
Chapter 2. Large-Scale Chemical Informatics on GPUs
Imran S. Haque and Vijay S. Pande
In this chapter we present the design and optimization of GPU implementations of two popular chemical similarity techniques: Gaussian shape overlay (GSO) and LINGO. GSO involves a data-parallel, arithmetically intensive iterative numerical optimization; we use it to examine issues of thread parallelism, arithmetic optimization, and CPU-GPU transfer overhead minimization. LINGO is a string similarity algorithm that, in its canonical CPU implementation, is bandwidth intensive and branch heavy, with limited data parallelism. We present an algorithmic redesign allowing GPU implementation of such a low arithmetic-intensity kernel and discuss techniques for memory optimization that enable large speedup. Source code for the programs described here is available online: PAPER (for Gaussian shape overlay) can be downloaded at
https://simtk.org/home/paper
under the GPL, and single-instruction, multiple LINGO (SIML) (for LINGO) at
https://simtk.org/home/siml
under a BSD license.
2.1. Introduction, Problem Statement, and Context
Chemical informatics uses computational methods to analyze chemical datasets for applications that include search and classification of known chemicals, virtually screening digital libraries of chemicals to find ones that may be active as potential drugs and predicting and optimizing the properties of existing active compounds. A common computational kernel in cheminformatics is the evaluation of a similarity (using various models of similarity) between a pair of chemicals. Such similarity algorithms are important tools in both academia and industry.
A significant trend in chemical informatics is the increasing size of chemical databases. Public databases listing known chemical matter exceed 30 million molecules in size, the largest exhaustive libraries (listing all possible compounds under certain constraints) are near 1 billion molecules, and virtual combinatorial libraries in use in industry can easily reach the trillions of compounds. Unfortunately, similarity evaluations are often slow, at or below 1000 evaluations/sec on a CPU. Adding to the problem, typical analyses in this field (such as clustering) must execute a number of similarity evaluations that are superlinear in the size of the database. The combination of rapidly growing chemical data-sets and computationally expensive algorithms creates a need for new techniques.
The massive data and task parallelism present in large-scale chemical problems makes GPU reimplementation an attractive acceleration method. In this chapter, we demonstrate 10-100× speedup in large-scale chemical similarity calculations, which allows the analysis of dramatically larger datasets than previously possible. Search problems that formerly would have required use of a cluster can now be efficiently performed on a single machine; alternatively, formerly supercomputer-scale problems can be run on a small number of GPUs. In particular, we show that two commonly used algorithms can be effectively parallelized on the GPU: Gaussian shape overlay (GSO)
[5]
, a three-dimensional shape comparison technique, and LINGO
[6]
, a string comparison method.
2.1.1. Gaussian Shape Overlay: Background
Gaussian shape overlay is an algorithm that measures the similarity of two molecules by calculating the similarity of their shapes in three-dimensional (3-D) space. In this method, a molecule is represented as a scalar field or function in 3-D space, where the value of the function at each point in space indicates whether the point is “inside” the molecule. Given a set of functions
ρ
Ai
(r
) that represent the density functions for each atom of a molecule (i.e., functions that are 1 inside the volume of an atom and 0 outside), the function for an entire molecule can be defined using one of the formulae in
Eq. 2.1
. The first computes interior points by taking the product of the complement of all atoms — defining an “exterior point” as one which is not inside any atom. The latter method uses the principle of inclusion-exclusion between sets to compute the same union of all atoms.
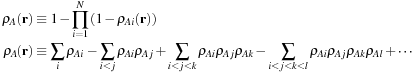
(2.1)
Equation 2.1
is motivated by considering each atom to be a set of points, and constructing the union of these sets. Defining atomic densities as indicator functions (one inside a given radius around a point and zero outside) generates the “hard-sphere” model of molecular shape. This model has several shortcomings (including nondifferentiability) that makes it difficult to use in computations. Consequently, Grant and Pickup developed the Gaussian model of molecular shape
[2]
, in which each atom's density function is defined not as a hard sphere, but as an isotropic spherical Gaussian function:

(2.2)
Such Gaussian functions are smooth and differentiable. Furthermore, simple closed-form expressions for the volumes, volume gradients (with respect to position), and Hessians of the product of an arbitrary number of such Gaussians are known
[2]
. This enables the calculation of the similarity of two molecules by calculating the maximum overlap possible between their shapes, maximized over all rigid-body transformations (translations and rotations). Mathematically, GSO seeks to maximize
Eq. 2.3
over all rigid body transformations. In this integral, the functions
ρA
and
ρB
are the density fields for each molecule, and the integral is taken over all space.
