
Chapter 4: Target identification and validation
Stephanie Kay Ashendena; Natalie Kurbatovab; Aleksandra Bartosikc a Data Sciences and Quantitative Biology, Discovery Sciences, R&D, AstraZeneca, Cambridge, United Kingdom
b Data Infrastructure and Tools, Data Science and Artificial Intelligence, R&D, AstraZeneca, Cambridge, United Kingdom
c Clinical Data and Insights, Biopharmaceuticals R&D, AstraZeneca, Warsaw, Poland
Abstract
Target identification and target validation are involved with the identification and validation of a target that can be modulated for a therapeutic purpose. Whilst there is much variation on what a target is and how to define one, we can consider a therapeutic target to be one that is involved with the mechanisms of a particular disease and modulation of it will affect the disease in a beneficial way. It is important to also note that when a drug fails, it tends to be due to safety concerns or lack of efficacy.
There are several computational methods that are applied within the target identification stages of drug discovery, including cheminformatic methods, such as similarity searching and network-based methods. However, much information is also contained within databases and scientific publications.
Keywords
Target identification; Target validation; Knowledge graphs; Target prediction; Gene prioritization; Natural language processing
Introduction
Target identification and target validation are involved with the identification and validation of a target that can be modulated for a therapeutic purpose. Whilst there is much variation on what a target is and how to define one, we can consider a therapeutic target to be one that is involved with the mechanisms of a particular disease and modulation of it will affect the disease in a beneficial way. It is important to also note that when a drug fails, it tends to be due to safety concerns or lack of efficacy.1
There are several computational methods that are applied within the target identification stages of drug discovery, including cheminformatic methods, such as similarity searching and network-based methods.2 However, much information is also contained within databases and scientific publications.
Whilst here we primarily focus on small molecules as the main drug modality, it must be noted, that there are wide variety of modalities beyond small molecules and antibodies, and therefore, it remains to be seen how much of the genome can be drugged effectively. Efforts to expand further into and utilize more of the druggable genome have been undertaken such as the initiative called Illuminating the Druggable Genome of which started in 2013.3 However, it is important to note the vast array of modalities that are now available. The druggable genome is the genes that encode proteins that can potentially be modulated. It is thought that of the approximately 20,000 genes that make up the human genome, approximately 3000 of these are considered part of the druggable genome despite only a few hundred targets being currently exploited.3 The program aims to create a resource for the study of protein families.3 The program along with Nature Reviews Drug Discovery publish content on understudied protein targets frequently and have included examples like Kna1.1 channels as potential therapeutic targets for early-onset epilepsy and PIP5K1A as a potential target for cancers that have KRAS or TP53 mutations.4 Again, it is important to note that developments in technology have allowed for new modalities to be used as therapeutic agents.5 Such an example is the approvals of ASOs (two) by the FDA in 2016.6
Animal models are another approach to understand human biology. A series of challenges named systems biology verification for Industrial Methodology for PROcess Verification in Research (sbv IMPROVER) was designed to address translatability between rodents and humans.7 This initiative saw scientists apply computational methodologies on datasets containing phosphoproteomics, transcriptomics, and cytokine data from both human and rat tissues that were exposed to various stimuli.7 A key finding was that even similar computational method had diverse performance and there was no clear “best practise”.7 Despite this, the computational methods were able to translate some of these stimuli and biological processes between the human and rat.7
Despite such successes in translatability, animal studies have been considered poor predictors of human responses for a very long time. Bracken explored this idea of why animal studies are often poor predictors of human reactions to exposure.8 Bracken summarized the reasoning as to why there is often a lack of translatability between animal studies and human trials is that many animal experiments do not appear to be designed and performed well.8 There may be other issues such as limitations in methods and biases.8 Despite this, Denayer and Van Roy explain that animal models allow for a greater understanding of biology, disease, and medicine.9
Targeting the wrong target can lead to failure of the development of the drug and therefore result in cost, not just financially, but in time as well.1 Failli and co-authors discuss that to improve this process, many developed platforms rely on estimates of efficacy that have been derived from computational methods that create scores of target-disease associations.1 Failli and co-authors also present novel methods that consider both efficacy and safety by using gene expression data, transcriptome based, as well as tissue disease-specific networks.1
Defining what a drug target is crucial but is not well defined.10 Santos and co-authors10 highlighted the need to keep accurate and current record of approved drugs and the target in which the drugs modulation results in therapeutic effects.10 The authors note several resources including databases and papers that contain this information but note that it is still difficult to find a complete picture.10 Owing to this, the authors attempt to annotate the current list of FDA-approved drugs (up to when published).
The understanding of what makes a good drug target is important in target identification and the drug discovery process as a whole. Gashaw and co-authors11 have previously discussed what a good drug target is and also detail what the target evaluation at Bayer HealthCare involved at time of writing. The authors highlight that first a disease is identified that does not currently have treatment and then the identification of a drug target follows. Target assessment follows comprising of experimental target assessment methods, theoretical drugability assessment, and thoughts on target-related/stratification biomarkers. Alongside this adverse event evaluation and intellectual property is carefully considered.11 These steps are important and investigate key areas of a target such as whether the target actually has a role within the disease of interest as well as whether the target can be modulated without adverse events.
Predicting drug target interactions is an area of great interest in the drug discovery process. Predicting such interactions can help to save time and money. The term drug prediction can be an ambiguous term. It can refer to the identification of a target of which will modulate the disease of interest (related to target focused approaches), but also can refer to the identification of a target that a drug is binding to (more related to phenotypic approaches). Unlike in targeted drug discovery approaches, phenotypic approaches do not have prior knowledge of a specific target or what role that target places within a disease.
