
7
Analysis of Frying
7.1 Introduction
Frying produces complex changes in the chemistry of both the food and the frying medium. Thermal oxidation, polymerization, hydrolysis, degradation, and Maillard reactions all produce several classes of new compounds. These compounds are analysed via a number of analytical methods, either qualitatively or quantitatively. The aim of analysis is to obtain composition data concerning the new compounds formed, in order to elucidate any nutritional changes they may have caused. This chapter, therefore, looks only at analytical methods related to composition studies, mostly chromatographic studies; spectroscopic studies are beyond its scope. The limitations of each method may warrant the development of new or improved methodology. The following limitations are considered while aiming to develop a new analytical method:
- The lower limit of detection or quantification.
- The high cost of the chemical or equipment.
- The reliability or reproducibility of the method.
- Low accuracy or validity.
- Low reproducibility.
- The complex nature of the frying medium and of the food.
- Different target analytes.
- The nature of the separation mode.
- The nature of the analytes.
Several review papers have aimed to collate the knowledge relevant to each topic or component of food frying. For example, Paul and Mittal (1997) reviewed the various regulations on the use of degraded oil or fats in the deep frying of foods; Hindra and Baik (2006) reviewed the kinetics of changes in the quality of the frying medium during food frying; Melton et al. (1994) reviewed the measurement of the stability of frying oils and of flavour; and Bansal et al. (2010) reviewed the rapid tests available to measure quality changes in frying oils and compared them with standard methods. Meanwhile, Zhang et al. (2015) reviewed the applications of different chromatographic techniques to the detection and identification of new constituents formed during food frying. Similarly, Zeb (2015a) reviewed the uses of different classes of chromatographic techniques, for the analysis of primary oxidation products of triacylglycerols (TAGs).
7.2 Analysis of Triacylglycerols
TAGs constitute the major part of the frying medium, and to a lesser extent of foods. Therefore, the majority of studies conducted to date have been based on the analysis of TAGs. Ruiz‐Gutierrez and Barron (1995) reviewed the different methods of analysing TAGs in a variety of matrices. They also summarized the most interesting biomedical applications of TAGs analysis. However, at that time, other chromatographic (especially liquid chromatography) and spectrometric techniques were not yet widely used.
Table 7.1 shows the different methods of TAG analysis developed since 2010. These methods were conducted in a number of different frying media, including virgin olive, sunflower, soybean, corn, fish, hazelnut, peanut, and Menhaden oil. Reversed‐phase liquid chromatography (RP‐LC) was found to be widely used in stationary phase, including C‐8 and C‐18. The majority of separations were achieved by gradient elution, mostly with acetonitrile, methanol, and 2‐propanol as the major solvents. Different detectors were used, including an evaporative light scattering detector (Adewuyi and Oderinde 2012; Solaesa et al. 2014; Beccaria et al. 2015; Abdallah et al. 2016; Tamba Sompila et al. 2017), ultraviolet (UV) detector (Gotoh et al. 2011; Lerma‐García et al. 2011; Abdallah et al. 2016), differential refractometric detector (Kıralan et al. 2015), and refractive index detector (Endo et al. 2011). These detectors have a low sensitivity, but are cheap and easy to use.
Table 7.1 Analytical methods of triacylglycerol (TAG) determination in different frying media and foods.
| Sample | Mode of separation | Detection system | Solvent system | Column | References |
| Several oils and fats | RP‐UHPLC | PDA‐ESI‐TOF‐MS | Isocratic: 2‐propanol : ACN : water (80 : 15 : 5) | Acquity BEHC‐18 (50 × 2.1 mm, 1.7 μm) | Pagliuca et al. (2018) |
| Extra virgin oil, sansa olive oil | RP | ESI‐Q‐ToF | Gradient: methanol/water (85 : 15) and 2‐propanol | Poroshell 120 EC‐C‐18 (3.0 × 50 mm, 2.7 μm) | La Nasa et al. (2018) |
| Sunflower, corn, and soybean oils | RP | ESI‐Q‐ToF | Gradient: methanol and CO2 | Two C‐18 columns | Gao et al. (2017) |
| Sesame Seed | RP‐UHPLC | APCI‐MS | Isocratic: acetonitrile/butanol (74/26) | Acquity UPLC HSS C‐18 (100 × 2.1 mm, 1.8 μm) | Ben Arfa et al. (2017) |
| Corn oil, lard, olive oil, palm oil, beef fat | RP | APCI‐MS | Gradient: ACN and 2‐propanol | Accucore C‐18 (2.1 × 100 mm) | Idrus et al. (2017) |
| Oils and fats | RP‐UHPLC | ELSD | Gradient: acetonitrile/2‐propanol | Six C‐18 columns | Tamba Sompila et al. (2017) |
| Olive oils | RP | ELSD, UV | Gradient: ACN and n‐pentanol | Kinetex C‐18 100 (150 × 4.6 mm, 2.6 μm) | Abdallah et al. (2016) |
| Marine foods | RP | APCI‐MS | Gradient: ACN and DCM | Luna C‐18 (2) (Phenomenex) columns (150, 2.1 mm, 3 μm) | Baiocchi et al. (2015) |
| Fish oil | Ag‐LC and RP‐LC | ELSD, APCI‐MS | Gradient: modified hexane for Ag‐LC and ACN, IPA | Nucleosil SA, 100 (150 × 1.0 mm, 5 μm) | Beccaria et al. (2015) |
| Hazelnut oil | RP | DFRD | Isocratic: ACN/2‐propanol/hexane (500 : 118 : 100) | Nucleosil 100 C‐18 (25 × 0.46 cm, 5‐μm) | Kıralan et al. (2015) |
| Sardine oil | RP | ELSD | Gradient: acetone : ACN | Lichrospher 100 RP 18, (25 x 4 mm, 5 μm) | Solaesa et al. (2014) |
| Brewer's yeast | RP | APCI‐MS | Gradient: ACN and 2‐propanol | HIRPB‐250AM (C‐8/C‐18 multi‐alkyl phase) | Řezanka et al. (2013) |
| Hazelnut oil | NP | APCI‐MS | Gradient: hexane and hexane‐2‐propanol (99 : 1) | Lux cellulose‐1 (250 × 4.6 mm, 3 μm) | Lísa and Holčapek (2013) |
| Peanut oil | RP | APCI‐MS | Gradient: ACN and 2‐propanol | Zorbax eclipse plus C‐18 (150 × 4.6 mm, 5 μm) | Hu et al. (2015) |
| Yeast | RP | APCI‐MS | Gradient: DCM/ACN | Alltima HP C‐18 hi‐load columns (150 × 2.1 mm, 3 μm) | Bhuiyan et al. (2013) |
| Luffa cylindrical and Adenopus breviflorus oils | RP | ELSD | Isocratic: acetone/2‐propanol (95 : 5) | SGERP‐C‐18 (250 × 4.6, 5 μm) | Adewuyi and Oderinde (2012) |
| Menhaden oil | RP | APCI‐IT‐TOF‐MS | Gradient: ACN and 2‐propanol | Ascentis express fused‐core C‐18 (150 × 4.6 mm, 2.7 μm) | Dugo et al. (2012) |
| Soybean, rapeseed, and palm oils | RP | RID | Gradient: acetone : ACN | C‐22 and C‐30 columns | Endo et al. (2011) |
| Fishes and marine mammals | RP | APCI‐MS, UV | Gradient: ACN/2‐propanol (3 : 7) | Intersil ODS, 250 × 10.0 mm, 10 μm) | Gotoh et al. (2011) |
| 36 Vegetable oils | RP | UV | Isocratic: ACN/n‐pentanol (70 : 30) | Kinetex C‐18 100 (150 × 4.6 mm, 2.6 μm) | Lerma‐García et al. (2011) |
| Rapeseed, sunflower, and palm oils | UHPLC | APCI‐MS/MS | Acetone: ACN | Acquity UPLC BEH C‐18 (100 × 2.1 mm, 1.7 mm) | Leskinen et al. (2010) |
| Corn, soybean, and sunflower oils | RP | ESI‐MS | Isocratic: methanol : 2‐Propanol (82 : 18) | Phenomenex C‐18 (150 x 3 mm) | Zeb and Murkovic (2010a) |
| Extra virgin olive oil, soybean oil, and blends | RP | APCI‐MS | Gradient: ACN and 2‐propanol | LiChrospher (ODS) (250 × 3 mm, 5 μm) | Fasciotti and Pereira Netto 2010 |
RP, reversed phase; NP, normal phase; HPLC, high‐performance liquid chromatography; UPLC, ultra‐pressure liquid chromatography; UHPLC, ultra high‐pressure liquid chromatography; PDA, photo diode array; ESI, electrospray ionization; APCI, atmospheric pressure chemical ionization; MS, mass spectrometry; ELSD, evaporative light‐scattering detector, Q‐TOF‐MS, quadrupole time‐of‐flight mass spectrometry; UV, ultraviolet; DFRD, differential refractometric detector; RID, refractive index detector; ACN, acetonitrile; DCM, dichloromethane.
Mass spectrometric detectors (MSDs) are widely used. They utilize different ionizations, such as electrospray ionization (ESI) (Zeb and Murkovic 2010a; Gao et al. 2017; La Nasa et al. 2018; Pagliuca et al. 2018) and atmospheric pressure chemical ionization (APCI) (Fasciotti and Pereira Netto 2010; Leskinen et al. 2010; Gotoh et al. 2011; Dugo et al. 2012; Bhuiyan et al. 2013; Lísa and Holčapek 2013; Řezanka et al. 2013; Zommick et al. 2013; Baiocchi et al. 2015; Beccaria et al. 2015; Ben Arfa et al. 2017; Syed Idrus et al. 2017). Most studies are based on APCI‐MS detection, with very few using ESI‐MS. In most cases, APCI‐MS do not give protonated molecular ions of TAG, so only adducts of the TAGs are widely used. In ESI‐MS, the molecular ion and its ammonium adducts – along with its diacylglycerol (DAG) or fatty acid (FA) moiety – are obtained as shown in Figure 7.1.
