
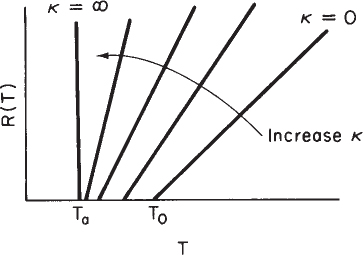
Vary Non-adiabatic Parameter κ. If one increases κ by either decreasing the molar flow rate, FA0, or increasing the heat-exchange area, A, the slope increases and for the case of Ta < T0 the ordinate intercept moves to the left, as shown in Figure 12-8.
κ=UACP0FA0Tc=T0+κTa1+κ
κ=0Tc=T0κ=∞Tc=Ta
On the other hand, if Ta > T0, the intercept will move to the right as κ increases.
12.5.2 Heat-Generated Term, G(T)
The heat-generated term, Equation (12-29), can be written in terms of conversion. [Recall that X = −rAV/FA0.]
G(T)=(-ΔHoRx)X (12-31)
To obtain a plot of heat generated, G(T), as a function of temperature, we must solve for X as a function of T using the CSTR mole balance, the rate law, and stoichiometry. For example, for a first-order liquid-phase reaction, the CSTR mole balance becomes
V=FA0XkCA=v0CA0XkCA0(1-X)
First-order reaction
X=τk1+τk (5-8)
Substituting for X in Equation (12-31), we obtain
G(T)=-ΔHoRxτk1+τk (12-32)
Finally, substituting for k in terms of the Arrhenius equation, we obtain
G(T)=−ΔH∘RxτAe−E/RT1+τAe−E/Rt(12−33)
Second-order reaction
Note that equations analogous to Equation (12-33) for G (T) can be derived for other reaction orders and for reversible reactions simply by solving the CSTR mole balance for X. For example, for the second-order liquid-phase reaction
X=(2τkCA0+1)-√4τkCA0+12τkCA0
the corresponding heat generated term is
G(T)=−ΔH∘Rx[(2τCA0Ae−E/RT+1)−√4τCA0Ae−E/RT+1]2τCA0Ae−E/RT(12−34)
Let’s now return to our first-order reaction, Equation (12-33), and examine the behavior of the G(T) curve. At very low temperatures, the second term in the denominator of Equation (12-33) for the first-order reaction can be neglected, so that G(T) varies as
Low T
G(T)=-ΔHoRxτAe-E/RT
(Recall that ΔHoRx
At very high temperatures, the second term on the right side in the denominator dominates in Equation (12-33) so that the τk in the numerator and denominator cancel, and G(T) is reduced to
High T
G(T)=-ΔHoRx
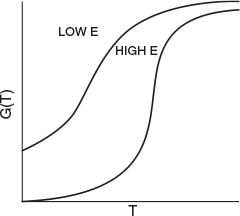
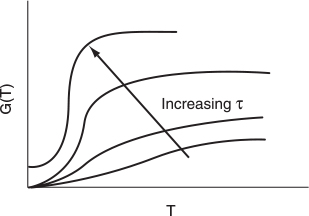
G(T) is shown as a function of T for two different activation energies, E, in Figure 12-9. If the flow rate is decreased or the reactor volume increased so as to increase τ, the heat-generated term, G(T ), changes, as shown in Figure 12-10.
Heat-generated curves, G (T)
Can you combine Figures 12-10 and 12-8 to explain why a Bunsen burner goes out when you turn up the gas flow to a very high rate?
12.5.3 Ignition-Extinction Curve
The points of intersection of R(T) and G(T) give us the temperature at which the reactor can operate at steady state. This intersection is the point at which both the energy balance, R(T), and the mole balance, G(T), are satisfied. Suppose that we begin to feed our reactor at some relatively low temperature, T01. If we construct our G(T) and R(T) curves, illustrated by curve y = G(T) and line a = R(T), respectively, in Figure 12-11, we see that there will be only one point of intersection, point 1. From this point of intersection, one can find the steady-state temperature in the reactor, Ts1, by following a vertical line down to the T-axis and reading off the temperature, Ts1, as shown in Figure 12-11.

If one were now to increase the entering temperature to T02, the G(T) curve, y, would remain unchanged, but the R(T) curve would move to the right, as shown by line b in Figure 12-11, and will now intersect the G(T) at point 2 and be tangent at point 3. Consequently, we see from Figure 12-11 that there are two steady-state temperatures, Ts2 and Ts3, that can be realized in the CSTR for an entering temperature T02. If the entering temperature is increased to T03, the R(T) curve, line c (Figure 12-12), intersects the G(T) curve three times and there are three steady-state temperatures, Ts4, Ts5, and Ts6. As we continue to increase T0, we finally reach line e, in which there are only two steady-state temperatures, a point of tangency at Ts10 and an intersection at Ts11. If the entering temperature is slightly increased beyond T05, say to T06, then there is a jump in reactor temperature, from Ts10 up to Ts12. This jump is called the ignition point for the reaction. By further increasing T0, we reach line f, corresponding to T06, in which we have only one reactor temperature that will satisfy both the mole and energy balances, Ts12. For the six entering temperatures, we can form Table 12-4, relating the entering temperature to the possible reactor operating temperatures.
