
Chapter 3. A Survey of Antibiotics
Each antibiotic has its own features that influence its suitability for a particular pathogen. This chapter introduces the major antibiotic classes. We then use fluoroquinolones to illustrate how antibiotics evolve. We also briefly consider antiseptics and disinfectants, because they help us kill pathogens on surfaces and because their use may contribute to the emergence of antibiotic resistance. If you are unfamiliar with the synthesis of DNA, RNA, and protein, you may find Appendix A, “Molecules of Life,” and Appendix B, “Microbial Life,” useful when considering processes blocked by antibiotics.
Antibiotics Are Selective Poisons
Antibiotics are relatively small molecules (about 20 to 100 times the size of a water molecule) that interfere with normal life processes of microbes and viruses. Human cells differ enough from pathogens for antibiotics to act selectively. For example, our cells lack walls whereas bacterial cells have them. Consequently, penicillin, which blocks cell wall synthesis, is specific to bacteria. Penicillin has adverse effects, but they arise from other properties. (Some people are allergic to the drug.)
Three general aspects of antibiotics are important when considering effectiveness. First, some antibiotics only block growth (static compounds), whereas others also kill cells (cidal or lethal compounds). Some drugs are static with one pathogen and lethal with another. For example, rifampicin kills Mycobacterium tuberculosis but blocks only the growth of Escherichia coli at concentrations usually used. The distinction is important, because static drugs allow the microbes to resume growth when the compound disappears from the body. Fortunately, the human immune system is effective at reducing the number of pathogens in an infection; consequently, static agents, such as tetracycline, can be effective treatments for some diseases. A second important feature is the molecular mechanism of antibiotic action. For example, agents that cause pathogens to break apart (lyse) cause the release of toxic microbial molecules into the patient. Those toxins can lengthen the time needed to recover from disease. A third issue is whether an antibiotic is a broad-spectrum agent or specialized for use with one pathogen species. For most treatment situations, the infecting pathogen is not identified. Treating with a broad-spectum agent allows effective treatment to begin immediately and often without the added expense of diagnostic tests. However, with diseases that require long treatment periods, such as tuberculosis, specialized agents that cause less damage to our normal bacterial flora are preferred. These narrow-spectrum agents are also less likely to select resistant mutants of other pathogens that may co-infect patients. Such benefits are not limited to tuberculosis; consequently, narrow-spectrum agents are likely to become more popular as rapid molecular diagnostic methods become more convenient.
Antibiotics Are Found in a Variety of Ways
Paul Ehrlich, Alexander Fleming, and Gerhard Domagk pioneered the development of antibiotics early in the Twentieth Century (see Box 3-1). Ehrlich made a variety of chemical derivatives that he examined to find ones that worked. Domagk followed in Ehrlich’s footsteps with the first agents that were widely used in clinical practice. Their general approach of testing many compounds has evolved into screening procedures that are now applied to hundreds of thousands of molecules. These methods, which are discussed in Chapter 9, “Making New Antibiotics,” are built on basic research that identifies potential drug targets.
Fleming found natural antibiotics. To obtain antibiotics from natural sources, samples from those sources are first incubated with a test microbe to determine whether the sample blocks growth. A positive sample is next split into parts with laboratory procedures that separate molecules into different test tubes for analysis. Eventually, the antibiotic molecules are isolated in pure form, and chemical analysis reveals the structure of the active form. Then medicinal chemists increase potency and safety by modifying the structure.
Occasionally, an antibiotic is discovered as a byproduct of efforts to find other agents, as was the case with salvorsan (refer to Box 3-1). In the early 1960s, scientists at Sterling Drug Company sought superior versions of quinine, a drug that had long been effective against the protozoan that causes malaria. Among the derivatives they synthesized was a compound that killed Gram-negative bacteria. This substance, called nalidixic acid, was used for many years as a treatment for urinary tract infections. The study of chemical derivatives of nalidixic acid led to the fluoroquinolones, a group of potent antibacterials. With drug discovery, scientists follow many leads that have dead ends. However, some lines of work yield successful products, as briefly described in the following pages and listed in Table 3-1.
