
Chapter 5. Computing GC Content: Parsing FASTA and Analyzing Sequences
In Chapter 1, you counted all the bases in a string of DNA.
In this exercise, you need to count the Gs and Cs in a sequence and divide by the length of the sequence to determine the GC content as described on the Rosalind GC page.
GC content is informative in several ways.
A higher GC content level indicates a relatively higher melting temperature in molecular biology, and DNA sequences that encode proteins tend to be found in GC-rich regions.
There are many ways to solve this problem, and they all start with using Biopython to parse a FASTA file, a key file format in bioinformatics.
I’ll show you how to use the Bio.SeqIO module to iterate over the sequences in the file to identify the sequence with the highest GC content.
You will learn:
-
How to parse FASTA format using
Bio.SeqIO -
How to read
STDIN(pronounced standard in) -
Several ways to express the notion of a
forloop using list comprehensions,filter(), andmap() -
How to address runtime challenges such as memory allocation when parsing large files
-
More about the
sorted()function -
How to include formatting instructions in format strings
-
How to use the
sum()function to add a list of numbers -
How to use regular expressions to count the occurrences of a pattern in a string
Getting Started
All the code and tests for this program are in the 05_gc directory.
While I’d like to name this program gc.py, it turns out that this conflicts with a very important Python module called gc.py which is used for garbage collection, such as freeing memory.
Instead, I’ll use cgc.py for calculate GC.
If I called my program gc.py, my code would shadow the built-in gc module, making it unavailable. Likewise, I can create variables and functions with names like len or dict which would shadow those built-in functions. This will cause many bad things to happen, so it’s best to avoid these names. Programs like pylint and flake8 can find problems like this.
Start by copying the first solution and asking for the usage:
$ cp solution1_list.py cgc.py $ ./cgc.py -h usage: cgc.py [-h] [FILE]Compute GC content positional arguments: FILE Input sequence file (default: <_io.TextIOWrapper
name='<stdin>' mode='r' encoding='utf-8'>) optional arguments: -h, --help show this help message and exit
Note that the positional
[FILE]is in square brackets to indicate that it is optional.This is a rather ugly message that is trying to explain that the default input is
STDIN.
As in Chapter 2, this program expects a file as input and will reject invalid or unreadable files.
To illustrate this second point, create an empty file using touch and then use chmod (change mode) to set the permissions to 000 (all read/write/execute bits off):
$ touch cant-touch-this $ chmod 000 cant-touch-this
Notice that the error message specifically tells me that I lack permission to read the file:
$ ./cgc.py cant-touch-this usage: cgc.py [-h] [FILE] cgc.py: error: argument FILE: can't open 'cant-touch-this': [Errno 13] Permission denied: 'cant-touch-this'
Now run the program with valid input and observe that the program prints the ID of the record having the highest percentage of GC:
$ ./cgc.py tests/inputs/1.fa Rosalind_0808 60.919540
This program can also read from STDIN.
Simply because I think it’s fun, I’ll show you how, in the bash shell, I can use the pipe operator (|) to route the STDOUT from one program to the STDIN of another program.
For instance, the cat program will print the contents of a file to STDOUT:
$ cat tests/inputs/1.fa >Rosalind_6404 CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC TCCCACTAATAATTCTGAGG >Rosalind_5959 CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT ATATCCATTTGTCAGCAGACACGC >Rosalind_0808 CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC TGGGAACCTGCGGGCAGTAGGTGGAAT
Using the pipe, I can feed this to my program:
$ cat tests/inputs/1.fa | ./cgc.py Rosalind_0808 60.919540
I can also use the < operator to redirect input from a file:
$ ./cgc.py < tests/inputs/1.fa Rosalind_0808 60.919540
To get started, remove this program and start over:
$ new.py -fp 'Compute GC content' cgc.py Done, see new script "cgc.py".
The following shows how to modify the first part of the program to accept a single positional argument that is a valid, readable file:
import argparse
import sys
from typing import NamedTuple, TextIO, List, Tuple
from Bio import SeqIO
class Args(NamedTuple):
""" Command-line arguments """
file: TextIO
def get_args() -> Args:
""" Get command-line arguments """
parser = argparse.ArgumentParser(
description='Compute GC content',
formatter_class=argparse.ArgumentDefaultsHelpFormatter)
parser.add_argument('file',
metavar='FILE',
type=argparse.FileType('rt'),
nargs='?',
default=sys.stdin,
help='Input sequence file')
args = parser.parse_args()
return Args(args.file)
The only attribute of the
Argsclass is a filehandle.Create a positional file argument that, if provided, must be a readable text file.
It’s rare to make a positional argument optional, but in this case, I want to either handle a single file input or read from STDIN.
