
Nanotechnologies for brain tumor therapy
G. Caruso, L. Merlo and M. Caffo, University of Messina, Messina, Italy
Abstract
This chapter introduces a potential definitive treatment in the struggle against malignant brain tumors. In spite of successful steps in biomedical research, brain glioblastomas remain a death sentence in all cases. The difficulty in overcoming the blood–brain barrier (BBB) is the biggest obstacle for chemotherapeutics, which cannot reach the tumor bed as efficacious doses. The advent of nanotechnology could revolutionize the approach to that. Nanoparticles are versatile, thanks to their particular chemical configuration, and are able to go through the BBB carrying drugs or genes targeted against brain tumors. This will improve the efficacy of treatments and reduce also timing and costs of oncological patients.
However the application in vivo of nanoparticles in the field of brain tumor treatment remains difficult to realize: their in vitro and in animal toxicity showed controversial results for the human health, and lacks also long-term follow-up.
Keywords
Blood–brain barrier; glioma; nanotechnology; neurosurgery; therapy
Overview on Brain Gliomas
Although cerebral gliomas represent only 2% of cancers, they account for about 45% of all primary central nervous system (CNS) tumors [1] being among the most deadly neoplasms.
In big series of patients harboring a malignant brain tumor, survival rate goes between 2% and 4% at 3–5 years from treatment [2,3]. This percentage goes up (until 11% of survival at 3 years) when considering smaller series [4].
Nowadays the great majority of patients harboring a malignant brain tumor receives the “Stupp protocol” (radiation+low-dose temozolomide (TMZ), followed by monthly TMZ) as initial treatment [5]. Survival data of 254 patients receiving this protocol showed that 16% of them were alive after 3 years, while only 9.8% after 5 years [6]. Although the 5-year survival rate was approximately double that of previous series, the survival curve showed no sign of plateau, which suggests that the asymptotic level of survival will eventually reach zero.
A different perspective has been provided by analyses examining the conditional probabilities of surviving in terms of additional years given different lengths of prior survival time. The largest conditional probability analysis comes from the American SEER database [7], which includes over 10,000 patients with glioblastoma (GBM) diagnosis from the years 1998 to 2008. The probabilities to survive an additional year were 53% from the time of diagnosis, 38% after 1 year of prior survival, 55% after 2 years, 70% after 3 years, 82% after 4 years, and 78% after 5 years. After 5 years of survival, the probability of surviving an additional 5 years was 45%. The percentage of patients surviving 5 years was 6.2, which implies that the percentage surviving 10 years was approximately 3. In this kind of analysis, variables predicting survival, such as tumor grade and patient age, lost their prognostic value after 3–4 years of prior survival.
The implication of these analyses is that a small percentage of patients survives at least 5–10 years, but there continues to be a substantial rate of death even after extended survival, although the death rate substantially decreases the longer the prior survival.
It is also important to recognize that some of the late “relapses” may not be recurrences of the original tumor but new disease induced by radiation treatment. Experimental work with animal models supports the reality of this risk [8].
Long-term survival can also be due to particular characteristics of patients themselves or of treatments they received. The great majority of long-term survivors (LTS) have had systemic chemotherapy, although this may simply reflect the fact that chemotherapy has been part of the standard treatment. In fact, there is one report of three LTS (11, 16, and 18 years) of 71 patients receiving only brachytherapy [9].
Patient characteristics most common among LTS are young age at the time of diagnosis, higher Karnofsky performance status (KPS), methylation of the MGMT promoter gene, and a complete surgical resection. However, there are LTS who are exceptions to each of these generalizations. The most extensive report of the characteristics of LTS comes from the German Glioma Network, which compared 69 patients who survived more than 36 months with 257 patients who survived less than 36 months [10]. LTS patients were younger but not significantly different with respect to KPS. They were also only marginally more likely to have complete resection at the time of initial surgery, but were significantly more likely to have two or more surgical interventions. In addition, LTS patients were significantly more likely to have methylation of MGMT promoter gene, but less likely to have EGFR amplification. The difference in p53 mutations was not significant. The most consistent variable associated with LTS was isocitrate dehydrogenase (IDH) mutations, as 33% of LTS patients had the mutation, compared to only 4% in control patients. Presence of MGMT methylation and IDH mutations were highly correlated but still partially dissociable. Specifically, patients with IDH mutations but without MGMT methylation had the same prognosis as patients without IDH mutations. It is also noteworthy that there was a significant number of LTS who had no IDH mutations.
As of now, we have only a minimal basis for predicting which patients will receive a significant benefit of treatment, and even less basis for choosing treatments that will be most successful.
The biology of gliomas can help for a targeted therapy.
Gliomas can originate from neural stem cells, progenitor cells, or from de-differentiated mature neural cells transformed into cancer stem cells [11]. Currently the most widely used gliomas histological and molecular classification, and grading system is that of the World Health Organization (WHO) [12] where astrocytomas are the most common subtype. Overall, GBM has the worst prognosis. According to the new molecular WHO classification, there are GBM IDH-wildtype and GBM IDH-mutant.
On the base of the course of the tumor, it is possible to distinguish other two subtypes of GBM: primary and secondary.
Primary GBM develops rapidly de novo in elderly patients, without clinical or histologic evidence of a less malignant precursor lesion and represents the vast majority of GBMs (~90%). Secondary GBMs progress from previous low-grade diffuse astrocytoma or anaplastic astrocytoma and manifest in younger patients, have a lesser degree of necrosis, are preferentially located in the frontal lobe, and carry a better prognosis. Histologically, primary and secondary GBMs are similar. They differ in their genetic and epigenetic profiles. Decisive genetic signposts of secondary GBMs are IDH1 mutations, which are absent in the primary and which are associated with a hypermethylation phenotype. IDH1 mutations are the earliest detectable genetic alterations in precursor low-grade diffuse astrocytomas and in oligodendrogliomas, indicating that these tumors derive from neural precursor cells that differ from those of primary GBMs. IDH1 mutations are diagnostic molecular markers of secondary GBMs and more reliable and objective than clinical criteria. Thus despite a similar histologic appearance, primary and secondary GBMs are distinct tumor entities that originate from different precursor cells and may require different therapeutic approaches [13].