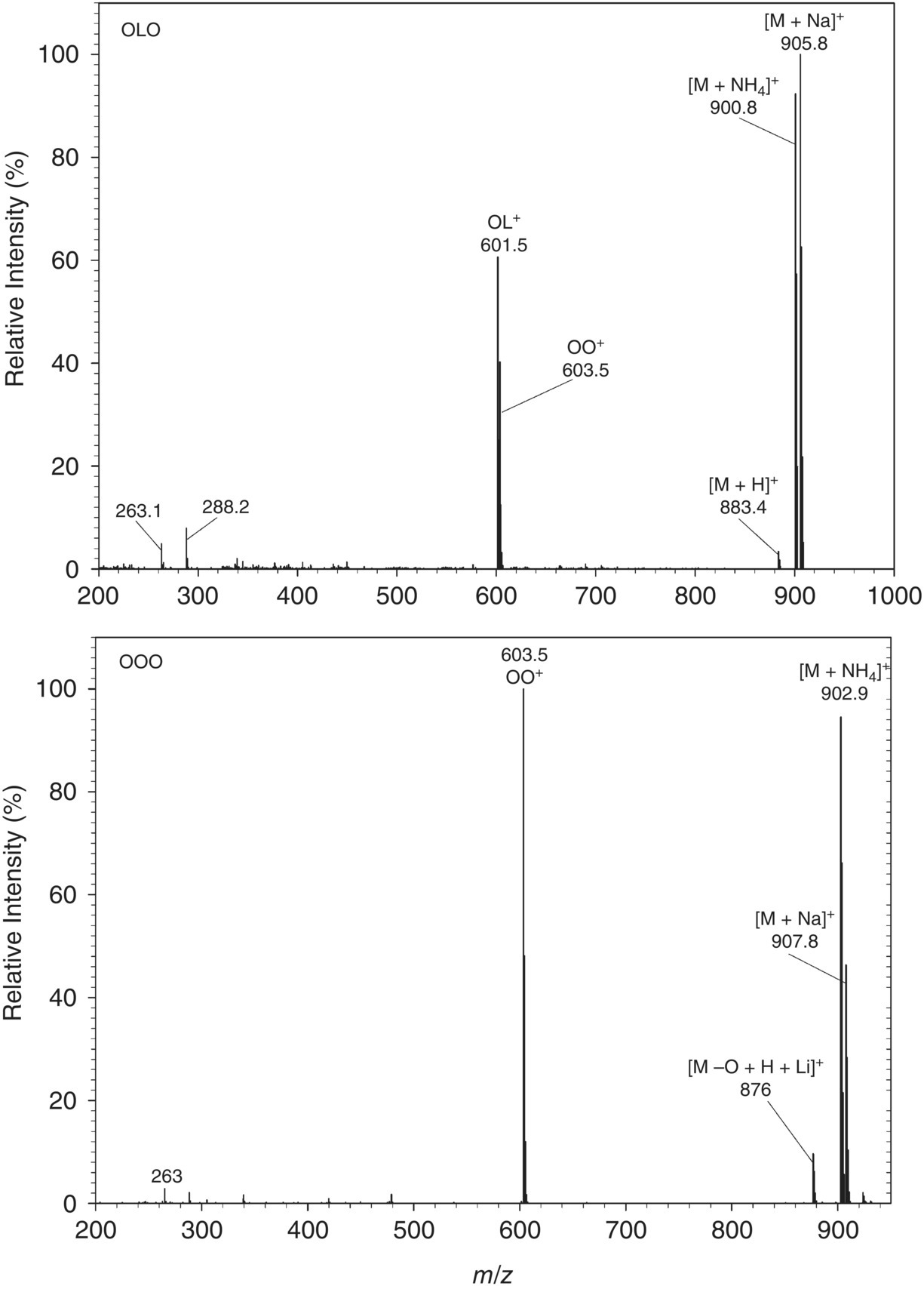
Figure 7.1 Representative electrospray ionization mass spectrometry (ESI‐MS) spectra of triacylglycerols (TAGs) (OLO and OOO) showing protonated molecular ions, sodium and ammonium adducts, diacylglycerol (DAG) ions, and fatty acid (FA) ions.
In liquid chromatography coupled to mass spectrometry (LC‐MS), the oils or extracted lipids from foods are usually not processed or derivatized. Sample preparation mostly involves the dissolution of a few milligrams of oil in high‐performance liquid chromatography (HPLC) solvent, mostly acetonitrile (ACN) or 2‐propanol. The samples are then injected into the system to obtain results. The separation of TAGs, however, depends on several factors, including stationary and mobile phase. Due to its high reliability and sensitivity, the use of HPLC‐MS is considered an absolute analytical tool, but the high cost of the MS device and the need for a skilled operator provide disadvantages to its use.
7.2.1 Analysis of Oxidized TAGS
During frying, TAGs are oxidized to form hydroperoxides, epoxide, and other oxidized species. These species are responsible for several properties of oils and fried foods. Therefore, analysis of oxidized TAGs is considered a prime target in food frying. This section summarizes the most widely used liquid chromatographic methods for the analysis of oxidized TAGs, as reproduced from Zeb (2015a) with the addition of recent literature.
7.2.1.1 Thin‐Layer Chromatography
Thin‐layer chromatography (TLC) is a widely used, fast, and relatively inexpensive chromatographic method for the separation of lipids, especially TAG or its oxidized products. TLC was used as starting method for the fractionation and isolation of TAGs and other lipids of biological significance (Zeb and Murkovic 2010c). Oette (1965) analysed several lipid peroxides using silica gel and various ratios of ethyl ether and petroleum ether as the solvent system. Perkins and Anfinsen (1971) reported a scheme for the extraction of nonvolatile oxidized compounds from the thermal oxidation of triolein, as described in Chapter 4. Different fractions of the oxidized compounds were separated using TLC. However, this method is very difficult to handle and requires extensive laboratory work.
Normal‐phase TLC can also be used to purify the oxidation products of the synthetic TAGs (Sjövall et al. 1997, 2001b) from the oils (Sjövall et al. 2001a), as well as from animal tissues (Skipski et al. 1968). For this purpose, a mobile phase of heptane/di‐isopropyl ether/acetic acid (60 : 40 : 4, v/v) solution was used. The band representing normal TAGs was eluted earlier, while the later band which contains the oxidized TAG molecules was scraped off from the plates. The selected lipids were recovered by extraction with chloroform/methanol (2 : 1, v/v) and washed with pure distilled water. The TAGs and their oxidation products were then detected in UV light after spraying with 2,7‐dichlorofluorescein. The same method was applied to the analysis of oxidized TAGs in serum low‐density lipoproteins (LDL). TAGs and their oxidized products, such as hydroxy, hydroperoxy, keto, and epoxy derivatives, were detected in UV light (Suomela et al. 2004a, 2004b, 2005). Table 7.2 shows the normal and reversed‐phase TLC methods for the analysis of oxidized TAGs (Sánchez‐Muniz et al. 1993; Chao et al. 2001).
Table 7.2 Thin‐layer chromatographic (TLC) methods for the analysis of oxidized triacylglycerols (TAGs).
Source: Reproduced with kind permission of Taylor & Francis (Zeb 2015a).
| Stationary phase | Solvent system | Solvent ratio (v/v) | Detection | References | |
| NP‐TLC | Silica gel | Toluene‐diethyl ether | 4 : 1 | Spraying of ferrous thiocynate | Peers and Coxon (1983) |
| Silica gel | Hexane‐diethyl ether | 3 : 2 | Spraying of ferrous thiocynate | Peers and Coxon (1983) | |
| Silica gel | Diethyl ether‐hexane | 40 : 60 | UV light | Neff et al. (1990) | |
| Silica gel F 60 | Hexane‐diethyl ether‐acetic acid | 80 : 20 : 1 | Iodine vapour | Sánchez‐Muniz et al. (1993) | |
| Silica gel H | Hexane‐isopropyl alcohol‐acetic acid | 60 : 40 : 4 | 2,4‐dinitropheny hydrazine | Sjövall et al. (2001a, 2001b) | |
| Silica gel G | Heptane‐isopropyl ether‐acetic acid | 60 : 40 : 4 | UV light | Suomela et al. (2005) | |
| Silica gel | Hexane‐diethyl ether | 80 : 20 | Iodine vapour | Ravi Kiran et al. (2015) | |
| Silica gel 60 F254 | Hexane‐diethyl ether‐acetic acid | 85 : 15 : 1 | Scanning densitometry | Correia et al. (2015) | |
| Silica gel 60 F254 | Hexane‐diethyl ether‐acetic acid | 30 : 70 : 1 | Primuline (Direct Yellow 59) and scanning densitometry | Schröter et al. (2016) | |
| Silica gel | Petroleum ether‐diethyl ether‐acetic acid | 70 : 40 : 1 | UV light | Karimi et al. (2017) | |
| RP‐TLC | RP‐18 | Methylene chloride‐acetonitrile | 30 : 70 | UV light | Neff et al. (1990) |
| RP‐18 | Acetonitrile‐methanol–water | 6 : 3 : 1 | Iodine vapour | Sánchez‐Muniz et al. (1993) | |
| RP‐18 | Either hexane‐diethyl ether‐acetic acid | 30 : 70 : 1 | Primuline (Direct Yellow 59) and scanning densitometry | Schröter et al. (2016) |
NP‐TLC, normal‐phase thin‐layer chromatopgraphy; UV, ultraviolet; RP‐TLC, reversed‐pahse thin‐layer chromatopgraphy.
Different solvent systems were used in normal‐phase TLC, such as diethyl ether‐hexane (40 : 60 or 80 : 20) by Neff et al. (1990) and Ravi Kiran et al. (2015); hexane‐diethyl ether‐acetic acid (80 : 20 : 1 or 85 : 15 : 1 or 30 : 70 : 1) by Sánchez‐Muniz et al. (1993), Correia et al. (2015), and Schröter et al. (2016); hexane‐isopropyl alcohol‐acetic acid (60 : 40 : 4) by Sjövall et al. (2001b, 2002) and Suomela et al. (2005); and petroleum ether‐diethyl ether‐acetic acid (70 : 40 : 1) by Karimi et al. (2017). In reversed‐phase TLC, methylene chloride‐acetonitrile (30 : 70) (Neff et al. 1990), acetonitrile‐methanol–water (6 : 3 : 1) (Chao et al. 2001), and petroleum ether‐diethyl ether‐acetic acid (70 : 40 : 1) (Karimi et al. 2017) were used. It was observed that normal‐phase TLC was more widely than reversed‐phase TLC. Diethyl ether‐hexane was the typical solvent for elution. All TLC methods reported were based on isocratic elution, but there is a lack of information on the gradient system that is today widely used in TLC (Stolyhwo and Privett 1973).
The disadvantages of TLC in the analysis of TAG oxidation products include the sensitivity of lipid peroxidation products to silica gel and oxygen in the air. An inert environment such as nitrogen is therefore necessary to use this method. From a biological perspective, another disadvantage is the relatively impossibility of separating a complex mixture of oxidized TAGs. Due to the advent of the novel, easy‐to‐use technologies of separation, it is therefore important to use other liquid chromatographic techniques such as HPLC with reliable methods of detections.
7.2.1.2 High‐Performance Liquid Chromatography
HPLC is one of the most reliable methods of separation for TAGs and their oxidation products. Kinter (1995) reviewed the different analytical techniques available for lipid oxidation products, showing liquid chromatography to be a reliable method of analysis of lipid oxidation chemistry. However, that review lacks information regarding the oxidation products of TAGs. On the basis of stationary phase, this chromatographic technique is classed into two main types: normal‐phase and reversed‐phase.
7.2.1.2.1 Normal‐Phase Liquid Chromatography
In NP‐LC, a polar stationary phase and a relatively nonpolar solvent system are used. NP‐LC has been used by a limited number of scientists for the study of oxidation compounds of TAGs. Table 7.3 lists some studies utilizing silica columns and nonpolar solvents. Peers and Coxon (1983) used an isocratic method of elution for the separation of hydroperoxide isomers produced by the auto‐oxidation of TAGs. The auto‐peroxidation products were purified by TLC and quantified through the UV detection of preparative normal‐phase HPLC. Similarly, another isocratic method was reported for the hydroperoxides produced in soybean seeds after storage (Clark and Snyder 1991). The hydroperoxides were eluted using 0.75% isopropyl alcohol in n‐hexane. The authors suggested that the methods will be helpful for monitoring seed germination and any changes during storage.
Table 7.3 Normal‐phase high‐performance liquid chromatographic (NP‐HPLC) methods for the analysis of oxidized triacylglycerols (TAGs).