Table 3-1. Antibiotic Classes and Resistance Mechanisms
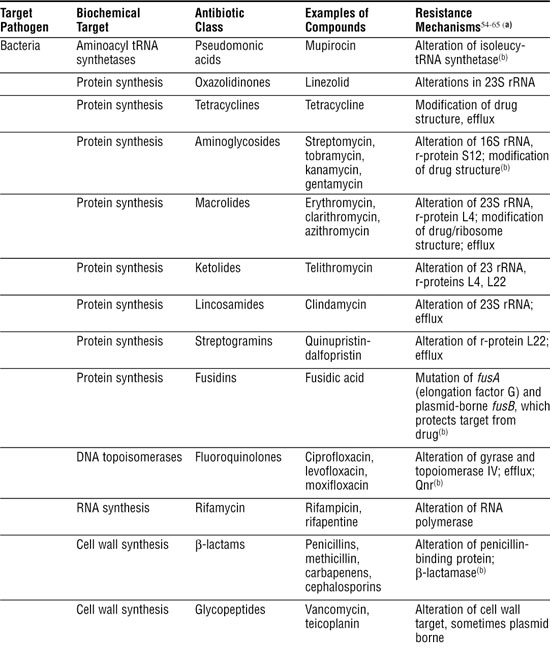
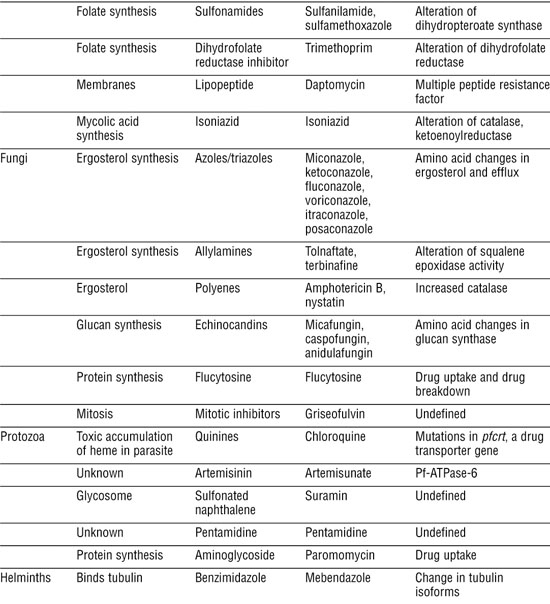
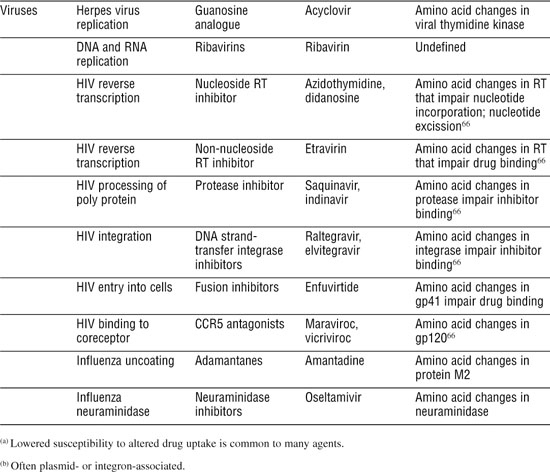
Antibacterial Agents Usually Attack Specific Targets
Much of our success with antibiotics has been with antibacterials, partly because bacterial biochemistry differs in many ways from human biochemistry and partly because bacteria are easy to grow and test for antibiotic susceptibility. Next, we briefly describe the major classes according to the processes they block.
Most of our antibacterial agents interfere with protein synthesis, a process that involves the interaction of many macromolecules. Protein synthesis can be quite sensitive to chemical inhibition. For example, the bioterror agent ricin, which is extremely toxic to humans, acts by inactivating ribosomes, the workbenches where proteins are made. Bacterial ribosomes are composed of two subunits (one called small or 30S and the other called large or 50S). The two ribosome subunits, which must come together for protein synthesis to occur, are composed of large RNA molecules (rRNA) and about 50 different proteins. As an early step in the process, more than 20 enzymes (aminoacyl tRNA synthetases) individually connect more than 20 transfer RNAs to 20 different amino acids. Each enzyme recognizes one type of tRNA and joins it to one type of amino acid to form an aminoacyl tRNA. Messenger RNA (mRNA) binds to ribosomes, aminoacyl tRNAs bind to mRNAs attached to ribosomes, amino acids covalently join, and newly made protein separates from ribosomes. Interference with any step can block bacterial growth.
The enzymes that join specific tRNA molecules to their cognate amino acids (aminoacyl tRNA synthetases) are inhibited by a drug called mupirocin. Because mupirocin has side effects when taken internally, it is generally restricted to external use. One of its current applications is clearing S. aureus from the noses of healthcare workers and newly admitted hospital patients during efforts to eradicate staphylococci and MRSA from hospitals.
Linezolid is a member of the oxazolidinones, the newest group of antibacterial agents. Linezolid blocks initiation of protein synthesis by binding to the small ribosome subunit, preventing it from joining with the large subunit. The drug is used mainly for treatment of infections caused by Gram-positive pathogens, because linezolid has almost no activity with Gram-negative bacteria. Linezolid received approval from the Food and Drug Administration in 2000, and in 2001 the first resistant mutant of S. aureus was reported. Linezolid use is carefully guarded to restrict the emergence of resistance.