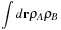
(2.3)
The computation is performed by numerical optimization to orient a pair of molecules in their optimally overlapping poses and then compute the overlap volume (Figure 2.1
)
[3]
. The objective function in such a calculation is typically a truncated form of
Eq. 2.1
, including only the single-overlap terms (see
Eq. 2.4
). Thus, both the objective and gradient calculations are arithmetically intensive and data parallel, involving a double loop over the atoms of each molecule and an exponential evaluation inside the loop body. These aspects make GPUs an attractive platform to implement GSO. In the first half of this chapter, we describe PAPER, our open-source implementation of GSO on NVIDIA GPUs
[5]
.
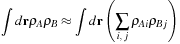
(2.4)
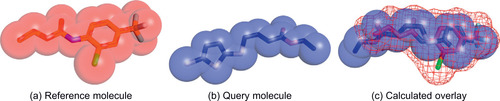 |
| Figure 2.1
A 3-D shape overlay of molecules. The reference and query molecules are depicted as sticks (to visualize bond structure) embedded within their space-filling representation. GSO rotates the query molecule to maximize its volume overlap (the spheres shown in b and c) with the volume of the reference (the mesh area shown in c).
|
2.1.2. LINGO: Background
Whereas GSO represents a molecule by its three-dimensional shape in space, the LINGO algorithm of Vidal, Thormann, and Pons takes a much simpler, text-based approach
[8]
. LINGO is a 2-D similarity method: one that operates on molecules as graphs (with vertices representing atoms and edges representing atomic bonds), ignoring their three-dimensional shape. Instead of operating directly on this molecular graph, LINGO processes a linear representation of the graph called a SMILES string, which is constructed by a depth-first traversal of the graph. The characters in the SMILES string represent various graph features, such as atom types, bond orders, ring openings and closings, and branching. Given a SMILES string, LINGO represents a molecule as the set of all overlapping
q
-character substrings in the SMILES (known as “Lingos,” as opposed to “LINGO” for the algorithm as a whole).
q
is typically set to 4 (i.e., Lingos have length 4), as this has been demonstrated to have superior performance in several applications.
Figure 2.2
presents an example of a molecule and its SMILES representation, and its LINGO substrings for
q
= 4.
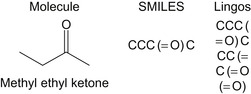 |
| Figure 2.2
Chemical graph structure of a common solvent, its SMILES representation, and its constituent Lingos.
|
The similarity between a pair of molecules A and B is defined by the following equation, where
N
x, i
represents the number of Lingos of type
i
in molecule
x
:
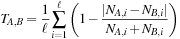
(2.5)
The canonical high-performance CPU implementation to rapidly evaluate this similarity on a CPU was presented by Grant
et al.
[4]
. This algorithm is optimized for the case in which many SMILES strings must all be compared against one query. The Lingos of the query string are inserted into a trie, a tree data structure allowing fast prefix search of strings. This trie is then converted into a deterministic finite state automaton (DFA)
[1]
; successive database strings can then be efficiently processed through this DFA. This algorithm suffers from a large amount of branching and poor memory locality in the simulation of the DFA, and it generally has poor data parallelism within each LINGO calculation. It is thus relatively unattractive for GPU implementation. In the latter half of this chapter, we discuss the algorithmic transformations and memory optimizations that enable the high-speed GPU implementation in SIML, our open-source package to calculate LINGO similarities on GPUs
[6]
.