Both the mole and energy balances are satisfied at the points of intersection or tangency.

Table 12-4 Multiple Steady-State Temperatures
Entering Temperature |
Reactor Temperatures |
||||
T01 |
Ts1 |
||||
T02 |
Ts2 |
Ts3 |
|||
T03 |
Ts4 |
Ts5 |
Ts6 |
||
T04 |
Ts7 |
Ts8 |
Ts9 |
||
T05 |
Ts10 |
Ts11 |
|||
T06 |
Ts12 |
||||
By plotting steady-state reactor temperature Ts as a function of entering temperature T0, we obtain the well-known ignition-extinction curve shown in Figure 12-13. From this figure, we see that as the entering temperature T0 is increased, the steady-state temperature Ts increases along the bottom line until T05 is reached. Any fraction-of-a-degree increase in temperature beyond T05 and the steady-state reactor temperature Ts will jump up from Ts10 to Ts11, as shown in Figure 12-13. The temperature at which this jump occurs is called the ignition temperature. That is, we must exceed a certain feed temperature, T05, to operate at the upper steady state where the temperature and conversion are higher.
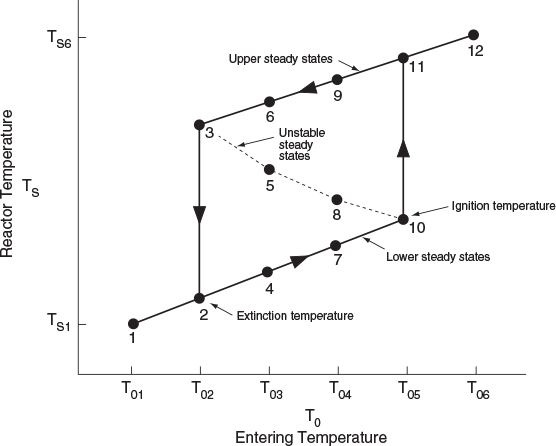
If a reactor were operating at Ts12 and we began to cool the entering temperature down from T06, the steady-state reactor temperature, Ts3, would eventually be reached, corresponding to an entering temperature, T02. Any slight decrease below T02 would drop the steady-state reactor temperature to immediately the lower steady-state value Ts2 . Consequently, T02 is called the extinction temperature.
The middle points 5 and 8 in Figures 12-12 and 12-13 represent unstable steady-state temperatures. Consider the heat-removed line d in Figure 12-12, along with the heat-generated curve y, which are replotted in Figure 12-14. If we were operating at the middle steady-state temperature Ts8, for example, and a pulse increase in reactor temperature occurred, we would find ourselves at the temperature shown by vertical line ②, between points 8 and 9. We see that along this vertical line ②, the heat-generated curve, y ≡ G(T), is greater than the heat-removed line d ≡ R(T) i.e., (G > R). Consequently, the temperature in the reactor would continue to increase until point 9 is reached at the upper steady state. On the other hand, if we had a pulse decrease in temperature from point 8, we would find ourselves on a vertical line ③ between points 7 and 8. Here, we see that the heat-removed curve d is greater than the heat-generated curve y (R > G), so the temperature will continue to decrease until the lower steady state is reached. That is, a small change in temperature either above or below the middle steady-state temperature, Ts8, will cause the reactor temperature to move away from this middle steady state. Steady states that behave in this manner are said to be unstable.
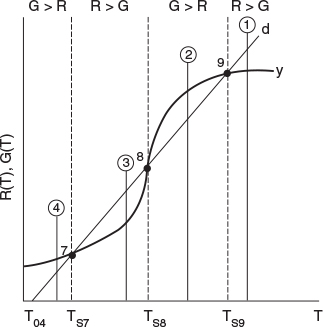
In contrast to these unstable operating points, there are stable operating points. Consider what would happen if a reactor operating at Ts9 were subjected to a pulse increase in reactor temperature indicated by line ① in Figure 12-14. We see that the heat-removed line d is greater than the heat-generated curve y (R > G), so that the reactor temperature will decrease and return to Ts9. On the other hand, if there is a sudden drop in temperature below Ts9, as indicated by line ②, we see the heat-generated curve y is greater than the heat-removed line d (G > R), and the reactor temperature will increase and return to the upper steady state at Ts9. Consequently, Ts9 is a stable steady state.
Next, let’s look at what happens when the lower steady-state temperature at Ts7 is subjected to pulse increase to the temperature shown as line ③ in Figure 12-14. Here, we again see that the heat removed, R, is greater than the heat generated, G, so that the reactor temperature will drop and return to Ts7. If there is a sudden decrease in temperature below Ts7 to the temperature indicated by line ④, we see that the heat generated is greater than the heat removed (G > R), and that the reactor temperature will increase until it returns to Ts7. Consequently, Ts7 is a stable steady state. A similar analysis could be carried out for temperatures Ts1, Ts2, Ts4, Ts6, Ts11, and Ts12, and one would find that reactor temperatures would always return to locally stable steady-state values when subjected to both positive and negative fluctuations.