The aminoglycosides bind to ribosomal proteins and ribosomal RNA, thereby blocking a variety of steps in protein synthesis. For example, streptomycin, one of our oldest antibiotics, attaches to a protein of the small ribosome subunit and locks the mRNA-ribosome complex in place. That prevents proper movement of mRNA across the ribosome and leads to incorporation of incorrect amino acids into new protein. Other members of this class are kanamycin, gentamycin, and tobramycin.
The tetracyclines first came to market in the 1940s, and over the years they cured many millions of infections. These compounds bind to the small subunit of ribosomes, but unlike streptomycin, tetracycline prevents aminoacyl-tRNA from properly binding. The tetracyclines have been used extensively in agriculture where they are sprayed on trees to cure fire blight and fed to cattle and hogs as growth promoters. One disadvantage of tetracycline is the permanent yellowing of teeth when used with children.
The macrolides are lethal agents that bind to the large ribosome subunit and prevent elongation of the new protein chain. These compounds include the old antibiotic erythromycin and two newer synthetic derivatives, clarithromycin and azithromycin. The macrolides are commonly used for bacterial pneumonia. Azithromycin is noteworthy because it persists for a long time in the body and need not be administered as often as other macrolides. A long antibiotic half-life was thought to be important for patients who have difficulty taking multiple pills each day, and the drug became popular. But long half-life is not necessarily good when resistance is considered, because selective pressure on the bacterial population is maintained for long times.
The large ribosomal subunit is also the target of the lincosamides. One of their members, clindamycin, controls infections caused by the anaerobic bacterium called Bacteroides. This normal inhabitant of the human digestive tract can multiply rapidly in tissue damaged by surgery or accident. Because rapid growth of Bacteroides causes serious infection, clindamycin occupies an important niche in the antibacterial armamentarium. However, the drug also kills other intestinal bacteria and permits the growth of Clostridium difficile. C. difficile causes serious intestinal problems, as discussed near the end of Chapter 5, “Emergence of Resistance.”
Another essential process in all organisms is DNA replication. The fluoroquinolones are the major inhibitors of this process. The targets of these drugs are two enzymes called DNA topoisomerases. (They change DNA topology by twisting/untwisting DNA and linking/unlinking DNA circles.) As a part of their reaction mechanism, topoisomerases break DNA, pull the broken ends apart, and pass another region of DNA or another DNA molecule through the gap. Then the topoisomerases seal the break. The fluoroquinolones trap the enzymes on DNA as drug-protein-DNA complexes in which DNA is broken. These complexes act as road blocks for the DNA replication machinery, thereby inhibiting cell division. The compounds appear to kill cells when the ends of the broken DNA are released from the drug-enzyme complexes, thereby fragmenting the bacterial chromosome. Because all bacteria are likely to contain DNA topoisomerases, any bacterium that takes up fluoroquinolone is expected to be susceptible.
RNA synthesis is also an essential process. Rifampicin is the most successful inhibitor of RNA synthesis. In the case of M. tuberculosis, rifampicin is highly lethal and serves as a major, first-line antituberculosis agent. Rifampicin is also active with S. aureus, and it is being drawn into clinical practice for MRSA. However, resistant mutants arise so often with S. aureus that rifampicin is rarely used in the absence of a second antibiotic.
Inhibitors of cell wall synthesis include the penicillins and their more recent derivatives, collectively called β-lactams. Treatment of growing bacteria with penicillin causes the cells to break apart: A turbid (cloudy) culture of susceptible bacteria will become clear. Bacteria that are not growing and making new cell wall material are generally not killed by penicillin. Four β-lactam classes have been developed: penicillins, cephalosporins, carbapenams, and monobactams.
Vancomycin is another cell-wall-synthesis inhibitor. It is an old drug, first approved for use in 1958. Vancomycin was originally isolated from a soil sample containing a species of Streptomyces that had been collected by a missionary in the interior jungles of Borneo. Vancomycin initially commanded considerable interest because S. aureus only rarely acquires resistance to the drug.67,68 Then methicillin and other new β-lactams displaced vancomycin, because the latter needs to be delivered intravenously. Eventually, resistance to the new β-lactams became widespread, and S. aureus acquired resistance to most other compounds. That led to the resurrection of vancomycin. For some infections caused by MRSA, vancomycin is now the only major agent available.
Daptomycin is another old compound initially obtained from Streptomyces. Early clinical trials revealed side effects at multiple, high doses, and the compound was shelved. (In the 1980s many other compounds were still available for S. aureus.) Daptomycin was brought back to the market when strains of S. aureus and S. pneumoniae became resistant to other agents. Daptomycin acts on membranes of Gram-positive cells; it has little effect on Gram-negative bacteria.