2.2. Core Methods
Large-scale chemical informatics calculations involve the calculation of many similarities at the same time, and so they have a large amount of task parallelism; we exploit this structure in both problems by calculating a large number of similarities at a time. The GSO problem in particular is arithmetic bound: its inner loop involves the calculation of
O
(MN
) exponential functions (where
M
and
N
are the number of atoms in the molecules being compared), and must be executed many times in a numerical optimization scheme (Eq. 2.4
). We make use of SIMD data parallelism and hardware evaluation of exponentials to maximize the arithmetic throughput of GSO on the GPU. LINGO, as implemented for high performance on the CPU, uses a deterministic finite-state automaton algorithm that exhibits large branch penalties and poor memory access locality on the GPU. We describe an algorithmic redesign for LINGO that minimizes branch divergence and makes special use of GPU hardware (texture caching) to maximize memory throughput.
2.3. Gaussian Shape Overlay: Parallelization and Arithmetic Optimization
The GSO calculation is a numerical maximization of
Eq. 2.4
over a seven-dimensional space (three translational coordinates and a four-dimensional quaternion parameterization of a rotation). In this section, we describe the design and optimization of PAPER, our GPU implementation of GSO
[5]
.
PAPER uses the BFGS algorithm
[7]
, a “pseudo-second-order” method that uses evaluations of the objective function and its first derivative (gradient), but no second-derivative evaluations. Because BFGS is a local optimizer and GSO is a global optimization problem, we start each optimization from several initial points. The key computational steps in this calculation are
1. repeatedly evaluate objective at test points along a search direction to find a “sufficiently” improved point (line search);
2. evaluate gradient at new point from line search;
3. update BFGS approximation to inverse Hessian matrix and use this to calculate new search direction.
The primary consideration in software architecture for the GPU is partition of work: what work should be done on the CPU vs. the GPU, how work should be partitioned among independent GPU cores (CUDA thread blocks or OpenCL work groups), and how work can be partitioned among threads or vector lanes in each core. PAPER makes use of the extensive task parallelism in chemical informatics to allocate work to GPU cores. Our target applications focus on searches with hundreds to thousands of comparisons to be performed. Because for each calculation we optimize from several (typically four) starting points, GSO yields a large number (# starting points ×# molecules) of independent problems, each of which can be assigned to an independent core (CUDA thread block or OpenCL work-group). Partitioning work among GPU threads and between the CPU and GPU is more involved and is the focus of this section.
2.3.1. Evaluation of the Data-Parallel Objective Function
Equation 2.4
decribes the objective function that must be implemented in GSO. It is inherently data parallel, containing a double loop over the
M
atoms of the reference molecule and
N
atoms of the query molecule with an arithmetically intensive loop body. This section describes the parallelization of the objective calculation (which has the same structure as the gradient) and presents several versions illustrating consecutive optimizations.
PAPER uses blocks of 64 threads to maximize the number of registers available to each thread. While it can be advantageous in some cases to use larger thread blocks to hide memory latency, in our application we typically have multiple thread blocks available on each multiprocessor. Since scheduling is done on a per-warp basis, using many smaller thread blocks is sufficient. In typical use cases, the number of terms to be calculated in the objective is larger than the number of available threads: typical molecules are 20-40 atoms, so that normal calculations will have 400-1600 terms. To parallelize the problem, we assign threads to calculate thread-block size “strips” of the interaction matrix consecutively, until the entire set of interactions has been evaluated. Instead of using atomic operations to accumulate the volume among threads, the function keeps an array in shared memory and does a parallel reduction at the end to sum the final overlap volume.
Figure 2.3
illustrates the order of the computation, and
Listing 2.1
provides code for a simple version of the objective evaluation.
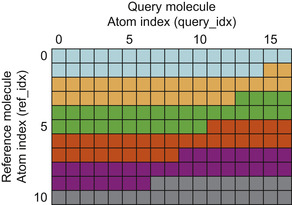 |
| Figure 2.3
Thread parallelization scheme in PAPER. Depicted is the full matrix of interactions between a reference molecule of 11 atoms and a query molecule of 17 atoms. Thread blocks of 32 threads process consecutive 32-element strips of the matrix. Each shade represents one iteration of a thread block. Note that no more than a thread block-sized strip of matrix elements must be materialized at any time during the computation.
|
We perform an addressing calculation (lines 12-13) at each loop iteration so that each thread evaluates the correct matrix element. Another strategy would be to disallow thread blocks to span rows of the interaction matrix; however, this has the potential to leave many threads idle, as there are usually more threads than the width of the matrix. Another option would be the use of a two-dimensional thread block. However, because the number of atoms varies by molecule in a batch, and because the number
of atoms is not likely to be a multiple of the GPU warp size, this also is likely to leave the GPU partially idle. Using the given loop structure ensures that all threads do useful work as long as there are terms left to compute.