The large number of resources available for aiding in the discovery of a drug target have been summarized by an online article by BiopharmaTrend of which listed 36 different web resources for identifying targets.12 This list includes databases such as WITHDRAWN,13 which is a resource for withdrawn and discontinued drugs, DisGeNET,14 which is focused on target-disease relationships as is Open Targets.15, 16
Open Targets uses human genetic and genomic data to aid in target identification and prioritization. The website describes that the Open Targets Platform has public data to aid in this task and the Open Targets Genetics Portal uses GWAS (Genome Wide Association Studies of which aims to associate particular variations with diseases) and functional genomics to identify targets. The data integration is complemented with large-scale systematic experimental approaches.15
Drug-target databases mentioned by Santos and co-authors included the Therapeutic Targets Database,17 of which focuses on known and explored therapeutic protein and nucleic acid targets, as well as the diseases, pathway information and any corresponding drugs. DrugBank,18 contains bioinformatic and cheminformatic resources for combining drug data and drug target information. Next, the authors mentioned SuperTarget19 which includes various drug information for target proteins. Finally, the authors mention the International Union of Basic and Clinical Pharmacology and British Pharmacology Society Guide to Pharmacology Database.20 This resource aggregates target information and separates targets into eight different target classes. Namely, GPCRs, Ion channels, Nuclear hormone receptors, Kinases, Catalytic receptors, Transporters, Enzymes, and other protein targets. It has been argued that focusing on a class of targets that are known to have key roles can provide beneficial outcomes in early drug discovery phases.21 The thought is that it will reduce changes for technical failure due to the diversification of the target space, and thus available binding pockets.21 This in turn increases potential clinical applications.21
These resources are important for understand the mechanisms of disease. Understanding the mechanism of disease is an important step in identifying the targets involved. However, a difficulty with understanding disease is that regardless of the technology used, capturing the complete biological picture, with all of its complexities, is not possible in most human diseases.22 The use of integrating multiple technologies such as multiomics is used to capture a more complete picture. For example, genetic data and transcriptomic data have been combined for the purpose of using gene expression profiles to identify disease-related genes for prioritization and drug repositioning.23, 24
Target identification predictions
As previously mentioned, target identification can relate to two distinct questions. First, we have a compound, what is the protein it is associated with where we have a linkage between a compound and a protein. Second, we have a disease what is the protein involved where there is a linkage between protein and disease.
Gene-disease association data have been used to predict therapeutic targets using a variety of classifiers and validating the results by mining scientific literature for the proposed therapeutic targets.25 Protein-protein interactions have also been used to predict genes involved in disease.26 Predicting mutations that are associated with disease is also of interest such as is demonstrated with the tool BorodaTM (Boosted RegressiOn trees for Disease-Associated mutations in TransMembrane) proteins.27
Several tools have been developed to aid in drug-target prediction with many different strategies in place. For example, Bayesian approaches, such as the tool BANDIT for predicting drug binding targets28 as well as deep learning approaches, such as the tool deepDTnet that is network based.29 Many methods have been summarized by Mathai and co-authors in their paper discussing validation strategies for target prediction methods.30 Here we have picked out a handful of key methods to discuss.
The SwissTargetPrediction tool uses multiple logistic regression to combine the different similarity values from shape and fingerprint compound similarities.31 HitPick is another online service32 for target prediction where a user uploads their compounds in SMILES format and uses 2D molecular similarity-based methods such as nearest neighbor similarity searching and machine learning methods that are based on naive Bayesian models (Laplacian modified).32 The chemical similarity is measured using Tanimoto coefficients. The tool SEA33 stands for the similarity ensemble approach and performs the predictions based on the chemical similarity of the compounds. The compounds have been annotated into sets (based on the receptor that the ligand modulates) and then the Tanimoto coefficient similarity score between these sets was calculated. Another method named SPiDER34 use self-organizing maps to identify targets. SLAP is a tool that uses a network approach and is able to make links between related data and these data can be of differing types.35, 36 Finally, PIDGIN,37, 38 which stands for Prediction Including Inactivity uses nearly 200 million bioactivity data points containing both active and inactive compounds from ChEMBL and PubChem for target prediction.
The validation strategies that are used in target prediction have been previously discussed and note that most strategies that are used are not accounting for biases in the data. The authors30 suggest external validation of the parameterized models and compared with the results from the internal validation.30
Mathai and co-authors30 summarized such validation strategies for target prediction methods. The authors note that it is important to understand how the data were validated, based on the data partitioning schemes (how the data is split into train and test sets) used, as well as how the performance was measured, based on the metrics used). The authors note that performance assessment for target prediction methods do not often take into account biases in the data and that new metrics are needed. However, they do suggest several steps that can be taken to represent a more realistic view on the prediction performance. Examples of their suggestions include using a combination of metrics and methods should be used, the minima, maxima, and distributions should be reported. The use of stratified sampling and external data should be used.30 The authors also discuss the validation strategies used by various tools including most of the examples shown above in Table 1.
