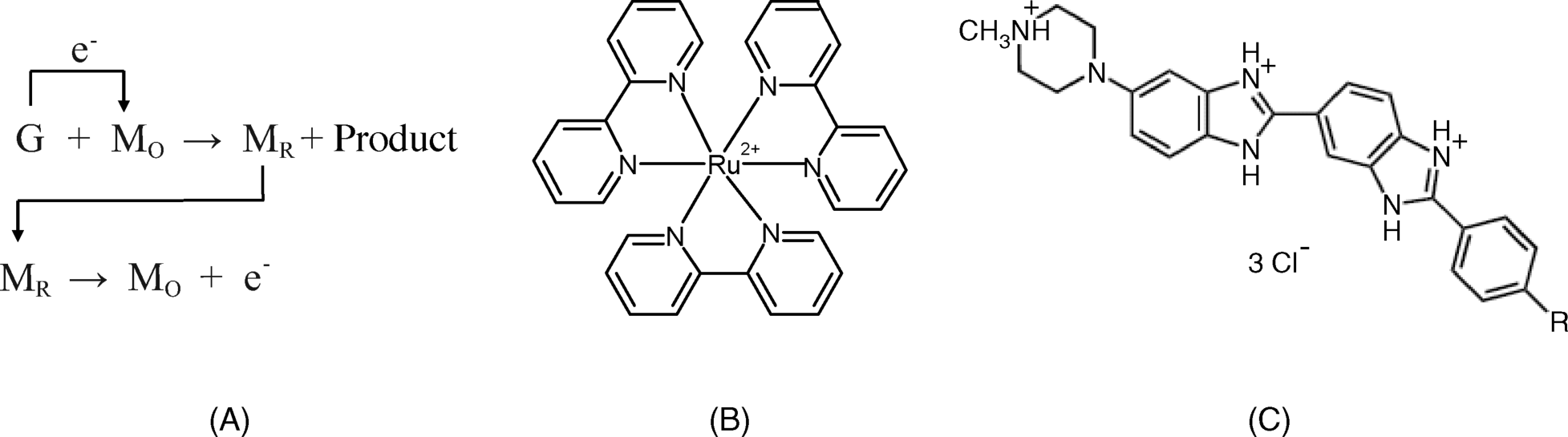
The main deficiency of direct guanine detection arises from the slow kinetics of the electrochemical reaction that results in relatively low peak currents and poor sensitivity with standard voltammetric methods. In order to alleviate this limitation, one can resort to a mediated oxidation scheme (Figure 16.14A). In this case, after hybridization, a positively charged redox mediator is attached to the double strand by electrostatic attraction. Suitable mediators are certain metal chelate complexes (such as ![]() [33],
[33], ![]() ,
, ![]() [34] (bypy = 2,2′-bipyridine; phen = ortho-phenanthroline) or the staining dye Hoechst 33258 (Figure 16.14). The mediator undergoes an electrochemical oxidation followed by its reaction with guanine, which is the final electron donor in the process [35]. The actual electrochemical reaction involves only the mediator and is therefore a fast electrochemical reaction. This brings about a considerable enhancement of sensitivity compared with direct guanine oxidation.
[34] (bypy = 2,2′-bipyridine; phen = ortho-phenanthroline) or the staining dye Hoechst 33258 (Figure 16.14). The mediator undergoes an electrochemical oxidation followed by its reaction with guanine, which is the final electron donor in the process [35]. The actual electrochemical reaction involves only the mediator and is therefore a fast electrochemical reaction. This brings about a considerable enhancement of sensitivity compared with direct guanine oxidation.
Figure 16.14 (A) Mediated electrochemical oxidation of guanine (G). ![]() and
and ![]() represent the redox mediator in the reduced and oxidized form, respectively. Typical mediators in guanine electrochemical oxidation: (B) tris(bipyridine)ruthenium(II) (
represent the redox mediator in the reduced and oxidized form, respectively. Typical mediators in guanine electrochemical oxidation: (B) tris(bipyridine)ruthenium(II) (![]() ); (C) the staining dye Hoechst 33258 (R = ߝOH).
); (C) the staining dye Hoechst 33258 (R = ߝOH).
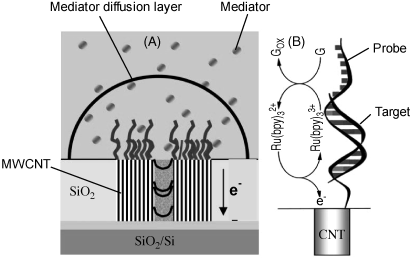
Carbon nanotubes offer considerable possibilities for improving the design of amperometric DNA sensors [36]. An example in this respect is represented by the application of carbon nanotubes as probe supports in a DNA sensor based on mediated guanine oxidation, as shown in Figure 16.15 [37]. In this approach, multiwalled carbon nanotubes have been grown on a Cr layer spotted with Ni catalyst sites. In order to impart chemical and mechanical stability to the resulting structure, nanotubes have been embedded in a ![]() layer formed by chemical vapor deposition. This dielectric layer allows each nanotube to act as an independent electrode in the array. Mechanical polishing, followed by electrochemical etching has then been applied to shorten and level nanotubes to the same plane as the
layer formed by chemical vapor deposition. This dielectric layer allows each nanotube to act as an independent electrode in the array. Mechanical polishing, followed by electrochemical etching has then been applied to shorten and level nanotubes to the same plane as the ![]() matrix. As a result, only the end of the nanotube, bearing carboxyl groups, remains exposed. About 70 nanotubes have been thus formed on a 20 × 20 μm metallic spot. Next, up to 900 probes have then been attached to each nanotube end by amide bonding to carboxyl groups. PCR amplicons of about 300 bases have been used as hybridization targets. Each target molecule contains about 70 intrinsic guanine bases and thus produces a sufficiently large oxidation current which allows the use of very low nanoelectrode densities. The transduction relies on mediated oxidation of guanine residues using tris(bipyridine)ruthenium(II) as mediator.
matrix. As a result, only the end of the nanotube, bearing carboxyl groups, remains exposed. About 70 nanotubes have been thus formed on a 20 × 20 μm metallic spot. Next, up to 900 probes have then been attached to each nanotube end by amide bonding to carboxyl groups. PCR amplicons of about 300 bases have been used as hybridization targets. Each target molecule contains about 70 intrinsic guanine bases and thus produces a sufficiently large oxidation current which allows the use of very low nanoelectrode densities. The transduction relies on mediated oxidation of guanine residues using tris(bipyridine)ruthenium(II) as mediator.
