1.2 Biological Networks
A network is a graph G(V, E) defined in terms of a set of vertices V and a set of edges E. In case of biological networks, a vertex ![]() is either a gene or protein encoded by an organism, and an edge e E joining two vertices
is either a gene or protein encoded by an organism, and an edge e E joining two vertices ![]() in the network represents biological properties connecting
in the network represents biological properties connecting ![]() and
and ![]() . A biological network can be directed or undirected depending on the biological relationship that used to join the pairs of vertices in the network. Both directed and undirected networks occur naturally in biological systems. Inference of these networks is a major challenge in systems biology. We briefly review two kinds of biological networks in the following sections.
. A biological network can be directed or undirected depending on the biological relationship that used to join the pairs of vertices in the network. Both directed and undirected networks occur naturally in biological systems. Inference of these networks is a major challenge in systems biology. We briefly review two kinds of biological networks in the following sections.
1.2.1 Directed Networks
In directed networks, each edge is identified as an ordered pair of vertices. According to the Central Dogma of Molecular Biology, genetic information is encoded in double-stranded DNA. The information stored in DNA is transferred to single-stranded messenger RNA (mRNA) to direct protein synthesis [42]. Signal transduction is the primary mean to control the passage of biological information from DNA to mRNA with mRNA directing the synthesis of proteins. A signal transduction event is usually triggered by the binding of external ligands (e.g., cytokine and chemokine) to the transmembrane receptors. This binding results in a sequential activation of signal molecules, such as cytoplasmic protein kinase and nuclear transcription factors (TFs), to lead to a biological end-point function [42]. A signaling pathway is composed of a web of gene regulatory wiring in response to different extracellular stimulus. Thus, signaling pathways can be viewed as directed networks containing all genes (or proteins) of an organism as vertices. A directed edge represents the flow of information from one gene to another gene.
1.2.2 Undirected Networks
Undirected networks differ from directed networks in that the edges in such networks are undirected. In other words, an undirected network can be viewed as a directed network by considering an undirected pair of vertices ![]() as two directed pairs (
as two directed pairs (![]() ) and (
) and (![]() ). Some biological networks are better suited for an undirected representation. Protein–protein interaction (PPI) network is an undirected network, where each protein is considered as a vertex and the physical interaction between a pair of proteins is represented as an edge [43].
). Some biological networks are better suited for an undirected representation. Protein–protein interaction (PPI) network is an undirected network, where each protein is considered as a vertex and the physical interaction between a pair of proteins is represented as an edge [43].
The past decade has witnessed a significant progress in the computational inference of biological networks. A variety of approaches in the form of network models and novel algorithms have been proposed to understand the structure of biological networks at both global and local level. While the grand challenge in a global approach is to provide an integrated view of the underlying biomolecular interaction mechanisms, a local approach focuses on identifying fundamental domains representing functional units of a biological network.
Both directed and undirected network models have been developed to reliably infer the biomolecular activities at a global level. As discussed above, directed networks represent an abstraction of gene regulatory mechanisms, while the physical interactions of genes are suitably modeled as undirected networks. Focus has also been on the computational inference of biomolecular activities by accommodating genome-wide data in diverse formats. In particular, gene set based approaches have gained attention in recent bioinformatics analysis [44, 45]. Availability of a wide range of experimental and computational methods have identified coherent gene set compendiums [46]. Sophisticated tools now exist to statistically verify the biological significance of a particular gene set of interest [46–48]. An emerging trend in this field is to reconstruct signaling pathways by inferring the order of genes in gene sets [19, 20]. There are several unique features associated with gene set based network inference approaches. In particular, such approaches do not rely on gene expression data for the reconstruction of underlying network.
The algorithms to understand biomolecular activities at the level of subnetworks have evolved over time. Community detection algorithms, in particular, originated with hierarchical partitioning algorithms that include the Girvan–Newman algorithm. Since these algorithms tend to produce a dendrogram as their final result, it is necessary to be able to rank the different partitions represented by the dendrogram. Modularity was introduced by Newman and Girvan to address this issue. Many methods have resulted with modularity at the core. More recently, though, it has been shown that modularity suffers from some drawbacks. While there have been some attempts to address these issues, newer methods continued to emerge such as Infomap. Research has also expanded to incorporate different types of biological networks and communities. Initially, only undirected and unweighted networks were the focus of study. Methods are now capable of dealing with both directed and weighted networks. Moreover, previous studies only concentrated on distinct communities that did not allow overlap. With the advent of the clique percolation method and other similar methods, overlapping communities are becoming increasingly popular. The aforementioned approaches have been used to identify the structural organization of a variety of biological networks including metabolic networks, PPI networks, and protein domain networks. Such networks have a power–law degree distribution and the quantitative signature of scale-free networks [49]. PPI networks, in particular, have been the subject of intense study in both bioinformatics and biology as protein interactions are fundamental for cellular processes [50].
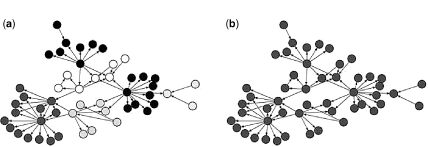
A common problem associated with the computational inference of a biological network is to assess the performance of the approach used in the inference procedure. It is quite assess as the structure of the true underlying biological network is unknown. As a result, one relies on biologically plausible simulated networks and data generated from such networks. A variety of in silico benchmark directed and undirected networks are provided by the dialogue for reverse engineering assessments and methods (DREAM) initiative to systematically evaluate the performance of reverse engineering methods, for example Refs. [37–41]. Figures 1.2 and 1.7 illustrate gold standard directed network, undirected network, and a network with community structure from the in silico network challenges in DREAM initiative.
Figure 1.2 (a) Example of a directed network. The figure shows Escherichia coli gold standard network from the DREAM3 Network Challenges [37–39]. (b) Example of an undirected network. The figure shows an in silico gold standard network from the DREAM2 Network Challenges [40, 41].
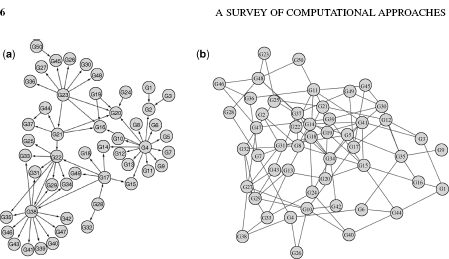
Figure 1.7 The E. coli network from the DREAM Initiative [39]. (a) The E. coli network is partitioned into six communities by ignoring edge direction. (b) The same E. coli network does not divide into any communities when edge direction is used. The disparity between the results is a strong indicator of the significance of edge direction. In both cases the appropriate version of Infomap was run for 100,000 iterations with a seed number of 1.