While the objective function in
Listing 2.1
is effectively parallelized across all GPU threads, several changes can be made to improve its performance. The first point of concern is the addressing calculation. Because the number of atoms in either molecule is unlikely to be a power of two, it is necessary to use a division to calculate the row index for each thread. However, integer division is a very expensive operation on current GPUs; when performed in the inner loop, as in the original objective, it adds significant overhead. It is possible to restructure the addressing such that the division is only performed once (Listing 2.2
; non-addressing computations have been elided). Restructuring the calculation in this way reduces the in-loop addressing overhead to two adds, a comparison, and two conditional adds, which are much cheaper than the integer division. In the full PAPER implementation, the objective is called multiple times per kernel invocation in the course of a line search; thus,
row_per_block
and
col_per_block
are precalculated at the start of the kernel invocation and stored in shared memory to amortize the cost of the division. Implementing this change to the objective and gradient functions leads to a measured 13% speedup in total optimization time.
With the integer division removed, the runtime becomes dominated by the evaluation of
Kij
and
Vij
at lines 18-19 in
Listing 2.1
, which involves two divides and two transcendental function evaluations — relatively expensive operations. The simplest optimization to apply here is the use of CUDA intrinsic functions for the division and transcendentals. CUDA intrinsics are low-level hardware operations that are often much faster than their library versions, but at the cost of accuracy. In the GSO calculation, experimentation showed that the reduced accuracy of intrinsic functions is not a problem.
Listing 2.3
shows the use of the
__expf
,
__powf
, and
__fdividef
intrinsics to speed up the slow operations in the objective core. Adding these three intrinsics more than doubles GSO performance with respect to the previous version (Listing 2.2
).

However, careful attention to instruction performance shows that this instruction stream can be further improved. In particular,
__powf
is expensive to evaluate, and it can be replaced in this case by cheaper CUDA intrinsics: reciprocal and reciprocal square root. As is often the case, this optimization produces results that are not numerically identical to the original; however, regression testing showed that the accuracy is sufficient for GSO. The final computational core is provided as
Listing 2.4
and is 10% faster than
Listing 2.3
.
Table 2.1
illustrates the performance gains from various tuning strategies on the overlap and gradient kernels, as measured by their effect on the overall program runtime (not just the overlap or gradient evaluation).
2.3.2. Kernel Fusion and CPU/GPU Balancing
It is common in multistage calculations such as GSO to have one or more steps that are not efficiently parallelized on the GPU. In the case of GSO, while the line search/objective evaluation and the gradient evaluation are very efficiently executed on the GPU (because of high data parallelism and arithmetic intensity), the BFGS direction update is not well parallelized. In PAPER, the BFGS update requires a large number of sequential low-dimensional (7-D) vector operations and small (7 × 7) matrix operations. These operations create a large amount of thread synchronization and many idle threads; it is thus not attractive to compute them on the GPU. However, moving them to the CPU also imposes a cost. In the case of the BFGS update, the coordinates, gradients, and objective values must be retrieved from the GPU to do the update, and the new direction uploaded to the GPU after the CPU has calculated the update.

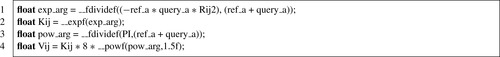 |
| Listing 2.3. |
| GSO core computation with CUDA intrinsics.
|
 |
| Listing 2.4. |
| GSO core computation with restructured CUDA intrinsics.
|
Using the CUDA Visual Profiler, it is possible to easily measure the overhead of the two strategies.
Table 2.2
shows the timings for various execution stages of PAPER on a 2000-molecule test set.