The sulfonamides (sulfa drugs) are bacteriostatic inhibitors of the enzyme dihydropteroate synthetase, which accelerates a key step of folate synthesis. (Folate is necessary for nucleic acid synthesis.) Because mammalian cells do not make folate (humans get folate from the diet), human cells are not affected by inhibitors of folate synthesis. In the late 1930s, a sulfa craze broke out, and thousands of derivatives were made. Some formulations were dangerous and led to the founding of the Food and Drug Administration (see Box 3-2). Sulfa drugs played a central role in preventing wound infections during World War II. (American soldiers were issued sulfa powder and instructed to sprinkle it on open wounds.) Allergic reactions are one of the problems with sulfa drugs; about 3% of the population experiences adverse reactions.
Antibacterial Agents May Have a Generalized Effect
Antibacterials act by binding specific targets and then corrupting specific biochemical processes. However, they also appear to stimulate a bacterial suicide process that amplifies the effect of the antibacterial. Recent work indicates that some lethal action arises through a response of bacteria to lethal stress in which peroxides accumulate and then decay to form toxic molecules called hydroxyl radicals.71,72,73 Hydroxyl radicals last only a fraction of a second, but during that time they can break nearby macromolecules. Small molecules are available that block formation of hydroxyl radicals. Treatment of E. coli with these agents almost completely protects the bacterium from the lethal action of oxolinic acid, an early type of quinolone. Thus, the idea is emerging that lethal signals caused by antibiotics trigger a cascade of reactive oxygen species that are responsible for much of the cell death.
Bacterial cells contain protective enzymes that break down peroxides, and when one of these enzymes is absent, several antibiotic types become more lethal.73 That raises the possibility for making inhibitors of the protective enzymes that will enhance the lethal activity of many antibiotics. Although such enhancers have not yet been developed, small-molecule additives have proven successful for β-lactams such as penicillin. Some bacteria make enzymes called β-lactamases that destroy β-lactams. When β-lactamase inhibitors are added to β-lactams, as is the case with Augmentin, the β-lactams are much more effective.
Most Antifungal Agents Attack Membranes and Cell Walls
All cells are surrounded by a semi-permeable membrane that contains a variety of proteins, lipids (fats), and sterols. Cholesterol is one of the sterol components of mammalian cell membranes. Fungal cells are biochemically similar to human cells, but their membranes contain ergosterol rather than cholesterol. The enzymes responsible for making ergosterol differ from those involved in making cholesterol; consequently, drugs that interfere with ergosterol formation are specific to fungal cells.
The yeast Candida albicans (see Figure 3-1) is a normal inhabitant of the mouth, throat, and vagina; it is kept under control by host defenses. However, C. albicans causes life-threatening systemic infections in persons undergoing cancer chemotherapy or otherwise experiencing immune suppression (see Box 3-3). Among the effective antifungals are triazoles, such as fluconazole, that target ergosterol. Triazoles weaken the membrane surrounding fungal cells and prevent fungal growth. Another effective drug is the polyene called amphotericin B. It binds to ergosterol and creates holes in fungal cell walls. Those holes then kill the fungus. Although amphotericin B is more active than triazoles, it has serious side effects that include kidney damage. Amphotericin B is generally not the first choice for fungal treatment.
Figure 3-1. Candida albicans. A photomicrograph of a dense culture shows hyphal-like growth characteristic of this organism that also has single, yeast-like cells.
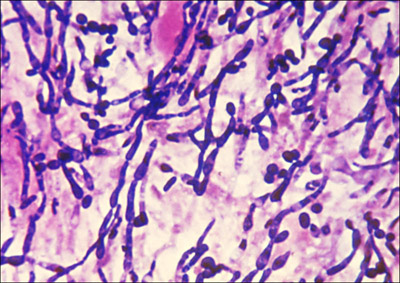
Public Health Image Library #2926
The echinocandins are a new class of antifungal compound that recently came to market in response to growing resistance among triazole antifungals. The echinocandins target an enzyme called glucan synthase, which is responsible for producing the main building block of the fungal cell wall.
Fungi are also attacked by flucytosine. This agent is structurally similar to the nucleotide subunits of DNA and RNA; when flucytosine incorporates into mRNA, it prevents synthesis of proteins by fungal ribosomes. In addition, enzymes in fungal cells chemically modify flucytosine, causing the drug to inhibit another enzyme (thymidylate synthetase) that is essential for DNA synthesis. Unfortunately, resistance emerges readily. Consequently, flucytosine has limited utility.