To do this, I use nargs='?' to indicate that the parameter should accept zero or one argument (see Table 2-2 in “Opening the Output Files”) and set default=sys.stdin.
In Chapter 2, I mentioned that sys.stdout is a filehandle that is always open for writing.
Similarly, sys.stdin is an open filehandle from which you can always read STDIN.
This is all the code that is required to make your program read either from a file or from STDIN, and I think that’s rather neat and tidy.
Modify your main() to print the name of the file:
def main() -> None:
args = get_args()
print(args.file.name)
Verify that it works:
$ ./cgc.py tests/inputs/1.fa tests/inputs/1.fa
Run pytest to see how you’re faring.
You should pass the first three tests and fail on the fourth:
$ pytest -xv
============================ test session starts ============================
....
tests/cgc_test.py::test_exists PASSED [ 20%]
tests/cgc_test.py::test_usage PASSED [ 40%]
tests/cgc_test.py::test_bad_input PASSED [ 60%]
tests/cgc_test.py::test_good_input1 FAILED [ 80%]
================================= FAILURES ==================================
_____________________________ test_good_input1 ______________________________
def test_good_input1():
""" Works on good input """
rv, out = getstatusoutput(f'{RUN} {SAMPLE1}')
assert rv == 0
> assert out == 'Rosalind_0808 60.919540'
E AssertionError: assert './tests/inputs/1.fa' == 'Rosalind_0808 60.919540'
E - Rosalind_0808 60.919540  E + ./tests/inputs/1.fa
E + ./tests/inputs/1.fa  tests/cgc_test.py:48: AssertionError
========================== short test summary info ==========================
FAILED tests/cgc_test.py::test_good_input1 - AssertionError: assert './tes...
!!!!!!!!!!!!!!!!!!!!!!!!! stopping after 1 failures !!!!!!!!!!!!!!!!!!!!!!!!!
======================== 1 failed, 3 passed in 0.34s ========================
tests/cgc_test.py:48: AssertionError
========================== short test summary info ==========================
FAILED tests/cgc_test.py::test_good_input1 - AssertionError: assert './tes...
!!!!!!!!!!!!!!!!!!!!!!!!! stopping after 1 failures !!!!!!!!!!!!!!!!!!!!!!!!!
======================== 1 failed, 3 passed in 0.34s ========================
The test is running the program using the first input file.
The output is expected to be the given string.
This is the expected string.
This is the string that was printed.
So far you have created a syntactically correct, well-structured, and documented program that validates a file input, all by doing relatively little work. Next, you need to figure out how to find the sequence with the highest GC content.
Get Parsing FASTA Using Biopython
The data from the incoming file or STDIN should be sequence data in FASTA format, which is a common way to represent biological sequences.
Let’s look at the first file to understand the format:
$ cat tests/inputs/1.fa >Rosalind_6404
A FASTA record starts with a
>at the beginning of a line. The sequence ID is any following text up to the first space.A sequence can be any length and can span multiple lines or be placed on a single line.
The header of a FASTA file can get very ugly, very quickly. I would encourage you to download real sequences from the National Center for Biotechnology Information (NCBI) or look at the files in the 17_synth/tests/inputs directory for more examples.
While it would be fun (for certain values of fun) to teach you how to manually parse this file, I’ll go straight to using Biopython’s Bio.SeqIO module:
>>> from Bio import SeqIO
>>> recs = SeqIO.parse('tests/inputs/1.fa', 'fasta')
The first argument is the name of the input file. As this function can parse many different record formats, the second argument is the format of the data.
I can check the type of recs using type(), as usual:
>>> type(recs) <class 'Bio.SeqIO.FastaIO.FastaIterator'>
I’ve shown iterators a couple of times now, even creating one in Chapter 4.
In that exercise, I used the next() function to retrieve the next value from the Fibonacci sequence generator.
I’ll do the same here to retrieve the first record and inspect its type:
>>> rec = next(recs) >>> type(rec) <class 'Bio.SeqRecord.SeqRecord'>
To learn more about a sequence record, I highly recommend you read the SeqRecord documentation in addition to the documentation in the REPL, which you can view using help(rec).
The data from the FASTA record must be parsed, which means discerning the meaning of the data from its syntax and structure.
If you look at rec in the REPL, you’ll see something that looks like a dictionary.
This output is the same as that from repr(seq), which is used to “return the canonical string representation of the object”:
SeqRecord(
seq=Seq('CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGC...AGG'),
id='Rosalind_6404',
name='Rosalind_6404',
description='Rosalind_6404',
dbxrefs=[])
The multiple lines of the sequence are concatenated into a single sequence represented by a
Seqobject.The
IDof a FASTA record is all the characters in the header starting after the>and continuing up to the first space.The
SeqRecordobject is meant to also handle data with more fields, such asname,description, and database cross-references (dbxrefs). Since those fields are not present in FASTA records, the ID is duplicated fornameanddescription, and thedbxrefsvalue is the empty list.