While these points are locally stable, they are not necessarily globally stable. That is, a large perturbation in temperature or concentration may be sufficient to cause the reactor to fall from the upper steady state (corresponding to high conversion and temperature, such as point 9 in Figure 12-14), to the lower steady state (corresponding to low temperature and conversion, point 7).
12.6 Nonisothermal Multiple Chemical Reactions
Most reacting systems involve more than one reaction and do not operate iso-thermally. This section is one of the most important, if not the most important, sections of the book. It ties together all the previous chapters to analyze multiple reactions that do not take place isothermally.
12.6.1 Energy Balance for Multiple Reactions in Plug-Flow Reactors
Energy balance for multiple reactions
In this section we give the energy balance for multiple reactions. We begin by recalling the energy balance for a single reaction taking place in a PFR, which is given by Equation (12-5)
dTdV=(-rA)[-ΔHRx(T)]-Ua(T-Ta)mΣj=1FjCPj (12-5)
When we have multiple reactions occurring, we have to account for, and sum up, all the heats of reaction in the reactor for each and every reaction. For q multiple reactions taking place in a PFR where there are m species, it is easily shown that Equation (12-5) can be generalized to (cf. P12-1(j))
i = Reaction number
j = Species
dTdV=qΣi=1(−rij)[−ΔHRxij(T)]−Ua(T−Ta)mΣj=1FjCPj(12−35)
Note that we now have two subscripts on the heat of reaction. The heat of reaction for reaction i must be referenced to the same species in the rate, rij, by which ΔHRxij. is multiplied, which is
[−rij][−ΔHRxij]=[Moles ofj reacted in reaction iVolume⋅time]×[joules "released" in reaction iMoles ofj reacted in reaction i]=[Joules "released" in reaction iVolume⋅time](12−36)
where the subscript j refers to the species, the subscript i refers to the particular reaction, q is the number of independent reactions, and m is the number of species. We are going to let
Qg=qΣi=1(−rij)[−ΔHRxij(T)]
Qr=Ua(T−Ta)
Then Equation (12-35) becomes
dTdV=Qg−QrmΣj=1FjCPj(12−37)
Equation (12-37) represents a nice compact form of the energy balance for multiple reactions.
Consider the following reaction sequence carried out in a PFR:
One of the major goals of this text is that the reader will be able to solve multiple reactions with heat effects, and this section shows how!
Reaction 1:Ak1→BReaction 2:Bk2→C
The PFR energy balance becomes
Qg=(−ΔHRx1A)(−r1A)+(−r2B)(−ΔHRx2B)Qr=Ua(T−Ta)
Substituting for Qg and -Qr in Equation (12-37) we obtain
dTdV=Ua(Ta-T)+(-r1A)(-ΔHRx1A)+(-r2B)(-ΔHRx2B)FACPA+FBCPB+FCCPC (12-38)
whereΔHRx1A=[J/mol of A reacted in reaction 1]andΔHRx2B=[J/mol of B reacted in reaction 2].
12.6.2 Parallel Reactions in a PFR
We will now give three examples of multiple reactions with heat effects: Example 12-5 discusses parallel reactions, Example 12-6 discusses series reactions, and Example 12-7 discusses complex reactions.
Living Example Problem
Example 12-5 Parallel Reactions in a PFR with Heat Effects
The following gas-phase reactions occur in a PFR:
Reaction 1:Ak1→B −r1A=k1ACA(E12-5.1)
Reaction 2:2Ak2→C −r2A=k2AC2A(E12-5.2)
Pure A is fed at a rate of 100 mol/s, at a temperature of 150°C and at a concentration of 0.1 mol/dm3. Neglect pressure drop and determine the temperature and molar flow rate profiles down the reactor.
Additional information
ΔHRx1A=−20,000J/(mol of A reacted in reaction 1)ΔHRx2B=−60,000J/(mol of B reacted in reaction 2)
CPA=90J/mol⋅C∘k1A=10exp[E1R(1300−1T)]s−1CPB=90J/mol⋅C∘E1/R=4000KCPC=180J/mol⋅C∘k2A=0.09 exp[E2R(1300−1T)]dm3mol⋅sUa=4000J/m3⋅s⋅C∘E2/R=9000KTa=100C (Constant)∘
