Université Sorbonne Paris Nord, LIMICS, Sorbonne Université INSERM, UMR 1142, Bobigny, France
In this chapter, we will see how to mix Python methods and OWL logical constructors within the same class.
10.1 Adding Python methods to OWL classes
With Owlready, OWL classes are Python classes (almost) like the others. It is therefore possible to include Python methods in these classes. Here is a simple example to calculate the price per tablet of a drug from its unit price (per box) and the number of tablets in the box:
... class price(Drug >> float, FunctionalProperty): ... pass
... class nb_tablet(Drug >> int, FunctionalProperty): ... pass
...
... class Drug(Thing):
... def get_price_per_tablet(self):
... return self.price / self.nb_tablet
Note that the Drug class is defined twice: the first definition is a forward declaration in order to be able to use the class in the definitions of the properties (see 5.8). Note also that, since we create a new ontology, we integrated the separator (here, #) at the end of the ontology IRI (see 5.1).
The method can then be called on the individuals of the class:
>>> my_drug = Drug(price = 10.0, nb_tablet = 5)
>>> my_drug.get_price_per_tablet()
2.0
In ontologies, it is common to use only classes and subclasses, instead of individuals (this is the case in, e.g., Gene Ontology), because the power of class representation is greater. In this case, Python allows you to define “class methods” which will be called on the class (or one of its subclasses) and which take this class (or subclass) as the first parameter.
Here is the same example as before, but using classes:
>>> with onto:
... class Drug(Thing): pass
...
... class price(Drug >> float, FunctionalProperty): ... pass
... class nb_tablet(Drug >> int, FunctionalProperty): ... pass
...
... class Drug(Thing):
... @classmethod
... def get_price_per_tablet(self):
... return self.price / self.nb_tablet
The method can then be called on the class and its subclasses:
>>> class MyDrug(Drug): pass
>>> MyDrug.price = 10.0
>>> MyDrug.nb_tablet = 5
>>> MyDrug.get_price_per_tablet()
2.0
Be careful, however, to make the two types of method (individual and class) coexist together; it is necessary to use different method names.
10.2 Associating a Python module to an ontology
When the ontologies are not created entirely in Python (as we did in the preceding example) but loaded from an OWL file, the Python methods can be defined in a separate Python module (.py file). This file can be imported manually or linked to the ontology via an annotation; in this case, Owlready will automatically import the Python module when the ontology is loaded.
For example, the following file, named “bacteria.py”, adds a method in the Bacterium and Staphylococcus classes of the bacteria ontology:
Note that we have not loaded the bacteria ontology (i.e., we have not called .load()) because it will be done by the main program. Note also that we have not indicated the superclass of Staphylococcus (which is Bacterium): indeed, it already appears in the OWL file, so there is no need to assert it a second time here! On the other hand, it is necessary to mention Thing as a superclass to state that the new class is an OWL class managed by Owlready and not a usual Python class. Generally, when creating a separate Python file with the methods, it is preferable to put only the methods inside and to keep the rest of the ontology (superclasses, properties, relations, etc.) in OWL to limit redundancy.
10.2.1 Manual import
We can then load the ontology and manually import the file “bacteria.py”:
>>> from owlready2 import *
>>> onto = get_ontology("bacteria.owl").load()
>>> import bacteria
Then, we create a Staphylococcus, and we call our method:
>>> my_bacterium = onto.Staphylococcus()
>>> my_bacterium.my_method()
It is a staphylococcus!
10.2.2 Automatic import
For this, it is necessary to edit the ontology with Protégé and add an annotation indicating the name of the associated Python module. This annotation is called python_module, and it is defined in the ontology “owlready_ontology.owl”, which it is necessary to import. Here are the steps:
1.
Launch Protégé and load the bacteria ontology.
2.
Go to the “Active Ontology” tab of Protégé.
3.
Import the ontology “owlready_ontology” by clicking the “+” button to the right of “Direct imports”. The ontology can be imported from the local copy which is in the installation directory of Owlready or from its IRI: www.lesfleursdunormal.fr/static/_downloads/owlready_ontology.owl.