If you print the sequence, this information will be stringified so it’s a little easier to read.
This output is the same as that for str(rec), which is meant to provide a useful string representation of an object:
>>> print(rec)
ID: Rosalind_6404
Name: Rosalind_6404
Description: Rosalind_6404
Number of features: 0
Seq('CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGC...AGG')
The most salient feature for this program is the record’s sequence.
You might expect this would be a str, but it’s actually another object:
>>> type(rec.seq) <class 'Bio.Seq.Seq'>
Use help(rec.seq) to see what attributes and methods the Seq object offers.
I only want the DNA sequence itself, which I can get by coercing the sequence to a string using the str() function:
>>> str(rec.seq) 'CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTT...AGG'
Note this is the same class I used in the last solution of Chapter 3 to create a reverse complement. I can use it here like so:
>>> rec.seq.reverse_complement()
Seq('CCTCAGAATTATTAGTGGGAGGGAAGGCCGGAAGCCTCAGAGAAACGGTTCTGG...AGG')
The Seq object has many other useful methods, and I encourage you to explore the documentation as these can save you a lot of time.1
At this point, you may feel you have enough information to finish the challenge.
You need to iterate through all the sequences, determine what percentage of the bases are G or C, and return the ID and GC content of the record with the maximum value.
I would challenge you to write a solution on your own.
If you need more help, I’ll show you one approach, and then I’ll cover several variations in the solutions.
Iterating the Sequences Using a for Loop
So far I’ve shown that SeqIO.parse() accepts a filename as the first argument, but the args.file argument will be an open filehandle.
Luckily, the function will also accept this:
>>> from Bio import SeqIO
>>> recs = SeqIO.parse(open('./tests/inputs/1.fa'), 'fasta')
I can use a for loop to iterate through each record to print the ID and the first 10 bases of each sequence:
>>> for rec in recs: ... print(rec.id, rec.seq[:10]) ... Rosalind_6404 CCTGCGGAAG Rosalind_5959 CCATCGGTAG Rosalind_0808 CCACCCTCGT
Take a moment to run those lines again and notice that nothing will be printed:
>>> for rec in recs: ... print(rec.id, rec.seq[:10]) ...
Earlier I showed that recs is a Bio.SeqIO.FastaIO.FastaIterator, and, like all iterators, it will produce values until exhausted.
If you want to loop through the records again, you will need to recreate the recs object using the SeqIO.parse() function.
For the moment, assume the sequence is this:
>>> seq = 'CCACCCTCGTGGTATGGCT'
I need to find how many Cs and Gs occur in that string.
I can use another for loop to iterate each base of the sequence and increment a counter whenever the base is a G or a C:
gc = 0
Initialize a variable for the counts of the G/C bases.
Iterate each base (character) in the sequence.
See if the base is in the tuple containing
GorC.Increment the GC counter.
To find the percentage of GC content, divide the GC count by the length of the sequence:
>>> gc 12 >>> len(seq) 19 >>> gc / len(seq) 0.631578947368421
The output from the program should be the ID of the sequence with the highest GC count, a single space, and the GC content truncated to six significant digits.
The easiest way to format the number is to learn more about str.format().
The help doesn’t have much in the way of documentation, so I recommend you read PEP 3101 on advanced string formatting.
In Chapter 1, I showed how I can use {} as placeholders for interpolating variables either using str.format() or f-strings.
I can add formatting instructions after a colon (:) in the curly brackets.
The syntax looks like that used with the printf() function in C-like languages, so {:0.6f} is a floating-point number to six places:
>>> '{:0.6f}'.format(gc * 100 / len(seq))
'63.157895'
Or, to execute the code directly inside an f-string:
>>> f'{gc * 100 / len(seq):0.06f}'
'63.157895'
To figure out the sequence with the maximum GC count, you have a couple of options, both of which I’ll demonstrate in the solutions:
-
Make a list of all the IDs and their GC content (a list of tuples would serve well). Sort the list by GC content and take the maximum value.
-
Keep track of the ID and GC content of the maximum value. Overwrite this when a new maximum is found.