Figure 16.15 Amperometric hybridization sensor based on multiwalled carbon nanoelectrodes with detection by ![]() mediated oxidation of guanine. (A) Schematic of sensor. Due to the relatively large distance between nanotubes, an individual, hemispherical diffusion layer is set up for each of them. (B) Reaction scheme showing the mediated electrochemical oxidation of guanine bases in the hybridized target. Adapted with permission from [37]. Copyright 2004 The Royal Society of Chemistry.
mediated oxidation of guanine. (A) Schematic of sensor. Due to the relatively large distance between nanotubes, an individual, hemispherical diffusion layer is set up for each of them. (B) Reaction scheme showing the mediated electrochemical oxidation of guanine bases in the hybridized target. Adapted with permission from [37]. Copyright 2004 The Royal Society of Chemistry.
The response signal of the sensor in Figure 16.15 has been obtained by multiscan alternating-current voltammetry, which yields a peak-shaped current potential curve. The peak current for the first scan, ![]() results from the joint contribution of two electrochemical processes: mediated guanine oxidation and oxidation of the free mediator. As guanine oxidation is an irreversible process, the second scan yields only the current associated with the free mediator reaction (
results from the joint contribution of two electrochemical processes: mediated guanine oxidation and oxidation of the free mediator. As guanine oxidation is an irreversible process, the second scan yields only the current associated with the free mediator reaction (![]() ). The actual response can therefore be obtained as
). The actual response can therefore be obtained as ![]() .
.
This approach resulted in a very sensitive sensor being obtained by conventional microfabrication technology. It can detect about 300 hybridized targets per spot, close to the detection limit of the fluorescence-based microarray technique. Such sensitivity makes it suitable for direct ![]() detection of mRNA.
detection of mRNA.
16.2.3 Intercalating Redox Indicators
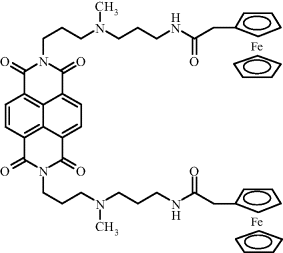
Intercalation of a redox indicator represents a simple procedure for labeling a duplex nucleic acid [38, 39]. A frequently used indicator of this type is daunomycin that undergoes facile electrochemical oxidation at 0.4–0.5 V vs. Ag|AgCl in neutral solutions. Figure 16.16 shows a purposely synthesized redox indicator, ferrocenyl naphthalene diimide. In this case, the flat, middle part of the molecule binds to DNA by intercalation, while the lateral chains bearing ferrocene active sites extend beyond the duplex structure.
Figure 16.16 Ferrocenyl naphthalene diimide, an intercalating redox probe. Adapted with permission from [39]. Copyright 2003 Wiley-VCH Verlag GmBH & Co. KGaA.
Single-use hybridization sensors based on intercalating indicators have been fabricated by adsorptive immobilization of the probe at carbon screen-printed electrodes [40]. The response has been recorded after daunomycin intercalation by reciprocal derivative chronopotentiometry using the integrated signal (peak area) as analytical signal. In order to avoid a spurious response, the sensor should be carefully rinsed after both hybridization and intercalation. The flow chart of such an assay is shown in Figure 16.17A. Testing of the sensor was carried out with a 21-mer micro-organism oligonucleotide as probe and a complementary target from the same organism as target. A significant response has been noted even in the presence of a noncomplementary target, probably due to nonintercalative binding of daunomycin to the probe layer. However, the response increases clearly with target concentration. In order to correct for the nonspecific response the posthybridization signal should be corrected by subtracting the signal obtained with a noncomplementary target.
Figure 16.17 (A) Main steps in electrochemical hybridization assays using screen-printed carbon electrodes and an intercalated redox indicator. After each step, the sensor should be carefully washed in order to remove remaining reagents. (B) Reciprocal derivative chronopotentiometry (RDCP) response of a hybridization electrochemical sensor to the concentration of DNA from Escherichia coli. Recorded after hybridization (2 min) at a probe-modified screen printed carbon electrode. Indicator: ![]() ; pH 7; 0.5 M NaCl; (a) RDCP curves with increasing target concentration (1.0 μg/ml steps). Dotted lines: blank response; solid lines: target added. (b) Calibration graph for target determination. (B) Adapted with permission from [29]. Copyright 1999 American Chemical Society.
; pH 7; 0.5 M NaCl; (a) RDCP curves with increasing target concentration (1.0 μg/ml steps). Dotted lines: blank response; solid lines: target added. (b) Calibration graph for target determination. (B) Adapted with permission from [29]. Copyright 1999 American Chemical Society.