Figure 10-1
“python_module” annotation in Protégé
4.
Add an annotation in the “Ontology header” section. The annotation type is “python_module”, and the value is the name of the module, here bacteria (see Figure 10-1). You may also use a Python package, for example, “my_module.my_package”.
Now, we no longer need to import the “bacteria” module: Owlready does this automatically each time the bacteria ontology is loaded. In the following example, we have saved the bacteria ontology (with the annotation “python_module”) in a new OWL file called “bacteria_owl_python.owl”:
We have seen in section 7.2 that, during the reasoning, the classes of individuals and the superclasses of classes could be modified. In this case, the available methods may change. In addition, in the case of polymorphism, that is to say, when several classes implement the same method differently, the implementation of the method for a given individual or class may change. This is “polymorphism with type inference”.
We created a bacteria. When we execute the method, it is the implementation of the class Bacterium which is therefore called. We will now call the reasoner.
>>> sync_reasoner()
The reasoner deduced that the bacterium is in fact a Staphylococcus (due to its relationships). Now, if we call the method, it is the implementation of the Staphylococcus class which is called:
>>> my_bacterium.my_method()
It is a staphylococcus!
10.4 Introspection
Introspection is an advanced object programming technique which consists in “analyzing” an object without knowing it, for example, in order to obtain the list of its attributes and their values.
For the introspection of individuals, the get_properties() method allows obtaining the list of properties for which the individual has at least one relation.
>>> onto.unknown_bacterium.get_properties()
{bacteria.has_shape,
bacteria.has_grouping,
bacteria.gram_positive,
bacteria.nb_colonies}
It is then possible to obtain and/or modify these relations. The getattr(object, attribute) and setattr(object, attribute, value) Python functions allow you to read or write an attribute of any Python object when the name of the attribute is known in a variable (see 2.9.4), for example:
>>> for prop in onto.unknown_bacterium.get_properties():
The returned values are the same as with the usual syntax “individual.property”: it is a single value for the functional properties and a list of values for the other properties. However, when doing introspection, it is often easier to treat all properties generically, whether they are functional or not. In this case, the alternative syntax “property[individual]” is preferable because it always returns a list of values, even when called on functional properties, for example:
>>> for prop in onto.unknown_bacterium.get_properties():
For class introspection, the get_class_properties() method works similarly to that of individuals. It returns the properties for which the class has at least one existential restriction (or universal, depending on the type of class property; see 6.3):
>>> onto.Pseudomonas.get_class_properties()
{bacteria.gram_positive,
bacteria.has_shape,
bacteria.has_grouping}
Owlready considers the parent classes, but also the equivalent classes. The syntax “property[class]” can be used to obtain and/or modify the existential restrictions of classes.
Finally, the INDIRECT_get_properties() and INDIRECT_get_class_properties() methods work in the same way, but also return indirect properties (i.e., inherited from a parent class).
In addition, the constructs() method allows you to browse all the constructors that refer to a class or a property. For example, we can look for the constructors referring to the InSmallChain class:
Here, we get only one construct, which is an intersection including an existential restriction with the class InSmallChain as value. We can then use this constructor’s subclasses() method to get a list of all the classes that use it:
We thus find the Streptococcus class in which we had placed this restriction (see 3.4.8).
10.5 Reading restrictions backward
The restrictions make it possible to define relationships at the level of the classes of the ontology, for example, “Pseudomonas has_shape some Rod”. Owlready provides easy access to these relationships with the syntax “Class.property” (see 4.5.4):
>>> onto.Pseudomonas.has_shape
bacteria.Rod
But how to read this existential restriction “backward”, that is to say, from the Rod class, go back to the Pseudomonas class? Even if we had defined the reverse property, which we could call “is_shape_of”, it would not answer our question, as the following example shows:
>>> with onto:
... class is_shape_of(ObjectProperty):
... inverse = onto.has_shape
>>> onto.Rod.is_shape_of
[]
Indeed, from a logical point of view, the following two propositions are different:
“Pseudomonas has_shape some Rod”
“Rod is_shape_of some Pseudomonas”
The first indicates that all Pseudomonas have a Rod shape, which is true. The second indicates that all Rod shapes are the shape of a Pseudomonas, which is not the same meaning (and is not true). For example, the Rod shape of a rugby ball is not the shape of a Pseudomonas.