I think that should be enough for you to finish a solution. You can do this. Fear is the mind killer. Keep going until you pass all the tests, including those for linting and type checking. Your test output should look something like this:
$ make test python3 -m pytest -xv --disable-pytest-warnings --flake8 --pylint --pylint-rcfile=../pylintrc --mypy cgc.py tests/cgc_test.py =========================== test session starts =========================== ... collected 10 items cgc.py::FLAKE8 SKIPPED [ 9%] cgc.py::mypy PASSED [ 18%] tests/cgc_test.py::FLAKE8 SKIPPED [ 27%] tests/cgc_test.py::mypy PASSED [ 36%] tests/cgc_test.py::test_exists PASSED [ 45%] tests/cgc_test.py::test_usage PASSED [ 54%] tests/cgc_test.py::test_bad_input PASSED [ 63%] tests/cgc_test.py::test_good_input1 PASSED [ 72%] tests/cgc_test.py::test_good_input2 PASSED [ 81%] tests/cgc_test.py::test_stdin PASSED [ 90%] ::mypy PASSED [100%] ================================== mypy =================================== Success: no issues found in 2 source files ====================== 9 passed, 2 skipped in 1.67s =======================
Solutions
As before, all the solutions share the same get_args(), so only the differences will be shown.
Solution 1: Using a List
Let’s look at my first solution.
I always try to start with the most obvious and simple way, and you’ll find this is often the most verbose.
Once you understand the logic, I hope you’ll be able to follow more powerful and terse ways to express the same ideas.
For this first solution, be sure to also import List and Tuple from the typing module:
def main() -> None:
args = get_args()
seqs: List[Tuple[float, str]] = []
for rec in SeqIO.parse(args.file, 'fasta'):
gc = 0
for base in rec.seq.upper():
if base in ('C', 'G'):  gc += 1
gc += 1  pct = (gc * 100) / len(rec.seq)
pct = (gc * 100) / len(rec.seq)  seqs.append((pct, rec.id))
seqs.append((pct, rec.id))  high = max(seqs)
high = max(seqs)  print(f'{high[1]} {high[0]:0.6f}')
print(f'{high[1]} {high[0]:0.6f}') 
Initialize an empty list to hold the GC content and sequence IDs as tuples.
Iterate through each record in the input file.
Initialize a GC counter.
Iterate through each sequence, uppercased to guard against possible mixed-case input.
Check if the base is a C or G.
Increment the GC counter.
Calculate the GC content.
Append a new tuple of the GC content and the sequence ID.
Take the maximum value.
Print the sequence ID and GC content of the highest value.
The type annotation List[Tuple[float, str]] on the seqs variable provides not only a way to programmatically check the code using tools like mypy but also an added layer of documentation. The reader of this code doesn’t have to jump ahead to see what kind of data will be added to the list because it has been explicitly described using types.
In this solution, I decided to make a list of all the IDs and GC percentages mostly so that I could show you how to create a list of tuples.
Then I wanted to point out a few magical properties of Python’s sorting.
Let’s start with the sorted() function, which works on strings as you might imagine:
>>> sorted(['McClintock', 'Curie', 'Doudna', 'Charpentier']) ['Charpentier', 'Curie', 'Doudna', 'McClintock']
When all the values are numbers, they will be sorted numerically, so I’ve got that going for me, which is nice:
>>> sorted([2, 10, 1]) [1, 2, 10]
Note that those same values as strings will sort in lexicographic order:
>>> sorted(['2', '10', '1']) ['1', '10', '2']
Now consider a list of tuples where the first element is a float and the second element is a str.
How will sorted() handle this?
By first sorting all the data by the first elements numerically and then the second elements lexicographically:
>>> sorted([(0.2, 'foo'), (.01, 'baz'), (.01, 'bar')]) [(0.01, 'bar'), (0.01, 'baz'), (0.2, 'foo')]
Structuring seqs as List[Tuple[float, str]] takes advantage of this built-in behavior of sorted(), allowing me to quickly sort the sequences by GC content and select the highest value:
>>> high = sorted(seqs)[-1]
This is the same as finding the highest value, which the max() function can do more easily:
>>> high = max(seqs)
high is a tuple where the first position is the sequence ID and the zeroth position is the GC content that needs to be formatted:
print(f'{high[1]} {high[0]:0.6f}')
Solution 2: Type Annotations and Unit Tests
Hidden inside the for loop is a kernel of code to compute GC content that needs to be extracted into a function with a test.
Following the ideas of test-driven development (TDD), I will first define a find_gc() function:
def find_gc(seq: str) -> float:
For now, I return
0. Note the trailing.tells Python this is afloat. This is shorthand for0.0.
Next, I’ll define a function that will serve as a unit test.
Since I’m using pytest, this function’s name must start with test_.
Because I’m testing the find_gc() function, I’ll name the function test_find_gc.
I will use a series of assert statements to test if the function returns the expected result for a given input.
Note how this test function serves both as a formal test and as an additional piece of documentation, as the reader can see the inputs and outputs:
def test_find_gc():
""" Test find_gc """
assert find_gc('') == 0.
assert find_gc('C') == 100.
assert find_gc('G') == 100.
assert find_gc('CGCCG') == 100.
assert find_gc('ATTAA') == 0.
assert find_gc('ACGT') == 50.
