
3.2.3 Reversible Reactions
All rate laws for reversible reactions must reduce to the thermodynamic relationship relating the reacting species concentrations at equilibrium. At equilibrium, the rate of reaction is identically zero for all species (i.e., –rA ≡ 0). That is, for the general reaction
![]()
the concentrations at equilibrium are related by the thermodynamic relationship for the equilibrium constant KC (see Appendix C).
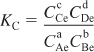
The units of the thermodynamic equilibrium constant, KC, are (mol/dm3)d + c – b – a.
To illustrate how to write rate laws for reversible reactions, we will use the combination of two benzene molecules to form one molecule of hydrogen and one of diphenyl. In this discussion, we shall consider this gas-phase reaction to be elementary and reversible:
![]()
or, symbolically,
![]()
The forward and reverse specific reaction rate constants, kB and k–B, respectively, will be defined with respect to benzene.
Benzene (B) is being depleted by the forward reaction
![]()
in which the rate of disappearance of benzene is
![]()
If we multiply both sides of this equation by –1, we obtain the expression for the rate of formation of benzene for the forward reaction:
![]()
For the reverse reaction between diphenyl (D) and hydrogen (H2),
![]()
the rate of formation of benzene is given as
![]()
Again, both the rate constants kB and k–B are defined with respect to benzene!!!
The net rate of formation of benzene is the sum of the rates of formation from the forward reaction [i.e., Equation (3-11)] and the reverse reaction [i.e., Equation (3-12)]:

Multiplying both sides of Equation (3-13) by –1, and then factoring out kB, we obtain the rate law for the rate of disappearance of benzene, –rB:
![]()
Replacing the ratio of the reverse to forward rate law constants by the reciprocal of the concentration equilibrium constant, KC, we obtain
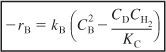
where
![]()
The equilibrium constant decreases with increasing temperature for exothermic reactions and increases with increasing temperature for endothermic reactions.
Let’s write the rate of formation of diphenyl, rD, in terms of the concentrations of hydrogen, H2, diphenyl, D, and benzene, B. The rate of formation of diphenyl, rD, must have the same functional dependence on the reacting species concentrations as does the rate of disappearance of benzene, –rB. The rate of formation of diphenyl is
![]()
Using the relationship given by Equation (3-1) for the general reaction
![]()
we can obtain the relationship between the various specific reaction rates, kB, kD:
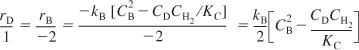
Comparing Equations (3-15) and (3-16), we see the relationship between the specific reaction rate with respect to diphenyl, kD, and the specific reaction rate with respect to benzene, kB, is
![]()
Consequently, we see the need to define the rate constant, k, with respect to a particular species.
Finally, we need to check to see if the rate law given by Equation (3-14) is thermodynamically consistent at equilibrium. Applying Equation (3-10) (and Appendix C) to the diphenyl reaction and substituting the appropriate species concentration and exponents, thermodynamics tells us that
![]()
Now let’s look at the rate law. At equilibrium, –rB ≡ 0, and the rate law given by Equation (3-14) becomes
![]()
Rearranging, we obtain, as expected, the equilibrium expression
![]()
which is identical to Equation (3-17) obtained from thermodynamics.
From Appendix C, Equation (C-9), we know that when there is no change in the total number of moles and the heat capacity term, ΔCP = 0, the temperature dependence of the concentration equilibrium constant is
![]()
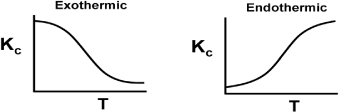
Therefore, if we know the equilibrium constant at one temperature, T1 [i.e., KC (T1)], and the heat of reaction, ![]() , we can calculate the equilibrium constant at any other temperature T. For endothermic reactions, the equilibrium constant, KC, increases with increasing temperature; for exothermic reactions, KC decreases with increasing temperature. A further discussion of the equilibrium constant and its thermodynamic relationship is given in Appendix C.
, we can calculate the equilibrium constant at any other temperature T. For endothermic reactions, the equilibrium constant, KC, increases with increasing temperature; for exothermic reactions, KC decreases with increasing temperature. A further discussion of the equilibrium constant and its thermodynamic relationship is given in Appendix C.
3.3 The Reaction Rate Constant
The reaction rate constant k is not truly a constant; it is merely independent of the concentrations of the species involved in the reaction. The quantity k is referred to as either the specific reaction rate or the rate constant. It is almost always strongly dependent on temperature. It also depends on whether or not a catalyst is present, and in gas-phase reactions, it may be a function of total pressure. In liquid systems it can also be a function of other parameters, such as ionic strength and choice of solvent. These other variables normally exhibit much less effect on the specific reaction rate than temperature does with the exception of supercritical solvents, such as supercritical water. Consequently, for the purposes of the material presented here, it will be assumed that kA depends only on temperature. This assumption is valid in most laboratory and industrial reactions and seems to work quite well.
It was the great Nobel Prize–winning Swedish chemist Svante Arrhenius (1859–1927) who first suggested that the temperature dependence of the specific reaction rate, kA, could be correlated by an equation of the type
![]()
where
A = pre-exponential factor or frequency factor
E = activation energy, J/mol or cal/mol
R = gas constant = 8.314 J/mol · K = 1.987cal/mol · K
T = absolute temperature, K
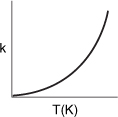
Equation (3-18), known as the Arrhenius equation, has been verified empirically to give the temperature behavior of most reaction rate constants within experimental accuracy over fairly large temperature ranges. The Arrhenius equation is derived in the Professional Reference Shelf 3.A: Collision Theory on the DVD-ROM.