Similarly, for the following two propositions:
“Nucleus is_part_of some Cell”
“Cell has_part some Nucleus”
The first indicates that every nucleus is part of a cell. The second indicates that every cell has a nucleus, which is different: in biology, the first proposition is true, while the second is false (e.g., bacteria are cells without nuclei).
However, it is sometimes useful to be able to read existential relationships backward. Owlready allows it: this can be done by combining the constructs() and subclasses() methods, as we did in the previous section. The inverse_restrictions() method automates this:
Note that we used set() to display the generator returned by inverse_restrictions(), after removing the duplicates.
10.6 Example: Using Gene Ontology and managing “part-of” relations
Gene Ontology (GO) is an ontology widely used in bioinformatics (see 4.7). GO consists of three parts: biological processes, molecular functions, and cell components. This third part describes the different elements of a cell: membranes, nucleus, organelles (such as mitochondria), and so on. It is particularly complex to manage because it includes both a hierarchy of “classic” inheritance using is-a relationships and a “part-of” relationship hierarchy. In this second hierarchy, called meronymy, the cell is decomposed into subparts, then into sub-subparts, and so on. The root of this hierarchy is therefore the entire cell, and the leaves the indivisible parts.
OWL and Owlready have relationships and methods to manage the inheritance hierarchy (subclasses(), descendants(), ancestors(), etc.; see 4.5.3). On the other hand, there is no standard OWL relation for meronymy nor specific methods in Owlready. We will see here how to add to the GO classes methods to access the subparts and super-parts, taking into account both the part-of and the is-a relations.
GO being quite large (almost 200 MB), loading takes several tens of seconds or even a few minutes, depending on the power of the computer and the download time of the OWL file. We will therefore load GO and store the Owlready quadstore in a file (see 4.7). In addition, we will use manual import here to associate our Python methods with OWL classes (see 10.2.1), so as not to have to modify GO by adding a “python_module” annotation.
GO uses arbitrary identifiers which are not directly understandable by humans. The following table summarizes the GO identifiers that we will need later:
This module defines four class methods in the class GO_0005575 (i.e., cellular_component). subparts() allows obtaining all the subparts of the component. This method takes into account the relationships BFO_0000051 (has-part) but also the relationships BFO_0000050 (part-of) read backward, contrary to what we would have obtained with .INDIRECT_BFO_0000051 (see 6.3). The transitive_subparts() method returns the subparts in a transitive manner, taking into account the child classes and the transitivity (if A is a subpart of B and B is a subpart of C, then A is also a subpart of C). The superparts() and transitive_superparts() methods work the same way for super-parts.
We can then import this module and access GO and “part-of” relationships. In the following example, we are looking at the part-of relationships of the nucleolus, which is a component located in the nucleus of the cell:
>>> from owlready2 import *
>>> from go_part_of import *
>>> nucleolus = go.search(label = "nucleolus")[0]
>>> print(nucleolus.subparts())
[GO_0005655:nucleolar ribonuclease P complex,
GO_0030685:nucleolar preribosome,
GO_0044452:nucleolar part,
GO_0044452:nucleolar part,
GO_0101019:nucleolar exosome (RNase complex)]
>>> print(nucleolus.superparts())
[GO_0031981:nuclear lumen]
Here, direct relationships (without taking transitivity into account) are not very informative. Transitive relationships are much richer:
>>> nucleolus.transitive_subparts()
{GO_0034388:Pwp2p-containing subcomplex of 90S preribosome,
GO_0097424:nucleolus-associated heterochromatin,
GO_0005736:DNA-directed RNA polymerase I complex,
GO_0005731:nucleolus organizer region,
GO_0101019:nucleolar exosome (RNase complex),
[...] }
>>> nucleolus.transitive_superparts()
{GO_0031981:nuclear lumen,
GO_0005634:nucleus,
GO_0043226:organelle,
GO_0044464:cell part,
GO_0005623:cell,
GO_0005575:cellular_component,
[...] }
10.7 Example: A “dating site” for proteins
Now, we will use the functionality of the “go_part_of.py” module to create a “dating site” for proteins. This site allows you to enter two protein names, and the site determines in which compartments of the cell they can meet (if an encounter is possible!). From a biological point of view, this is important because two proteins that do not have a common “meeting site” cannot interact together.