Conventional brain tumor treatments include surgery, radiation therapy, and chemotherapy [14–16]. Each of them has its own disadvantages. Surgical treatment usually represents the first approach: it is invasive but warrants a histological diagnosis. Radiation and chemotherapy are less invasive and can be used alone or often as adjuvant therapy to obtain a prolonged progression-free survival. Radiotherapy gives a better control of seizures with no substantial differences in overall survival [17] but higher risk to develop post-radiation leukoencephalopathy [18]. The effectiveness of systemic chemotherapy is limited by toxic effects on healthy cells, generally resulting in morbidity or mortality of the patient. Moreover the presence of the blood–brain barrier (BBB) limits the passage of a wide variety of anticancer agents [19]. Several drugs possess poor solubility, high toxicity and high dosage, nonspecific delivery, in vivo degradation, and short circulating half-lives.
Despite intensive research for more effective treatments, aggressive multimodal protocols have extended the median survival but often with a significant impairment in the quality of life.
These disappointing results might be due to tumor invasion into functional brain tissue, lack of chemosensitivity, and shortcomings of the systemic delivery [20]. Some forms of systemic chemotherapy partially able to cross the BBB (nitrosoureas such as BCNU and lomustine, and alkylating agents such as TMZ) provide only modest advantages when added to radiotherapy [21]. Moreover, low-molecular-weight chemotherapeutics do not achieve and maintain effective steady-state concentrations within GBM cells because of short blood half-lives [22]. Currently, most therapeutic agents targeting brain tumors are delivered systemically by intravenous and oral route that have limitations reducing the effectiveness of these drugs. The limited success in treating brain tumors results from the tumor cell chemoresistance (natural or acquired), poor selectivity of the antitumor drugs, and presence of the BBB. Multidrug resistance (MDR) is ascribed to ineffective drug delivery to tumor tissues and tumor cells. The advance of the drug delivery to the tumor sites and the decrease of the MDR-based drug efflux can represent ideal solution [23].
Devices that are able to deliver efficacious drugs’ concentrations into the brain tumor cells are nanoparticles-derived.
Nanotechnology is becoming always more diffuse and used in lot of fields: from electronics to medicine. Nanodevices are sometimes very expensive.
Analyzing the cost of treatment of a brain tumor patient, the items to consider are bed days, investigations, surgery, radiotherapy, chemotherapy, and outpatient follow-up. In a British database the mean costs for each of these components were 442 Pounds for neuroradiological investigations, 2,407 Pounds for neurosurgical bed days, 2,068 Pounds for neurosurgery, 434 Pounds for neuropathology, 8,832 Pounds for radiotherapy, 440 Pounds for chemotherapy, and 1,078 Pounds for outpatients follow-up. Considering the total treatment costs per patient, the range goes from 1,978 to 26,980 Pounds [24].
Large-scale manufacturing remains a costly and challenging aspect in the clinical translation of biotargeted nanomedicines. At present, there are few cost–benefit studies available for nanomedicine products. As an example the cost-effectiveness of Doxil (Janssen Biotech Inc., PA, USA; PEGylated liposomal doxorubicin) and Abraxane (Celgene Corporation, NJ, USA; nano-albumin bound paclitaxel) compared with their conventional standard-of-care generic alternatives, doxorubicin, and paclitaxel, respectively. In 2009 the average cost per dose of Doxil was US$5,594 compared with $62–162 for doxorubicin, and the average cost per dose for Abraxane was $5,054 compared with $90–454 for paclitaxel. Notable health- and cost-related benefits of Doxil and Abraxane are lower cardiac toxicity and reduced vehicle toxicity, respectively. Although neither nanomedicine products have shown an increase in overall patient survival, the reduction in toxicities and their associated cost have largely justified the higher cost [25]. Increasing pressure to reduce healthcare costs puts an even greater burden on the nanomedicine innovator to justify the real cost-to-benefit ratio.
The manufacturing also changes the price of nanomaterials. For example, graphene can be synthesized either through chemical vapor deposition or from the liquid-phase exfoliation of graphite, the latter being the most widely adopted procedure because technologically simple, highly efficient, and low-cost [26]. Such optimized technique of synthesis caused its overall costs dramatically to drop: while in 2008 graphene was one of the most expensive materials, costing about $100 million per cm2, now reduced graphene oxide costs approximately $270 per gram [27].
In order to lower the costs, many of the companies that are currently developing biotargeted cancer therapies are smaller start-ups featuring pipelines based on technologies originally developed in academic laboratories, rendering high development costs even more daunting. There is a movement toward collaborative efforts between large and small pharma, government, nonprofit agencies, and venture capital firms to defray costs of new therapeutic development [25].
Blood–Brain Barrier
The brain is a unique organ highly protected by two major barriers, the largest BBB and the blood–cerebrospinal fluid barrier. Overcoming the BBB would improve significantly the delivery of targeted drugs to malignant glioma cells.