Source: Reproduced with kind permission of Taylor & Francis (Zeb 2015a).
| Sample | Type of oxidized TAG | Column, size | Solvent system | Detector | References |
| Purified TAG mixtures | Isomers of hydroperoxides | Partisil 5, 25 × 0.94 mm | Isocratic, ethanol (0.4%) in hexane | UV | Peers and Coxon (1983) |
| Soybean seeds | Hydroperoxides | Ultrasphere silica column | Isocratic, 0.75% isopropyl alcohol in hexane | UV | Clark and Snyder (1991) |
| Pure TAGs | Isomers of hydroperoxides | Supelcosil LC‐Si, 250 × 2.1 mm | Isocratic, hexane‐diethyl ether (500 : 34) | DAD/FL | Ohshima et al. (1996) |
| Triolein, peanuts | Primary hydroperoxides | Nucleosil 100 Si, 250 × 4.0 mm | Isocratic, 2% propanol in hexane | UV | Gladovič et al. (1997) |
| Vegetable oils | Hydroperoxides, epoxides, oxo products | Waters S3W (15 × 2.1 mm) and waters diol (20 × 3.0 mm), and waters S5W (250 × 2.1 mm) | Gradient, hexane and hexane‐MTBE | DAD | Steenhorst‐Slikkerveer et al. (2000) |
| Sunflower oils | Hydroperoxides, hydroxides, keto | Silica 60 column | Isocratic, n‐heptane‐diethyl ether (82 : 18) | UV | Morales et al. (2010, 2012b) |
| Edible oils | Cis/trans epoxides, hydroperoxides, hydroxides, ketodienes | Silica 60 column | Isocratic, n‐heptane‐diethyl ether (82 : 18) | UV/ELS | Morales et al. (2012b) |
An HPLC method utilizing chemiluminescence (CL) detection was used for the determination of lipid hydroperoxides in LDL present in human blood plasma (Miyazawa et al. 1990). This method involved the separation of LDL‐total lipids with normal‐phase silica gel HPLC. The elution was carried out using isocratic methanol‐chloroform‐1‐propanol‐water (9 : 1 : 2 : 0.1) with the post‐column reaction of hydroperoxides with cytochrome c‐haem monitored by CL. The authors suggested that the method can be used in patients with atherosclerosis and hyperlipidaemia. Similarly, Noguchi et al. (1998) studied the hydroperoxides in the human blood plasma LDL. Separation was carried out using gradient elution with hexane‐2‐propanol (90 : 10) and hexane‐t‐butyl alcohol (70 : 30) with the help of silica column. Akasaka et al. (1993) developed an HPLC post‐column detection method for the determination of hydroperoxides in human plasma. The separation was carried out using gradient elution with hexane and hexane‐1‐butanol. The mechanism of the method is the post‐column reaction of hydroperoxides with diphenyl‐l‐pyrenylphosphine (DPPP) (Akasaka et al. 1992; Akasaka and Ohrui 2000). The phenyl phosphine reacts with hydroperoxides and forms an oxide (Figure 7.2), which exhibits fluorescence and thus can be detected using a fluorescence detector. The hydroperoxides produced by the oxidation of a pure TAG or pure FA mixture were measured accurately by HPLC with post‐column detection using DPPP (Ohshima et al. 1996). This method was used for the quantitative and qualitative determination of isomeric lipid hydroperoxides using isocratic hexane‐diethyl ether (500 : 34) as solvent.

Figure 7.2 Reaction of triphenylphosphine with lipid hydroperoxide. This reaction is mostly used as a post‐column derivatization.
In another study, the hydroperoxides from the triolein in peanuts were determined using silica column and isocratic elution with 2% propanol in hexane (Gladovič et al. 1997). This paper describes the uses of UV detection, fast atom bombardment mass spectrometry (FAB‐MS), and nuclear magnetic resonance (NMR) spectroscopy for the structural elucidation of novel hydroperoxides. The method is applicable for samples with a peroxide value (PV) more than 10 meq kg−1; it is thus of the limited application with respect to samples containing a small amount of peroxides, such as biological samples. A much more sensitive method, such as a diode array detector (DAD), can also be used. Steenhorst‐Slikkerveer et al. (2000) reported an HPLC‐DAD method for the analysis of hydroperoxides in vegetable oils. The elution was carried out using normal‐phase column. The authors also used MS detection for the characterization of oxidation compounds. They reported hydroperoxides, epoxy and oxo TAGs, mono‐ and dihydroxy TAGs, and other oxo products. They suggested that their method is very suitable for the characterization and quantitation of nonvolatile TAG oxidation products in reference samples or real oxidized samples of vegetable oils and other fat‐based food products. Cis/trans epoxides, hydroperoxydienes, ketodienes, and hydroxydienes could be measured during autoxidation at 40 and 80 °C of FA methyl esters produced from edible oils with different degrees of unsaturation, such as high linoleic sunflower oil and high oleic sunflower oil (Morales et al. 2010, 2012a, 2012b).
In conclusion, the NP‐LC methods reported here were mostly isocratic elution with hexane as major solvent, while the UV detector was the most used method of detection. Separation and structural elucidation, however, were not possible without the use of other spectroscopic techniques, such as NMR or MS. The major disadvantages of normal‐phase LC for the determination of oxidation products of TAGs include the sensitivity of the oxidized compounds, the high retention in the column, and a possible oxidation. In order to avoid such cases, either pre‐column derivatization or reversed‐phase columns are preferred.
7.2.1.2.2 Reversed‐Phase Liquid Chromatography
RP‐LC employs nonpolar stationary phases and relatively polar solvents. Dorsey and Dill (1989) reviewed in detail the molecular mechanism underlying the separation on reversed‐phase stationary phases. Later, Claessens and van Straten (2004) extensively reviewed the chemical and thermal stability of stationary phases for the preparation of reversed‐phase chromatography. Rafferty et al. (2007) recently updated their review on the molecular mechanism of separation of reverse‐phase chromatography. These reviews, however, lack information on the applications of RP‐LC to the individual class of compounds. RP‐LC has been used extensively in lipid analysis, but the corresponding review (Ruiz‐Gutierrez and Barron 1995) focused on the reversed‐phase methodologies used for the analysis of oxidized TAGs. A discussion of the molecular mechanism of the separation of oxidized TAGs on the reversed phase is beyond the scope of this section. Table 7.4 shows several RP‐HPLC methods reported for the analysis of oxidized TAGs.
Table 7.4 Reversed‐phase high‐performance liquid chromatographic (RP‐HPLC) methods for the analysis of oxidized triacylglycerols (TAGs).
Source: Reproduced with kind permission of Taylor & Francis (Zeb 2015a).
| Column, size | Solvent system | Detection | Type of oxidized TAGs | Sample | References |
| Inertsil C‐8, 150 × 4.6 mm | Isocratic, MeOH‐water | UV | Hydroperoxides, hydroxyl products | Pure TAGs | Terao et al. (1988) |
| Zorbax C‐18, 25 × 0.46 cm | Isocratic, ACN‐MC‐MeOH (85 : 15 : 1) | UV/FID | Hydroperoxides, hydroperoxides, epidioxides | Pure trilinolein and trilinolenin | Miyashita et al. (1990) |
| Dynamax C‐18, 25 × 2.14 cm | Isocratic, MC‐ACN (30 : 70) | RI/UV | Mono‐, bis‐, tris‐hydroperoxides | Pure trilinolein | Neff et al. (1990) |
| Dynamax C18, 25 × 2.14 cm | Isocratic, MC‐ACN (30 : 70) | RI/UV | Mono‐, bis‐, tris‐hydroperoxides | Pure trilinolenin | Frankel et al. (1990, 1992) |
| Nova‐Pak C‐18, 150 × 3.9 mm | Isocratic, ACN‐DCM‐MeOH (80 : 10 : 10) | UV/ELSD | Total oxidized TAGs | Pure triolein, trilinolein, and trilinolenin | Viinanen and Hopia (1994) |
| Inertsil C‐8, 150 × 4.6 mm | Isocratic, MeOH‐water (98 : 2) | UV | Hydroperoxides, hydroxyl products | Human plasma | Araujo et al. (1995) |
| Ultrastyragel, 25 × 0.94 cm | Isocratic, THF | RI | Oxidized TAG momers | Frying oils | Márquez‐Ruiz et al. (1993) |
| Finepak SIL C‐18, 250 × 4.6 mm | Isocratic MeOH or MeOH‐EtOH (5 : 1) | CL/UV | Mono‐, bis‐, tris‐hydroperoxides | Vegetable oils | Miyazawa et al. (1995) |
| Nova‐Pak C‐18, 150 × 3.9 mm Spherisorb S5 ODS2, 250 × 4.6 mm | Isocratic, MeOH‐2‐propanol (90 : 10) and MeOH:2‐propanol‐DCM (80 : 10 : 10) | CL/UV/ELSD | Hydroperoxides | Purified TAGs | Mäkinen et al. (1996) |
| Supelcosil LC‐18 DB, | Isocratic, T‐Butanol‐MeOH (1 : 1) | CL | Hydroperoxides | Butter and dairy spreads | Christensen and Holmer (1996) |
| Vydac ODS, 250 × 4.6 mm | Isocratic, ACN‐MC‐MeOH (90 : 5 : 5) | UV | Mono‐, bis‐, tris‐hydroperoxides | Triolein, trilinolein, and trilinolenin | Neff and Byrdwell (1998) |
| AsahiPak C8P50, 150 × 4.6 mm | Gradient, water‐ACN‐MeOH (45 : 35 : 20) and ACN‐MeOH (80 : 20) | UV | Hydroperoxides | Pure TAG | Sugino (1999) |
| Spherisorb S5W ODS2, 200 × 4.6 mm | Gradient, ACN, and MTBE | UV/ELSD | Mono‐, bis‐hydroperoxides, hydroxides | Vegetable oils | Bauer‐Plank and Steenhorst‐Slikkerveer (2000) |
| Prevail C‐18, 250 × 4.6 mm | Isocratic, ACN‐DCM (65 : 35) | DAD/ELSD | TAG oxidation | Pure TAGs | Christensen and Holmer (1996) |
| Phenogel 300, 300 × 7.5 mm | Isocratic, THF | ELSD/RI | Polar TAGs as oxidized TAGs | Frying oils | Caldwell et al. (2011) |
Pure TAGs were oxidized and their oxidation products were separated on isocratic methanol‐water and detected using electrochemical (Yamada et al. 1987) or UV detectors (Terao et al. 1988). The reversed‐phase separation and UV detections revealed hydroperoxides and hydroxides as major classes of oxidized TAGs. Similarly, pure synthetic trilinolein and trilinolenin were auto‐oxidized and analysed using RP‐HPLC (Miyashita et al. 1990). The separation was carried using an isocratic elution with ACN‐MC‐MeOH (85 : 15 : 1) with UV and flame ionization detector (FID) detection. The oxidation products were TAG hydroperoxides, hydroperoxides, and epidioxides. Mono‐, bis‐, and tris‐hydroperoxides were reported to form in the pure trilinolein oxidation (Neff et al. 1990). Similar products were also reported from the oxidation of pure trilinolenin and other TAGs using the same method (Frankel et al. 1990, 1992). Similar studies reported the oxidation products of TAG pure standards such as trilinolenin, trilinolein, and triolein, as well as rapeseed oil TAG (Viinanen and Hopia 1994). Novapak C‐18 column and isocratic elution with ACN‐DCM‐MeOH (80 : 10 : 10) was used with UV and evaporative light‐scattering detectors (ELSDs). Neff and Byrdwell (1998), with the help of the previously mentioned elution system, reported mono‐, bis‐, and tris‐hydroperoxides from the same TAG standard. However, these authors reported the structure of these oxidized compounds with the help of MS. Other studies also reported the oxidation of pure TAG mixtures producing hydroperoxides with the help of gradient elution with water‐ACN‐MeOH (45 : 35 : 20) and ACN‐MeOH (80 : 20) (Sugino 1999), isocratic (ACN‐DCM, 65 : 35) (Wijesundera et al. 2008), and two isocratic systems of MeOH‐2‐Propanol (90 : 10) and MeOH‐2‐propanol‐DCM (80 : 10 : 10) (Mäkinen et al. 1996). The latter study suggested that CL and UV detectors are more suitable during early stages of oxidation, due to their good sensitivity and selectivity as compared with ELSD.