If a function accepts a
str, I always start by testing with the empty string to make sure it returns something useful.A single
Cshould be 100% GC.Same for a single
G.Various other tests mixing bases at various percentages.
It’s rarely possible to exhaustively check every possible input to a function, so I often rely on spot-checking.
Note that the hypothesis module can generate random values for testing.
Presumably, the find_gc() function is simple enough that these tests are sufficient.
My goal in writing functions is to make them as simple as possible, but no simpler. As Tony Hoare says, “There are two ways to write code: write code so simple there are obviously no bugs in it, or write code so complex that there are no obvious bugs in it.”
The find_gc() and test_find_gc() functions are inside the cgc.py program, not in the tests/cgc_test.py module.
To execute the unit test, I run pytest on the source code expecting the test to fail:
$ pytest -v cgc.py ============================ test session starts ============================ ... cgc.py::test_find_gc FAILED [100%]
The unit test fails as expected.
The first test passes because it was expecting
0.This test fails because it should have returned
100.
Now I have established a baseline from which I can proceed.
I know that my code fails to meet some expectation as formally defined using a test.
To fix this, I move all the relevant code from main() into the function:
def find_gc(seq: str) -> float:
""" Calculate GC content """
if not seq:
return 0
gc = 0
for base in seq.upper():
if base in ('C', 'G'):
gc += 1
return (gc * 100) / len(seq)
This guards against trying to divide by 0 when the sequence is the empty string.
If there is no sequence, the GC content is 0.
This is the same code as before.
Then I run pytest again to check that the function works:
$ pytest -v cgc.py ============================ test session starts ============================ ... cgc.py::test_gc PASSED [100%] ============================= 1 passed in 0.30s =============================
-
Define a function to test.
-
Write the test.
-
Ensure the function fails the test.
-
Make the function work.
-
Ensure the function passes the test (and all your previous tests still pass).
If I later encounter sequences that trigger bugs in my code, I’ll fix the code and add those as more tests.
I shouldn’t have to worry about weird cases like the find_gc() function receiving a None or a list of integers because I used type annotations.
Testing is useful.
Type annotations are useful.
Combining tests and types leads to code that is easier to verify and comprehend.
I want to make one other addition to this solution: a custom type to document the tuple holding the GC content and sequence ID.
I’ll call it MySeq just to avoid any confusion with the Bio.Seq class.
I add this below the Args definition:
class MySeq(NamedTuple):
""" Sequence """
gc: float
name: str
The GC content is a percentage.
I would prefer to use the field name
id, but that conflicts with theid()identity function which is built into Python.
Here is how it can be incorporated into the code:
def main() -> None:
args = get_args()
seqs: List[MySeq] = []
for rec in SeqIO.parse(args.file, 'fasta'):
seqs.append(MySeq(find_gc(rec.seq), rec.id))
high = sorted(seqs)[-1]
print(f'{high.name} {high.gc:0.6f}')
Use
MySeqas a type annotation.Create
MySequsing the return value from thefind_gc()function and the record ID.This still works because
MySeqis a tuple.Use the field access rather than the index position of the tuple.
This version of the program is arguably easier to read. You can and should create as many custom types as you want to better document and test your code.
Solution 3: Keeping a Running Max Variable
The previous solution works well, but it’s a bit verbose and needlessly keeps track of all the sequences when I only care about the maximum value. Given how small the test inputs are, this will never be a problem, but bioinformatics is always about scaling up. A solution that tries to store all the sequences will eventually choke. Consider processing 1 million sequences, or 1 billion, or 100 billion. Eventually, I’d run out of memory.
Here’s a solution that would scale to any number of sequences, as it only ever allocates a single tuple to remember the highest value:
def main():
args = get_args()
high = MySeq(0., '')
for rec in SeqIO.parse(args.file, 'fasta'):
pct = find_gc(rec.seq)
if pct > high.gc:
high = MySeq(pct, rec.id)
print(f'{high.name} {high.gc:0.6f}')
Initialize a variable to remember the highest value. Type annotation is superfluous as
mypywill expect this variable to remain this type forever.Calculate the GC content.
See if the percent GC is greater than the highest value.
If so, overwrite the highest value using this percent GC and sequence ID.
Print the highest value.
For this solution, I also took a slightly different approach to compute the GC content:
def find_gc(seq: str) -> float:
""" Calculate GC content """
return (seq.upper().count('C') +
seq.upper().count('G')) * 100 / len(seq) if seq else 0
Use the
str.count()method to find theCs andGs in the sequence.Since there are two conditions for the state of the sequence—the empty string or not—I prefer to write a single
returnusing anifexpression.
I’ll benchmark the last solution against this one.