The BBB is responsible for maintenance of neuronal microenvironment, tissue homeostasis, vasotonous regulation, fibrinolysis and coagulation, and blood cell activation and migration during physiological and pathological processes. Physiologically, BBB is composed of an inner endothelial cell layer forming the wall of the capillary and contains tight junctions (TJs) and a basement membrane upon which pericytes and astrocytic feet processes lie [28]. The BBB endothelial cells differ from endothelial cells in the rest of the body by the absence of fenestrations, more extensive TJs, and sparse pinocytic vesicular transport. Endothelial cells TJs limit the paracellular flux of hydrophilic molecules across the barrier (Fig. 8.1).
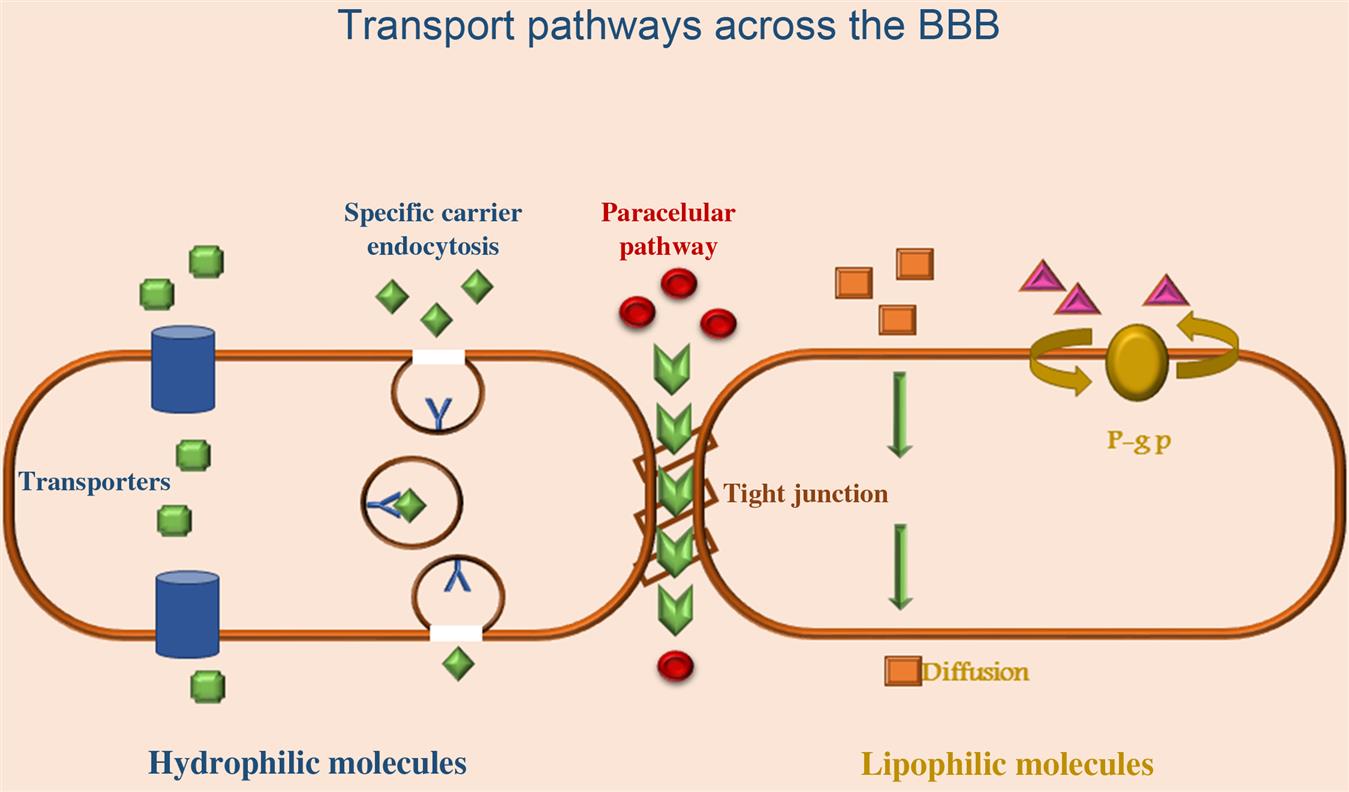
The TJs are located on the apical region of endothelial cells and are formed by a complex network made of a series of parallel, interconnected, transmembrane, and cytoplasmatic strands of proteins [29]. They consist of three integral membrane proteins, namely, claudin, occludin, and junction adhesion molecules, and a number of cytoplasmic accessory proteins. The tightness of the BBB is due to the physical complexity of its junctional structure and the molecular substructure, in particular, the presence of transmembrane proteins, which help to seal the intercellular cleft. Adherens junctions (AJs) located below the TJs in the basal region of the lateral plasmamembrane are composed of transmembrane glycoproteins (cadherins) linked to the cytoskeleton by cytoplasmatic proteins, thus providing an additional tightening structure between the adjacent endothelial cells at the BBB [30].
Despite the rapid development in understanding the molecular structure and receptor expressed of the BBB, many of the CNS-associated diseases remain undertreated by effective therapies. The majority of drugs and large-molecular-weight particulate agents such as recombinant proteins, peptides, monoclonal antibodies, small-interfering RNA (siRNA), and gene therapeutics do not readily permeate into the brain parenchyma.
There are different mechanisms by which molecules can cross the BBB. Passive diffusion is dependent on concentration gradient, lipophilicity, and molecular weight that allows molecules to move between cells (paracellular way) or across cells (transcellular way) down their electrochemical gradient without the requirement of metabolic energy. For example, small water-soluble molecules simply diffuse through the TJs. Small lipid-soluble substances like alcohol and steroid hormones penetrate transcellularly by dissolving in their lipid plasmamembrane. However the majority of small-molecule drugs have a higher molecular weight or current water solubility preventing their simple diffusion across the barrier. For almost all other substances, including essential materials such as glucose and aminoacids, transport proteins (carriers), specific receptor-mediated, or vesicular mechanisms (adsorptive transcytosis) are required to pass the BBB.