Measurements examine two versions of PAPER: one in which the gradient kernel only calculates the gradient, with BFGS updates on the host, and one with a “fused” gradient kernel, which both calculates the gradient and does the (poorly parallelized) BFGS update on the GPU. In both versions, a small amount of data is copied from the GPU to the CPU on each iteration to check for convergence.
The results in
Table 2.2
demonstrate that on this problem, it is extremely advantageous to keep some poorly parallelized work on the GPU. While the BFGS update takes a significant amount of GPU time (over 12% of the total kernel time, despite having much less arithmetic work than the gradient itself), it is much cheaper than moving the necessary data back to the CPU. The results also show that fusing the line search and gradient+BFGS kernels is unlikely to lead to significant gains: the 10 μs data-transfer overhead in copying completion flags is dominated by the total kernel execution time. This was borne out in testing: a single-kernel version (with all operations in the same kernel call) had essentially identical performance to the two-kernel version.
2.4. LINGO: Algorithmic Transformation and Memory Optimization
Unlike GSO, which is an arithmetic-bound computation with extensive internal data parallelism, LINGO has few arithmetic operations per memory access and has poor data parallelism. Furthermore, while the GSO algorithm for the GPU is essentially a parallelized version of the CPU GSO algorithm (BFGS), the canonical CPU algorithm for high-performance LINGO is ill suited to the GPU. This algorithm
[4]
compiles reference strings into a deterministic finite state automaton, which is simulated for each query string. To implement the DFA state transitions requires either significant amounts of branching, or a moderately sized, randomly accessible lookup table (LUT); neither option is good for
GPUs. Branch-heavy code will pay a significant penalty in warp divergence. Worse, none of the memory spaces in the CUDA memory model are well suited to implement a high-performance LUT of the type required: global memory requires aligned, coherent access; texture memory requires spatial locality; constant memory requires that all threads in a warp access the same element for high performance; and shared memory is limited in size. Global, texture, and constant memory are inappropriate for a random-access LUT because different threads are unlikely to use coherent accesses; shared memory is small and may not be able to hold the LUT while maintaining reasonable multiprocessor occupancy (necessary to hide the latency of streaming database Lingos in from global memory). In this section, we discuss the design and optimization of SIML, our algorithm to calculate LINGOs efficiently on the GPU
[6]
.
Consequently, an algorithmic transformation is necessary to implement LINGO efficiently on the GPU. The standard LINGO equation (Eq. 2.5
) can be recast into a different form:
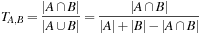
(2.6)
In this equation, each molecule (A or B) is represented as a multiset, a generalization of a set in which each element can have cardinality greater than one; the multiset for a molecule contains its Lingos and their counts. The cardinality of each element in a multiset intersection is the minimum of its counts in either set (or maximum, for a multiset union). SIML thus represents each molecule as a pair of integer vectors: one containing the Lingos in sorted order, and one with corresponding counts. Here, the optimality of
q
= 4 is fortuitous, as it allows us to trivially pack a 4-character Lingo into a 32-bit integer (each character is 8 bits). SIML also precalculates the cardinality of each set independently; thus, the only quantity needed to calculate the similarity between two SMILES strings is the size of their multiset intersection. This can be efficiently calculated using the algorithm in the following listing, similar to merging sorted lists (Listing 2.5
).
While the simple structure of the algorithm makes it attractive on the GPU, it is not optimal for the CPU.
Table 2.3
compares the performance of the SIML multiset algorithm on the CPU against a DFA-based LINGO implementation. The DFA method has nearly twice the throughput of the multiset method. This reflects a common theme in GPU programming — algorithms optimal for the GPU may in fact represent de-optimization for the CPU.
2.4.1. SIML GPU Implementation and Memory Tuning
Because of the poor data parallelism in any single LINGO similarity calculation, we use an individual CUDA thread per similarity; each thread in a block calculates the similarity of its query molecule against a common reference molecule for the entire block. While this approach makes good use of task parallelism in large-scale LINGO calculations, simply running
Listing 2.5
per-thread on the GPU results in very poor performance. The first optimization is to load the block's reference molecule into shared memory, rather than global memory. Since all threads access the same reference molecule, this significantly reduces global memory traffic. However, this is sufficient only to reach approximate performance parity with the CPU.