Table 1
| Tool | Notes | Website | Validation strategy | References |
|---|---|---|---|---|
| SwissTargetPrediction | Online tool using both 2D and 3D similarity | http://www.swisstargetprediction.ch/ | Cross validation | 31,39,40 |
| HitPick | Online tool molecular similarity-based methods | http://mips.helmholtz-muenchen.de/hitpick/ | Large data set | 32 |
| SEA | Online tool using chemical similarity to a set of reference molecules | http://sea.bkslab.org/ | Experimental validation | 33 |
| SPiDER | Uses self-organizing maps along with CATS pharmacophore and molecular operating environment descriptors | http://modlabcadd.ethz.ch/software/spider/ | Cross validation | 34 |
| SLAP | Network-based approach links different but related types of information | http://cheminfov.informatics.indiana.edu:8080/slap/ | Large data set | 35,36 |
| PIDGIN | Bernoulli Naive Bayes algorithm | https://github.com/lhm30/PIDGIN | Cross validation and other datasets | 37,38 |
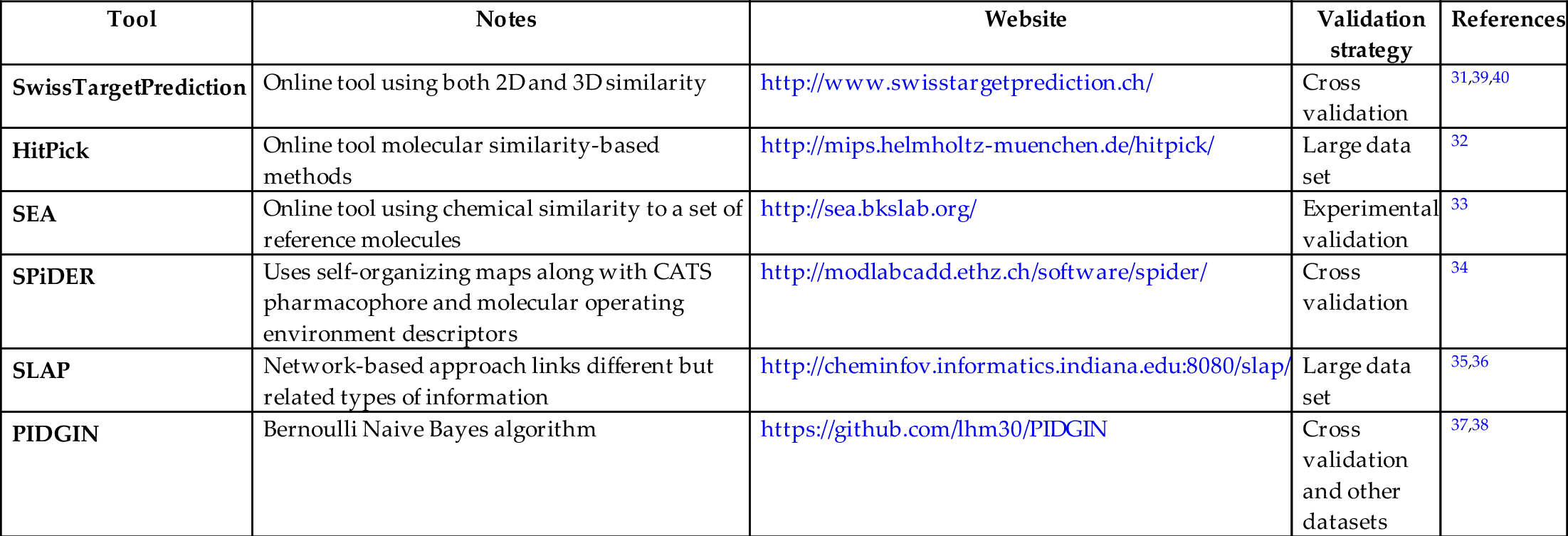
When validating targets, methods are required to identify the most complexing targets that have been identified. MANTIS_ML41 is a tool that uses genetic screens from HTS that uses machine learning to rank disease-associated genes where the authors showed good predictive performance when applied to chronic kidney disease and amyotrophic lateral sclerosis. The tool works by compiling features from multiple sources, followed by an automated machine learning preprocessing step with exploratory data analysis and finally stochastic semisupervised learning.41 These features are derived from sources such as disease-specific resources such as CKDdb,42 a database containing multiomic studies of chronic kidney disease, data filtered by tissue and disease such as from the GWAS catalog43 and resources that are more generic like the Genome Aggregation Database (GnomAD)44 and the Molecular Signatures database (MSigDB),45 of which has the purpose of combining exome and genome sequencing data from large-scale sequencing projects.41
In some cases, a compound will have been discovered to modulate a protein for a particular disease, but the actual target is not known. For example, the observed change in protein expression that is the result of a target upstream interacting with a compound. The structure of the target can also be predicted, and deep learning has been applied to such effect. The distance between pairs of residues can be predicted via neural networks even with fewer homologous sequences.46
Gene prioritization methods
After identifying a list of genes, it is often useful to prioritize this list to focus the search, ideally starting with the most likely target gene. Methods for gene prioritization vary in their approaches from network-based methods to matrixes. Moreau and Tranchevent47 previously reviewed computational tools for prioritizing candidate genes and noted that the overall aim is to ingrate data for the purpose of identifying the most promising genes for validation. It often utilizes prior knowledge such as in the form of keywords that describe the phenotype or known gene associations to a particular process or phenotype. Furthermore, with a number of gene prioritization tools available, methods to benchmark such gene prioritization tools have been undertaken with the authors48 (Guala and Sonnhammer) recommending the use of Gene Ontology49 data alongside the tool FunCoup.50, 51 Because of the cost of identifying and validating new genes that are involved in diseases, many of the algorithms have had the performance measured by using known disease genes.52
Many of these tools rely on different types of data and algorithms to perform gene prioritization. For example, the use of genomic data fusion, such as is the case with the gene prioritization tool ENDEAVOUR.53, 54 Other approaches include Beegle,55 of which mines the literature to find genes that have a link to the search query. It then uses ENDEAVOUR to perform the gene prioritization step. Cross-species approaches have been proposed such as in the case of GPSy.56 The use of Bayesian matrix factorization using both genomic and phenotypic information has also been implemented.57 Network based approaches are also used in gene prioritization tasks.