Different substances are transported through free diffusion mechanism either paracellularly or transcellularly. Paracellular diffusion is a nonsaturable and noncompetitive movement of compounds between cells. It occurs to a limited extent at the BBB, due to the TJs. Transcellular diffusion (transcytosis) is a nonsaturable and noncompetitive movement across cells of lipophilic substances. Facilitated diffusion is a form of carrier-mediated endocytosis in which solute molecules bind to specific membrane protein carriers that trigger a conformational change in the protein. This results in a carrying through of the substance to the other side of the membrane, from high to low concentration (passive diffusion). This mechanism contributes to the transport of various substances including aminoacids, nucleoside, small peptide, monocarboxylates, and glutathione.
Carrier-mediated transport (CMT) or carrier-mediated influx processes involve putative proteins that facilitate the movement of poorly permeable solutes across cellular membranes. The CMT system is expressed on both the luminal and abluminal membranes of the brain capillary endothelium and operates in both directions. CMT systems can be exploited for brain drug-delivery after reformulating the drug in such a way that the drug assumes a molecular structure mimicking that of the endogenous ligand. If compounds need to be moved against a concentration gradient, ATP may provide the energy to facilitate the process.
The uptake of nutrients from blood into the brain is facilitated by the solute carrier transporter families. These influx carriers are involved in the transport of a broad range of substrates including glucose, amino acids, nucleosides, fatty acids, minerals, and vitamins in various human tissues, including the brain.
The active efflux transport is responsible for extruding drugs from the brain and this mechanism is a major obstacle for the accumulation of a wide range of biologically active molecules in the brain. The ATP-binding cassette (ABC) transporter P-glycoprotein and multidrug-resistant protein represent the principle efflux mechanism of these agents [31]. The most abundantly present component of this system is efflux P-glycoprotein, which is a product of the ABCB1 gene. Inhibition of P-glycoprotein in preclinical studies has enhanced the penetration of paclitaxel into the brain, indicating the feasibility of achieving improved drug delivery to the brain by suppression of P-glycoprotein [32].
Endocytosis and transcytosis allow the internalization, sorting and trafficking of many plasma macromolecules. Endocytosis is a process where molecules from the circulation are internalized in vesicles and are directed to endosomes or lysosomes within the cell. Endocytosis can be isolated into bulk-phase (fluid phase or pinocytosis) endocytosis and mediated endocytosis (receptor and absorptive mediated). Bulk-phase endocytosis is the noncompetitive, nonsaturable, temperature- and energy-dependent nonspecific uptake of extracellular fluids. Transcytosis refers to the transcellular movement of molecules.
Receptor-mediated endocytosis or clathrin-dependent endocytosis provides for a highly specific and energy-mediated transport enabling eukaryotic cells to selective uptake macromolecules as specific cargo. Cells have different receptors for the uptake of many different types of ligands, including hormones, growth factors, enzymes, and plasma proteins. This process occurs at the brain for macromolecular substances, such as transferrin, insulin, leptin, IGF-I, and IGF-II, and is a highly specific type of energy-dependent transport [33].
Adsorptive endocytosis/transcytosis facilitates the transport of large peptides such as IgG, histone, albumin, native ferritin, horseradish peroxidase, and dextran. Adsorptive-mediated endocytosis is characterized by an electrostatic interaction between a positively charged substance and the negatively charged sites on the brain endothelial cell surface (e.g., glycoprotein) [34]. Adsorptive processes largely depend upon electrostatic interactions that allow the positively charged moiety of the substrate to bind to the negatively charged cell membrane. Receptor-mediated transport is mainly employed in the transport of macromolecules like peptides and proteins across the BBB, by conjugating the substance with ligands such as lactoferrin, transferrin, and insulin. It is an important transport mechanism of predominant interest in drug delivery.
Cell-mediated transcytosis is a recently identified route of drug transport across the BBB [35]. This transport route relies on immune cells such as monocytes or macrophages to cross the intact BBB. Unlike the aforementioned transport pathways, which normally permit only solute molecules with specific properties, cell-mediated transcytosis is unique in that it can be used for virtually any type of molecule or material as well as particulate carrier systems.
Thanks to the abovementioned mechanisms, under physiologic conditions, the BBB is relatively impermeable. Under pathologic conditions, a number of chemical mediators are released that increase BBB permeability. Several of these mediators of BBB opening have been studied in both in vivo and in vitro experiments and include glutamate, aspartate, taurine, ATP, endothelin-1, ATP, NO, MIP-2, tumor necrosis factor-α (TNF-α), MIP2, and IL-h, which are produced by astrocytes [36]. Other humoral agents reported to increase BBB permeability are bradykinin, 5-HT, histamine, thrombin, UTP, UMP, substance P, quinolinic acid, plateletactivating factor, and free radicals [37]. The source of these BBB-modulating mediators is of interest. Some of these agents are released by endothelium and endothelium itself responds to the released agents. For example, endothelin (ET-1) acts on ETA receptors. Under physiologic conditions, nerve terminals of neurons running close to blood vessels release mediators, such as histamine, substance P, and glutamate, which influence BBB permeability.
The BBB is poorly developed in brain tumor leading to increased vascular permeability [38]. Investigations have shown that there is opening of interendothelial TJ in human gliomas [39] and metastatic adenocarcinoma [40]. The expression of the TJ protein claudin-1 is lost in the microvessels of GBM, whereas claudin-5 and occludin are significantly downregulated and ZO-1 expression is unaffected [41]. A loss of 55 kDa occluding expression in microvessels, observed in astrocytoma and metastatic adenocarcinoma, may also contribute to endothelial TJ opening [42].