For this, we will use
The Flask Python module to make a dynamic website (see 4.12).
The MyGene Python module to perform searches on the MyGene server and retrieve the GO concepts associated with each of the two proteins. This module allows you to do search on genes (and the proteins they code). MyGene is used as follows:
import mygene
mg = mygene.MyGeneInfo()
dico = mg.query(’name:"<gene_name>"’,
fields = "<searched fields>",
species = "<species>",
size = <number of genes to search for>)
The call to MyGene returns a dictionary which itself contains lists and other dictionaries. For example, we can search for all of the GO terms associated with insulin as follows:
>>> import mygene
>>> mg = mygene.MyGeneInfo()
>>> dict = mg.query(’name:"insulin"’,
... fields = "go.CC.id,go.MF.id,go.BP.id,"
... species = "human",
... size = 1)
>>> dict
{’max_score’: 13.233688, ’took’: 17, ’total’: 57,
’hits’: [{’_id’: ’3630’, ’_score’: 13.233688,
’go’: {’BP’: [{’id’: ’GO:0002674’},
{’id’: ’GO:0006006’}, [...] ]}}]}
“Go.CC.id”, “go.MF.id”, and “go.BP.id” represent the three main parts of GO (cellular components, molecular functions, and biological process, respectively). For our dating site, we will only use “CC”. Although they originate from Gene Ontology, these actually describe the localization in the cell of the gene product, that is, the protein (in general), and not the gene itself (the genes normally remain in the nucleus, for eukaryotic cells).
More information is available on the MyGene website:
Owlready and Gene Ontology (GO) to make the semantic intersection of the GO terms describing the cellular compartments of the two proteins. A “simple” intersection (in the set sense of the term) is not sufficient: the intersection must take into account both the “is-a” relations of inheritance and the “part-of” relations. For example, a protein A present only in the membranes and a protein B present only in the mitochondria may meet in the membrane of the mitochondria. Indeed, the mitochondria membrane is a membrane, and it is a part of the mitochondria, as shown in the following diagram:
The following program describes the protein dating site. It begins by importing and initializing all of the modules:
Owlready
The “go_part_of” module that we created in the previous section
Flask
MyGene
Then, the search_protein() function is defined. It takes as input a protein name (in English), such as “insulin”, and returns all of the GO terms of the cellular component type (“CC”) associated with it in MyGene. For this, we check that at least one result (hit in English) is found, and then we get the “CC”. If only one CC is found, MyGene returns it; otherwise, it is a list. To facilitate processing, we systematically create a list called cc. Then we go through this list and extract the GO identifier. The identifiers returned by MyGene are of the form “GO: 0002674” and not “GO_0002674” as in the OWL version of GO. So we replace all “:” with “_”. Finally, we recover the concept of the corresponding ontology using the obo namespace (which was imported from the go_part_of module).
The semantic_intersection() function performs the semantic intersection of two sets containing GO concepts of cellular components in four steps:
1.
We create two sets, subparts1 and subparts2, containing the components associated with each of the two proteins as well as their subparts in a transitive way. For this, we reuse the static method transitive_subparts() that we defined in the module go_part_of.py in the previous section. We then have the sets of all the components where each of the two proteins can be encountered, taking into account the is-a and part-of relations.
2.
We compute the intersection of these two sets with the operator “&” (see 2.4.7 for sets in Python), and we call the result common_components.
3.
We now have to simplify the common_components set. Indeed, it includes the concepts that we are looking for, but also all their descendants and their subparts (in the previous example with “membrane” and “mitochondria”, we therefore have “membrane of the mitochondria” but also “inner membrane of the mitochondria” and “outer membrane of the mitochondria”). In order to speed up the processing of the next step, we first create a cache (using a dictionary). This cache matches each GO concept in common_components with all of its (transitive) subparts.