Such network-based tools included Rosalind which uses knowledge graphs to predict linkages between disease and genes that uses data integration (in graph format) and relational inference.58 MaxLink, which is a guilt-by-association network which aims to identify and rank genes in relation to a given query list.59 NetRank is based on the algorithm used in the ToppGene Suite60, 61 and uses the Page Rank with priors algorithm where a random surfer is more likely to end up in nodes that are initially relevant.60, 62 Other algorithms include, NetWalk, which is based on the random walk with restarts algorithm which operates by walking from a node to another randomly but at any time the walk can be restarted (based on probability).52Using not only the shortest path interaction but also taking into the global interactome structure has been shown to have performance advantages.52 NetProp is based on the network propagation algorithm of which was designed to address the issue of many approaches using only local network information.
GUILD63 (Genes Underlying Inheritance Linked Disorders), uses four network-based disease-gene prioritization algorithms, namely, NetShort (considers node importance for a given phenotype will have a shorter distance to other seed nodes within the network), NetZcore (normalizes the scores of nodes in the network in relation to a set of random networks with similar topology and therefore considers the relevance of the node for a given phenotype), NetScore (considers multiple shortest paths from source to target) and NetCombo (averaged normalization score for each of the other three prioritization algorithms). GUILD also includes NetRank, NetWalk, NetProp, and fFlow of which is based on the FunctionalFlow algorithm.60, 64 This algorithm generalizes the guilt-by-association method by considering groups of proteins that may or may not physically interact with each other.64 In fFlow, the scores annotated from nodes with higher scores “flow” toward nodes with lower scores depending on the edge capacity.60 The authors who describe the FunctionalFlow algorithm compare it with three others, Majority64, 65 (consider all neighboring proteins and calculate how many times each annotate occurs for each protein), Neighborhood64, 66 (considers all proteins within a radius and then for each function consider if it is over-represented), and GenMultiCut. The authors note there are different considerations of GenMultiCut with two instances agreeing that functional annotations on interaction networks should be made with the purpose of reducing the occurrences of different annotations that are associated with neighboring proteins.64
Machine learning is also used in gene prioritization tasks. In the case of Amyotrophic Lateral Sclerosis, Bean and co-authors published a knowledge-based machine learning approach that used a variety of available data to predict new genes involved in Amyotrophic Lateral Sclerosis.67 Isakov and co-authors used machine learning gene prioritization tasks to identify risk genes for inflammatory bowel disease and the authors performed a literature search to identify whether particular genes had been published alongside inflammatory bowel disease.68 Prioritizing loci from genome wide association studies (GWAS) studies is another important prioritization task that machine learning has aided in.69 Nicholls and co-authors reviewed the machine learning approaches used in loci prioritization and noted that GWAS results require aid in sorting through noise as well and understanding which are the most likely causal genes and variants.
Machine learning and knowledge graphs in drug discovery
Introduction
The rapid accumulation of unstructured knowledge in the biomedical literature and high throughput technologies are primary sources of information for modern drug discovery.
Knowledge graph concept allows linking of vast amounts of biomedical data with ease. It provides many algorithmic approaches, including machine learning, to answer pharmaceutical questions of drug repositioning, new target discovery, target prioritization, patient stratification, and personalized medicine.70–73
There is no single definition of a knowledge graph. The term in its modern meaning appeared in 2012 when Google announced its new intelligent model of the search algorithm. From the list of definitions, the most descriptive one is the following:
“A knowledge graph acquires and integrates information into an ontology and applies a reasoner to derive new knowledge”.74
A knowledge graph is a graph in its classical graph theory definition. The underlying mathematical graph concept distinguishes the knowledge graph from other knowledge-based concepts. Besides, it is a semantic graph when we encode the meaning of the data alongside the data in the form of ontology. The main idea here is that we are collecting data into the self-descriptive graph by integrating data and metadata. In all applications and domains, the knowledge graph is a flexible structure allowing to change, delete, and add new data. This dynamism requires an accurate gathering of provenance and versioning information.
Reasoning and the fact that knowledge graph being a graph in the mathematic sense, are fundamental characteristics of the knowledge graph.
Knowledge graphs are usually large structures. For example, Google’s Knowledge graph by the end of 2016 contained around seventy thousand millions of connected facts.75 Typical knowledge graph in biopharmaceutical domain consists of millions of nodes and a thousand-million of edges. Significant volumes of data are applicable for reasoning with the help of machine learning techniques. The bonus of the graph structure is the set of graph theory algorithms.
Graph theory algorithms
Graph theory is a well-established mathematical field that started in the 18th century with Leonhard Euler paper on the Seven Bridges of Königsberg.
Mathematically, we are defining a graph as a set of nodes connected by edges: G = (N, E), where N is a set of nodes also called vertices, and E is a set of two-nodes, whose elements are called edges. An undirected graph is a graph where all the edges are bidirectional. In contrast, a graph where the edges point in a direction is called a directed graph.
Both nodes and edges can have properties that play the role of weights in different algorithms (Fig. 1).
In the context of knowledge graphs, the important graph theory algorithms that are used either directly or as feature extraction techniques for machine learning:
- • connected components,
- • shortest path,
- • minimum spanning tree,
- • variety of centrality measures, and
- • spectral clustering.