The key to performance in the SIML kernel is memory layout. If multisets were laid out in “molecule-major” order (all elements for a single molecule stored contiguously, followed by the next molecule, etc.), as would be appropriate for the CPU implementation of SIML, consecutive GPU threads would read from global memory with a stride equal to the length of the largest multiset (Figure 2.4a
). This stride is typically larger than the global memory request size, so a separate read transaction must be dispatched for every thread. This becomes the critical bottleneck: with molecule-major multiset layout, the CUDA Visual Profiler indicates that the kernel's arithmetic throughput is only 15% of peak on a GeForce GTS 250.
 |
| Figure 2.4
“Molecule-major” and “Lingo-major” layouts for storing the Lingos of multiple molecules in memory. Dark gray squares indicate the memory addresses read by consecutive threads. “MX LY” indicates the Yth Lingo of the Xth molecule.
|

Transposing the multiset layout, such that all multisets’ first elements are contiguous, followed by the second elements, and so on, nearly solves the problem (Figure 2.4b
). However, each thread maintains its own index into its query multiset (a row index in the “Lingo-major” multiset matrix); if these indices differ, then memory access will be uncoalesced. One option is to have a shared row index among all threads: each thread increments its query multiset pointer as far as possible, and then waits at a barrier (__syncthreads
) before the block moves on to the next multiset row. While this solution ensures that all global loads are coalesced, it has a relatively high overhead in threads that must wait idle at the barrier; it is able to achieve 80% of peak arithmetic throughput. The best option
on GPU hardware is to associate a two-dimensional texture with the multiset matrix and use texture loads instead of uncached global loads. Because the texture cache is optimized for spatial locality, it is able to absorb the overhead of the misalignment in row indices between threads. The SIML kernel using a 2-D texture for global memory access is able to achieve 100% of peak single-issue arithmetic throughput (as measured by CUDA Visual Profiler), demonstrating that careful optimization of memory layout and access method can turn a problem traditionally considered to be memory bound into one that is arithmetic bound. An implementation of this transposed, textured kernel is provided as
Listing 2.6
.
2.5. Final Evaluation
Figure 2.5
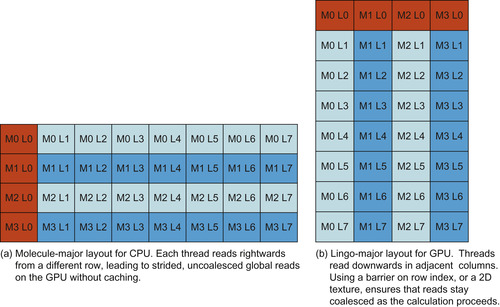
compares the performance of the tuned PAPER implementation against OpenEye ROCS, a commercial implementation of Gaussian shape overlay. ROCS supports various “modes,” which represent different approximations to the GSO objective function. The objective implemented in PAPER is equivalent to the “Exact” mode in ROCS. ROCS performance was measured on one core of an Intel Core i7-920; PAPER was run on an NVIDIA GeForce GTX 480. Because the complexity of the GSO kernel varies by the size of the molecules being compared, we present speedup plots for small (10 atoms), medium (22 atoms), and large (44 atoms) molecules. The chosen “medium” size corresponds to the average heavy atom (non-hydrogen) count in a popular chemical screening library.
 |
| Figure 2.5
PAPER performance versus OpenEye ROCS.
|

Two trends are immediately obvious from the graph. First, PAPER requires a large number of molecules to be optimized at the same time for effective speedup. This is typical for GPU algorithms, especially those relying on task parallelism. Because PAPER dispatches only one optimization per thread block, and the GPU can run multiple thread blocks per GPU core (streaming multiprocessor, or SM, in NVIDIA terminology), it is necessary to dispatch many optimizations before the GPU is fully loaded. Performance continues to improve past this lower bound (present around 25 optimizations in the plot) because CPU-GPU copy overhead and kernel dispatch latency can be more effectively amortized with larger batch sizes. The second trend is that the GPU is less effective for very small molecules, which achieve only slightly more than 10× speedup, rather than the 90 − 100× possible on larger molecules. The number of interaction terms is very small for such molecules, so that kernel setup, kernel dispatch time, and idle threads come to dominate performance. Ultimately, however, PAPER is able to demonstrate two orders of magnitude speedup on problem sizes typical in our application domain.