Fungal infections range from being a nuisance to being life threatening. A variety of skin infections, such as ringworm, athlete’s foot, and jock itch are caused by fungi. Skin infections are often controlled by topical creams containing tolnaftate, an inhibitor of ergosterol synthesis. Tolnaftate and related compounds are generally ineffective against fungal infections that grow under the nails of toes and fingers, because nails serve as tough barriers to drug entry. However, nail infections can be treated with another compound, griseofulvin, which is administered systemically. Many months of treatment are required to permit the nails to grow out fungus-free. Among the side effects of griseofulvin treatment are liver problems; consequently, liver function is usually monitored during treatment.
Antiprotozoan Agents Tend to Be Disease-Specific
Protozoan diseases are considered individually, because the antibiotics used are specific to a particular pathogen. (Many antibacterial and antifungal agents are broad spectrum.) At the top of the list is malaria, which is caused by members of the genus Plasmodium. In 2004, the World Health Organization estimated that 300 to 500 million episodes of malaria occur each year and that about 1 million persons die of the disease annually. Malaria is out of control in parts of India and Africa. Quinine, which was discovered to have antimalarial properties by ancient Peruvians (see Box 3-4), served as the main antimalarial agent for many years. Potent derivatives, such as chloroquine and quinacrine, gradually replaced quinine. Chloroquine is thought to act by accumulating in the malaria parasite and causing the buildup of a metabolic product that kills the parasite. Another antimalaria drug, artemisinin (see Box 3-4), is extracted from the wormwood plant, a native of China. Artemisinin derivatives are 10 to 100 times more effective than chloroquine at clearing the malaria parasite from humans. How artemisinin works is not known.
Trypanosomes represent another group of medically important protozoa. They cause sleeping sickness in Africa; in parts of South and Central America they cause leishmaniasis and Chagas disease. These diseases are spread by insects and can be fatal, particularly to children. Trypanosomes contain an organelle called a kinetoplast that is absent from human cells. (Kinetoplasts are mitochondria that contain a large mass of circular, interlinked DNA molecules.) The activity of the kinetoplast is blocked by a compound called pentamidine. Trypanosomes are also unusual in containing an organelle called a glycosome where many enzymes involved in the breakdown of sugars are located. Possessing a glycosome makes trypanosomes highly sensitive to a drug called suramin.
The protozoans Giardia and Cryptosporidium are less life threatening than trypanosomes, but they occasionally cause a form of diarrhea that is difficult to cure. Both protozoans occur in water supplies, with Giardia found even in crystal-clear mountain streams. In 1993, Cryptosporidium reached the Milwaukee, Wisconsin, water supply and sickened 403,000 persons.74 Paromycin, which is also an antibacterial agent, interferes with Giardia protein synthesis. Another effective compound is metronidazole. This antibiotic is converted into a DNA-breaking agent by enzymes that are active mainly when oxygen concentration is low.
Antihelminth Agents Are Used with a Wide Variety of Worms
Humans carry worms of many types that cause a variety of symptoms ranging from chronic fatigue to death. Helminths are multicellular and have distinct organ systems (for example, muscular, nervous, digestive, and reproductive). Mebendazole is a leading deworming agent. It binds to tubulin, the protein subunit of microtubules, primarily in cells of the worm gut and worm surface. (Microtubules are long structural elements of eukaryotic cells.) Disruption of microtubules leads to reduced uptake of nutrients and starvation of the worms. Another drug, levamisole, is sometimes used with mebendazole. Levamisole paralyzes the worms, and they are expelled alive.
Antiviral Agents Are Often Narrow Spectrum
Understanding antiviral agents often requires knowledge of the particular virus being controlled, because antivirals tend to be specific to one or a few closely related viruses. The many viruses fall into two general categories: those that use DNA as their genetic material (DNA viruses) and those that use RNA (RNA viruses). All viruses, without exception, are obligate intracellular pathogens. Consequently, most antiviral agents must cross the plasma membrane of human cells to access replicating virus. In contrast, many other pathogens grow outside their host cells, making many antibiotics effective without having to enter human cells. This feature means that antivirals may have more safety problems.
Viruses that affect humans bind to proteins on the surface of our cells that serve as viral receptors. Virus particles then enter the cells, some of the viral components separate, and transcription of the viral genome begins. The cellular ribosomes make viral proteins, and replication of the viral genome occurs. (Some viruses replicate in the nucleus of the host cell, whereas other virus types replicate in the cytoplasm.) As viral components accumulate, they assemble into progeny virus. With many viruses, the genes are expressed in a tightly coordinated order to assure that the correct proteins are present at specified times.