Araujo et al. (1995) reported an isocratic method with MeOH‐water (98 : 2) as eluent, Inertsil C‐8 column, and UV detection. Hydroperoxides and hydroxyl products were detected in the human plasma of a patient with hyperlipidaemia. The oxidative stress caused by these oxidized compounds was contributing to the hyperlipidaemia. The eating of a high‐fat diet or thermally oxidized lipids is a contributing factor in this condition. Recent studies showed that frying oils or fats are major contributors to the development of fatty liver and hyperlipidaemia (Zeb and Mehmood 2012). They contain significant amounts of oxidized TAGs, which are digested through food and thus contribute to increases in reactive oxygen species (ROS) and oxidized LDL (Márquez‐Ruiz et al. 1993; Dobarganes and Márquez‐Ruiz 2003; Velasco et al. 2004, 2005). Frying oils have been found to contain oxidized compounds as polar compounds (Márquez‐Ruiz et al. 1995). These polar compounds were separated using isocratic elution with THF on the reversed‐phase column and detected using a refractive index (RI) detector. Auto‐ or photo‐oxidation of oils produces mono‐, bis‐, and tris‐hydroperoxides, which were detected using UV and CL detection (Miyazawa et al. 1995). These compounds were separated by isocratic elution with MeOH or MeOH‐EtOH (5 : 1) on Finepak SIL C18 column. The CL detector was very helpful to the hydroperoxides produced in the butter and dairy spreads, as reported by Christensen and Holmer (1996). These authors used an isocratic, tert‐Butanol‐MeOH (1 : 1) as solvent system and CL as the detection method. The method was sensitive, fast, and reliable for the detection of primary oxidation compounds in dairy products. Bauer‐Plank and Steenhorst‐Slikkerveer (2000) also reported the use of RP‐HPLC for the detection of TAG hydroperoxides and primary oxidation products. The gradient elution was carried out using ACN and MTBE, while oxidation compounds were detected using UV and ELSD. A linear correlation was obtained by plotting the PV and peak area of hydroperoxides, which shows that the method was in line with the standard iodometric method. However, as with other methods, it did not show the individual oxidation compounds. Recently, Caldwell et al. (2011) reported a rapid method for the determination of the total polar compounds (TPC) in several used frying oils. The authors concluded that the method was very similar to the official American Oil Chemists' Society (AOCS) method (Cd 22–91), and was capable of separating and quantifying polymerized TAG.
In reviewing all the reported RP‐LC methods for the determination of oxidized TAGs, it was found that the majority used the isocratic method of elution with methanol and ACN as major solvents and a UV detector as detection methods. These reported methods were mostly focused on the standard TAGs or frying oils and fats, and limited papers showed oxidation of TAGs in other biological samples. The disadvantage of these methods, which is significant in terms of the real chemistry of oxidized TAGs, is the use of detection systems such as UV, RI, DAD, and ELSD‐CL, which are not as easy to use in the study of the oxidation and formation of a single oxidized compound as novel MS detection systems.
7.2.1.3 Liquid Chromatography Mass Spectrometry
As evident from the literature discussed so far, only MS provides sufficient information about the structural elucidation of oxidized products produced by the oxidation of TAGs in various biological systems. MS is the most used method of detection for the analysis of lipids, especially TAGs (Dugo et al. 2005; Beermann et al. 2007; Zeb and Murkovic 2010a; Beccaria et al. 2014) and oxidized glycerolipids (Cheung et al. 1994; Harrison et al. 2000; Davis et al. 2006; Fang et al. 2010; Picariello et al. 2010; Choi et al. 2011; Hui 2014). The use of MS for the analysis of lipids or their products is thus classed as ‘lipidomics’ (Gross et al. 2005; Armstrong 2009; Blanksby and Mitchell 2010; Bou Khalil et al. 2010; Ekroos et al. 2010; Gonzalez‐Covarrubias 2013).
Easy ambient sonic‐spray ionization mass spectrometry (EASI‐MS) was used to authenticate oxidation in edible oils (Simas et al. 2010). Similarly, transmission‐mode direct analysis in real time ionization coupled with high‐resolution mass spectrometry (TM‐DART–HRMS) was used to monitor chemical changes in several vegetable oils during their thermal oxidation (Vaclavik et al. 2009, 2013). Table 7.5 shows the LC‐MS methods reported for the determination of oxidation products of TAG in various biological samples. Two main types of ionization of MS – atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI) – are widely reported. These methods are discussed in this section, irrespective of the stationary phase classification.
Table 7.5 High‐performance liquid chromatography coupled with mass spectrometry (HPLC‐MS) for the analysis of oxidized triacylglycerols (TAGOXs).
Source: Reproduced with kind permission of Taylor & Francis (updated and modified from Zeb 2015a).
| Column, size | Solvent system | Detection | Type of oxidized TAGs | Sample | References |
| Inerstil ODS2, 250 × 4.6 mm | Gradient: ACN and DCM | ELSD/APCI‐MS | Mono‐, bis‐, tris‐hydroperoxides, hydroperoxy epidioxides | Pure triolein, trilinolein, and trilinolenin | Neff and Byrdwell (1998) |
| Dynamax C‐18, 30 × 2.25 cm | Isocratic: ACN‐DCM (40 : 60) | RI/APCI‐MS | Hydroperoxides, epoxy ketones | Triolein model TAGs | Byrdwell and Neff (1999) |
| Inerstil ODS2, 25 × 4.6 mm | Gradient: ACN and DCM | ELSD/APCI‐MS | Mono‐, bis‐hydroperoxides, epoxides, epidioxides | Canola oil | Byrdwell and Neff (2001) |
| Inerstil ODS‐80‐A, 25 × 4.6 cm | Gradient: ACN and DCM | APCI‐MS | Hydroperoxides, epoxides, ketones, and dimeric products | Pure triolein and trilinolein | Warner et al. (2001) |
| Inerstil ODS3, 25 × 4.6 mm | Gradient: ACN and DCM | ELSD/APCI and ESI‐MS | Mono‐, bis‐hydroperoxides, epoxides, epidioxides | Canola oil | Byrdwell and Neff (2002) |
| Nova‐Pak C‐18, 150 × 3.9 mm | Isocratic: MeOH‐Hexane (85 : 15) | APCI‐MS | Hydroperoxides, epoxides | Pure Triolein TAG | Zhang et al. (2002) |
| Supelcosil LC‐18, 250 × 4.6 mm | Gradient: 2‐propanol‐MeOH, 20–80% | ELSD/ESI‐MS | Hydroperoxides, diepoxides, hydroxides | Corn oil, sunflower oil | Sjövall et al. (1997, 2001a, 2001b, 2003) |
| Inerstil ODS3, 25 × 4.6 mm | Gradient: ACN and DCM | ESI‐MS | Oxidized tristearin monomers, dimers, chain addition/shortening products | Triolein | Byrdwell and Neff (2004) |
| Ultra RP 18, 250 × 2.1 mm | Gradient: ACN and chloroform | ESI‐MS | Hydroperoxides, epoxides | Pure TAGs | Giuffrida et al. (2004) |
| Discovery HS C18, 250 × 4.6 mm | Gradient: 2‐propanol‐MeOH, 20–80% | ELSD / ESI‐MS | Hydroperoxides, epoxides, hydroxides, and keto products | Sunflower oil | Suomela et al. (2005) |
| Acquity UPLC BEH 150 × 1 mm | Gradient: ACN‐MeOH‐water (19 : 19: 2) and Isopropanol | ESI‐MS | Hydroperoxides | Mouse liver | Ikeda et al. (2009) |
| Phenomenex C‐18, 250 × 4.6 mm | Gradient: 2‐propanol‐MeOH | ESI‐MS | Mon‐, di‐, tri‐hydroxides | Castor oil | Lin and Chen (2010) |
| Nucleodur ISIS C‐18, 250 × 4.6 mm | Gradient: ACN‐water‐formic acid (50 : 50 : 0.1) and isopropanol‐acetic acid‐formic acid (90 : 10 : 1) | ELSD/ESI‐MS | Mon‐, di‐, tri‐, tetrahydroperoxides | Pure TAGs, rapeseed oil, and fish | Tarvainen et al. (2010, 2015) |
| Kinetex C‐18, 120 × 2.1 mm | Gradient: ACN and acetone‐ACN | ESI‐MS/MS | Hydroperoxides,hydroxides, diepoxides | Pure TAGs | Suomela et al. (2011) |
| Phenomenx C‐18, 150 × 3.0 mm | Isocratic: 2‐propanol‐MeOH (18 : 82) | ESI‐MS | Mono‐, bis‐hydroperxides, mono‐, bis‐epoxides, epoxy epidioxides, hydroxyl epidioxides | Model TAGs, edible oils | Zeb and Murkovic (2010b) |
| PL gel MIXED‐E SEC, 7.5 × 300 mm | Isocratic: THF | TM‐DART–HRMS | Oxidized TAG, TAG oligomers | Edible oils | Vaclavik et al. (2009, 2013) |
7.2.1.3.1 HPLC APCI‐MS
It is clear from Table 7.5 that the initial work reported was mostly focused on the oxidation of pure standard TAGs. For example, Neff and Byrdwell (1998) used a gradient method of elution (ACN and DCM) to auto‐oxidize triolein, trilinolein, and trilinolenin in the dark at 50–60 °C until the oxidation products reached about 30% of the original samples. They then analysed the oxidation products using RP‐HPLC‐APCI‐MS. Mono‐, bis‐, and tris‐hydroperoxides, hydroperoxy epidioxides, and epoxides were found to be the major oxidation products. A similar gradient method of ACN and DCM was used to study the oxidation compounds produced by pure triolein and trilinolein (Warner et al. 2001). APCI‐MS revealed the formation of hydroperoxides, epoxides, ketones, and dimeric products. The authors were also able to study the secondary oxidation compounds formed from hydroperoxides. This study was helpful in explaining the source of the deep‐fried flavour that is characteristic of high linoleic frying oils. Similarly, the oxidation products produced by the pure triolein were reported by Zhang et al. (2002). The separation was carried out using Novapak C‐18 column and isocratic elution with MeOH‐Hexane (85 : 15). The major oxidation products were monohydroperoxides, bishydroperoxides, and epoxides. This paper shows the first comprehensive investigation into the interaction between Laccase enzymes and lipids containing unsaturated FAs. Similarly, Byrdwell and Neff (1999) determined mono‐ and bis‐hydroperoxides, epoxides, and epidioxides from the oxidation of triolein at 190 °C. The oxidation products were separated using isocratic elution with ACN‐DCM (40 : 60) and quantified using APCI‐MS. Byrdwell and Neff (2001) also showed the uses of gradient elution with ACN and DCM for the determination of TAG oxidation products formed by the auto‐oxidation of normal and genetically modified canola oils. Mono‐ and bis‐hydroperoxides, epoxides, and epidioxides were detected and quantified using RP‐LC‐APCI‐MS. The APCI spectra revealed the protonated molecular ions [M + H] +, [M‐18] +, and [M + 90] + adducts.