The explanation for loss of TJ molecules in brain tumor microvessels is not clear. However, VEGF, cytokines [43], and Scatter factor or hepatocyte growth factor [44] secreted by astrocytoma and other brain tumors may be involved in downregulating TJ molecules leading to TJ opening, increased vascular permeability, and cerebral edema. It is also possible that poorly differentiated neoplastic astrocytes do not release factors necessary for BBB function.
Since cerebral edema is an important consequence of brain tumor, water channel molecule, AQP4, has been examined in brain tumor by several investigators. AQP4 is massively upregulated in astrocytoma and metastatic adenocarcinoma and this correlates with BBB opening assessed by contrast-enhanced computed tomograms [45]. Mice deficient in AQP4 have a much better survival than wild-type mice in a model of brain edema caused by acute water intoxication. Upregulation of AQP4 has also been noted in rat models of ischemia [46] and brain injury [47]. Thus, it seems that breakdown of the BBB associated with brain tumors and other forms of brain injury increases the expression of AQP4. However the exact mechanism of AQP4 upregulation in different clinical situations is not known.
Nanoparticles
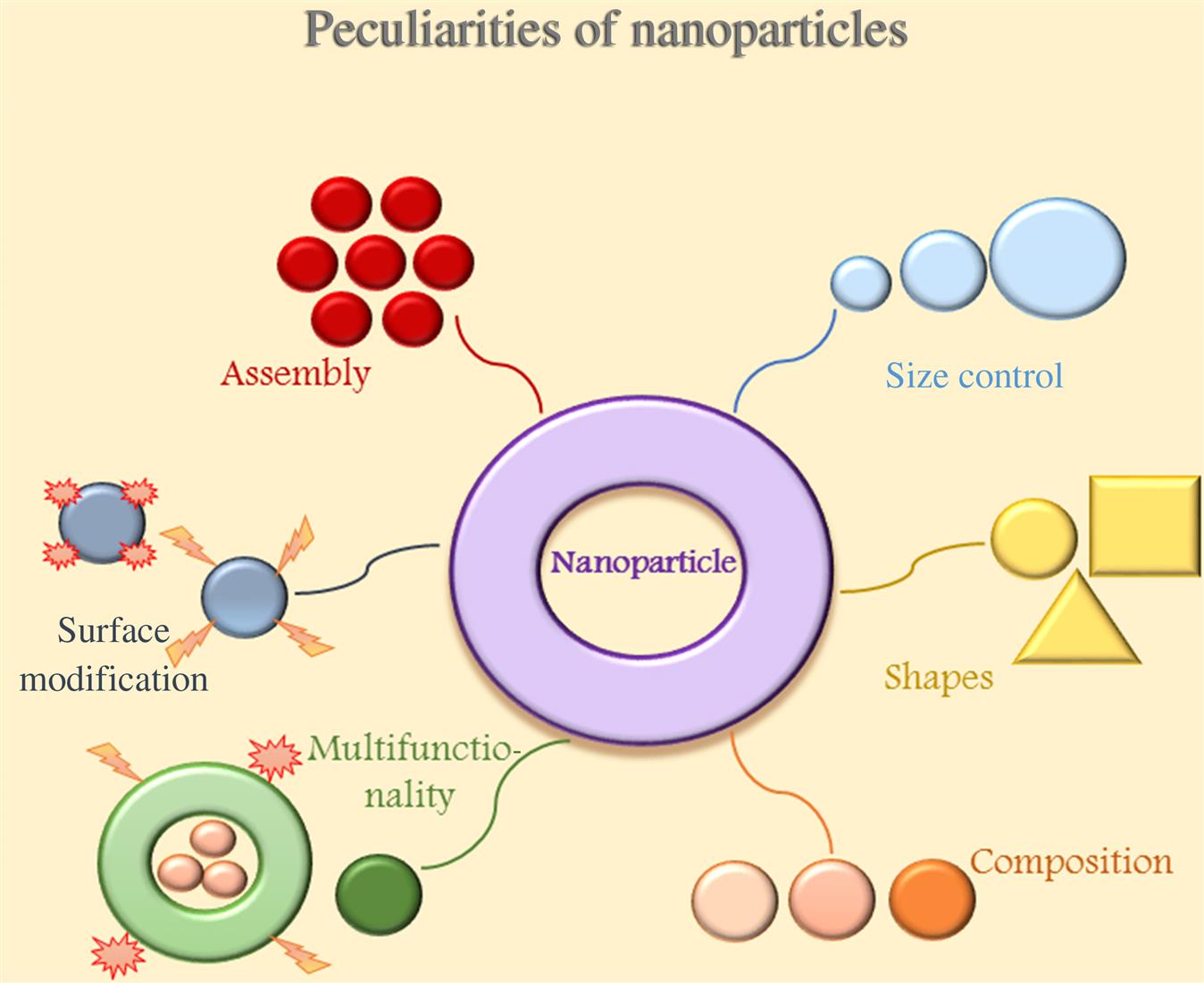
Nanoscience has a huge potential to bring benefits in areas as diverse as drug development, water decontamination, information and communication technologies, and the production of stronger, lighter materials. Nanotechnology is an emerging field that deals with interactions between cells and engineered molecules. The “nano” concept is also applied to the field of medicine, being nanomedicine used in drug delivery systems, and in cancer diagnosis and therapy. A nanoparticle is the smallest unit (10−9 m) that can still behave as a whole entity in terms of properties and transport and is able to cross biological membranes and access cells, tissues, and organs (Fig. 8.2) [48]. Thus, the use of nanoparticles in brain tumors treatment seems to be very promising because it has the ability to overcome the obstacle represented by the BBB. It also adds specificity and selectivity to the drugs delivered across the BBB focusing only to cancer cells, so avoiding unwilling side effects in the surrounding healthy tissues [49]. The first application of a targeted nanosystem for drug delivery concerned liposomes and it was reported in 1980 [50].