First I need to generate an input file with a significant number of sequences, say 10K.
In the 05_gc directory, you’ll find a genseq.py file similar to the one I used in the 02_rna directory.
This one generates a FASTA file:
$ ./genseq.py -h
usage: genseq.py [-h] [-l int] [-n int] [-s sigma] [-o FILE]
Generate long sequence
optional arguments:
-h, --help show this help message and exit
-l int, --len int Average sequence length (default: 500)
-n int, --num int Number of sequences (default: 1000)
-s sigma, --sigma sigma
Sigma/STD (default: 0.1)
-o FILE, --outfile FILE
Output file (default: seqs.fa)
Here’s how I’ll generate an input file:
$ ./genseq.py -n 10000 -o 10K.fa Wrote 10,000 sequences of avg length 500 to "10K.fa".
I can use that with hyperfine to compare these two implementations:
$ hyperfine -L prg ./solution2_unit_test.py,./solution3_max_var.py '{prg} 10K.fa'
Benchmark #1: ./solution2_unit_test.py 10K.fa
Time (mean ± σ): 1.546 s ± 0.035 s [User: 2.117 s, System: 0.147 s]
Range (min … max): 1.511 s … 1.625 s 10 runs
Benchmark #2: ./solution3_max_var.py 10K.fa
Time (mean ± σ): 368.7 ms ± 3.0 ms [User: 957.7 ms, System: 137.1 ms]
Range (min … max): 364.9 ms … 374.7 ms 10 runs
Summary
'./solution3_max_var.py 10K.fa' ran
4.19 ± 0.10 times faster than './solution2_unit_test.py 10K.fa'
It would appear that the third solution is about four times faster than the second running on 10K sequences. You can try generating more and longer sequences for your own benchmarking. I would recommend you create a file with at least one million sequences and compare your first solution with this version.
Solution 4: Using a List Comprehension with a Guard
Figure 5-1 shows that another way to find all the Cs and Gs in the sequence is to use a list comprehension and the if comparison from the first solution, which is called a guard.
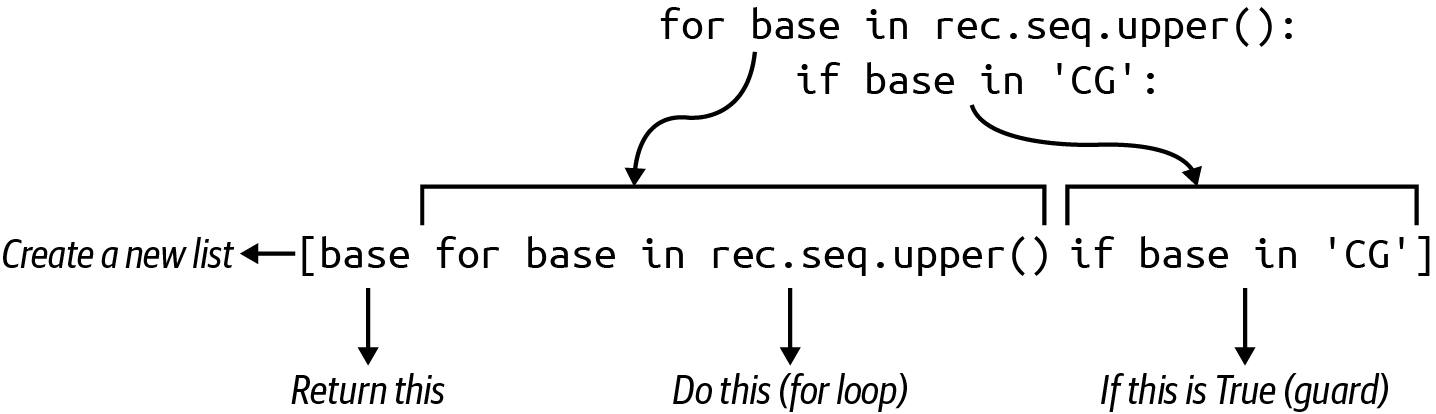
Figure 5-1. A list comprehension with a guard will select only those elements returning a truthy value for the if expression
The list comprehension only yields those elements passing the guard which checks that the base is in the string 'CG':
>>> gc = [base for base in 'CCACCCTCGTGGTATGGCT' if base in 'CG'] >>> gc ['C', 'C', 'C', 'C', 'C', 'C', 'G', 'G', 'G', 'G', 'G', 'C']
Since the result is a new list, I can use the len() function to find how many Cs and Gs are present:
>>> len(gc) 12
I can incorporate this idea into the find_gc() function:
def find_gc(seq: str) -> float:
""" Calculate GC content """
if not seq:
return 0
gc = len([base for base in seq.upper() if base in 'CG'])
return (gc * 100) / len(seq)
Another way to count the Cs and Gs is to select them using a list comprehension with a guard.