The major disadvantages of the use of APCI‐MS in oxidation studies include aldehydes, which do not produce abundant protonated molecular ions in APCI‐MS. The hydroxyl containing TAG is not detected as well as with hydroperoxides, while the protonated molecular ions of nearly all normal or oxidized TAGs are found in very low abundance.
7.2.1.3.2 HPLC ESI‐MS
ESI‐MS is more widely used in TAG oxidation studies than is APCI‐MS, as shown in Table 7.5. Byrdwell and Neff (2002) used both APCI‐MS and ESI‐MS for the analysis of oxidation products of canola oil TAGs. The gradient elution was carried out with ACN and DCM using Inertsil ODS‐3 column. The major oxidation products were mono‐ and bis‐hydroperoxides, epoxides, and epidioxides. The authors showed that the product ion mass spectra produced by the ESI‐MS/MS were very similar to the conventional APCI‐MS spectra. Both systems allowed the correct identification of positional isomers based on the ratios of fragment ions of DAGs. Another study by the same authors showed that oxidized tristearin monomers, dimers, and chain addition or shortening products were oxidized products of triolein at the frying temperature (Byrdwell and Neff 2004). Another gradient method of elution (ACN and chloroform) was reported by Giuffrida et al. (2004). These authors identified epoxy‐ and hydroperoxy‐TAGs, which were formed by the air and 18O2 oxidation of 1,2‐dipalmitoyl‐3‐oleoyl‐glycerol (PPO) and 1,3‐dipalmitoyl‐2‐oleoyl‐glycerol (POP). The analyses were carried out using RP‐LC‐ESI‐MS. The MS was a triple quadruple mass analyser in positive ion mode. In the MS spectra, the fragmentation of hydroperoxy‐TAGs was distinct from their epoxy‐TAGs homologues and consisted of simultaneous losses of hydrogen peroxide and water.
The frying of sunflower and corn oils was reported to produce hydroperoxides, diepoxides, and hydroxides (Sjövall et al. 1997, 2001a, 2001b, 2002, 2003). These studies showed a gradient elution with 2‐propanol‐MeOH (20–80%) on the Supelcosil LC‐18 column. The oxidized products were hydroperoxides, diepoxides, and hydroxides. Sjövall et al. (2003) showed that the rapid peroxidation with tert‐butyl hydroperoxide was an effective LC‐ESI‐MS method of enriching natural oils and fats in TAG core aldehydes for biochemical and metabolic testing. Suomela et al. (2005) reported a method based on RP‐HPLC and ESI‐MS for the analysis of oxidized TAGs in chylomicrons and very low‐density lipoproteins (VLDLs) of growing pigs. The gradient elution with 2‐propanol‐MeOH (20–80%) was used on Discovery HS C18 column. The reported oxidation compounds were hydroperoxides, epoxides, hydroxides, and keto products. Recently, Ikeda et al. (2009) reported an effective method for the global analysis of TAG molecular species from complex lipid mixtures of mouse liver and white adipose tissue using RP‐LC coupled with ESI‐quadruple/time‐of‐flight hybrid mass spectrometer (QTOF‐MS). The gradient elution was based on ACN‐MeOH‐water (19 : 19 : 2) and isopropanol. The authors showed that several new oxidized TAGs were detected in mouse adipose tissue, along with non‐oxidized TAGs in mouse liver.
A Phenomenex C‐18 column and gradient elution with 2‐propanol‐MeOH was used along with ESI‐MS detection for the determination of hydroxides in castor oil (Lin and Chen 2010). A model TAG mixture was thermally oxidized at 110 °C, and the products were studied with the help of an isocratic method consisting of 2‐propanol‐MeOH (18 : 82) (Zeb and Murkovic 2010b). Identification and quantification of the oxidized species were carried out using ESI‐MS. The same method was applied to different edible oils with similar (Zeb and Murkovic 2011) and different (Zeb and Murkovic 2013a, 2013b) TAG compositions as compared to the model TAGs in the presence of synthetic antioxidants and thermal stress. It was found that the major oxidation compounds of the TAG mixtures were mono‐ and bis‐hydroperoxides, mono‐ and bis‐epoxides, epoxy epidioxides, and hydroxyl epidioxides. TAGs with similar FA compositions showed similar oxidation compounds, irrespective of the oxidation treatment applied (Zeb 2012). The advantage of this method was that it required only a single run to provide a complete profile. However, it was unable to separate the isomers of individual oxidized TAG.
A gradient elution with ACN‐water‐formic acid (50 : 50 : 0.1) and isopropanol‐acetic acid‐formic acid (90 : 10 : 1) was used to separate and identify oxidized molecular species of pure TAGs, rapeseed oil, and fish samples using UHPLC (Tarvainen et al. 2010, 2015). ESI‐MS spectra revealed the presence of mono‐, di‐, tri‐, and tetra‐hydroperoxides and vast amounts of several oxidation products. This UHPLC‐ESI‐MS method proved to be fast, highly selective, and sensitive, consuming one‐tenth as many solvents as a previously used high‐performance LC‐ESI‐MS method (Kuksis et al. 2009a, 2009b). Recently, Suomela et al. (2011) showed the separation of region‐isomers of oxidized compounds of TAGs using gradient elution with ACN and acetone‐ACN. The analysis was carried out using UHPLC coupled with tandem MS. It was found that the method was helpful in the separation of region‐isomers of oxidized TAGs.
Ito et al. (2017a) developed a chiral stationary phase for separation of oxidized TAGs using LC‐MS. The authors proved that the combination of chiral stationary phase coupled to an LC‐MS/MS method with lipase enzyme was a powerful tool for evaluating the mechanisms of lipid oxidation during food deterioration. The downsides included the high cost of MS, the need for a highly skilled worker to conduct the analysis of oxidized lipids (especially oxidized TAGs), and the unavailability of a spectral database.
7.2.2 Quantification of Oxidized TAGs
The oxidized products of TAGs can be quantified using different liquid chromatographic methods. Sjövall et al. (2001a) quantitatively estimated the residual TAGs and mono‐, di‐, and tri‐tertiary butyl hydroperoxide (TBHP) adducts from the TLC band using mol% of the total ion current. The mono‐TBHP was 11.7 mol%, di‐TBHP was 8.7 mol%, while tri‐, tetra‐, and penta‐TBHP adducts were present in trace amounts. This study showed that TBHP adducts of the TAG oxidation products can be quantified easily using TLC with MS. Our previous study showed that hydroxy hydroperoxides, hydroxy epidioxides, epoxy epidioxides, hydroperoxides, and epoxides were formed in amounts less than 5.0% in the Rancimat at 110 °C (Zeb and Murkovic 2011), with epoxy epidioxides produced (about 8%) in greater amounts than the previously mentioned oxidation products. The addition of synthetic antioxidant was found to produce significant changes in the formation of these products. Another study (Zeb 2012) showed the formation and quantification of auto‐oxidation products of TAG in Camellia oil. The amount of epoxy hydroperoxides was 1.12%, that of epoxy epidioxides 5.5%, and that of epoxides 6.89%. MS was highly useful for the quantification of individual oxidation compounds in the simple or complex system. However, the high cost of MS and the difficult interpretation of individual MS spectra of oxidized products provide disadvantages to such studies. Therefore, UV and other detection systems are used for quantification and detection purposes. Morales et al. (2014) showed that oxidized products of fatty acid methyl esters (FAME) can be analysed using HPLC‐UV. These authors showed that when high linoleic sunflower oil was oxidized for 35 days at 40 °C, 24.12 mg g−1 of hydroperoxy dienes were produced. Similar amounts of hydroperoxides (25.25 mg g−1) were also produced along with relatively small amounts of keto‐ and hydroxy‐dienes. The autoxidation of high oleic sunflower oil produced 12.21 mg g−1 of hydroperoxy dienes, 13.27 mg g−1 of hydroperoxides, and a small amount of keto‐ and hydroxy‐dienes. These results clearly show that the previously mentioned oxidized products were produced in double amounts when another double bond was present in the FA. It is clear that the process of oxidation and the compositions of TAGs play a key role in the formation of oxidation compounds.
7.3 Analysis of FA Oxidation Products
FAs produced by the hydrolysis of TAGs form several minor oxidation products. The details given here are reproduced from Xia and Budge (2017), who reviewed the analysis of epoxides, alcohols, and ketones. For example, Velasco et al. (2004) determined monoepoxy FAs in oils heated at the same temperature and for the same time as reported by Berdeaux et al. (1999). The total amounts of the six common monoepoxy FAs were 14.24 and 9.44 mg g−1 in olive and sunflower oils, respectively (Velasco et al. 2004). The authors determined methyl epoxystearates and methyl epoxyoleates in 10 used frying oils and concluded that: (i) more trans epoxides than cis epoxides were produced in monounsaturated oils; and (ii) a greater concentration of epoxides was found in monounsaturated oils than in polyunsaturated oils, consistent with Berdeaux et al. (1999); this may be attributed to a lower tendency of epoxystearates to form polymers and to participate in further oxidative reactions (Velasco et al. 2004).
In addition to frying oils, epoxy FAs have been determined at much lower concentrations (approximately 1000‐fold lower) in fresh oil, with cis‐9,10‐epoxystearate, cis‐12,13‐epoxyoleate, and cis‐9,10‐epoxyoleate being the most prominent (Mubiru et al. 2013). Trans epoxy FAs were generally present at lower levels than cis epoxy FAs in fresh oils. For example, in sunflower oils, individual cis epoxy FAs ranged from 55 to 1430 μg g−1, while trans epoxy FAs ranged from 4 to 33 μg g−1. Moreover, a strong correlation was reported between the ratio of epoxystearate to epoxyoleate and the ratio of 18 : 1–18 : 2 FAs.
Mubiru et al. (2014) measured the six common monoepoxy FAs in food matrices including biscuits, meat, butter, and nuts. A range of 3–171 μg g−1 total epoxy FAs was determined in the foods, and no relationship was found between the sample type and the total epoxy FA concentrations. A large amount of trans‐9,10 epoxystearate (53.66 μg g−1) was reported for the butter sample; the other food matrices had lower concentrations, ranging from 0.75 to 17.33 μg g−1.
Considering the locations of the epoxy groups in the six most measured monoepoxy FAs, in which the epoxy groups take the place of the original double bond in position 9 or 12 in methyl oleate and linoleate, these epoxy FAs seem to be formed by peroxide addition to double bonds. Positional isomers, such as 8, 9‐epoxystearate and 10, 11‐epoxystearate (Frankel 1984), may be formed by the cyclization of alkoxy radicals.