The peculiarities of nanoparticles (NPs) (size, electrostatic charge, and lipophilic characteristics) allow them to enter the brain tissue freely, bypassing the BBB and carrying drugs. Tumor angiogenesis possesses leaky vasculature that impedes drug delivery: an abnormal basement membrane and fissures between the endothelial cells due to an absent pericytes lining. That together with a poor lymphatic drainage system causes a differential interstitial pressure at the center of tumors compared with that at the periphery. Thus, molecules ranging from approximately 10 to 100 nm accumulate in the tumor and are retained for longer, unlike the uncoated smaller drugs cleared by kidneys. This is called “enhanced permeability and retention” (EPR) effect. The retention time of drugs packed in NPs is 10× higher than that of unpacked drugs [51]. Hence the EPR effect attributed to the leaky vasculature is considered a boon for drug-delivery systems within the nanosize range.
When administered systemically, nanoparticles (NPs) can protect the loaded drugs from degradation. Small therapeutic molecular agents that are normally poorly distributed can be incorporated into NPs via a variety of chemical methods, including encapsulation, adsorption, and covalent linkage, while macromolecules can be attached to the surface of NPs to improve targeting. Reduction of toxicity to peripheral organs can also be achieved with these systems [14]. Targeted drug-delivery systems can convey drugs more effectively and conveniently, increase patient compliance, and reduce healthcare costs.
The surface of the nanocarrier can be engineered to increase the blood circulation half-life and influence the bio-distribution, while attachment of targeting ligands to the surface can result in enhanced uptake by target tissues. The net result of these properties is to lower the systemic toxicity of the therapeutic agent, while increasing the concentration of the agent in the area of interest, resulting in a higher therapeutic index for the therapeutic agent. In addition to therapeutic drugs, imaging agents can also incorporated into nanocarriers to improve tumor detection and imaging. Finally, nanoparticles can be engineered to be multifunctional with the ability to target diseased tissue, carry imaging agents for detection, and deliver multiple therapeutic agents for combination therapy. Prolonged circulation properties are ideal for slow or controlled release of therapeutic agents into the blood to treat vascular disorders. Long circulating particles may have application in vascular imaging, or even act as artificial nanoscale red blood cells. Recent advances in synthetic polymer chemistry afford precise control over the architecture and polydispersity of polymers, polymer-conjugates, and block copolymers. Some of these novel materials can form sterically stabilized nanoscale self-assembling structures with macrophage-evading properties. A unique attribute of nanoplatform-based delivery systems is their multifunctionality, characterized by multiple components, which include imaging agents, therapeutic agents, targeting ligands, and “cloaking” agents that avoid interference with the immune system. Nanotheranostic platforms are powerful tools for imaging and treatment of cancer. Multifunctionality of these systems offers a number of advantages over conventional agents. These include targeting to a diseased site thereby minimizing systemic toxicity, the ability to solubilize hydrophobic or labile drugs leading to improved pharmacokinetics and their potential to image, treat and predict therapeutic response. Targeted nanoparticle-based treatment technologies with diagnostic capabilities are referred to as theranostic agents as they form a class of agents, which can serve diagnostic and therapeutic functions simultaneously. In the current state of technology, tumor detection and therapy are performed separately. A more efficient and effective method can be achieved with theranostic nanoparticles, which would integrate the efforts for detection, treatment and follow-up monitoring of tumor response, and assist in the decision-making process for the need for further treatment.
Nanomaterial-based agents that are specific for nanomedicine include mainly polymer- or lipid-based carriers. Drugs can be absorbed onto the surface, entrapped inside, or dissolved within the matrix of these vehicles. Nanocarriers can be also combined into cells both using transfection agents, often toxic and not clinically approved, and without that agents [52]. Actual research has developed multifunctional NPs able to respond to the environment, so facilitating a more effective drug delivery. The diversity of delivery systems allows NPs to have different arrays of shapes, sizes, and components tailored for specific applications.
NPs have been shown to enter inside cells via passive transport [53] and active endocytosis [54–56]. Once inside the cells, NPs are transported to the endo-lysosomal system, where they are usually destroyed. Anyway, polylactide (PLA) and polyglycolide (PLGA)-NPs as well as lipid nanocapsules are able to escape the lysosomal compartment by disrupting the integrity of the lysosome membrane [57–59]. This endo-lysosomal escape leads to NP accumulation in the cytoplasm whose effects are unknown yet.
Among the numerous molecules studied, liposomes, gold-NP, and graphene-NP-derived seem to be among the most promising.
Liposomes are vesicles made up of a lipid bilayer, resembling a cell membrane. The lipids form a bilayer based on hydrophobic interactions in continuous parallel packing, with the hydrophilic head groups positioned toward the aqueous environment. They possess advantages of carrying hydrophilic, lipophilic, as well as amphoteric drug molecules, either entrapped inside it or on its micellar surface. Liposomes prove ideal carriers for biological agents such as siRNA because of their stable aqueous core. Moreover, it is possible to combine RNA-interfering strategies with traditional chemotherapeutics. One example is the Raf/MEK/extracellular signal-related kinase (ERK) pathway, which is essential for cellular proliferation, and found to be aberrant in several cancers [60]. As a result, several inhibitors of key proteins in the cascade have been developed as potential chemotherapeutics. Recently, it has been demonstrated that liposomes encapsulating an Mcl1-specific siRNA (siMcl1) and a chemical MEK inhibitor (PD0325901) showed a valid antitumor efficacy in vitro and in vivo [61].