“Connected components” is a collective name for algorithms which find clusters/islands. Within the biomedical domain, this class of algorithms operates to identify protein families, to detect protein complexes in protein-protein interactions networks, to identify technological artefacts.76
"Shortest path" is an algorithm to find the shortest path along edges between two nodes. The most famous example of this class is Dijkstra's algorithm.77 In biological knowledge graphs, this algorithm is used to calculate network centralities.
“Minimum Spanning tree” is an algorithm that helps to find a subset of the edges of a connected, edge-weighted undirected graph that connects all the nodes, without any cycles and with the minimum possible total edge weight. The greedy algorithm, commonly in use is Kruskal's algorithm.78 In the drug discovery domain, this class of graph theory algorithms is used to define disease hierarchies, cluster nodes, compare biological pathways, and subgraphs.
A widely accepted fact in biological data analyses is that in most graphs, some nodes or edges are more important or influential than others. This importance can be quantified using centrality measures.79 We often use centrality measures as features for machine learning models. Here is the list of commonly used centrality measures:
- • Degree centrality is simply the number of connections for a node.
- • Closeness centrality indicates how close a node is to all other nodes in the graph.80
- • Betweenness centrality quantifies how many times a particular node comes in the shortest chosen path between two other nodes.81
- • Eigenvector centrality is a measure of the influence of a node in a network. It assigns relative scores to all nodes in the network based on the concept that connections to high-scoring nodes contribute more to the score of the node in question than equal connections to low-scoring nodes.82 Popular Google's Pagerank algorithm is a variant of the eigenvector centrality (Fig. 2).83
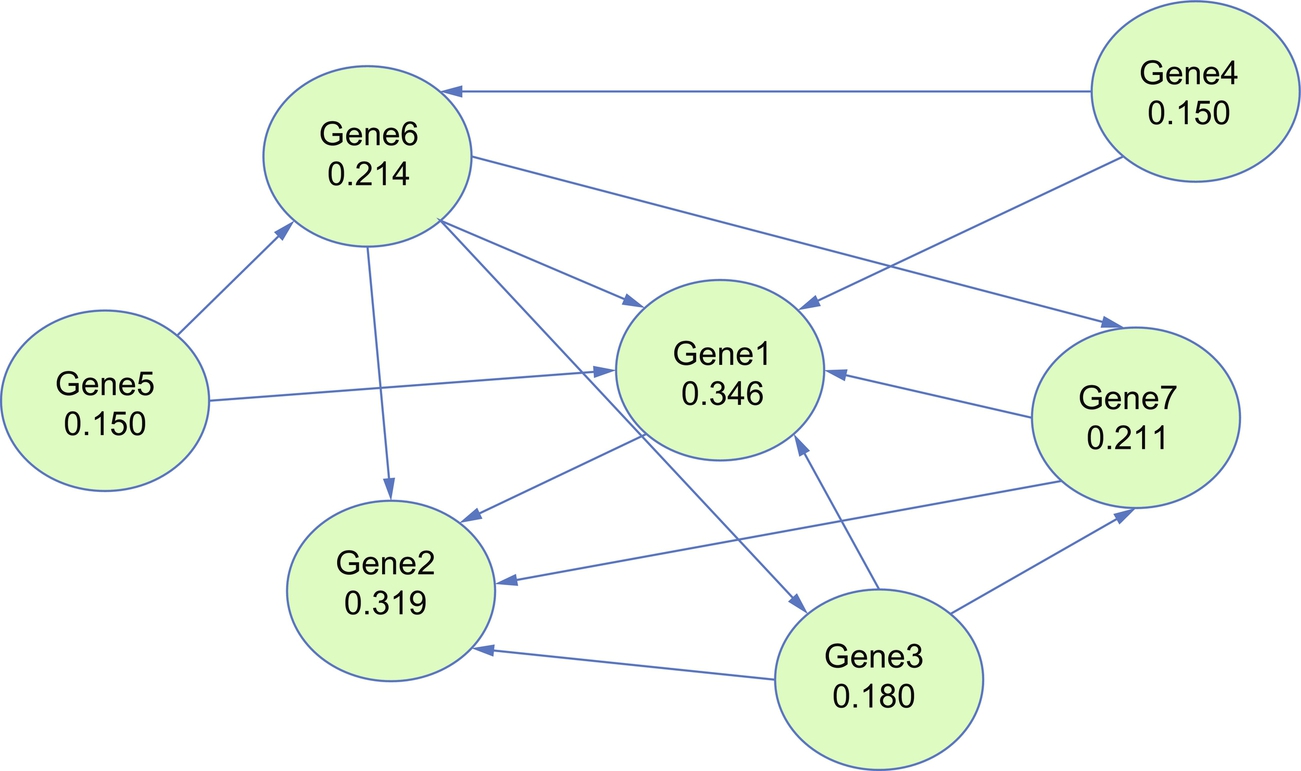
Fig. 2 ArticleRank (the variant of the PageRank algorithm) results. Gene1 has the highest centrality score.
We apply centrality measures for nodes prioritization in drug discovery knowledge graphs, for instance, to find the essential proteins/genes for a particular disease, and to find the artefacts.
Spectral clustering algorithm has three primary stages:
- • construction of a matrix representation of the graph (Laplacian matrix),
- • computation of eigenvalues and eigenvectors of the matrix and mapping of each point to a lower-dimensional representation based on one or more eigenvectors, and
- • finally, clustering of points based on the new representation.
Spectral clustering is used as an unsupervised feature learning technique for machine learning models.
Graph-oriented machine learning approaches
In general, machine learning approaches on graph structures can be divided into two broad categories:
- • feature extraction from the graph followed by an application of machine learning and
- • graph-specific deep network architectures that directly minimize the loss function for a downstream prediction task.