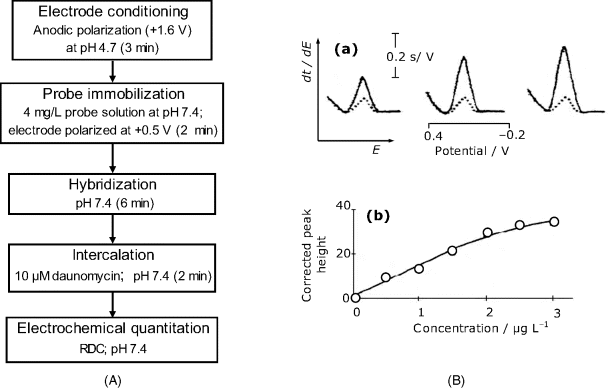
A typical response of a redox intercalator-based sensor is shown in Figure 16.17B(a). This sensor has been built up on a screen-printed carbon electrode using ![]() as intercalated indicator. Note the relatively large response for the blank sample (dotted lines) that should be subtracted from the overall signal obtained with target-containing samples (solid lines). Figure 16.17B(b) presents the plot of the corrected response vs. target concentration; this calibration graph is linear over a limited concentration range.
as intercalated indicator. Note the relatively large response for the blank sample (dotted lines) that should be subtracted from the overall signal obtained with target-containing samples (solid lines). Figure 16.17B(b) presents the plot of the corrected response vs. target concentration; this calibration graph is linear over a limited concentration range.
16.2.4 Covalently Bound Redox Indicators in Sandwich Assays
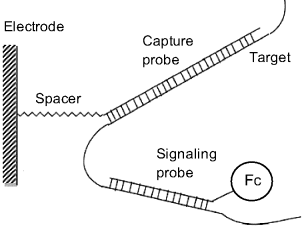
Principles of sandwich DNA sensors have been introduced in Section 7.5.2. This method involves a capture probe and a labeled signaling probe. After the hybridization of the capture probe with the target, the overhanging target fragment is conjugated by hybridization with the signaling probe that is tagged with a ferrocene carboxylic acid residue in order to perform amperometric transduction (Figure 16.18). A sensor thus configured requires the nonhybridized signaling probe in the solution to be washed out prior to the transduction step. This can be avoided by careful engineering of the sensing layer, as in the case of the sensor presented in ref. [41]. In this example the capture probe is integrated in a composite self-assembled monolayer at a gold electrode along with molecular wires and insulating alkane molecules terminated by polyethylene glycol moieties. A molecular wire is a molecule that includes an extended conjugated ![]() -orbital system that allows electrons flowing from the gold electrode across the alkane layer. The role of the external polyethylene glycol sheet is to block the access of electroactive species in solution (including free signaling probes) to the terminus of molecular wires. After hybridization, ferrocene tags that are attached to the signaling probe by sufficiently long alkane fragments, protrude from the surface layer and reach the molecular wires that enable electron transfer between ferrocene and the electrode. This is not possible for ferrocenes linked to signaling probe molecules in the solution phase.
-orbital system that allows electrons flowing from the gold electrode across the alkane layer. The role of the external polyethylene glycol sheet is to block the access of electroactive species in solution (including free signaling probes) to the terminus of molecular wires. After hybridization, ferrocene tags that are attached to the signaling probe by sufficiently long alkane fragments, protrude from the surface layer and reach the molecular wires that enable electron transfer between ferrocene and the electrode. This is not possible for ferrocenes linked to signaling probe molecules in the solution phase.
Figure 16.18 An electrochemical DNA sensor based on sandwich assay. Fc stands for ferrocene. Adapted with permission from [38]. Copyright 2001 Chemical Society of Japan.
The use of a redox probe brings about improved transduction characteristics as the redox probe reaction is fast and occurs at relatively low potentials. This results in a convenient signal/background ratio and allows the detection to be performed by means of very sensitive electrochemical methods, such as differential pulse voltammetry or alternating-current voltammetry. Moreover, the sandwich configuration with a thoroughly engineered sensing layer lessens the risk of interferences. Electrochemical DNA sensors based on sandwich assays are available on a commercial basis as disposable chips.
16.2.5 Covalently Bound Redox Indicators in Spatially Resolved Transduction
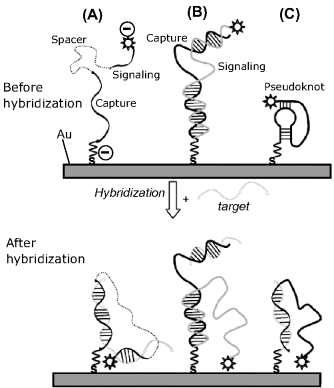
An important recognition–transduction approach relies on the hybrid configuration-dependent response (Figure 16.19). Thus, case A in Figure 16.19 corresponds to a sandwich assay in which the capture and signaling probes are connected through a non-nucleotide spacer. The negative redox marker is repelled by the negatively charged probe at the electrode surface. Target hybridization with both capture and signaling probes gives rise to a ternary complex that shifts the label to near the electrode surface and allows the electrochemical reaction to proceed. In case B, the capture and signaling strands form a discontinuous duplex. By hybridization, the target displaces the tagged end of the signaling probe and allows the redox label to approach the electrode surface. Finally, in case C a labeled probe is shaped as a pseudoknot. It changes its configuration by hybridization with the target and the label shifts as a consequence to the electrode surface. All the above methods are of the “signal on” type because the hybridization event displaces the label to the electrode surface from a position far from the electrode. In this way, the response current increases with respect to the background current. Conversely, “signal-off”-type methods are based on label displacement from the electrode surface to a location far from the electrode in response to the hybridization event. As a result, the current decreases after hybridization. “Signal-off” methods are less sensitive owing to limitations imposed by the background current.
Figure 16.19 Hybridization sensors with a “signal-on” architecture. (A) Sandwich assay using capture and signaling probes connected through a linker; (B) Target induced strand displacement in a discontinuous duplex between capture and signaling probes; (C) target-induced disruption of a pseudoknot. Adapted with permission from [42]. Copyright 2009 Wiley-VCH Verlag GmBH & Co. KGaA.