Glioma cells show an upregulation of expression of IL-13 receptor α2 on their surface cells. In a recent study the improvement of internalization of doxorubicin-loaded nanoliposomes, targeted with conjugated IL-13, and cytotoxicity in U251 glioma cells has been shown. In an in vivo animal model the authors demonstrated the inhibition of the growth of subcutaneously implanted gliomas [62]. In anticancer gene therapy the efficiency of liposomes has been increased through surface ligand targeting, via monoclonal antibodies to specific receptors up-regulated in glioma cells surface such as transferring receptors, LDL receptors, and IL-13 receptors.
For glioma gene therapy, viral vectors have been used to deliver suicide genes, pro-apoptotic genes, p53, cytokines, and caspases. These studies have shown promising preclinical results, but clinical trials have been limited by the fact that transduced cells were found only within a very short distance of the delivery site [63].
A study by Jaworski and colleagues [64] revealed that graphene platelets are toxic to U87 and U118 glioma cell lines, indicating their potential therapeutic applicability. Because of their large surface area, graphene platelets did not enter into glioma cells, but adhered to them: graphene electrons interacted with cell membranes and receptors blocking the supply of nutrients, inducing stress and activating apoptosis. Graphene is a carbon allotrope with a bidimensional hexagonal structure and with its derivatives, such as its oxide (GO), has shown potentials in numerous fields [65]. The importance of the functionalization of GO has been the key for the biological and biomedical applications of graphene which range from targeting controlled drug/gene delivery, photothermal and photodynamic cancer therapy, biological sensing and imaging, to multifunctional nanoplatforms [66]. The photothermal activity of graphene has also been investigated in the treatment of brain tumors. A recent study combined the chemo-photothermal targeted therapy of glioma within one novel multifunctional drug delivery system using a targeting peptide (IP)-modified mesoporous silicacoated graphene nanosheet (GSPI). Doxorubicin was conjugated with the GSPI-based system (GSPID), showing synergistic chemo-photothermal properties. Cytotoxicity experiments demonstrated a higher rate of death of glioma cells [67].
Tian et al. [68] showed the feasibility of encapsulating the temozolomide [(3,4-dihydro-3-methyl-4-oxoimidazo [5,1-d]-as-tetrazine-8-carboxamide (TMZ)], an alkylating agent, into polybutylcyanoacrylate (PBCA) polymeric nanoparticle NPs by polymerization. Based on the pattern of distribution in body organs, higher concentrations of TMZ were detected in the brain after binding to PBCA nanoparticles coated with polysorbate-80, which may be more useful for treating brain tumors.
Thermosensitive magnetic NPs were prepared with the magnetic NPs covered by a thermosensitive polymer with a critical temperature of 40–45°C. These systems were suitable for hyperthermia treatment of cancers such as brain cancer. The thermosensitive polymer shell could be ruptured at the site of action by applying an external magnetic field to increase the temperature of the inner magnetic core [69]. The NanoTherm therapy, also termed magnetic fluid hyperthermia, combined with fractionated stereotactic radiotherapy, is a new local heat treatment of solid tumors (such as glioblastoma multiforme and prostate carcinoma). Three major components are required for NanoTherm therapy: NanoTherm, Nanoplan, and NanoActivator F100 (MagForce Nanotechnologies AG, Berlin, Germany). NanoTherm is a magnetofluid consisting of superparamagnetic iron oxide nanoparticles (SPIONs), which are colloidally dispersed in water with a high iron concentration. The iron oxide magnetite (Fe3O4) core is approximately 12 nm in diameter and coated with an aminosilane-type shell. Due to their aminosilane coating, these small magnets can be finely dispersed in water, forming a colloidal solution that is dispensable with a syringe. Once inside the alternating magnetic field applicator, NanoActivator, these specifically designed nanoparticles are responsible for the production of warmth. Through this high-frequency magnetic field, the nanoparticles begin to oscillate and warmth is produced from directly within the tumor tissue. Depending on the temperature reached and length of treatment, the tumor cells are either directly destroyed or sensitized for the accompanying chemotherapy or radiation. NanoTherm therapy has been used successfully to treat glioblastoma multiforme in a Phase II GBM trial [70].
Nanoparticles can also be of aid in diagnosis.
Gold nanoparticles (GNPs) exhibit unique physicochemical properties, including the ability to bind amine and thiol groups, allowing surface modification and use in biomedical applications. GNPs are used to prepare nanoshells composed of gold and copper, or gold and silver to function as contrast agents in MRI, and gold–silica for photothermal ablation of tumor cells. Classically, GNPs enter into cells with a nonspecific receptor-mediated endocytosis mechanism [71]. In vivo GNPs passively accumulate at tumor sites that have leaky immature vasculature with wider fenestrations than normal mature blood vessels.
Thanks to its unique properties as contrast enhancement medium, graphene reveals accuracy in brain and spinal cord imaging. Sheets of nano-graphene are intrinsically photoluminescent, thus able to provide in vivo high-resolution live cell imaging with high quality and low cytotoxicity [72]. In addition, thanks to this property, graphene can be used also intraoperatively to obtain fluorescent imaging both during tumor and aneurysm surgery [73].
Actual molecular diagnosis avails itself of detecting the presence of specific sequences of DNA, RNA, or proteins by using biosensors. Applying engineered nanosensors, namely, nanopores, for this purpose, gives faster results than previous methods [74]. The versatility of graphene confers the capacity to detect not only single nucleotides, but also neurotransmitters, opening the possibility to its employment also for molecular diagnosis [75,76]. The affinity showed with fluorescent molecules gives space to test graphene biosensors based on fluorescence resonance energy transfer, which provide quantitative and real-time imaging of intra- and intercellular proteins’ interactions, DNA, and miRNA [77–79]. In addition, it is possible to use graphene as a nanoscaffold for devices that improve the sensitivity and specificity of current biosensors.