Feature extraction from graph
Feature extraction or feature learning from the graph can be done in a supervised, semisupervised, and unsupervised manner using classic graph algorithms and machine learning techniques. Our ultimate goal in the feature learning from a graph is to extract the information encoded in graph structure to cover diverse patterns of graphs.
One of the examples of unsupervised feature learning for feature extraction from a graph is the application of spectral clustering algorithms. This explicit deterministic approach provides mathematically defined features that cover graph patterns, but it is computationally expensive and hard to scale. Another example of the usage of graph algorithms for feature extraction is centrality measures (Table 2).
Table 2
| Gene | Centrality measure (ArticleRank) |
|---|---|
| Gene1 | 0.346 |
| Gene2 | 0.319 |
| Gene3 | 0.180 |
| Gene4 | 0.150 |
| Gene5 | 0.150 |
| … |
As an alternative to the direct application of graph algorithms, representation learning approaches started to win the race of feature extraction from graphs.73, 84–86, This type of representation learning is a semisupervised method with an objective to represent local neighborhoods of nodes in the created features.
A broadly used representation learning algorithm is node2vec.84 Given any graph, it can learn continuous feature representations for the nodes, which can then be used for various downstream machine learning tasks. The algorithm creates new features also called embeddings, or compressed representations by using random walks together with graph theory connected components, algorithmic ideas and simple neural network with one layer. The algorithm node2vec consists of three steps:
- • Sampling. A graph is sampled by random walks with two parameters p and q that can be tuned specifically for the downstream analysis task. Parameter q controls the discovery of the broader neighborhood, and parameter p controls the search of the microscopic view around the node.
- • Training. The method uses the skip-gram network that accepts a node from the sampling step as a one-hot vector as an input and maximizes the probability for predicting neighbor nodes using backpropagation on just single hidden-layer feedforward neural network.
- • Computing embeddings. Embeddings are the output of a hidden layer of the network (Fig. 3).
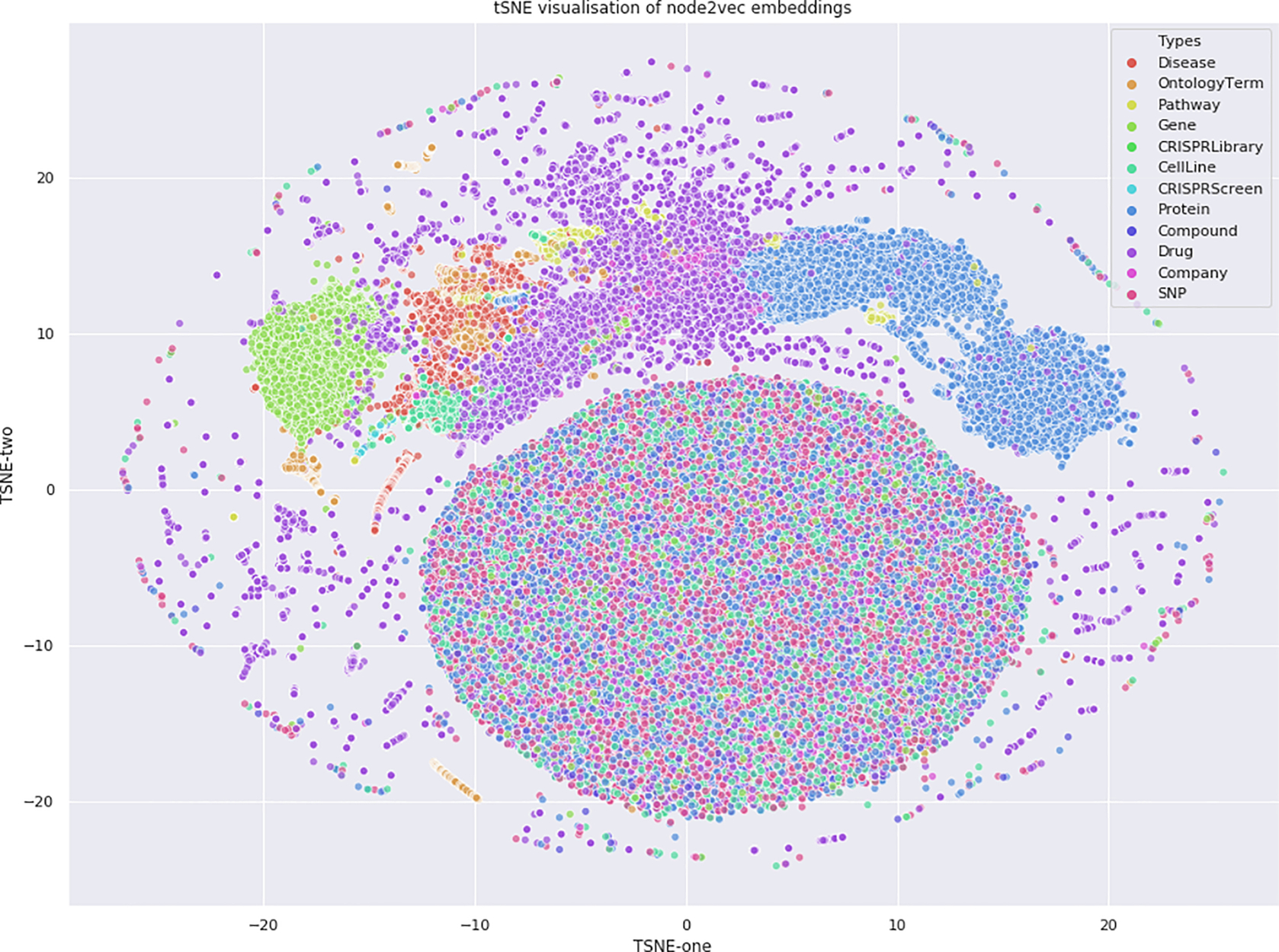
Fig. 3 node2vec embeddings used for tSNE plot. Shows the clustering of the node types in embedded space.