4.
We create a new set, largest_common_components, which is empty at the beginning. We add to it all the concepts of common_components which is not a subpart of another concept in common_components. Note the use of “else” in the “for” loop, which allows you to execute instructions when the loop has iterated over all items (that is to say, no “break” has been encountered; see 2.6). Finally, we return largest_common_components.
The rest of the program defines two web pages with Flask. The first (path “/”) is a basic form with two text fields to enter the names of the proteins and a button to validate. The second (path “/result”) computes and displays the result. It first calls the search_protein() function twice, once for each protein, then the semantic_intersection() function. Finally, it generates a web page displaying the components associated with the first protein, the second, and the components where they are likely to meet.
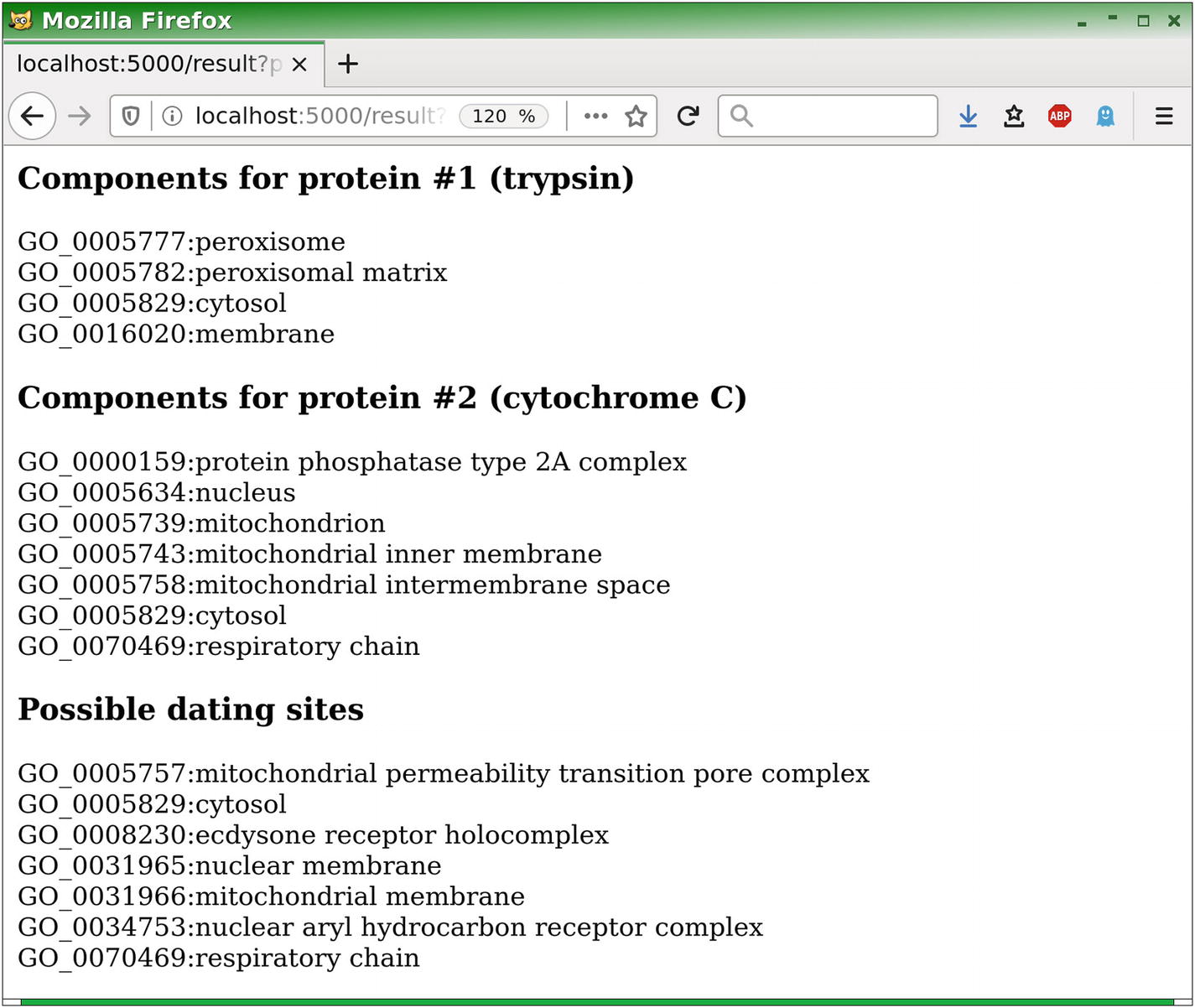
In order to test our dating site, here are some examples of protein names: trypsin, cytochrome C, insulin, insulin-degrading enzyme, insulin receptor, glucagon, hemoglobin, elastase, granzyme B, decorin, beta-2-microglobulin, and so on.