Solution
0. Number each reaction. This step was given in the problem statement.
(1)Ak1A→B(2)2Ak2A→C
1. Mole balances:
dFAdV=rA (E12–5.3)
dFBdV=rB (E12–5.4)
dFCdV=rC (E12–5.5)

Following the Algorithm
2. Rates:
Rate laws
r1A=-k1CAA (E12–5.1)
r2A=-k2AC2A (E12–5.2)
Relative rates
Reaction 1:r1A-1=r1B1;r1B=-r1A=k1CAAReaction 2:r2A-2=r2C1;r2C=-12r2A=k2A2C2A
Net rates
rA=r1A+r2A=-k1CAA-k2CA2A (E12–5.6)
rB=r1B=k1CAA (E12–5.7)
rC=r2C=12k2CA2A (E12–5.8)
3. Stoichiometry (gas-phase but ΔP = 0, i.e., p = 1):
CA=CT0(FAFT)(T0T) (E12–5.9)
CC=CT0(FCFT)(T0T) (E12–5.11)
FT=FA+FB+FC (E12–5.12)
klA=10exp[4000(1300-1T)]s-1 (E12–5.13)
(T in K)
k2A=0.09exp[9000(1300-1T)]dm3mo1⋅s (E12–5.14)
Major goal of CRE: Analyze Multiple Reactions with Heat Effects
4. Energy balance:
The PFR energy balance [cf. Equation (12-35)]
dTdV=Ua(Ta-T)+(-r1A)(-ΔHRx1A)+(-r2A)(-ΔHRx2A)FACPA+FBCPB+FCCPC (E12–5.15)
becomes
dTdV=4000(373-T)+(-r1A)(20,000)+(-r2A)(60,000)90FA+90FB+180FC (E12–5.16)
5. Evaluation:
The Polymath program and its graphical outputs are shown in Table E12-5.1 and Figures E12-5.1 and E12-5.2.
Living Example Problem

Why does the temperature go through a maximum value?
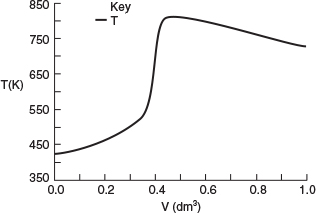
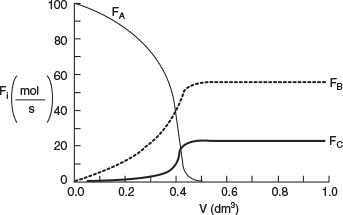
Analysis: From Figure E12-5.1 we see that the temperature increases slowly up to a reactor volume of 0.4 dm3 then suddenly jumps up (ignites) to a temperature of 850 K. Correspondingly, from Figure E12-5.2 the reactant molar flow rate decreases sharply at this point. The reactant is virtually consumed by the time it reaches a reactor volume V - 0.45 dm3; beyond this point, Qr > Qg, and the reactor temperature begins to drop. In addition, the selectivity ˜SB/C=FB/FC=55/22.5=2.44
12.6.3 Energy Balance for Multiple Reactions in a CSTR
Recall that for the steady-state mole balance in a CSTR with a single reaction [−FA0X = rAV,] and that ΔHRx(T)=ΔHoRx+ΔCP(T-TR)
˙Q-˙WS-FA0ΣΘjCPj(T-T0)+[ΔHRx(T)][rAV]=0 (11-27A)
Again, we must account for the “heat generated” by all the reactions in the reactor. For q multiple reactions and m species, the CSTR energy balance becomes
Energy balance for multiple reactions in a CSTR
˙Q−˙Ws−FA0mΣj=1ΘjCPj(T−T0)+VqΣi=1rijΔHRxij(T)=0(12−39)
Substituting Equation (12-20) for ˙Q
UA(Ta−T)−FA0mΣj=1CPjΘj(T−T0)+VqΣi=1rijΔHRxij(T)=0(12−40)
For the two parallel reactions described in Example 12-5, the CSTR energy balance is
UA(Ta-T)-FA0∑mj=1ΘjCPj(T-T0)+Vr1AΔHRxlA(T)+Vr2AΔHRx2A(T)=0 (12-41)
Major goal
As stated previously, one of the major goals of this text is to have the reader solve problems involving multiple reactions with heat effects (cf. Problems P12-23C, P12-24C, P12-25C, and P12-26B). That’s exactly what we are doing in the next two examples!
The elementary liquid-phase reactions
Ak1→Bk2→C
take place in a 10-dm3 CSTR. What are the effluent concentrations for a volumetric feed rate of 1000 dm3/min at a concentration of A of 0.3 mol/dm3? The inlet temperature is 283 K.