Figure 2.6
illustrates the performance of SIML on three generations of NVIDIA GPU, compared with the performance of a DFA-based LINGO implementation (contributed by NextMove Software) running on one core of an Intel Core i7-920. The benchmark problem for both datasets was the computation of an all-vs.-all similarity matrix on 32,768 molecules. As shown in
Table 2.3
, the multiset-based algorithm runs at about half the speed of the DFA algorithm on a CPU. However, the multiset algorithm performs very well on a GPU. SIML achieves over 11× greater throughput than the CPU DFA LINGO implementation on a G92-architecture GeForce GTS 250 and over 23× higher throughput on a GF100-based GeForce GTX 480.
 |
| Figure 2.6
SIML performance versus DFA-based LINGOs.
|
We are investigating possible optimizations to both PAPER and SIML. In the PAPER objective/gradient computational core (Listing 2.4
), a significant amount of time is spent calculating functions of the reference and query radii that are invariant over the course of the optimization. In particular, the reciprocal and reciprocal square root functions together are as expensive as the following exponential evaluation. One possible option is to precalculate the relevant functions of the radii (ref_a * query_a * inv
and
8 * PIRTPI * rsq * inv
) and store them in lookup tables in shared memory. This approach has the potential to significantly reduce the number of operations in the core computation, but at the cost of higher memory usage.
The SIML kernel is extremely sensitive to the design of the memory subsystem of the underlying hardware. The version presented has been optimized for the G80/G92 and GT200 NVIDIA architectures, for which texture reads are the only cached reads from global memory. However, the recent GF100 (Fermi) architecture features, in addition to the texture cache, L1 and L2 caches for global memory. It is possible that tuning access methods (such as using non-textured global memory reads) or block sizes (to better fit cache sizes) may significantly affect performance. In general, because LINGO is a memory-sensitive kernel, investigating cache tuning beyond the simple texturing done here is an interesting avenue for future work.
Acknowledgments
We thank Roger Sayle of NextMove Software for his contribution of a DFA-based LINGO implementation for benchmarking and comparison.
[1]
A.V. Aho, M.J. Corasick,
Efficient string matching: an aid to bibliographic search
,
Commun. ACM
18
(6
) (1975
)
333
–
340
.
[2]
J.A. Grant, B.T. Pickup,
A Gaussian description of molecular shape
,
J. Phys. Chem.
99
(1995
)
3449
.
[3]
J.A. Grant, M.A. Gallardo, B.T. Pickup,
A fast method of molecular shape comparison: a simple application of a Gaussian description of molecular shape
,
J. Comput. Chem.
17
(1996
)
1653
.
[4]
J.A. Grant, J.A. Haigh, B.T. Pickup, A. Nicholls, R.A. Sayles,
Lingos, finite state machines, and fast similarity searching
,
J. Chem. Inf. Model.
46
(2006
)
1912
–
1918
.
[5]
I.S. Haque, V.S. Pande,
PAPER — accelerating parallel evaluations of ROCS
,
J. Comput. Chem.
31
(1
) (2010
)
117
–
132
.
[6]
I.S. Haque, V.S. Pande, W.P. Walters,
SIML: a fast SIMD algorithm for calculating LINGO chemical similarities on GPUs and CPUs
,
J. Chem. Inf. Model.
50
(4
) (2010
)
560
–
564
.
[7]
W.H. Press, S.A. Teukolsky, W.T. Vetterling, B.P. Flannery,
Numerical Recipes in C
.
second ed.
(1992
)
Cambridge University Press
,
Cambridge
.
[8]
D. Vidal, M. Thormann, M. Pons,
LINGO, an efficient holographic text based method to calculate biophysical properties and intermolecular similarities
,
J. Chem. Inf. Model.
45
(2005
)
386
–
393
.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.