A variety of chemical strategies are used to selectively kill viruses. One involves interference with viral replication using nucleoside analogues. To the virus the analogue appears to be a normal component of DNA, but when incorporated into a new DNA strand, the analogue blocks further DNA synthesis. This process, which is called chain termination, also occurs in human cells, but many of these cells are not replicating their DNA, especially in adults. In general, inhibitors of DNA replication are well tolerated, at least when short-term toxicity is monitored. Another strategy is to prepare short pieces of DNA or RNA that form strong complementary base pairs with viral RNA or DNA. The resulting hybrids block viral replication and in some cases cause cleavage of viral nucleic acids. These agents, which are called antisense oligonucleotides and interference RNA, have good potential as antiviral agents when methods are developed for placing them at the site of infection. A third strategy is to administer proteins that resemble host cell receptors for a given virus. Viruses bind to these decoys rather than the proper cell receptors. Other inhibitors block viral uncoating and the assembly of new virus particles. In principle, five steps of the virus life cycle can be targeted: viral entry, viral uncoating, genome replication, virion assembly, and viral exit.
Ribavirin is an exception to the rule that antivirals are specific to particular viruses—this broad-spectrum agent is active against a variety of RNA viruses. Ribavirin is a nucleoside analog that forms base pairs with cytosine and uracil, creating mutations when viral replication occurs. Accumulation of many mutations incapacitates the virus. Viruses that are not actively replicating are generally not susceptible to ribavirin-type antiviral agents. Among the pathogens normally treated with ribavirin are Lassa fever virus, respiratory syncytial virus, and hepatitis C virus. With the latter, ribavirin is combined with interferon-alpha, a natural antiviral agent made by our bodies.
Foscarnet is another antiviral agent with activity against a variety of viruses. This drug binds to the polymerases of hepatitis B, HIV, and some herpes viruses. Unfortunately, the high doses that must be used cause serious side effects. Next, we describe several virus-specific agents arranged by virus type.
Human Immunodeficiency Virus (HIV)
HIV is an RNA virus that has a complex life cycle with many steps that are vulnerable to chemical interference (see Figure 3.2). The virus enters human CD4+ lymphocytes, cells that are part of the immune defense system, by first binding to a cell surface receptor and co-receptor. When inside the cell, a viral enzyme called reverse transcriptase converts the RNA form of the viral genome into a DNA form. (This reverse flow of genetic information, from RNA to DNA, led to the name retrovirus for HIV and related viruses.) Viral DNA, through the action of another viral protein (integrase), inserts itself into a human chromosome. From its position in a human chromosome, the virus instructs the human cell to make new copies of viral RNA and proteins, which assemble to form immature progeny viruses that move to the edge of the human cell. There, additional viral proteins insert into the cell membrane. Membrane buds containing viral material break off and release virus particles, each surrounded by a coat of host cell membrane decorated with viral protein. One of the interesting features of HIV and related viruses is the production of several proteins as a single, long protein molecule that gets cut into individual proteins by a viral protease. If this cutting fails to occur, the virus cannot reproduce.
Figure 3-2. Human immunodeficiency virus. Transmission electron micrograph showing HIV that has budded out of infected cells.
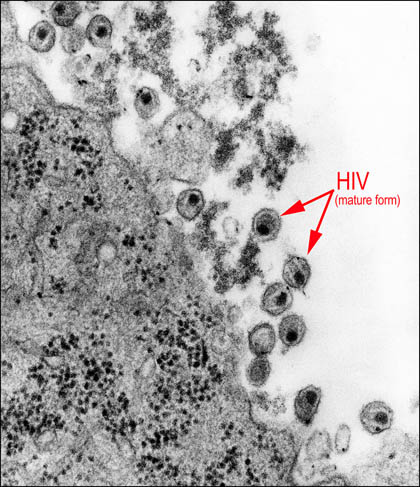
Public Health Image Library # 8254
Azidothymidine (AZT) was one of the earliest inhibitors of HIV. This agent blocks the action of reverse transcriptase, thereby preventing the virus from converting its RNA genome into the DNA form. AZT is a nucleoside analogue that is incorporated into the growing DNA chain, acting as a chain terminator. Host cells that are actively replicating, such as those lining the digestive tract and some blood cells, are not as sensitive to AZT as the virus. Nevertheless, serious side effects can arise.
Other anti-HIV drugs include several protease inhibitors, non-nucleoside polymerase inhibitors, and integrase inhibitors. Because persons infected with HIV contain large swarms of virus that readily mutate, treatment with a single agent leads to drug-resistant virus. Consequently, persons infected with HIV are treated with a cocktail of several agents.