Solution 5: Using the filter() Function
The idea of a list comprehension with a guard can be expressed with the higher-order function filter().
Earlier in the chapter I used the map() function to apply the int() function to all the elements of a list to produce a new list of integers.
The filter() function works similarly, accepting a function as the first argument and an iterable as the second.
It’s different, though, as only those elements returning a truthy value when the function is applied will be returned.
As this is a lazy function, I will need to coerce with list() in the REPL:
>>> list(filter(lambda base: base in 'CG', 'CCACCCTCGTGGTATGGCT')) ['C', 'C', 'C', 'C', 'C', 'C', 'G', 'G', 'G', 'G', 'G', 'C']
So here’s another way to express the same idea from the last solution:
def find_gc(seq: str) -> float:
""" Calculate GC content """
if not seq:
return 0
gc = len(list(filter(lambda base: base in 'CG', seq.upper())))
return (gc * 100) / len(seq)
Use
filter()to select only those bases matching C or G.
Solution 6: Using the map() Function and Summing Booleans
The map() function is a favorite of mine, so I want to show another way to use it.
I could use map() to turn each base into a 1 if it’s a C or G, and a 0 otherwise:
>>> seq = 'CCACCCTCGTGGTATGGCT' >>> list(map(lambda base: 1 if base in 'CG' else 0, seq)) [1, 1, 0, 1, 1, 1, 0, 1, 1, 0, 1, 1, 0, 0, 0, 1, 1, 1, 0]
Counting the Cs and Gs would then be a matter of summing this list, which I can do using the sum() function:
>>> sum(map(lambda base: 1 if base in 'CG' else 0, seq)) 12
I can shorten my map() to return the result of the comparison (which is a bool but also an int) and sum that instead:
>>> sum(map(lambda base: base in 'CG', seq)) 12
Here is how I could incorporate this idea:
def find_gc(seq: str) -> float:
""" Calculate GC content """
if not seq:
return 0
gc = sum(map(lambda base: base in 'CG', seq.upper()))
return (gc * 100) / len(seq)
Transform the sequence into Boolean values based on their comparison to the bases C or G, then sum the
Truevalues to get a count.
Solution 7: Using Regular Expressions to Find Patterns
So far I’ve been showing you multiple ways to manually iterate a sequence of characters in a string to pick out those matching C or G.
This is pattern matching, and it’s precisely what regular expressions do.
The cost to you is learning another domain-specific language (DSL), but this is well worth the effort as regexes are widely used outside of Python.
Begin by importing the re module:
>>> import re
You should read help(re), as this is a fantastically useful module.
I want to use the re.findall() function to find all occurrences of a pattern in a string.
I can create a character class pattern for the regex engine by using square brackets to enclose any characters I want to include.
The class [GC] means match either G or C:
>>> re.findall('[GC]', 'CCACCCTCGTGGTATGGCT')
['C', 'C', 'C', 'C', 'C', 'C', 'G', 'G', 'G', 'G', 'G', 'C']
As before, I can use the len() function to find how many Cs and Gs there are.
The following code shows how I would incorporate this into my function.
Note that I use the if expression to return 0 if the sequence is the empty string so I can avoid division when len(seq) is 0:
def find_gc(seq: str) -> float:
""" Calculate GC content """
return len(re.findall('[GC]', seq.upper()) * 100) / len(seq) if seq else 0
Note that it’s important to change how this function is called from main() to explicitly coerce the rec.seq value (which is a Seq object) to a string by using str():
def main() -> None:
args = get_args()
high = MySeq(0., '')
for rec in SeqIO.parse(args.file, 'fasta'):
pct = find_gc(str(rec.seq))
if pct > high.gc:
high = MySeq(pct, rec.id)
print(f'{high.name} {high.gc:0.6f}')
Coerce the sequence to a string value or the
Seqobject will be passed.
Solution 8: A More Complex find_gc() Function
In this final solution, I’ll move almost all of the code from main() into the find_gc() function.
I want the function to accept a SeqRecord object rather than a string of the sequence, and I want it to return the MySeq tuple.
First I’ll change the tests:
def test_find_gc() -> None:
""" Test find_gc """
assert find_gc(SeqRecord(Seq(''), id='123')) == (0.0, '123')
assert find_gc(SeqRecord(Seq('C'), id='ABC')) == (100.0, 'ABC')
assert find_gc(SeqRecord(Seq('G'), id='XYZ')) == (100.0, 'XYZ')
assert find_gc(SeqRecord(Seq('ACTG'), id='ABC')) == (50.0, 'ABC')
assert find_gc(SeqRecord(Seq('GGCC'), id='XYZ')) == (100.0, 'XYZ')
These are essentially the same tests as before, but I’m now passing SeqRecord objects.