16.2.6 Enzyme Labels in Amperometric Nucleic Acid Sensors
Enzymes are widely used as labels in affinity sensors and can therefore also act as transduction labels in nucleic acids sensors [19, 39, 43]. Inexpensive enzymes with a high turnover number, such as horseradish peroxidase, glucose oxidase and alkaline phosphatase have been used to this end. A proper calibration graph is obtained when the response current is proportional to the concentration of the hybrid-bonded enzyme, that is, when the enzyme assembly operates under kinetic control.
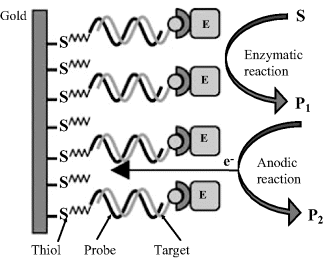
An enzyme-linked DNA sensor can be developed according to the scheme in Figure 16.20. In this approach, an ethanethiol-terminated probe has been attached by self-assembly onto a gold screen-printed electrode while the target has been tagged with biotin at the 5′ end. Hybrid labeling has been effected with a streptavidin-conjugated enzyme (alkaline phosphatase) that binds to the hybridized target through streptavidin–biotin affinity reaction. After washing out the solution enzyme, the substrate (![]() -naphthyl phosphate) is left to undergo enzymatic hydrolysis to the electrochemically active product
-naphthyl phosphate) is left to undergo enzymatic hydrolysis to the electrochemically active product ![]() . Electrochemical oxidation of this product to P2 provides the response current that has been monitored by differential pulse voltammetry. This scheme needs the target to be tagged with an affinity label. Tagging is effected during the prior polymerase chain reaction by adding a tagged nucleotide to the reaction mixture.
. Electrochemical oxidation of this product to P2 provides the response current that has been monitored by differential pulse voltammetry. This scheme needs the target to be tagged with an affinity label. Tagging is effected during the prior polymerase chain reaction by adding a tagged nucleotide to the reaction mixture.
Figure 16.20 Configuration of an electrochemical enzyme-linked DNA sensor based on affinity coupling of the enzyme to the target. E stands for enzyme and S is the substrate. Adapted with permission from [44]. Copyright 2004 Elsevier.
The principles of the mediated enzyme sensor can also be applied for the purpose of transduction in DNA biosensors. An easy way in this respect relies on attaching a mediator (e.g., ferrocene) to the target. By hybridization, the mediator is brought into the surface layer. A suitable enzyme and its substrate are then added to the solution afterwards and the response current is recorded.
Wiring of the enzyme with the electrode through a redox polymer brings about noticeable advantages, particularly due to the fact that the redox polymer protects the electrode surface against the contact with electroactive species in the solution and thus eliminates the need for washing out or separating them before the transduction step. Such a sensor is presented in Figure 16.21. The sensing layer has been formed on a gold ultramicroelectrode (diameter ≤100 μm). First, cysteamine has been self-assembled at the gold electrode surface and then, an -NH2 terminated target ((![]() ): NH2-C6-ATTCGACAGGGATAGTTCGA) has been coupled to the -NH2 groups in cysteamine using polyoxyethylene bis(glycidyl ether) as the linker. An osmium redox hydrogel layer was then grown over so as to include the probe. A biotin-tagged sequence from Listeria monocytogenes ((
): NH2-C6-ATTCGACAGGGATAGTTCGA) has been coupled to the -NH2 groups in cysteamine using polyoxyethylene bis(glycidyl ether) as the linker. An osmium redox hydrogel layer was then grown over so as to include the probe. A biotin-tagged sequence from Listeria monocytogenes ((![]() ): biotin-ATTCGACAGGGATAGTTCGA) has been used as probe, whereas the control oligonucleotide sequence consisted of (
): biotin-ATTCGACAGGGATAGTTCGA) has been used as probe, whereas the control oligonucleotide sequence consisted of (![]() ): biotin-ATTCGACAGGGATAGTTCGA. The enzyme was glucose oxidase tagged with biotin. The hybridization occurs within the matrix formed by the redox polymer and brings the biotin-tagged target into the surface layer. Subsequently, this tag binds the avidin-tagged enzyme to the hybrid.
): biotin-ATTCGACAGGGATAGTTCGA. The enzyme was glucose oxidase tagged with biotin. The hybridization occurs within the matrix formed by the redox polymer and brings the biotin-tagged target into the surface layer. Subsequently, this tag binds the avidin-tagged enzyme to the hybrid.
Figure 16.21 A redox hydrogel-based enzyme-linked hybridization sensor. Adapted with permission from [45]. Copyright 2009 Wiley-VCH Verlag GmBH & Co. KGaA.
In order to assess the hybridization, the substrate (glucose) is then added to the sample solution. Enzymatic oxidation of glucose releases electrons that are transferred to the negatively charged electrode through the redox polymer layer and gives rise to the response current. Clearly, the response is a function of the amount of available hybrid and, consequently, of the concentration of the target in the sample.
For calibration and assay, the steady-state current has been recorded at a constant potential (−0.35 V vs. Ag/AgCl, in samples of pH 7.4 containing 20 mM glucose and 0.15 M NaCl. The calibration graph plotted as current vs. the logarithm of the target concentration is linear from ![]() to
to ![]() M. The very small size of the sensor allows to assay samples of about 10 μL volume.