Influenza Virus
Influenza is a disease for which effective vaccines can be produced, although new ones are needed every year. Antibiotics are useful for persons who fail to mount a strong protective immune response (infants and the elderly). Influenza virus (see Figure 3-3) normally gains access to human cells by binding to the cell surface. Then the plasma membrane attached to the virus retracts (invaginates) into the cell. As the membrane passes into the cytoplasm of the host cell, it forms a hollow ball called a vesicle. Virus particles are inside the vesicles. When a vesicle reaches the cytoplasm, its interior becomes acidic, which enables the outer layer of viral protein to detach from the rest of the virus. The remainder of the virus then escapes from the vesicle and begins to replicate. Amantadine interferes with the removal of viral surface proteins, thereby trapping the virus inside the vesicles. Oseltamivir (Tamiflu) is a neuraminidase inhibitor that prevents active virus from emerging from infected cells and spreading. (Neuraminidases are enzymes that clip terminal parts off glycoproteins and glycolipids, thereby enabling virus to exit from human cells.)
Figure 3-3. Influenza virus. Transmission electron micrograph of H1N1 swine flu virions.
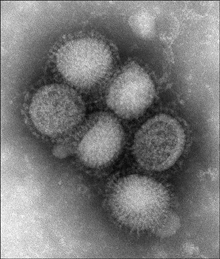
Public Health Image Library #11212; photo credit, C. S. Goldsmith and A. Balish.
Herpes Virus
Herpes viruses are large DNA viruses (see Figure 3-4) categorized into eight distinct types that replicate in skin cells where the virus causes lesions. In the case of herpes simplex, the lesions are often called “cold sores” or “fever blisters.” These sores heal within a couple of weeks, but they recur, often when a person is under stress. A variety of stressors, such as sunlight, activate the virus. The source of recurring outbreaks appears to be infected nerve cells that harbor the virus in a dormant state and protect it from attack by the human immune system. Acyclovir is an antiherpes agent that acts as a chain terminator, thereby blocking herpes virus DNA replication. In this case, conversion of acyclovir to its active form is carried out efficiently by a viral enzyme but not by host enzymes. This selectivity has made acyclovir very useful. Because dormant virus does not replicate extensively, it is unaffected by the drug. Consequently, acyclovir reduces only the symptoms of infection, and then only if taken early in the outbreak cycle. (Herpes drugs do not clear latent infection.)
Figure 3-4. Herpes virus. Transmission electron micrograph of herpes simplex virus.
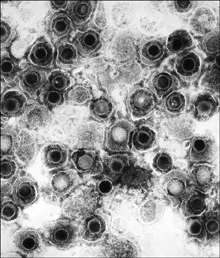
Public Health Image Library #10235
Antibiotic Classes Evolve
Antibiotic classes evolve when selective forces favor some compounds over others. The selective forces for antibiotics are the intrinsic activity of the compounds, the commercial market (some agents are more successful because their owners have better sales organizations), toxic side effects (side effects can cause a very effective compound to be quickly pulled from the market), and the emergence of resistance. The evolution of the fluoroquinolones serves as an example. Nalidixic acid, the prototype compound, was first reported in the early 1960s. (For structures, see Figure 3-5.) With most bacterial diseases, nalidixic acid is not very effective, and resistant bacteria are recovered from many patients who fail therapy. In the late 1970s, an effort to find more potent derivatives produced norfloxacin. This agent contains a fluorine and several other chemical groups that make norfloxacin effective with Gram-negative bacteria. Within a few years, an even better compound, ciprofloxacin, came along. At about the same time, ofloxacin was synthesized. Ofloxacin is a mixture of right-handed and left-handed forms (mirror images) in which only the left-handed one is active. By purifying the left-handed form (levofloxacin), chemists increased activity by a factor of two. Consequently, levofloxacin replaced ofloxacin in most markets. Ciprofloxacin and levofloxacin proved effective with Gram-negative bacteria. Moreover, both developed excellent safety records, although occasional problems with connective tissue damage kept them from being used with children. (In adults, drug treatment causes an increased incidence of Achilles’ tendon rupture, which can be severe.) Both compounds are still among the market leaders.
Figure 3-5. Chemical structures of quinolone-class molecules.