The following screenshots show the dating site obtained and its use:
10.8 Summary
In this chapter, you have learned how to mix Python and OWL in order to associate Python methods with an OWL class having a rich semantics. We have also seen how to perform introspection on OWL classes and entities.

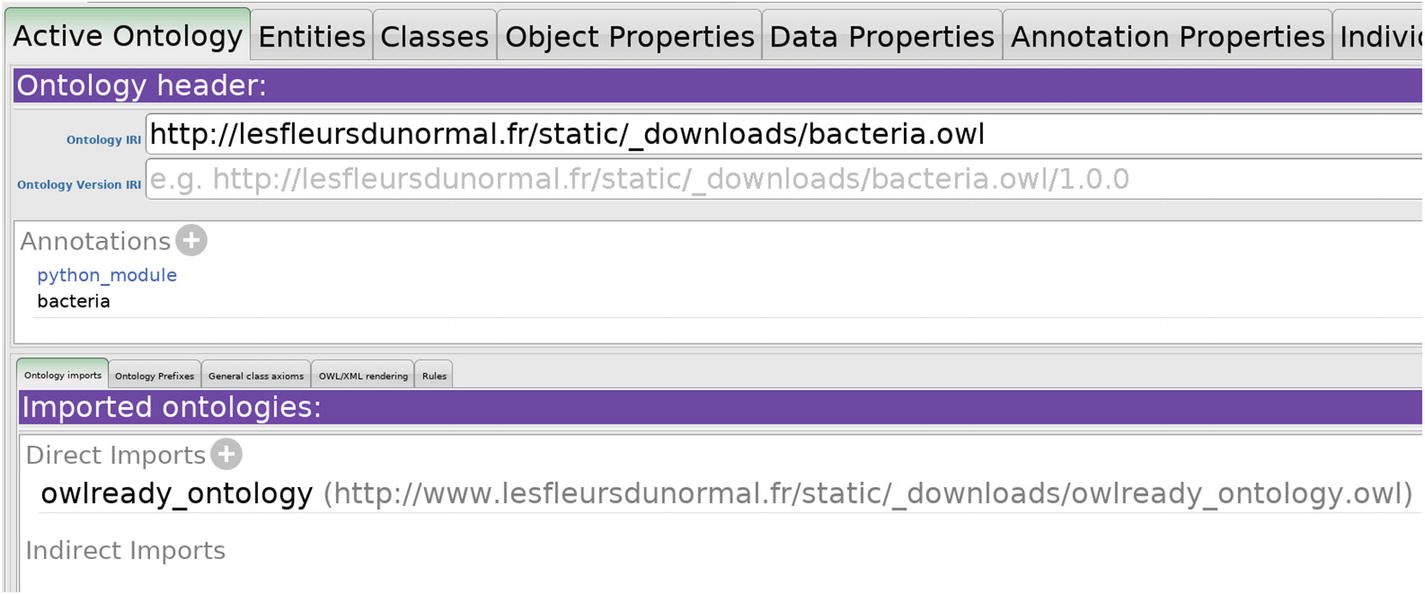 Figure 10-1
Figure 10-1