The preceding results reported for epoxides were determined by gas chromatography (GC); therefore, individual epoxy FAs were identified and quantified. In contrast, NMR methods determine total epoxide content. For instance, Goicoechea and Guillen (2010) determined monoepoxides and diepoxides in sunflower oils heated at 70 and 100 °C with aeration. At 100 °C, both monoepoxides and diepoxides reached a maximum value at 161 and 107 mmol L−1 oil (approximately 56 and 39 mg g−1) and showed a declining trend afterwards; at 70 °C, they reached 41 and 123 mmol L−1 oil (approximately 14 and 45 mg g−1).
Available data on the occurrence of alcohols and ketones are scarcer than for epoxides. Hydroxy FAs were first quantified in hydrogenated groundnut oil, and individual C18 hydroxy FAs were found to be present at between 2 and 494 μg g−1, with a total amount of 1422 μg g−1 (Wilson et al. 1997). In fried sunflower oil (180 °C for 10 hours), total C18 hydroxy FAME ranged between 1.9 and 5.5 mg g−1 oil; C18 keto FAME and C18 epoxy FAME were also measured, determined at 0.5–2.5 and 1.3–4.4 mg g−1 oil, respectively (Marmesat et al. 2008). In extra virgin olive oil heated at 190 °C for 7.5–32 hours, primary alcohols were determined by NMR at 2–5 mmol mol−1 TAG (approximately 0.7–1.7 mg g−1 TAG), while secondary alcohols were between 4 and 20 mmol mol−1 TAG (approximately 1.4–7.0 mg g−1 TAG) (Martínez‐Yusta and Guillen 2014a). In this experiment, (E)‐ and (Z)‐epoxides reached concentrations of 20 and 12 mmol mol−1 TAG (approximately 7.0 and 4.2 mg g−1), respectively. Three other food types, Spanish doughnut, pork adipose tissue, and farmed salmon fillets, were exposed to the same experimental conditions and did not show great differences in alcohol or epoxide content. However, in a later study, the same authors (Martínez‐Yusta and Guillén 2014b) compared the formation of primary alcohols in fried soybean oils and the same three food types (doughnuts, adipose tissue, and salmon fillets) and found that primary alcohols had much higher concentrations in the salmon fillets than the other three media. The authors also reported that the primary alcohols began to be detectable after 7.5 hours of frying in soybean oil and Spanish doughnut and after 10 hours of frying in pork adipose tissue and salmon fillet.
Hydroxy dienes and keto dienes were monitored during oxidation of sunflower oil‐derived FAME at 40 °C (Morales et al. 2010). Due to the limits of quantification, which were 0.05 mg g−1 for hydroxydienes and 0.02 mg g−1 for ketodienes, hydroxydienes and ketodienes were detectable but not quantifiable at the beginning of the experiments and increased in concentration over time. After 91 hours of heating, 3.1 mg g−1 of ketodienes and 1.3 mg g−1 of hydroxy dienes were found in the FAME derived from high linoleic sunflower oil. Under the conditions of the study, more ketodienes than hydroxydienes were found in FAME derived from both high linoleic and high oleic sunflower oils throughout the oxidation period. For high linoleic sunflower oil oxidized under similar conditions, the same group found concentrations of ketodienes and hydroxydienes up to 1.6 and 0.7 mg g−1, respectively (Morales et al. 2014).
Steenhorst‐Slikkerveer et al. (2000) determined the concentrations of epoxy‐, oxo‐, and hydroxy‐TAGs in oxidized rapeseed oil and linseed/safflower oil. The oils were analysed when received, without any artificial oxidation. Epoxy‐ and oxo‐TAGs co‐eluted, so they were determined together, at 2734 ppm (approximately 2.7 mg g−1) in rapeseed oil and 28 701 ppm (approximately 28.7 mg g−1) in linseed/safflower oil. The concentrations of hydroxy TAGs were 8759 and 3549 ppm (approximately 8.8 and 3.5 mg g−1) in rapeseed oil and linseed/safflower oil, respectively.
Quantitative results on the occurrence of epoxides, alcohols, and ketones are summarized in Table 7.6. It should be noted that possible discrepancies exist among the results, due to the different techniques and oxidation conditions employed. The quantitative data show clear trends, where most oxidized oils have epoxy, hydroxy, and keto compounds at concentrations in the range of mg g−1. In un‐oxidized oils, these oxygenated compounds were quantifiable using chromatographic techniques but were present at much lower levels, in the range of μg g−1.
Table 7.6 Occurrence of epoxides, alcohols, and ketones in FAME, TAGs, oils, and foods with or without artificial oxidation.
Source: Data from Xia and Budge (2017).
| Sample | Oxidation condition | Quantitative technique | Analyte | Range of quantity | References |
| FAME | |||||
| Methyl oleate | Heated at 180 °C for 5–15 hours | GC‐FID | Monoepoxy FAs | 14.2–35.2 mg g−1 | Berdeaux et al. (1999) |
| Methyl linoleate | Heated at 180 °C for 5–15 hours | GC‐FID | Monoepoxy FAs | 9.2–19.1 mg g−1 | Berdeaux et al. (1999) |
| FAME derived from high linoleic sunflower oil | Heated at 40 °C for 0–91 hours | HPLC‐UV | Ketodienes | Up to 3.094 mg g−1 | Morales et al. (2010) |
| HPLC‐UV | Hydroxydienes | Up to 1.338 mg g−1 | Morales et al. (2010) | ||
| FAME derived from high oleic sunflower oil | Heated at 40 °C for 0–192 hours | HPLC‐UV | Ketodienes | Up to 1.726 mg g−1 | Morales et al. (2010) |
| HPLC‐UV | Hydroxydienes | Up to 0.590 mg g−1 | Morales et al. (2010) | ||
| FAME derived from high linoleic sunflower oil | Heated at 80 °C for 0–6 hours | HPLC‐UV | Ketodienes | Up to 0.38 mg g−1 | Morales et al. (2012a) |
| HPLC‐UV | Hydroxydienes | Up to 0.46 mg g−1 | Morales et al. (2012a) | ||
| FAME derived from high oleic sunflower oil | Heated at 80 °C for 0–17 hours | HPLC‐UV | Ketodienes | Up to 0.78 mg g−1 | Morales et al. (2012a) |
| HPLC‐UV | Hydroxydienes | Up to 0.31 mg g−1 | Morales et al. (2012a) | ||
| HPLC‐ELS | Methyl trans‐epoxystearate | Up to 1.33 mg g−1 | Morales et al. (2012a) | ||
| HPLC‐ELS | Methyl cis‐epoxystearate | Up to 1.52 mg g−1 | Morales et al. (2012a) | ||
| TAGs | |||||
| Triolein | Heated at 180 °C for 5–15 hours | GC‐FID | Monoepoxy FAs | 13.3–35.7 mg g−1 after CH3ONa‐catalysed transmethylation | Berdeaux et al. (1999) |
| Trilinolein | Heated at 180 °C for 5–15 hours | GC‐FID | Monoepoxy FAs | 6.9–18.3 mg g−1 after CH3ONa‐catalysed transmethylation | Berdeaux et al. (1999) |
| TAG mixture | Heated at 110 °C for 1–14 hours | HPLC‐ESI‐MS | Oxidized TAGs | Zeb and Murkovic (2010c) | |
| Oils | |||||
| Hydrogenated ground nut oil | — | GC‐MS‐SIM | C‐18 hydroxy FA isomers (C‐6–C‐17 substitution) | 2–494 μg g−1 | Wilson et al. (1997) |
| Olive oil | Heated at 180 °C for 15 hours | GC‐FID | Monoepoxy FAs | 13.52 mg g−1 with on‐column injection, 13.72 mg g−1 with split injection | Velasco et al. (2002) |
| Sunflower oil | Heated at 180 °C for 15 hours | GC‐FID | Monoepoxy FAs | 10.88 mg g−1 with on‐column injection, 10.87 mg g−1 with split injection | Velasco et al. (2002) |
| Olive oil | Heated at 180 °C for 5–15 hours | GC‐FID | Monoepoxy FAs | 4.29–14.24 mg g−1 | Velasco et al. (2004) |
| Sunflower oil | Heated at 180 °C for 5–15 h | GC‐FID | Monoepoxy FAs | 5.10–9.44 mg g−1 | Velasco et al. (2004) |
| Used frying oils | — | GC‐FID | Monoepoxy FAs | 3.37–14.42 mg g−1 | Velasco et al. (2004) |
| Edible oils | — | GC‐FID | cis‐epoxy‐oleic acid | 110–2300 μg g−1 | Fankhauser‐Noti et al. (2006) |
| GC‐FID | cis‐epoxy‐linoleic acid | 150–1550 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| GC‐FID | Diepoxy linoleic acid | 0.2–1.5 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| Frying oils | — | GC‐FID | cis‐epoxy‐oleic acid | 1900–7300 μg g−1 | Fankhauser‐Noti et al. (2006) |
| GC‐FID | trans‐epoxy‐oleic acid | 2800–16 400 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| GC‐FID | cis‐epoxy‐linoleic acid | 1400–2000 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| GC‐FID | trans‐epoxy‐linoleic acid | 1800–3300 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| GC‐FID | Diepoxy linoleic acid | 4.9–16.0 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| Sunflower oils | Heated at 180 °C for 10 hours | GC‐FID | Epoxy FAs | 1.3–4.4 mg g−1 | Marmesat et al. (2008) |
| GC‐FID | Keto FAs | 0.5–2.5 mg g−1 | Marmesat et al. (2008) | ||
| GC‐FID | Hydroxy FAs | 1.9–5.5 mg g−1 | Marmesat et al. (2008) | ||
| Fresh oils | — | GC‐FID | Epoxy FAs | 0.03–2 mg g−1 | Mubiru et al. (2013) |
| Used frying fat/oil | — | GC‐FID | Epoxy FAs | 0.05–16.57 mg g−1 | Brühl et al. (2016) |
| Pumpkin seed oil | — | GC‐FID | Epoxy FAs | 1.12–5.10 mg g−1 | Brühl et al. (2016) |
| Sweet almond oil | — | GC‐FID | Epoxy FAs | 0.72–2.65 mg g−1 | Brühl et al. (2016) |
| Sunflower oil | — | GC‐FID | Epoxy FAs | 0.06–1.90 mg g−1 | Brühl et al. (2016) |
| Groundnut oil | — | GC‐FID | Epoxy FAs | 0.10–7.25 mg g−1 | Brühl et al. (2016) |
| Olive oil | — | GC‐FID | Epoxy FAs | 0.10–0.32 mg g−1 | Brühl et al. (2016) |
| Rapeseed oil | — | HPLC‐MS | Epoxy‐ and oxo‐TAG | 2734 ppm (2734 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) |
| HPLC‐MS | Oxo‐21/2‐glycerides | 506 ppm (506 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| HPLC‐MS | Hydroxy‐TAG | 8759 ppm (8759 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| HPLC‐MS | Dihydroxy‐TAG | 1049 ppm (1049 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| Linseed/safflower oil | — | HPLC‐MS | Epoxy‐ and oxo‐TAG | 28 701 ppm (28 701 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) |
| HPLC‐MS | Oxo‐21/2‐glycerides | 2146 ppm (2146 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| HPLC‐MS | Hydroxy‐TAG | 3549 ppm (3549 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| HPLC‐MS | Dihydroxy‐TAG | 1823 ppm (1823 μg g−1)a | Steenhorst‐Slikkerveer et al. (2000) | ||
| Sunflower oil | Heated at 100 °C with aeration | 1H NMR | Monoepoxides | Maximum level at 161 mmol L−1 (56 mg g−1)b | Goicoechea and Guillen (2010) |