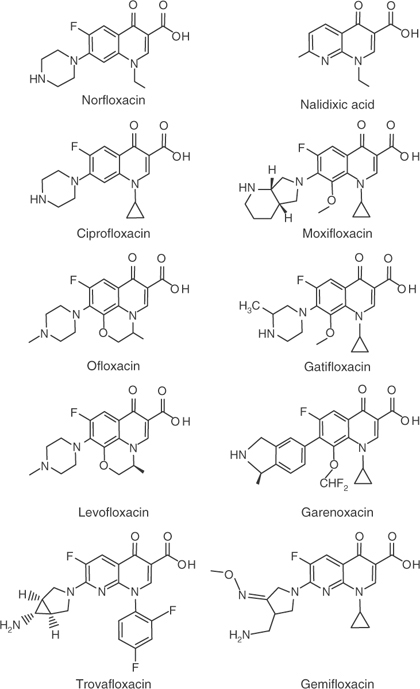
By the 1980s, the penicillins began to lose their effectiveness with important Gram-positive bacteria, mainly S. aureus and S. pneumoniae. Ciprofloxacin was tried with MRSA, but resistance emerged quickly: Within a few years, fluoroquinolone-resistant MRSA spread through hospitals worldwide.75 Ciprofloxacin was also tried in the S. pneumoniae market, and again resistance developed.76 Meanwhile, an effort was mounted to develop new fluoroquinolones that would control Gram-positive pathogens. A compound called trovafloxacin was brought to market in 1998. The compound was highly touted, and a year later, it was prescribed at a rate of about 300,000 prescriptions per month. Then 14 patients suffered acute liver failure, and six died. Trovafloxacin was quickly removed from use for most clinical indications, and it became “extinct.” As penicillin and erythromycin resistance grew among isolates of S. pneumoniae, physicians turned increasingly to levofloxacin, even though it had only modest activity. Levofloxacin had been used safely with millions of patients, and this safety record became a major marketing asset. Soon two other fluoroquinolones, gatifloxacin and moxifloxacin, entered the Gram-positive market. Both were more active with S. pneumoniae than levofloxacin, but the levofloxacin sales force continued to increase its market share. In 2006 an analysis of Canadian patients connected gatifloxacin with problems of sugar metabolism.77 Gatifloxacin was pulled from the pneumonia market, leaving it only as a cure for eye infections. That left the major respiratory market to levofloxacin and moxifloxacin. Two other compounds, gemifloxacin and garenoxacin, came along, but at this writing they have not displaced levofloxacin and moxifloxacin.
A new chapter in fluoroquinolone history is being opened with tuberculosis. Resistance to the two main antituberculosis agents, rifampicin and isoniazid, is growing. Consequently, replacements for these agents need to be found. Because moxifloxacin and gatifloxacin are more active with M. tuberculosis than other quinolones, they were chosen for clinical trials to evaluate fluoroquinolone effectiveness. Using a fluoroquinolone for long-term treatment of tuberculosis is problematic, because treatment selects resistant mutants among other bacteria that co-infect or normally live in tuberculosis patients. For example, fluoroquinolone-resistant pneumococci have been attributed to fluoroquinolone use for tuberculosis.78 The reverse is also true: Treatment of S. pneumoniae with fluoroquinolones can lead to fluoroquinolone-resistant M. tuberculosis.79 Thus, adding fluoroquinolones to first-line anti-TB therapies requires consideration of quinolone resistance.80
Quinolone-like compounds continue to evolve in research laboratories where efforts are being made to identify derivatives that restrict the emergence of resistance.81,82 Without such work, the class is likely to die due to resistance. Resistance has already eliminated fluoroquinolone effectiveness with two major pathogens, MRSA and Neisseria gonorrhoeae. With two others, pathogenic E. coli and Pseudomonas aeruginosa, the prevalence of resistance is approaching the point of serious concern.
Antiseptics and Disinfectants Decontaminate Surfaces
Antiseptics and disinfectants kill microbes, but they are not considered antibiotics because they are too toxic for internal use. They damage pathogens in a variety of ways, from breaking macromolecules to creating holes in cell membranes. Antiseptics are agents applied to the skin. Alcohol and compounds commonly included in hand-washing lotions are considered to be antiseptics. Disinfectants are agents used on inanimate surfaces. Bleach and formaldehyde fall in this category. Some agents, such as iodine and quaternary ammonium compounds, are in both categories. Antiseptics and disinfectants are especially important in hospitals prior to surgery, because microbes present on skin can enter wounds and establish serious infections.
Perspective
Five factors greatly reduce our fear of pathogens. One is sanitation. By purifying water and protecting food supplies, we reduce deaths to diseases such as cholera and typhoid. Vaccines constitute a second factor. They prime our immune systems to quickly rid our bodies of specific pathogens. The third is good nutrition, because that improves immune status. A fourth factor is insect control. Malaria and yellow fever, once common in the southern United States and Central America, are now uncommon. (Insect-borne diseases are still rampant in Africa.) Antibiotics are the fifth factor. They are especially important when one or more of the first four is absent. For example, antibacterials can be crucial for survival of immunodeficient persons, such as newborns, persons infected with HIV, or patients treated with immunosuppressants. The next chapter discusses how antibiotics are used to cure disease.