Tensor decomposition is another embedding technique we are using in knowledge graphs since graph can be represented as a third-order binary tensor, where each element corresponds to a triple, 1 indicating a true fact and 0 indicating the unknown.
There are many approaches to factorize the third-order binary tensor,87–89 amongst them relatively complex nonlinear convolutional models,90, 91 and simple linear models like RESCAL87 and TuckER.92 The latest is based on Tucker decomposition93 of the binary tensor of triples into a core tensor multiplied by a matrix along with each mode. Rows of the matrices contain entity and relation embeddings, while entries of the core tensor determine the level of interaction between them. This approach uses a mathematically principled widely studied model. TuckER’s results for edge prediction are compatible with the nonlinear models. Besides, tensor decomposition model can serve as a baseline for more elaborate models, for instance by incorporating graph algorithms.
We use node2vec, tensor decomposition and other embedding techniques94–96 in pharmaceutical knowledge graphs to predict the most probable labels of the nodes, for instance, functional labels of genes/proteins, and possible side effects.
Another application of embedding techniques is edge prediction, when we wish to predict whether a pair of nodes in a graph should have an edge connecting them, for instance, to discover novel interactions between genes.
After the feature extraction, the standard preprocessing ML methods can be used to reduce dimensionality following by preferable ML classification or regression methodology for the downstream task.
Graph-specific deep network architectures
There are supervised machine learning approaches specially developed for graph structures.
These methods base on graph-specific deep network architectures that match the structure of the graphs and directly minimize the loss function for a downstream prediction task using several layers of nonlinear transformations. The basic idea is the creation of a special graph neural network layer that is able to interact with the knowledge graph directly. It takes a graph as input and performs computations over the structure.97 By propagating information between nodes principally using the graph edges, it is possible to maintain the relational inductive biases present in the graph structure.98 The relational inductive bias term means that we are forcing an algorithm to prioritize structural, relational solutions, independent of the observed data. A generic example of inductive bias in machine learning techniques is a regularization term.
Drug discovery knowledge graph challenges
As we mentioned before the volumes of structured data that are coming from -omics technologies and natural language processing give us an opportunity to link together masses of research papers and experimental results to build knowledge graphs that connect genes, diseases, cell lines, drugs, side effects, and other interconnected biomedical information in a systematic semantic manner.
However, there are a lot of challenges to deal with: trustworthiness of the resources the original data are coming from, different experimental designs and technological biases, noisy data, usage of different ontologies that do not always have cross-references, and ascertainment biases. Some of these challenges have solutions in the field of data science dealing with data unification.99 Data integration for biomedical knowledge graph requires assigning of weights for the data that are coming from different sources and have different levels of trust. Both data unification and data integration are extremely time-consuming tasks.
Even having an ideal drug discovery knowledge graph with ideally unified and integrated data, we would have computational challenges as well. Most of the techniques mentioned in the previous sections have been developed for the social networks, paper references, and web pages ranking, where there are limited types of nodes and relations. Biomedical graphs differ from these original domains significantly—we are dealing with the diversity of node and edge types.
Besides, in the real world, we have a lot of noise amongst biomedical data, lack of negative experimental results, biases toward particular disease areas. All mentioned earlier biomedical data challenges requires careful, systematic testing of different methods, including the search processes of the most relevant subgraphs for a particular drug discovery task and methods for the evaluation and prioritization of the resources data are coming from.
Data, data mining, and natural language processing for information extraction
What is natural language processing
Natural language processing (NLP) is ultimately about accessing information fast and finding the relevant parts of the information. It differs from text mining in that if you have a large chunk of text, in text mining you could search for a specific location such as London. In text mining, you would be able to pull out all the examples of London being mentioned in the document. With NLP, rather than asking it to search for the word London, you could ask it to bring back all mentions of a location or ask intelligent questions such as where an individual lives or which English cities are mentioned in the document. It takes into consideration the surrounding information as well. To summarize, natural language processing is concerned with processing the interactions between source data, computers, and human beings.
How is it used for drug discovery and development
Implementing an NLP pipeline can be completed with packages such as NLTK (Natural Language Toolkit)100 and spacy101 in python102 and there are many tutorial on line.103 This tutorial is summarized here. The first step with natural language processing is to process sentences one word at a time. This is known as word tokenization. For example, take the sentence “What drugs were approved last year?”, you would split this into individual word tokens; “what,” “drugs,” “were,” “approved,” “last,” “year” and “?”. We can then remove punctuation which is often useful as it is treated as a separate token and you may find punctuation being highlighted as the most common token. However, punctuation can really affect the meaning of a sentence. It is also possible to remove stop words which are those that occur frequently such as “and” or “the” and therefore can be beneficial to remove to focus on the more informative text. Next, we predict the parts of speech for example determining the type of word (noun, adjective, etc.) and how each word is separated. Following this, lemmatization is performed where we reduce a word to its simplest form. For example, run, runner and running would be reduced to the lemma formation run. These all are referring to the same concept and reducing to the lemma formation helps a computer interpret as such. The next stage is known as dependency parsing which works out how all the words within a sentence relate to each other. Following this, there is a range of analysis we can perform next such as named entity recognition where a model predicts what each word (or if you have done further processing steps to identify phrases rather than single words) refers to such as names, locations, organizations, and objects. Steps like coreference resolution (identifying expressions that are related to a particular entity)104 can be used to identify what we are talking about even after the name has been mentioned for example: “Random forest is a popular machine learning algorithm. It can perform both classification and regression tasks” The “it” on its own may be difficult to determine what we are talking about but taking the whole two sentences we can understand that in this case “it” refers to the random forest algorithm. This step is difficult and as technology improves—we should expect to see this improve.