| Diepoxides | Maximum level at 107 mmol L−1 (39 mg g−1)b | Goicoechea and Guillen (2010) | |||
| Sunflower oil | Heated at 70 °C with aeration | 1H NMR | Monoepoxides | Maximum level at 41 mmol L−1 (14 mg g−1)b | Goicoechea and Guillen (2010) |
| Diepoxides | Maximum level at 123 mmol L−1 (44 mg g−1)b | Goicoechea and Guillen (2010) | |||
| Extra virgin olive oil | Heated at 190 °C for periods of 8 hours day−1 for 5 days | 1H NMR | (E)‐9,10‐epoxystearic acyl groups | 22.8 mmol L−1 (7.9 mg g−1)b | Guillén and Uriarte (2012) |
| 1H NMR | (Z)‐9,10‐epoxystearic acyl groups | 14.5 mmol L−1 (5.0 mg g−1)b | Guillén and Uriarte (2012) | ||
| Soybean oil | Heated at 100 °C for 4–20 days | 1H NMR | Epoxides | 8.4–90.7 mmol kg−1 (2.6–28.3 mg g−1)b | Xia et al. (2016) |
| High oleic sunflower oil | Heated at 40 °C for 0–86 days | HPLC‐UV | Ketodienes | 0.13–1.34 mg g−1 | Morales et al. (2012b) |
| HPLC‐UV | Hydroxydienes | 0.13–0.72 mg g−1 | Morales et al. (2012b) | ||
| High linoleic sunflower oil | Heated at 40 °C for 0–41 days | HPLC‐UV | Ketodienes | 0.12–0.79 mg g−1 | Morales et al. (2012b) |
| HPLC‐UV | Hydroxydienes | 0.36–1.12 mg g−1 | Morales et al. (2012b) | ||
| Soybean oil | Heated at 40 °C for 0–53 days | HPLC‐UV | Ketodienes | 0.07–2.97 mg g−1 | Morales et al. (2014) |
| HPLC‐UV | Hydroxydienes | 1.02–3.53 mg g−1 | Morales et al. (2014) | ||
| Rapeseed oil | Heated at 40 °C for 0–46 days | HPLC‐UV | Ketodienes | Up–2.1 mg g−1 | Morales et al. (2014) |
| HPLC‐UV | Hydroxydienes | Up–3.71 mg g−1 | Morales et al. (2014) | ||
| Foods | |||||
| Infant foods | — | GC‐FID | Epoxidized soybean oil | 9–86 μg g−1 | Fankhauser‐Noti et al. (2005) |
| Oily sauces | — | GC‐FID | Epoxidized soybean oil | 47–580 μg g−1 | Fankhauser‐Noti et al. (2005) |
| Products in oil | — | GC‐FID | Epoxidized soybean oil | 85–350 μg g−1 | Fankhauser‐Noti et al. (2005) |
| Bakery foods | — | GC‐FID | cis‐epoxy oleic acid | 150–4240 μg g−1 | Fankhauser‐Noti et al. (2006) |
| GC‐FID | cis‐epoxy linoleic acid | 60–3460 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| GC‐FID | Diepoxy linoleic acids | 0.5–5.3 μg g−1 | Fankhauser‐Noti et al. (2006) | ||
| Products in oil | — | GC‐FID/GC–MS | Epoxidized soybean oil | 36–374 μg g−1 (GC‐FID) and 42–363 μg g−1 (GC‐MS) | Biedermann‐Brem et al. (2007) |
| Biscuits | — | GC‐FID | Epoxy FAs | 46.95–74.49 μg g−1 | Mubiru et al. (2014) |
| Mayonnaise | — | GC‐FID | Epoxy FAs | 32.6–49.16 μg g−1 | Mubiru et al. (2014) |
| Nuts | — | GC‐FID | Epoxy FAs | 48.88–170.78 μg g−1 | Mubiru et al. (2014) |
| Butter | — | GC‐FID | Epoxy FAs | 115.34 μg g−1 | Mubiru et al. (2014) |
| Meat | — | GC‐FID | Epoxy FAs | 3.24–5.58 μg g−1 | Mubiru et al. (2014) |
| Chocolate, cocoa butter | — | GC‐FID | Epoxy FAs | 0.57–3.38 mg g−1 | Brühl et al. (2016) |
| Extra virgin olive oil fried with doughnut/pork/salmon fillets | Heated at 190 °C for 7.5–3.02 hours | 1H NMR | (E)‐9,10‐epoxystearic acyl groups | 4–20 mmol mol−1 TAG (1.4–7.0 mg g−1)c | Martínez‐Yusta and Guillén (2014b) |
| (Z)‐9,10‐epoxystearic acyl groups | 4–12 mmol mol−1 TAG (1.4–4.2 mg g−1)c | Martínez‐Yusta and Guillén (2014b) | |||
| Primary alcohols | 2–5 mmol mol−1 TAG (0.7–1.7 mg g−1)c | Martínez‐Yusta and Guillén (2014b) | |||
| Secondary alcohols | 4–20 mmol mol−1 TAG (1.4–7.0 mg g−1)c | Martínez‐Yusta and Guillén (2014b) | |||
a Values in units of ppm were converted into μg g−1 using 1 : 1 ratio.
b Values in units of mmol kg−1 or mmol L−1 were converted into mg g−1 for comparison, using the molecular weights of methyl epoxystearate and methyl diepoxystearate and assuming that all monoepoxides were methyl epoxystearate and all diepoxides were methyl diepoxystearate. The density of oil was assumed at 0.9 kg L−1 for calculation.
c Values in units of mmol mol−1 TAG were converted into mg g−1 for comparison, using the molecular weights of methyl epoxystearate for epoxides and methyl hydroxystearate for alcohols. The molecular weight of TAG was assumed at 900 g mol−1 for calculation.
—, information not available; GC, gas chromatography; FID, flame ionization detector; FA, fatty acid; FAME, fatty acid methyl esters; HPLC, high‐performance liquid chromatography; UV, ultraviolet; ELS, evaporative light scattering; TAG, triacylglycerol; ESI, electrospray ionization; MS, mass spectrometry; SIM; NMR, nuclear magnetic resonance.
7.3.1 Quantification of Oxygenated FAs
While GC‐MS may help to identify the structures of oxygenated FAs, standards of commercial FAs are used as references to determine retention times and response factors in GC‐FID. The major challenge in measuring oxygenated FAs in oils is that they are present at low concentrations. In order to make them detectable, it is necessary to extract and concentrate them from large amounts of the unaltered FAs. One useful approach to such separation employs solid‐phase extraction (SPE) with a silica column as the stationary phase. When this technique was applied to oxidized fats and oils directly, the relatively polar compounds, including DAGs, monoacylglycerols (MAGs), and free fatty acids (FFAs), were separated from TAGs (Dobarganes et al. 2000). For SPE separation between non‐oxygenated and oxygenated FAME, the first elution solvent, comprising a less polar solvent mixture such as hexane : ethyl acetate 98 : 2 (v/v) (Jenske and Vetter 2008), hexane : diethyl ether 95 : 5 (v/v) (Velasco et al. 2002), and hexane : diethyl ether 98 : 2 (v/v) (Marmesat et al. 2008), removes unaltered FAME, so oxygenated FAME are retained. The second elution solvent, normally ethyl acetate or diethyl ether, is sufficiently polar to elute the oxygenated FAME from the silica column. By removing the unaltered FAME, SPE acts as a concentration step and has been useful in quantifying trace levels of oxygenated FAs (Velasco et al. 2002; Jenske and Vetter 2008; Marmesat et al. 2008). The SPE step can also be modified for specific purposes. For example, Wilson et al. (1997) used three elutions on a silica column to separate monohydroxy FAs from dihydroxy FAs and other more polar compounds. Such SPE separations can be particularly useful in improving resolution with GC. For instance, Mubiru et al. (2013) also used three elutions on a silica column to elute epoxy FAs with n‐hexane : diethyl ether (90 : 10, v/v), and successfully removed co‐eluting hydroxy FA interferences from monoepoxy FAs. The pretreatment methods and detection conditions are summarized in Table 7.7.
Table 7.7 Gas chromatographic (GC) quantitative methods and corresponding pretreatment and detection conditions for analysis of oxygenated fatty acids (FAs) in foods.
Source: Reproduced with kind permission of the Institute for Food Technologists (Xia and Budge 2017).
| Analytes | Samples | Pretreatment | Extraction condition | Detection | Column | LOD/LOQ | References |
| Monohydroxy (plus hydroperoxy) FAs | Nut oil | Hydrogenation, transmethylation, SPE, and methylation of OH group | SPE: 1st elution: 95 : 5 hexane : ethyl acetate; 2nd elution: 80 : 20 hexane : ethyl acetate | EI‐MS‐SIM | CP‐Sil 19 fused silica (25 m × 0.25 mm i.d.) | LOD = 0.2 ng (injection volume = 1 μL) | Wilson et al. (1997) |
| Mono epoxy FAs | Thermoxidized olive and sunflower oils | Transmethylation and SPE | SPE: 1st elution: 95 : 5 hexane : diethyl ether; 2nd elution: diethyl ether | FID | DB‐Wax fused silica (30 m × 0.25 mm i.d.) | Not reported | Velasco et al. (2002) |
| Mono epoxy FAs | Thermoxidized olive and sunflower oils/used frying oils | Transmethylation and SPE | SPE: 1st elution: 95 : 5 hexane : diethyl ether; 2nd elution: diethyl ether | FID | HP‐Innowax fused silica (30 m × 0.25 mm i.d.) | Not reported | Velasco et al. (2004) |
| Methyl diepoxy linoleate | Oil/sauce/infant food | Transmethylation, NPLC extraction | NPLC column: packed with a cyano phase (25 cm × 0.2 mm i.d.) Mobile phase: 20% MTBE/ pentane | FID | A column coated with a 0.2 mm film of PS‐255, a methyl polysiloxan (30 m × 0.25 mm i.d.) | LOD = 2 mg kg−1 extract and LOQ = 6 mg kg−1 | Fankhauser‐Noti et al. (2005, 2006) |
| Monoepoxy FAs | Oils/snacks | Transmethylation | N/A | FID | SP‐2560 (100 m × 0.25 mm i.d.) | Not reported | Fankhauser‐Noti et al. (2006), Biedermann‐Brem et al. (2007) |
| Methyl mono−/di‐epoxy linoleate | Oily foods | Transmethylation | N/A | EI/CI‐MS | Rtx 2330 (20 m × 0.25 mm i.d.) | LOD < 1 mg kg−1 (CI) LOQ = 3 mg kg−1 (EI) | Biedermann‐Brem et al. (2007) |
| Epoxy, keto, and hydroxy FAs | Thermoxidized sunflower oils | Transmethylation, SPE, and hydrogenation | SPE: 1st elution: 98 : 2 hexane : diethyl ether; 2nd elution: diethyl ether | FID | DB‐Wax fused silica (60 m × 0.25 mm i.d.) | LOQ = 1.6–2.1 μg mL−1 | Marmesat et al. (2008) |
| Keto and hydroxy FAs | Milk fat | Transmethylation and SPE | SPE: 1st elution: 98 : 2 hexane : diethyl ether; 2nd elution: diethyl ether | FID | DB‐Wax fused silica (30 m × 0.32 mm i.d.) | Not reported | Márquez‐Ruiz et al. (2011) |
| Mono epoxy FAs | Arachid/colza/corn/ frying/ olive/soya/ sunflower/ salad/mixed/ soya oils | Transmethylation and SPE | SPE: 1st elution: 98 : 2 hexane : diethyl ether; 2nd elution: 90 : 10 hexane : diethyl ether; 3rd elution: 70 : 30 hexane : diethyl ether | FID | CP‐Sil 88 (60 m × 0.25 mm i.d.) | LOD = 1.45 μg g−1 and LOQ = 2.9 μg g−1 | Mubiru et al. (2013) |
| Mono epoxy FAs | Oil/crisps/pork/milk powder | Modified Bligh and Dyer method, transmethylation, and SPE | SPE: 1st elution: 98 : 2 hexane : diethyl ether; 2nd elution: 90 : 10 hexane : diethyl ether | FID | CP‐Sil 88 (50 m × 0.25 mm i.d.) | LOD = 1.7–10.2 μg g−1 and LOQ = 3.3–20.5 μg g−1 | Mubiru et al. (2014) |
RSD, relative standard deviation; LOD, limit of detection; LOQ, limit of quantification.