M. The very small size of the sensor allows to assay samples of about 10 μL volume.
The previously outlined approaches rely on fully dedicated technologies leading to a device specific to a well-defined target. In order to increase the throughput of the sensor fabrication process, it is advantageous to resort to a generic platform that can be readily adapted to various applications. Such a platform has been constructed using an avidin-containing redox polymer layer that has been coated on a carbon electrode [46]. The biotinylated capture probe was thereafter attached to the previously formed layer through biotin–avidin binding. Target determination has been carried out by the sandwich assay method, using a peroxidase-tagged signaling probe. In order to perform the assay, the electrode is incubated with the tested solution which contains the analyte nucleic acid, the signaling probe and ![]() . The hybridization of the signaling probe sets up an electrical contact between the redox polymer and the peroxidase label. Enzymatic reduction of
. The hybridization of the signaling probe sets up an electrical contact between the redox polymer and the peroxidase label. Enzymatic reduction of ![]() proceeds and causes electrons to flow through the redox polymer towards the immobilized enzyme. Remarkably, the test could be conducted in the presence of extraneous DNA and serum because the surface redox polymer blocks the access of interferents to the electrode surface. A screen-printing fabrication technology based on these principles has been devised [47].
proceeds and causes electrons to flow through the redox polymer towards the immobilized enzyme. Remarkably, the test could be conducted in the presence of extraneous DNA and serum because the surface redox polymer blocks the access of interferents to the electrode surface. A screen-printing fabrication technology based on these principles has been devised [47].
The advantage of this design results from the possibility of readily attaching various biotinylated capture probes to the same platform in order to obtain sensors for various nucleic acids.
In order to enhance the response current, the enzyme has been tagged with several complementary probes [48]. One of them binds to the immobilized target whilst the remaining ones attach by hybridization to other enzyme molecules. The result is a molecular dendritic structure including several enzymes molecules attached to each probe–target hybrid.
Nonbiotic materials can also be used as catalyst labels. Thus, in a sandwich assay, platinum nanoparticles have been attached to the signaling probe. Platinum is a catalyst for electrochemical reduction of ![]() and the resulting current allows the hybridization event to be traced [49].
and the resulting current allows the hybridization event to be traced [49].
16.2.7 Electrochemical DNA Arrays
Due to the simplicity of the transduction and, implicitly, of the measuring equipment, as well as to the possibility of performing measurements on turbid samples, electrochemical DNA arrays have been subjected to intensive research effort [50]. Thus, a DNA array has been fabricated by electrochemical polymerization of polypyrrole in the presence of capture-probe-tethered pyrrole at gold microelectrodes arrayed on a silicon chip [51]. This process has been performed by means of microcells actuated by a micromanipulator or by scanning electrochemical microscopy. Spots (50 × 50 μm) have been fabricated in this way on 128 microelectrodes arrays. Amperometric signal transduction has been performed by means of a redox intercalator.
The commercially available ElectraSense® platform takes advantage of the enzyme amplification effect. This platform can test up to 12 000 probes in less than 25 s [52]. It is based on a biotin-labeled target sample which after hybridization is tagged with avidin-conjugated peroxidase. In the presence of ![]() , the substrate (3, 3′, 5, 5′-tetramethylbenzidene) is converted to a reducible product that is detected by means of its cathodic current.
, the substrate (3, 3′, 5, 5′-tetramethylbenzidene) is converted to a reducible product that is detected by means of its cathodic current.
16.2.8 Nucleic Acids as Recognition Materials for Non-Nucleotide Compounds
As the toxic action of many pollutants (mutagens and carcinogens) is related to their interaction with DNA, it is reasonable to exploit this property by using dsDNA as recognition material in pertinent biosensors for pollution monitoring. To this end, a DNA layer is immobilized at the surface of an electrode. After incubating the sensor with the sample, possible alterations in DNA structure can be noted as modifications in the electrochemical response. The most straightforward method for detecting DNA interaction with the analyte is based on changes in the electrochemical signal of guanine oxidation. In this respect, a thorough investigation of various low molecular compounds of analytical concern has been carried out using both ds- and ssDNA [40]. It has been proved that some compounds that interact with DNA by intercalation reduce the guanine signal, whereas other compounds with similar action do enhance it. As expected, the effect is stronger with dsDNA. This implies that a DNA sensor cannot discriminate between various pollutants but can be used as a warning device that indicates the presence of harmful compounds in environmental samples. A preconcentration step of natural water samples is necessary to this end. Compared with microbiological genotoxicity tests, the DNA sensor assay is faster and is suitable for field-analysis applications.
Another alternative approach uses the electrochemical reactivity of the analyte itself. Thus, pollutants like aromatic amines or phenols undergo electrochemical oxidation at carbon electrodes after being accumulated at the surface by interaction with a previously attached DNA layer [34]. The response due to the accumulated analyte comes in addition to the guanine signal that is also altered by analyte intercalation. Detection limits close to 0.1 μM have been reported for some polycyclic aromatic amines when the analyte electrochemical signal has been recorded.
16.2.9 Aptamer Amperometric Sensors
Aptamers, that are specific receptors for proteins, exhibit particular advantages with respect to antibodies and have therefore been intensively investigated for the purpose of amperometric biosensor development [42, 53–56]. Various amperometric transduction methods for such sensors have been advanced using either redox indicators or redox enzymes.
16.2.9.1 Aptamer Sensors Based on Redox Indicators
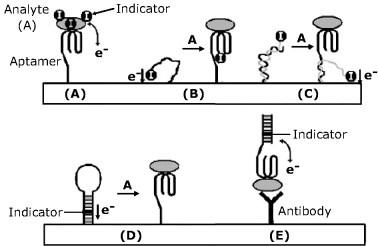
In order to perform amperometric transduction, a redox indicator can be attached to the protein-analyte by various methods. A simple labeling procedure is based on the electrostatic attraction between a negatively charged protein and a cationic indicator (such as methylene blue). Specific binding of the protein to the electrode attached aptamer brings the indicator to a position where it can undergo an electrochemical reaction yielding the response current (Figure 16.22A). Figure 16.22B shows a signal-off approach, in which the indicator is linked to the aptamer that is unfolded in the free form and allows electrons to flow between the redox indicator and the electrode. Analyte binding induces a drastic change in the aptamer conformation that cuts off the electron flow. In Figure 16.22C the aptamer is hybridized with an indicator-bearing oligonucleotide so that the indicator is kept far from the surface. This duplex is partially disrupted by the analyte–aptamer interaction which lets the indicator undergo an electrochemical reaction (signal on approach). An application of a hair pin structured aptamer is shown in Figure 16.22D. In the free form, the duplex moiety of the aptamer is labeled by intercalation with a redox indicator close to the electrode surface. Analyte binding to the aptamer breaks down the duplex and the indicator shifts away by diffusion (signal off). A mixed sandwich approach is shown in Figure 16.22E where the analyte recognition is performed by an antibody, whereas the labeled indicator reacts with the analyte at another site so as to bring the indicator close to the electrode. Care should be taken to prevent a positively-charged indicator from binding to the antibody protein. This can be achieved by blocking the nonspecific sites on the antibody.