Additional information
CPA=CPB=CPC=200J/mol⋅Kk1=3.3min−1at 300 K, with E1=9900cal/molk2=4.58min−1at 500 k, with E2=27,000cal/molΔHRx1A=−55,000J/mol A UA=40,000J/min⋅K with Ta=57∘CΔHRx2B=−71,500J/mol B
Solution
The Algorithm:
0. Number Each Reaction
Reaction (1)Ak1→BReaction (2)Bk2→C
1. Mole Balance on Every Species
Species A: Combined mole balance and rate law for A
V=FA0-FA-rA=v0[CA0-CA]-r1A=v0[CA0-CA]k1CA (E12–6.1)
Solving for CA gives us
CA=CA01+τk1 (E12–6.2)
Species B: Combined mole balance and rate law for B
V=0-CBv0-rB=CBv0rB (E12–6.3)
2. Rates
The reactions follow elementary rate laws
(a)Laws(b)Relative Rates(c)Net Ratesr1A=−k1ACA≡−k1CAr1B=−r1ArA=r1Ar2B=−k2BCB≡−k2CBr2C=−r2BrB=r1B+r2B
Substituting for r1B and r2B in Equation (E12-6.3) gives
V=CBv0k1CA-k2CB (E12–6.4)
Solving for CB yields
CB=τk1CA1+τk2=τk1CA0(1+τk1)(1+τk2) (E12–6.5)
-r1A =k1CA=k1CA01+τk1(E12−6.6)
-r2B=k2CB=k2τk1CA0(1+τk1)(1+τk2)(E12−6.7)
4. Energy Balances
Applying Equation (12-41) to this system gives
[r1AΔHRxlA+r2BΔHRx2B]V-UA(T-Ta)-FA0CPA(T-T0)=0 (E12–6.8)
Substituting for FA0 = υ0CA0, r1A, and r2B and rearranging, we have
G(T)︷[-ΔHRx1Aτk11+τk1-τk1τk2ΔHRx2B(1+τk1)(1+τk2)]=R(T)︷CPA(1+K)[T-Tc](E12-6.9)
G(T)=[-ΔHRx1Aτk11+τk1-τk1τk2ΔHRx2B(1+τk1)(1+τk2)](E12-6.10)
R(T)=CPA(1+k)[T−Tc](E12-6.11)
5. Parameter Evaluation
κ=UAFA0CPA=40,000J/min⋅K(0.3mol/dm3)(1000dm3/min)200J/mol⋅K=0.667 (E12–6.12)
Tc=T0+κTa1+κ=283+(0.666)(330)1+0.667=301.8K (E12–6.13)
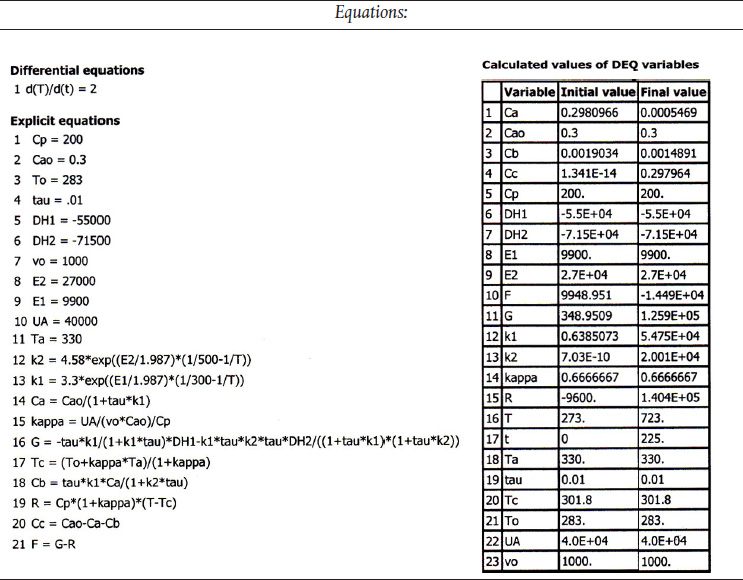
We are going to generate G(T) and R(T) by "fooling" Polymath to first generate T as a function of a dummy variable, t. We then use our plotting options to convert T(t) to G(T) and R(T). The Polymath program to plot R(T) and G(T) vs. T is shown in Table E12-6.1, and the resulting graph is shown in Figure E12-6.1 (http://www.umich.edu/~elements/5e/tutorials/Polymath_fooling_tutorial_Example-12-6.pdj).
Incrementing temperature in this manner is an easy way to generate R(T) and G(T) plots.
Table E12-6.1 Polymath: Program and Uutput
Living Example Problem
When F = 0, then G(T) = R(T) and the steady states can be found.
Wow! Five (5) multiple steady states!
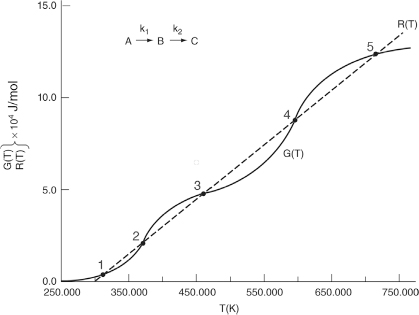
Analysis: Wow! We see that five steady states (SS) exist!! The exit concentrations and temperatures listed in Table E12-6.2 were determined from the tabular output of the Polymath program. Steady states 1, 3, and 5 are stable steady states, while 2 and 4 are unstable. The selectivity at steady state 3 is ˜SB/C=0.2650.002=132.5
Table E12-6.2 Effluent Concentrations and Temperatures
SS |
T(K) |
CA (mol/dm3) |
CB (mol/dm3) |
CC (mol/dm3) |
1 |
310 |
0285 |
0.015 |
0 |
2 |
363 |
0.189 |
0.111 |
0.0 |
3 |
449 |
0.033 |
0.265 |
0.002 |
4 |
558 |
0.0004 |
0.163 |
0.132 |
5 |
677 |
0.001 |
0.005 |
0.294 |
12.6.5 Complex Reactions in a PFR
The following complex gas-phase reactions follow elementary rate laws
(1)000A+2B→C000-r1A=klACAC2B0000ΔHRxlB=-15,000cal/mole0B
(2)0002A+3C→D000-r2C=k2CC2AC3C0000ΔHRx2A=-10,000cal/mole0A
and take place in a PFR. The feed is stoichiometric for reaction (1) in A and B with FA0 = 5 mol/min. The reactor volume is 10 dm3 and the total entering concentration is CT0 = 0.2 mol/dm3. The entering pressure is 100 atm and the entering temperature is 300 K. The coolant flow rate is 50 mol/min and the entering coolant fluid has a heat capacity of CPc0 = 10 cal/mol · K and enters at a temperature of 325 K.