The use of ontologies (relationship structure within a concept) decrease linguistic ambiguity by mapping words to standardized terms and to establish a hierarchy of lower and higher-level terms105 MedDRA is an example of the medical dictionary used by HCPs, pharmaceutical companies and regulators.106 Another example includes SNOMED CT, which standardized clinical terminology with the use of concept codes, descriptions, relationships, and reference sets.107 One can download more ontologies dedicated to drug safety-related NLP from online repositories, for example, BioPortal or via NCBO Ontology Recommender Service.
Several NLP techniques have become standard tools available in text processing tools. For example, a search engine such as Apache Solr has built-in functions that allow for: tokenization (splitting text into tokens), stemming (removing stems from words), ignoring diacritics (which sometimes is helpful for non-English texts), conversion to lowercase words, applying phonetic algorithms like Metaphone. Omics technologies generate a lot of data that needs computational approaches to pull out the useful information, by means of data mining.23 These data can be used to help identify and validate potential drug targets.
Another essential NLP method in NLP is building the n-gram model of a corpus (a collection of texts). N-grams are tuples (2-gram is a doublet, 3-gram is triplet, and 4-gram is a quadruplet) of letters or words that appear consecutively in the original text, for example, when the text is "GAATTC," the 3-grams are "GAA," "AAT," "ATT," and "TTC". N-grams are most commonly used in machine translations and computational biology, especially in analyzing DNA and RNA sequences (e.g., finding sequences with similar n-gram profiles). A generalization of an n-gram is a skip-gram in which the elements constituting a group do not have to be next to each other in the original sequence [e.g., grouping Nth word with (N + 2)th word]. Google OpenRefine free online tool allows user to apply n-gram fingerprints for noisy data curation, transformation, and mining.
N-gram is a special case of Markov models. A simple Markov model called Markov chain is a set of possible states together with probabilities of transitioning between two states (every pair A, B of states has attached probabilities of the transition A → B and B → A). Markov models identified high cumulative toxicity during a phase II trial ifosfamide plus doxorubicin and granulocyte colony-stimulating factor in soft tissue sarcoma in female patients.108
Another NLP technique which has become popular in recent years is word2vec. It relies on word n-grams or skip-grams to find which words have a similar meaning. word2vec relies on the distributional hypothesis: that words with similar distributions in sentences have similar meanings. The internal representation of every word in such word2vec model is a numerical vector, usually with hundreds or more dimensions (hence the "vec" part in the name of the method). An interesting property of this method is that the word vectors can be assessed for their similarity (e.g., by computing a dot product of two vectors), or even added or subtracted (e.g., a word2vec model can be trained to compute that "king − man + woman = queen" which in other words could be expressed as "king is to man what queen is to woman"). This model has lots of potential in natural language applications, but there are also other applications such as BioVec, an application of word2vec to proteins and gene vectors.109 Extracting drug-drug interactions110 and drug repurposing111 are two more examples of word2vec application.
Where is it used in drug discovery and development (and thoughts on where it is going at the end)
NLP has applications in summarizing documents, speech recognition, and chatbots. A recent patent112 has been filled which details a system and a method for delivering targeted advertising by receiving verbal requests, processing, and interpreting the utterance and selecting advertisement based on the determined context.
Apache Solr has used for indexing both structured and unstructured data from studies in DataCelerate platform as a part of BioCelerate’s Toxicology Data Sharing Initiative113 launched in May 2018. The platform aims to integrate high-quality toxicology data from various sources and different pharmaceutical companies and scientific institutions. Linguamatics IQVIA I2E AMP 2.0 tool also has been used to search and analyze ADRs and drug-drug interactions.114 Chillbot115 online NLP app integrates gene names with PubMed publications used to discover relations with other genes. Not only literature search can be automated but also the experimental design itself. Hybrow (Hypothesis Browser) developed in Nigam Shah lab is a system for computer-aided hypothesis evaluation, experimental design and model building.116
BioBERT (Bidirectional Encoder Representations from Transformers for Biomedical Text Mining)117 is described by the authors as a “pretrained biomedical language representation model” that can be used for text mining of biomedical content. It is based on the pretrained BERT model (Bidirectional Encoder Representations from Transformers model)118 which allows for tokens to be analyzed in a multidirectional (left to right and right to left) manner which can help with understanding context).
BeFree is a system that was developed to identify relationships between entities such as drugs and genes with diseases.119 The authors found that only some of the gene-disease associations that were discovered were also collected in curated databases highlighting the need to find alternative ways of curating information rather than just manually. DigDee is another example of a tool using NLP for data extraction of disease–gene relationships. This method also takes into account biological events such as phosphorylation and mutations and the authors argue this may represent abnormal behavior caused by the disease.120
Adverse event detection is another area of use for NLP in the drug discovery and understanding pipeline. This has been an area of consideration for some time and in one study, Melton and Hripcsak used NLP to assess discharge summaries of patients and was found to be an effective method.121
Another proposed use for natural language processing relates to mining historic clinical narratives which could reveal important information. In the study,122 the authors focus on chronic diseases, stating that globally there are growing incidences of chronic illnesses and electronic health records are used to analyze patient data that are used to aid chronic disease risk prediction. Historic data can be used to support these predictions.