Figure 16.22 Amperometric aptamer sensors using a redox indicator. Adapted with permission from [53]. Copyright 2009 Wiley-VCH Verlag GmBH & Co. KGaA.
16.2.9.2 Aptamer Sensors Based on Catalytic Reactions
Various strategies for enzyme-linked transduction have been tested with aptamer-based thrombin sensors [57], as emphasized in Figure 16.23. Thus, the method (A) in this figure relies on the ability of thrombin to catalyze the conversion of peptide-bound p-nitroaniline to free p-nitroaniline. This compound undergoes electrochemical reduction in both forms, but at different potentials, which allows the detection of the free form by differential pulse voltammetry.
Figure 16.23 Possible configurations of enzyme-linked amperometric aptamer sensors. (A) The analyte (thrombin) catalyzes the formation of an electrochemically active compound; (B) sandwich assay using a second, enzyme-labeled aptamer; (C) the analyte is adsorbed at the electrode surface and then coupled with an enzyme-labeled aptamer. Adapted with permission from [53]. Copyright 2009 Wiley-VCH Verlag GmBH & Co. KGaA.
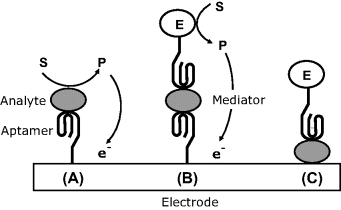
Figure 16.23B shows a sandwich assay involving a second aptamer that is linked to a redox enzyme (for example, peroxidase). In the presence of the substrate and a suitable mediator, the enzymatic reaction gives rise to the response current. In both previous approaches, the aptamer receptor has been attached to a gold electrode by self-assembly via a thiol spacer. Next, this layer has been incubated with mercaptoethanol that adsorbs strongly on the gold free surface and displaces the aptamer molecules randomly adsorbed. In addition, adsorbed mercaptoethanol molecules block any exposed surface site and prevent spurious adsorption of sample components.
In the third alternative (Figure 16.23C) the thrombin analyte has been adsorbed on the mercaptoethanol-modified gold surface and then the resulting layer has been exposed to the enzyme-labeled aptamer. As in case (B), the transduction has been achieved by monitoring the current flowing in the presence of the enzyme substrate and a mediator.
Among these three alternatives, the second one achieved the best sensitivity due to the high turnover number of the label enzyme. In the third alternative, nonspecific adsorption of the labeled aptamer gives rise to a significant background current with a negative effect on the detection limit. Further improvements are needed in order to render such sensors compatible with real biological samples.
16.2.10 Outlook
Hybridization sensors allow simple and fast identification of harmful microorganisms in environment sample and in foods and can be used to monitor biological warfare agents by detecting microorganism nucleic acids contained in such organisms.
Various transduction strategies for amperometric DNA sensors have been developed. The label-free method relies on the anodic reaction of the guanine base included in the nucleic acid sequence. A more general method is based on redox indicators intercalated in the double-strand hybrid. Alternatively, redox indicators can be covalently attached to a signaling probe so as to place them either near to or far from the electrode surface. By hybridization, the indicator is shifted to be far from or near to the electrode, respectively. The first alternative is of the “signal-off” type; the second is a “signal-on” technique.
Much improvement in sensitivity is brought about by utilization of enzyme labels that perform a chemical amplification of the response current. The enzyme produces an electrochemically active compound that generates the response current. Redox enzymes are particularly advantageous because they allow coupling with a redox hydrogel in order to perform wired electron transfer to the electrode. In this way, the enzyme present in the bulk of the solution is not involved in the electrochemical reaction which eliminates the need for washing out before the detection step.
When compared with other transduction methods, electrochemical transduction renders a DNA sensor relatively simple, less costly and more amenable for miniaturization and mass production. A critical issue with such sensors is their relatively poor detection limits. That is why preliminary amplification by the polymerase chain reaction is usually needed [58]. Much improvement in sensitivity is expected from the use of carbon nanotubes [59].
Hand-held analyzers based on nucleic acid sensors have also been developed allowing field analysis to be conducted with obvious advantages [60].
Amperometric aptamer sensors appear as an alternative to standard immunological assay and open new possibilities for detecting proteins. Transduction in amperometric aptamer sensors can be achieved by means of redox indicators, but best sensitivity is achieved by means of enzyme labels.
1. Centi, S., Laschi, S., and Mascini, M. (2009) Strategies for electrochemical detection in immunochemistry. Bioanalysis, 1, 1271–1291.
2. Fowler, J. M., Wong, D. K. Y., Halsall, H. B. et al. (2008) Recent developments in electrochemical immunoassays and immunosensors, in Electrochemical Sensors, Biosensors, and Their Biomedical Applications (eds X. Zhang, H. Ju and J. Wang) Academic Press, Amsterdam, pp. 115–143.
3. Warsinke, A., Benkert, A., and Scheller, F.W. (2000) Electrochemical immunoassays. Fresenius J. Anal. Chem., 366, 622–634.
4. Yakovleva, J. and Emneus, J. (2008) Electrochemical immunoassay, in Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications (ed. P.N. Bartlett), John Wiley & Sons, Chichester, pp. 377–410.