To make this work in the REPL, you will need to import a couple of classes:
>>> from Bio.Seq import Seq
>>> from Bio.SeqRecord import SeqRecord
>>> seq = SeqRecord(Seq('ACTG'), id='ABC')
If you look at the object, it looks similar enough to the data I’ve been reading from input files because I only care about the seq field:
SeqRecord(seq=Seq('ACTG'),
id='ABC',
name='<unknown name>',
description='<unknown description>',
dbxrefs=[])
If you run pytest, your test_find_gc() function should fail because you haven’t yet changed your find_gc() function.
Here’s how I wrote it:
def find_gc(rec: SeqRecord) -> MySeq:
The function accepts a
SeqRecordand returns aMySeq.Initialize this to a floating-point
0..This syntax is new as of Python 3.8 and allows variable assignment (first) and testing (second) in one line using the walrus operator (
:=).This is the same code as before.
Return a
MySeqobject.
The walrus operator := was proposed in PEP 572, which notes that the = operator allows you to name the result of an expression only in a statement form, “making it unavailable in list comprehensions and other expression contexts.” This new operator combines two actions, that of assigning the value of an expression to a variable and then evaluating that variable. In the preceding code, seq is assigned the value of the stringified sequence. If that evaluates to something truthy, such as a nonempty string, then the following block of code will be executed.
This radically changes the main() function.
The for loop can incorporate a map() function to turn each SeqRecord into a MySeq:
def main() -> None:
args = get_args()
high = MySeq(0., '')
for seq in map(find_gc, SeqIO.parse(args.file, 'fasta')):
if seq.gc > high.gc:
high = seq
print(f'{high.name} {high.gc:0.6f}')
Initialize the
highvariable.Use
map()to turn eachSeqRecordinto aMySeq.Compare the current sequence’s GC content against the running high.
Overwrite the value.
Print the results.
The point of the expanded find_gc() function was to hide more of the guts of the program so I can write a more expressive program.
You may disagree, but I think this is the most readable version of the program.
Benchmarking
So which one is the winner?
There is a bench.sh program that will run hyperfine on all the solution*.py with the seqs.fa file.
Here is the result:
Summary
'./solution3_max_var.py seqs.fa' ran
2.15 ± 0.03 times faster than './solution8_list_comp_map.py seqs.fa'
3.88 ± 0.05 times faster than './solution7_re.py seqs.fa'
5.38 ± 0.11 times faster than './solution2_unit_test.py seqs.fa'
5.45 ± 0.18 times faster than './solution4_list_comp.py seqs.fa'
5.46 ± 0.14 times faster than './solution1_list.py seqs.fa'
6.22 ± 0.08 times faster than './solution6_map.py seqs.fa'
6.29 ± 0.14 times faster than './solution5_filter.py seqs.fa'
Going Further
Try writing a FASTA parser. Create a new directory called faparser:
$ mkdir faparser
Change into that directory, and run new.py with the -t|--write_test option:
$ cd faparser/ $ new.py -t faparser.py Done, see new script "faparser.py".
You should now have a structure that includes a tests directory along with a starting test file:
$ tree
.
├── Makefile
├── faparser.py
└── tests
└── faparser_test.py
1 directory, 3 files
You can run make test or pytest to verify that everything at least runs.
Copy the tests/inputs directory from 05_gc to the new tests directory so you have some test input files.
Now consider how you want your new program to work.
I would imagine it would take one (or more) readable text files as inputs, so you could define your arguments accordingly.
Then what will your program do with the data?
Do you want it to print, for instance, the IDs and the length of each sequence?
Now write the tests and code to manually parse the input FASTA files and print the output.
Challenge yourself.
Review
Key points from this chapter:
-
You can read
STDINfrom the open filehandlesys.stdin. -
The
Bio.SeqIO.parse()function will parse FASTA-formatted sequence files into records, which provides access to the record’s ID and sequence. -
You can use several constructs to visit all the elements of iterables, including
forloops, list comprehensions, and the functionsfilter()andmap(). -
A list comprehension with a guard will only produce elements that return a truthy value for the guard. This can also be expressed using the
filter()function. -
Avoid writing algorithms that attempt to store all the data from an input file, as you could exceed the available memory on your machine.
-
The
sorted()function will sort homogeneous lists of strings and numbers lexicographically and numerically, respectively. It can also sort homogeneous lists of tuples using each position of the tuples in order. -
Formatting templates for strings can include
printf()-like instructions to control how output values are presented. -
The
sum()function will add a list of numbers. -
Booleans in Python are actually integers.
-
Regular expressions can find patterns of text.
1 As the saying goes, “Weeks of coding can save you hours of planning.”