Parameters
0000KlA=40(dm3mo1)2/min00at00300K00with00E1=8,000cal/mol0000K2C=2(dm3mo1)4/min00at00300K00with00E2=12,000cal/molCPA=10cal/mol/K00Ua=80ca1m3⋅min⋅KCPB=12cal/mol/K00Ta0=325KCPC=14cal/mol/K00.m=50mo1/minCPD=16cal/mol/K00CPC0=10cal/mol/K
Plot FA, FB, FC, FD, p, T, and Ta as a function of V for
(a) Co-current heat exchange
(b) Countercurrent heat exchange
(c) Constant Ta
(d) Adiabatic operation
Gas Phase PFR No Pressure Drop (p = 1)
1. Mole Balances:
(1)dFAdV=rA (FA0=50mol/min) (E12-7.1)(2)dFBdV=rB(FB0=100mol/min)(E12-7.2)(3)dFCdV=rCVf=100dm3(E12-7.3)(4)dFDdV=rD(E12-7.4)
Following the Algorithim

2. Rates:
2a. Rate Laws
(5)r1A=−k1ACAC2B(E12-7.5)(6)r2C=−k2CC2AC3C(E12-7.6)
2b. Relative Rates
(7)r1B=2r1A(E12-7.7)(8)r1C=−r1A(E12-7.8)(9)r2A=23r2C=−23k2CC2AC3C(E12-7.9)(10)r2D=−13r2C=−13k2CC2AC3C(E12-7.10)
2c. Net Rates of reaction for species A, B, C, and D are
(11)rA=r1A+r2A=-k1ACAC2B-23k2CC2AC3A(12)rB=r1B=-2klACAC2B(13)rc=r1C+r2C=klACAC2B-k2CC2AC3c(14)rD = r2D = 13 k2C C2A C3c(E12-7.11)(E12-7.12)(E12-7.13)(E12-7.14)
Major goal of CRE
3. Selectivity:
At V = 0, FD = 0 causing SC/D to go to infinity. Therefore, we set SC/D = 0 between v = 0 and a very small number, say, v = 0.0001 dm3 to prevent the ODE solver from crashing.
(15)000SC/D=if0(V>0.0001)0then0(FCFD)else(0)000000000(E12-7.15)
4. Stoichiometry:
(16)0000CA=CT0(FAFT)P(T0T)000000000000000000000000000000(E12-7.16)(17)0000CB=CT0(FBFT)P(T0T)00000000000000000000000000000(E12-7.17)
(18)0000CC=CT0(FCFT)P(T0T)000000000000000000000000000000(E12-7.18)(19)0000CD=CT0(FDFT)P(T0T)00000000000000000000000000000(E12-7.19)
(20)0000P=100000000000000000000000000000000000000000000000(E12-7.20)(21)0000FT=FA+FB+FC+FD00000000000000000000000000000(E12-7.21)
5. Parameters:
(22)0000K1A=40exp[E1R(1300−1T)](dm3/mol)2/min0000000000000000000000000(E12-7.22)(23)0000K2C=2exp[E2R(1300−1T)](dm3/mol)4/min0000000000000000000000000(E12-7.23)
(24) CA0 = 0.2mol/dm3
(25) R= 1.987cal/mol/K
(26) E1 = 8,000cal/mol
(27) E2 = 12,000cal/mol
other parameters are given in the problem statement, i.e., Equations (28) to (34) below.
(28) CpA, (29) CpB, (30) Cpc, (31) ˙mc
6. Energy Balance:
Recalling Equation (12-37)
(35)000dTdV=Qg−QrΣFjCPj00000000000000000000000(E12-7.35)
The denominator of Equation (E12-7.36) is
(36)000ΣFjCPj=FACPA+FBCPB+FCCPC+FDCPD00000000000000000000000(E12-7.36)
The “heat removed” term is
Qr=Ua(T−Ta)
The “heat generated” is
(38) Qg=ΣrijΔHRxij=r1BΔHRx1B+r2AΔHRx2A (E12-7.38)
(a) Co-current heat exchange
The heat exchange balance for co-current exchange is
(39)000dTadV=Ua(T−Ta)˙mcCpCD00000000000000000000000(E12-7.39)
Part (a) Co-current flow: Plot and analyze the molar flow rates, and the reactor and coolant temperatures as a function of reactor volume. The Polymath code and output for co-current flow are shown in Table E12-7.1.