5. Belluzo, M.S., Ribone, M.E., and Lagier, C.M. (2008) Assembling amperometric biosensors for clinical diagnostics. Sensors, 8, 1366–1399.
6. Kalab, T. and Skladal, P. (1995) A disposable amperometric immunosensor for 2,4-dichlorophenoxyacetic acid. Anal. Chim. Acta, 304, 361–368.
7. Skladal, P. and Kalab, T. (1995) A multichannel immunochemical sensor for determination of 2,4-dichlorophenoxyacetic Acid. Anal. Chim. Acta, 316, 73–78.
8. Campbell, C.N., de Lumley-Woodyear, T., and Heller, A. (1999) Towards immunoassay in whole blood: separationless sandwich-type electrochemical immunoassay based on in-situ generation of the substrate of the labeling enzyme. Fresenius J. Anal. Chem., 364, 165–169.
9. Liu, G.D. and Lin, Y.H. (2007) Nanomaterial labels in electrochemical immunosensors and immunoassays. Talanta, 74, 308–317.
10. Das, J., Aziz, M.A., and Yang, H. (2006) A nanocatalyst-based assay for proteins: DNA-free ultrasensitive electrochemical detection using catalytic reduction of p-nitrophenol by gold-nanoparticle labels. J. Am. Chem. Soc., 128, 16022–16023.
11. Kuramitz, H. (2009) Magnetic microbead-based electrochemical immunoassays. Anal. Bioanal. Chem., 394, 61–69.
12. Wang, J., Liu, G.D., and Jan, M.R. (2004) Ultrasensitive electrical biosensing of proteins and DNA: Carbon-nanotube derived amplification of the recognition and transduction events. J. Am. Chem. Soc., 126, 3010–3011.
13. Yu, X., Munge, B., Patel, V. et al. (2006) Carbon nanotube amplification strategies for highly sensitive immunodetection of cancer biomarkers. J. Am. Chem. Soc., 128, 11199–11205.
14. Blanco-Lopez, M.C., Lobo-Castanon, M.J., Miranda-Ordieres, A.J. et al. (2004) Electrochemical sensors based on molecularly imprinted polymers. TrAC-Trends Anal. Chem., 23, 36–48.
15. Suryanarayanan, V., Wu, C.T., and Ho, K.C. (2010) Molecularly Imprinted Electrochemical Sensors. Electroanalysis, 22, 1795–1811.
16. Blanco-Lopez, M.C., Lobo-Castanon, M.J., Miranda-Ordieres, A.J. et al. (2003) Voltammetric sensor for vanillylmandelic acid based on molecularly imprinted polymer-modified electrodes. Biosens. Bioelectron., 18, 353–362.
17. Blanco-Lopez, M.C., Gutierrez-Fernandez, S., Lobo-Castanon, M.J. et al. (2004) Electrochemical sensing with electrodes modified with molecularly imprinted polymer films. Anal. Bioanal. Chem., 378, 1922–1928.
18. de-los-Santos-Alvarez, P., Lobo-Castanon, M.J., Miranda-Ordieres, A.J. et al. (2004) Current strategies for electrochemical detection of DNA with solid electrodes. Anal. Bioanal. Chem., 378, 104–118.
19. Sassolas, A., Leca-Bouvier, B.D., and Blum, L.J. (2008) DNA biosensors and microarrays. Chem. Rev., 108, 109–139.
20. Labuda, J., Brett, A.M.O., Evtugyn, G. et al. (2010) Electrochemical nucleic acid-based biosensors: Concepts, terms, and methodology (IUPAC Technical Report). Pure Appl. Chem., 82, 1161–1187.
21. Pale![]() ek, E. (1960) Oscillographic polarography of highly polymerized deoxyribonucleic acid. Nature, 188, 656–657.
ek, E. (1960) Oscillographic polarography of highly polymerized deoxyribonucleic acid. Nature, 188, 656–657.
22. Pale![]() ek, E. and Fojta, M. (2001) Detecting DNA hybridization and damage. Anal. Chem., 73, 74A–83A.
ek, E. and Fojta, M. (2001) Detecting DNA hybridization and damage. Anal. Chem., 73, 74A–83A.
23. Pale![]() ek, E. (2009) Fifty years of nucleic acid electrochemistry. Electroanalysis, 21, 239–251.
ek, E. (2009) Fifty years of nucleic acid electrochemistry. Electroanalysis, 21, 239–251.
24. Pale![]() ek, E., and Fojta, M. (2012) Nucleic Acid Electrochemistry: Basics and Applications, Springer, Berlin.
ek, E., and Fojta, M. (2012) Nucleic Acid Electrochemistry: Basics and Applications, Springer, Berlin.
25. Oliveira-Brett, A.M. (2008) Electrochemical DNA assays, in Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications (ed. P.N. Bartlett), John Wiley & Sons, Chichester, pp. 411–442.
26. Brabec, V., Vetterl, V., and Vrana, O. (1996) Electroanalysis of biomolecules, in Experimental Techniques in Bioelectrochemistry (eds V. Brabec, G. Milazzo, and D. Walz), Birkhäuser, Basel, pp. 287–359.
27. Dryhurst, G. (1977) Electrochemistry of Biological Molecules, Academic Press, New York.
28. Brabec, V. (1980) Electrochemical oxidation of nucleic acids and proteins at graphite electrode – qualitative aspects. Bioelectrochem. Bioenerget., 7, 69–82.
29. Wang, J., Bollo, S., Paz, J.L.L. et al. (1999) Ultratrace measurements of nucleic acids by baseline-corrected adsorptive stripping square-wave voltammetry. Anal. Chem., 71, 1910–1913.
30. Wu, K.B., Fei, J.J., Bai, W. et al. (2003) Direct electrochemistry of DNA, guanine and adenine at a nanostructured film-modified electrode. Anal. Bioanal. Chem., 376, 205–209.