Although promising in vitro results have been reported, it remains unclear how effective such a system would be due to intra- and interindividual patient heterogeneity considering also that toxicity of NPs in humans has not been tested yet. The majority of studies are performed in experimental settings, both in vitro and in animal models, but their translation into the clinics remains difficult due to human variability and safety-related issues.
Conclusion and Critical Issues
As summarized in the previous chapters, nanomedicine seems the efficacious answer to brain tumor treatment. It reduces hospital stays, lowers morbidity and mortality, and in a prospective view large diffusion can reduce health costs for long-term patients.
However, nanomedicine encounters difficulties in becoming widely used. It is a not completely clarified field. Toxic effects of the interactions between NPs and biological systems are not definitively assessed in humans.
There are people daily exposed to NPs originating from combustion, welding, and biomedical applications, e.g., workers in industries such as cars, aerospace, electronics, or chemical. Notwithstanding, a complete evaluation of interactions between NPs and biological systems is lacking.
It is known that NPs exhibit toxic manifestations and can result in allergy, fibrosis, organ failure, and various kind of toxicities (nephro-, hematological, neuro-, hepatic, splenic, and pulmonary) [80,81].
In addition, NPs surfaces are involved in catalytic and oxidative processes potentially cytotoxic. High dose of single-walled carbon nanotubes origins reactive oxygen species (ROS), mitochondrial dysfunction, oxidative stress, and changes in cell morphology when incubated with keratinocytes and bronchial epithelial cells. Intratracheal instillation of carbon nanotubes in rodents caused chronic lung inflammation with foreign body granuloma formation and interstitial fibrosis [82].
It is also not exactly known how long NPs remains into the body and if, during this time they can interact with healthy cells. Carbon nanotubes and quantum dots, e.g., are long-lasting remaining in the body for a long period, this make them potentially toxic and limits their use for reiterated treatments. Copper, iron, gold, iron, manganese, titanium, silica, and other carbon-based nanomaterials are some of the NPs to which humans are exposed significantly and may cause several health-related problems including neurotoxicity [83]. The effect of NPs on the cell membrane may be due to their direct toxicity, or indirectly, they may induce some cascade mechanism that disrupts the tight junctions in the BBB or alters the permeability of the membrane. It has been shown that intravenous, intraperitoneal, or intracerebral administration of Ag, Cu, or Al NPs disrupts the BBB, as indicated by staining with albumin-bound Evans blue [84].
Another example is nanoparticulate TiO2, used to develop cosmetics, foodstuffs, toothpaste, sun blocks, printing ink, car materials, cleaning products, materials for industrial photocatalytic applications including solar cells, and catalysts for remediation of organic matter in wastewater. TiO2 NPs accumulate in the brain and induce structural changes in the neuronal structure [85,86]. Altered gene expression was detected for prenatal TiO2 NP exposure, which was involved in cell death, brain development, and response to oxidative stress in newborn pups [87].
Iron oxide or SPIONs are small NPs composed of a Fe3O4 (magnetite) or Fe2O3 (maghemite) core. Their potential application ranges from biomedical imaging (magnetic resonance imaging, positron emission tomography, or ultrasound as contrast agent), gene and drug delivery, tissue regeneration, hyperthermia in cancer treatment, catalysis, and magnetic storage. They are extensively used specifically for brain imaging or brain-targeted drug and gene delivery, due to their ability to move across the BBB [88]. SPIONs shown a potential toxicity that can lead to altered gene expression, actin modulation, interference with cell cycle regulation and signaling pathways, excessive ROS generation, and disruption of iron homeostasis. Fe3O4 NPs also had a substantial cytotoxic effect on PC12 cells by modulating the cell cycle and inducing apoptosis [89].
Concerning carbon-based nanomaterials, they have a potential use in a variety of biomedical applications, including early diagnosis of cancer, imaging, targeted photothermal therapy, drug delivery, and tissue engineering. Studies of carbon nanomaterials have indicated the potential neurotoxic effects after inhalation or systemic exposure. A study has shown that oxidative stress is involved in this toxic pathway, with surface coating playing an important role [90]. In vivo studies demonstrate systemic bio-distribution and biopersistence of graphene following intravenous delivery. Similar to other foreign bodies, graphene has the potential to induce foreign body tumors, thus long-term adverse health impacts has to be considered. The limited literature on in vitro toxicity suggests that graphene can be either benign or toxic to cells, its biological response depending on layers number, lateral size, stiffness, hydrophobicity, surface functionalization, and dose. Direct or indirect generation of ROS unbalanced by the cellular antioxidant enzymes is currently the main mechanism proposed for graphene toxicity [91]. It has been observed that GO directly interacts with the cell membranes leading to their physical damage [92]. In vitro studies agree that GO promotes cytotoxicity mainly generating ROS in a dose-dependent manner [93]. Graphene is also genotoxic, penetrating into the cells and causing damage of DNA such as fragmentation and/or chromosomal aberrations even for low doses at short time of exposure [94].
Notwithstanding, there are others potential risks related with this novel approach. Some cancer cell types could develop drug resistance making ineffective the drugs released from the targeted NPs. Moreover, NPs might change stability, solubility, and pharmacokinetic properties of the carried drugs. In addition, some materials used to create NPs possess low toxicity but degrade quickly and do not circulate in tissues long enough to warrant a sustained drug/gene delivery.
Objects of debate are the results about the long-term effects of interactions between NPs and coating of molecules and target cells. In order for this promising field to rapidly progress, focus must be placed on elucidating the safety of these novel materials.