Table E12-7.1 Polymath Program and Output for Co-current Exchange

Co-current heat exchange
Living Example Problem
The temperature and molar flow rate profiles are shown in Figure E12-7.1.
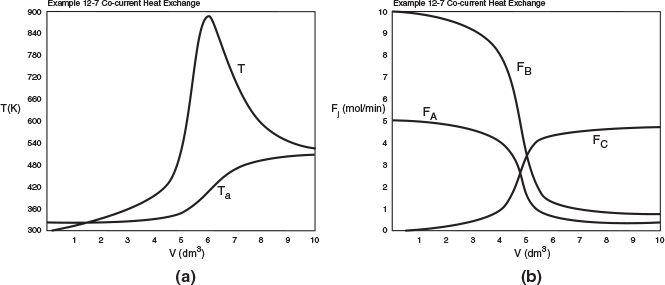
Figure E12-7.1 Profiles for co-current heat exchange; (a) temperature (b) molar flow rates. Note: The molar flow rate FD is very small and is essentially the same as the bottom axis.
Analysis: Part (a): We also note that the reactor temperature, T, increases when Qg > Qr and reaches a maximum, T = 930 K, at approximately V = 5 dm3. After that, Qr > Qg the reactor temperature decreases and approaches Ta at the end of the reactor. For co-current heat exchange, the selectivity ˜Sc/D=4.630.026=178
Part (b) Countercurrent heat exchange: We will use the same program as part (a), but will change the sign of the heat-exchange balance and guess Ta at V = 0 to be 507 K and see if the value gives us a Ta0 of 325 K at V = Vf.
dTadV=-Ua(T-Ta)˙mcCPCool
We find our guess of 507 K matches Ta0 = 325 K. Are we lucky or what?!
Table E12-7.2 Polymath Program and Output for Countercurrent Exchange
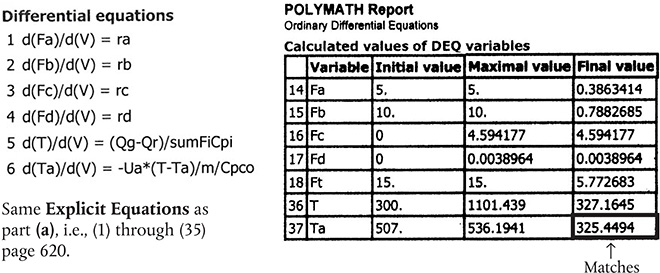
Countercurrent heat exchange
Living Example Problem

Analysis: Part (b): For countercurrent exchange, the coolant temperature reaches a maximum at V = 1.3 dm3 while the reactor temperature reaches a maximum at V = 2.7 dm3. The reactor with a countercurrent exchanger reaches a maximum reactor temperature of 1100 K, which is greater than that for the co-current exchanger, (i.e., 930 K). Consequently, if there is a concern about additional side reactions occurring at this maximum temperature of 1100 K, one should use a co-current exchanger or if possible use a large coolant flow rate to maintain constant Ta in the exchanger. In Figure 12-7.2 (a) we see that the reactor temperature approaches the coolant entrance temperature at the end of the reactor. The selectivity for the coun-tercurrent systems ˉSC/D=175,
Part (c) Constant Ta: To solve the case of constant heating-fluid temperature, we simply multiply the right-hand side of the heat-exchanger balance by zero, i.e.,
dTadV=-Ua(T-Ta).mcCP*0
and use Equations (E12-7.1) through (E12-7.39).
Table E12-7.3 Polymath Program and Output for Constant Ta

Constant Ta

Analysis: Part (c): For constant Ta, the maximum reactor temperature, 870K, is less than either co-current or countercurrent exchange while the selectivity, ˉSC/D=252.9
Part (d) Adiabatic: To solve for the adiabatic case, we simply multiply the overall heat transfer coefficient by zero.
Ua=80*0
Table E12-7.4 Polymath Program and Output for Adiabatic Operation

Living Example Problem
Adiabatic operation

Analysis: Part (d): For the adiabatic case, the maximum temperature, which is the exit temperature, is higher than the other three exchange systems, and the selectivity is the lowest. At this high temperature, the occurrence of unwanted side reactions certainly is a concern.
Overall Analysis Parts (a) to (d): Suppose the maximum temperature in each of these cases is outside the safety limit of 750 K for this system. Problem P12-2A (i) asks how you can keep the maximum temperature below 750 K.
12.7 Radial and Axial Variations in a Tubular Reactor
COMSOL application

In the previous sections, we have assumed that there were no radial variations in velocity, concentration, temperature, or reaction rate in the tubular and packed-bed reactors. As a result, the axial profiles could be determined using an ordinary differential equation (ODE) solver. In this section, we will consider the case where we have both axial and radial variations in the system variables, in which case we will require a partial differential (PDE) solver. A PDE solver such as COMSOL will allow us to solve tubular reactor problems for both the axial and radial profiles, as shown in the Radial Effects COMSOL Web module on the CRE Web site (http://www.umich.edu/~elements/5e/web_mod/radialeffects/index.htm.7 In addition, a number of COMSOL examples associated with and developed for this book are available from the COMSOL Web site (www.comsol.com/ecre).