31. Wang, J., Cai, X.H., Tian, B.M. et al. (1996) Microfabricated thick-film electrochemical sensor for nucleic acid determination. Analyst, 121, 965–969.
32. Wang, J., Kawde, A.N., Erdem, A. et al. (2001) Magnetic bead-based label-free electrochemical detection of DNA hybridization. Analyst, 126, 2020–2024.
33. Napier, M.E., Loomis, C.R., Sistare, M.F. et al. (1997) Probing biomolecule recognition with electron transfer: Electrochemical sensors for DNA hybridization. Bioconjugate Chem., 8, 906–913.
34. Wang, J., Rivas, G., Cai, X. et al. (1997) DNA electrochemical biosensors for environmental monitoring. A review. Anal. Chim. Acta, 347, 1–8.
35. Popovich, N.D., Eckhardt, A.E., Mikulecky, J.C. et al. (2002) Electrochemical sensor for detection of unmodified nucleic acids. Talanta, 56, 821–828.
36. He, P.A., Xu, Y., and Fang, Y.Z. (2006) Applications of carbon nanotubes in electrochemical DNA biosensors. Microchim. Acta, 152, 175–186.
37. Koehne, J., Li, J., Cassell, A.M. et al. (2004) The fabrication and electrochemical characterization of carbon nanotube nanoelectrode arrays. J. Mater. Chem., 14, 676–684.
38. Takenaka, S. (2001) Highly sensitive probe for gene analysis by electrochemical approach. Bull. Chem. Soc. Jpn., 74, 217–224.
39. Takenaka, S. (2003) Electrochemical detection of DNA with small molecules, in Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes (eds M. Demeunynck, C. Bailly, and W. D. Wilson), Wiley-VCH, Weinheim, pp. 224–245.
40. Mascini, M., Palchetti, I., and Marrazza, G. (2001) DNA electrochemical biosensors. Fresenius J. Anal. Chem., 369, 15–22.
41. Umek, R.M., Lin, S.W., Vielmetter, J. et al. (2001) Electronic detection of nucleic acids – A versatile platform for molecular diagnostics. J. Mol. Diagnos., 3, 74–84.
42. Miranda-Castro, R., de-los-Santos-Alvarez, N., Lobo-Castanon, M.J. et al. (2009) Structured nucleic acid probes for electrochemical devices. Electroanalysis, 21, 2077–2090.
43. Willner, I., Shlyahovsky, B., Willner, B. et al. (2009) Amplified DNA biosensors, in Functional Nucleic Acids for Analytical Applications (eds Y. Li and Y. Lu), Springer, New York, pp. 199–252.
44. Carpini, G., Lucarelli, F., Marrazza, G. et al. (2004) Oligonucleotide-modified screen-printed gold electrodes for enzyme-amplified sensing of nucleic acids. Biosens. Bioelectron., 20, 167–175.
45. Hajdukiewicz, J., Boland, S., Kavanagh, P. et al. (2009) Enzyme-amplified amperometric detection of DNA using redox mediating films on gold microelectrodes. Electroanalysis, 21, 342–350.
46. Campbell, C.N., Gal, D., Cristler, N. et al. (2002) Enzyme-amplified amperometric sandwich test for RNA and DNA. Anal. Chem., 74, 158–162.
47. Dequaire, M. and Heller, A. (2002) Screen printing of nucleic acid detecting carbon electrodes. Anal. Chem., 74, 4370–4377.
48. Dominguez, E., Rincon, O., and Narvaez, A. (2004) Electrochemical DNA sensors based on enzyme dendritic architectures: An approach for enhanced sensitivity. Anal. Chem., 76, 3132–3138.
49. Polsky, R., Gill, R., Kaganovsky, L. et al. (2006) Nucleic acid-functionalized Pt nanoparticles: Catalytic labels for the amplified electrochemical detection of biomolecules. Anal. Chem., 78, 2268–2271.
50. Dill, K. and Ghindilis, A. (2009) Electrochemical Detection on Microarrays, in Microarrays (eds K. Dill, P. Grodzinski, and R.H. Liu), Springer, New York, pp. 25–34.
51. Szunerits, S., Bouffier, L., Calemczuk, R. et al. (2005) Comparison of different strategies on DNA chip fabrication and DNA-sensing: Optical and electrochemical approaches. Electroanalysis, 17, 2001–2017.
52. ElectraSense ® 12K/4X2K microarrays: hybridization and electrochemical detection http://www.combimatrix.com/products_electrasense.htm.
53. Hianik, T. and Wang, J. (2009) Electrochemical aptasensors – recent achievements and perspectives. Electroanalysis, 21, 1223–1235.
54. Sassolas, A., Blum, L.J., and Leca-Bouvier, B.D. (2009) Electrochemical aptasensors. Electroanalysis, 21, 1237–1250.
55. Xu, Y., Cheng, G.F., He, P.G. et al. (2009) A review: Electrochemical aptasensors with various detection strategies. Electroanalysis, 21, 1251–1259.
56. Willner, I. and Zayats, M. (2007) Electronic aptamer-based sensors. Angew. Chem. Int. Ed., 46, 6408–6418.
57. Mir, M., Vreeke, M., and Katakis, L. (2006) Different strategies to develop an electrochemical thrombin aptasensor. Electrochem. Commun., 8, 505–511.
58. Pedrero, M., Campuzano, S., and Pingarron, J.M., (2011) Electrochemical genosensors based on PCR strategies for microorganisms detection and quantification. Anal. Methods, 3, 780–789.
59. Teles, F.R.R. and Fonseca, L.R. (2008) Trends in DNA biosensors. Talanta, 77, 606–623.
60. Lee, T.M.H. and Hsing, I.M. (2006) DNA-based bioanalytical microsystems for handheld device applications. Anal. Chim. Acta, 556, 26–37.