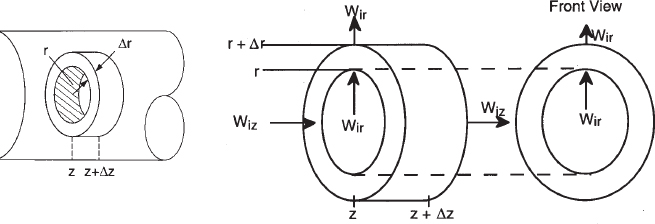
We are going to carry out differential mole and energy balances on the differential cylindrical annulus shown in Figure 12-15.
7 An introductory webinar on COMSOL can be found on the AIChE webinar Web site: http://www.aiche.org/resources/chemeondemand/webinars/modeling-non-ideal-reactors-and-mixers.
12.7.1 Molar Flux
In order to derive the governing equations, we need to define a couple of terms. The first is the molar flux of species i, Wi (mol/m2 • s). The molar flux has two components, the radial component, Wir, and the axial component, Wiz. The molar flow rates are just the product of the molar fluxes and the cross-sectional areas normal to their direction of flow Acz. For example, for species i flowing in the axial (i.e., z) direction
Fiz = Wiz Acz
where Wiz is the molar flux in the axial direction (mol/m2/s), and Acz (m2) is the cross-sectional area of the tubular reactor.
In PDF Chapter 14 on the Web site (http://www.umich.edu/~elements/5e/14chap/Fogler_Web_Ch14.pdf we discuss the molar fluxes in some detail, but for now let’s just say they consist of a diffusional component, -De(∂Ci/∂z), and a convective-flow component, UzCi, so that the flux in the axial direction is
Wiz=-De∂Ci∂z+UZCi00000000000000000(12-42)
where De is the effective diffusivity (or dispersion coefficient) (m2/s), and Uz is the axial molar average velocity (m/s). Similarly, the flux in the radial direction, r, is
Wir=-De∂Ci∂r+UrCi00000000000000000(12-43)
Radial direction
where Ur (m/s) is the average velocity in the radial direction. For now, we will neglect the velocity in the radial direction, i.e., Ur = 0. A mole balance on species A (i.e., i = A) in a cylindrical system volume of length Δz and thickness Δr as shown in Figure 12-15 gives
Mole Balances on Species A
(Moles0of0Ain0at0r)0=0WAr⋅(Cross-sectional0areanormal0to0radial0flux)=WAr⋅2πrΔz(Moles0of0Ain0at0z)0=0WAz⋅(Cross-sectional0areanormal0to0axial0flux)=WAz⋅2πrΔr(Moles0of0Ain0at0r)−(Moles0of0Aout0at0(r+Δr))+(Moles0of0Ain0at0z)−(Moles0of0Aout0at0(z+Δz))+(Moles0of0Aformed)=(Moles0of0Aaccumulated)
WAr2πrΔz|r-WArAπrΔz|r+Δr+WAz2πrΔr|Z-WAz2πrΔr|z+Δz+rA2πrΔrΔz=∂CA(2πrΔrΔz)∂t
Dividing by 2πrΔrΔz and taking the limit as Δr and Δz → 0
-1r∂(rWAr)∂r-∂WAz∂z+rA=∂CA∂t
Similarly, for any species i and steady-state conditions
Using Equations (12-42) and (12-43) to substitute for Wiz and Wir in Equation (12-44) and then setting the radial velocity to zero, Ur = 0, we obtain
-1r∂∂r[(-De∂Ci∂rr)](-∂∂z[-De∂Ci∂z+UzCi])+ri0=00
For steady-state conditions and assuming Uz does not vary in the axial direction
This equation will also be discussed further in PDF Chapter 17 on the Web site.
12.7.2 Energy Flux
When we applied the first law of thermodynamics to a reactor to relate either temperature and conversion or molar flow rates and concentration, we arrived at Equation (11-10). Neglecting the work term we have for steady-state conditions
In terms of the molar fluxes and the cross-sectional area, and (q = Q/Ac )
Ac[q + (ΣWi0Hi0 - ΣWiHi)] = 0 (12-47)
The q term is the heat added to the system and almost always includes a conduction component of some form. We now define an energy flux vector, e, (J/m2 · s), to include both the conduction and convection of energy.
e = energy flux J/s·m2
e = Conduction + Convection
where the conduction term q (kJ/m2 · s) is given by Fourier’s law. For axial and radial conduction, Fourier’s laws are
qZ0=-ke∂T∂z0000and0000qr0=-ke∂T∂r
where ke is the thermal conductivity (J/m·s·K). The energy transfer (flow) is the flux vector times the cross-sectional area, Ac, normal to the energy flux
Energy flow = e · Ac