With appropriate pretreatment, including transmethylation and SPE, GC‐FID has been reported to be a powerful tool in simultaneously determining epoxy, hydroxy, and keto FAs (Marmesat et al. 2008), as well as monoepoxy FAs alone (Berdeaux et al. 1999; Velasco et al. 2002). In these studies, hydrogenation led to simpler chromatograms and better peak shapes for epoxy FAs. This step was particularly important for the resolution of keto and hydroxy FAs by GC, as their peaks severely co‐eluted in their unsaturated forms. An obvious disadvantage is that hydrogenation resulted in a loss of information about the number and position of double bonds in the hydroxy and keto FAs. Unsaturated oxygenated FAs have been discussed, but the limited information concerning their structures in nonmodel systems has prevented easy quantification by GC. Measurements of unsaturated monoepoxy FAs in fresh oils have been conducted by Mubiru et al. (2013). The limit of detection (LOD) and limit of quantification (LOQ) were 1.45 and 2.9 μg /g of oil, respectively. This method has also been applied to monitor epoxy FA formation in fats, oils, and chocolates (Brühl et al. 2016).
Methoxy derivatives of C‐18 hydroxy FAME isomers were employed as a marker of lipid peroxidation (Wilson et al. 1997), where hydrogenation was conducted to reduce hydroperoxy groups to hydroxy groups and to eliminate double bonds; thus, this approach did not differentiate between hydroxy and hydroperoxy FAs. Two ion fragments resulted from cleavage at each side of the OCH3 group, giving the location of the original hydroxy/hydroperoxy group and enabling quantification in selective ion monitoring (SIM) mode. This approach has been applied to food and oil samples, with a detection limit of 0.2 ng when monitoring a single isomer. It was further developed for simultaneous determination of hydroxy and epoxy FAs in human plasma, where such FAs were converted into methoxy and dimethoxy derivatives, respectively (Wilson and Lyall 2002); however, no applications have been reported for food.
Fluorinated reagents, such as fluorinated anhydride and pentafluorobenzoyl chloride, have been used to analyse hydroxy FAME in biological samples where hydroxy groups were derivatized into fluorinated groups. The utilization of fluorinated derivatives with NCI increased sensitivity and selectivity for hydroxy FAs (Stan and Scheutwinkel‐Reich 1980; Jenske and Vetter 2008). A recent study reported the detection limits to be 300 fg–2 pg for 2‐hydroxy FAME and 50–500 fg for 3‐hydroxy FAME in bovine milk fat (Jenske and Vetter 2008). However, since fluorinated derivatives of hydroxy FAME have seldom been used for the study of lipid oxidation, their applicability to unsaturated FAs needs verification. Like hydroxy FAME, the use of fluorinated reagents facilitated the quantification of keto FAME. With improved sensitivity in negative CI (NCI) mode, PFB derivatives have been mainly applied for quantification, with a detection limit of 0.1 pg (Hachey et al. 1991). No applications of these fluorinated derivatives have been reported for the study of lipid oxidation in food and oil.
In summary, GC has shown excellent sensitivity for the determination of oxygenated FAs as minor products of FAs oxidation, but there remain unresolved problems in co‐elution and identification of unknown compounds. Co‐elution results in even fewer characteristic spectra and further difficulties in the identification of the peaks. Instead of traditional one‐dimensional GC, two‐dimensional GC, employing two columns of distinct polarity, has also been used to analyse food FAs, which significantly improved the separations of both the major and the minor components (Hyötyläinen et al. 2004). This technique has potential in obtaining improved separations of complex oxygenated oxidation products in GC.
7.4 Analysis of Sterol Oxidation
Guardiola (2002) published a very exciting book on the analysis, occurrence, and biological effects of cholesterol and phytosterol oxidation products. The advent of modern analytical instrumentation and techniques, however, has given rise to the most precise, correct, and reproducible results on sterols and their oxidation products during frying. It is worth mentioning that not all of the scientific reporting on this subject is based on frying alone, and studies on similar food compositions and frying conditions can be used to evaluate data or extrapolate them to the real food matrix during frying.
7.4.1 Analysis of Cholesterol and COPs
Foods of animal origin are largely formed of cholesterol, which is oxidized to form cholesterol oxidation products (COPs). Dinh et al. (2011) reviewed the cholesterol contents of different meat and poultry products. They found that much of the early data (through the early 1980s) was produced using either spectrophotometric, gravimetric, or enzymatic methods, many of which were originally developed to measure cholesterol in blood serum (Caldironi and Manes 2006). Cholesterol in foods has been determined gravimetrically by precipitation using digitonin or tomatin, with digitonin the more common (Bragagnolo 2009). Digitonin is washed and the precipitated cholesterol is weighed and calculated using several common factors to account for the proportion of cholesterol in the precipitate. A series of sample preparation steps can be followed from lipid extraction to purification, or direct precipitation of cholesterol from the extract can be performed.
Enzymatic determination of cholesterol has been used for food samples, but it is much more common in clinical laboratories (Bragagnolo 2009). Enzymatic methods employ cholesterol esterase to cleave any ester bond and cholesterol oxidase to oxidize cholesterol to peroxide. The peroxide then reacts with peroxidase and 4‐aminophenazone (or other reagents) to produce a pigment that can be measured spectrophotometrically (Rambaldi et al. 2009). The enzymatic method has been used commonly for dairy products and occasionally for other foods, including meats (Caldironi and Manes 2006). It was found to yield comparable results to the GC technique when used to determine the cholesterol content of milk (not statistically tested) and processed foods containing primarily animal fat or fat of animal origin (Ulberth and Reich 1992). Nonetheless, this method lacks specificity, because other sterols with a 3β‐OH group, including phytosterols, can also be oxidized to form similar pigments. Therefore, in processed foods containing primarily lipids of vegetable origin, the enzymatic method overestimates cholesterol content compared with GC. Jiang et al. (1991) evaluated four methods of cholesterol determination in egg yolk: colourimetric, enzymatic, GC, and HPLC. Results with the enzymatic method were found to be similar to those with GC and HPLC. All three achieved excellent precision; colourimetry alone did not.
In colour‐based methods, the application of the Liebermann–Burchard reaction or other colour development reactions is usually the key step after extraction and de‐esterification. Colouring reagents include acetic acid and concentrated sulfuric acid, which react in solvents such as chloroform and ether (Liebermann–Burchard reaction); para‐toluene sulfonic acid or similar reagents containing glacial acetic acid and concentrated sulfuric acid; and iron salt (iron chloride or iron sulfate) in sulfuric acid and glacial acetic acid (Tonks 2006). Colour‐producing reagents are added after extraction, saponification, and purification, or sometimes directly to the extract. Colourimetric methods usually suffer various interferences from unsaturated FAs, proteins, and other steroids, or even from vitamin A. Cholesterol esters, if not hydrolysed, can react with colour reagents in different ways. The colour reactions depend on a double bond system, other functional groups, and the presence of a nonpolar side chain. Colourimetric methods have commonly been used to measure cholesterol in meats; however, the lack of specificity and colour stability, the issue of temperature dependency, and the turbidity of the final colour mean there are grave concerns about their accuracy (Tonks 2006). Without saponification, Bohac et al. (1988) showed that the colourimetric method overestimated the cholesterol content of meats. Even with saponification, the presence of more unsaturated FAs (more double bonds) yielded greater cholesterol concentrations without antioxidant protection. The cholesterol contents of saponified samples with an antioxidant were similar to those analysed by GC. Gravimetric, colourimetric, and enzymatic methods require strict control of analytical conditions in order to provide precise and accurate results (Bragagnolo 2009).
7.4.1.1 Gas Chromatography
GC and HPLC have been studied extensively in recent years. Chromatography is preferred for cholesterol analysis in foods because of its specificity in separating cholesterol from other unsaponifiable compounds based on their differences in terms of physical and chemical properties and their interactions with stationary and mobile phases. Processed meats with non‐meat ingredients contain not only cholesterol but also plant sterols, tocopherol, tocotrienol, saturated hydrocarbons, squalene, aliphatic alcohols, terpene alcohols, triterpene alcohols, and stearoyl esters, all of which are unsaponifiable (Fenton 1992). Recently, HPLC has been used more than GC, as it is thought to decrease cholesterol oxidation through its lower operational temperature and it allows for a nondestructive separation; however, GC is still preferred for its higher sensitivity (Bragagnolo 2009).
Because of the interference of other unsaponifiable materials and the cost and availability of highly specific detectors, the effectiveness of the chromatographic separation and quantification of cholesterol greatly depends on the heart of the chromatographic system: the column. GC is still the most commonly used technique for the determination of sterols, including cholesterol – especially when cholesterol is the sole compound of interest (Fenton 1992; Abidi 2001). GC columns have evolved dramatically, from simple packed columns to capillary columns with wide variation in polarity. Ubhayasekera et al. (2004) evaluated three methods of saponification and trans‐esterification of tallow using GC and GC‐MS, and showed that GC‐MS is more sensitive than GC. Yen et al. (2010) developed a GC‐MS method for the separation of cholesterol and eight COPs. Cardenia et al. (2012) developed a fast GC‐MS method for the analysis of COPs in fried fish. They separated all COPs studied in 3.6 minutes with very good separation, as shown in Figure 7.3. The separated compounds were 7α‐HC, 19‐HC, 7β‐HC, β‐CE, α‐CE, triol, 25‐HC, and 7‐KC.

Figure 7.3 Fast GC‐MS trace (TIC) of TMS derivatives of a standard COP mixture, obtained with a 10‐m RTX‐5 column. Peak identification: 1, 7α‐HC; 2, 19‐HC; 3, 7β‐HC; 4, β‐CE; 5,α‐CE; 6, triol; 7, 25‐HC; 8, 7‐KC.
Source: Reproduced with kind permission from Wiley‐VCH Verlag GmbH & Co. KGaA. (Cardenia et al. 2012).
The authors evaluated the sensitivity of the method in a standard COP mixture by defining the LOD and LOQ of each oxysterol. The LOD and LOQ of fast GC‐MS method varied from 2.18 to 5.07 and from 6.93 to 16.90 ng mL−1, respectively (Table 7.8). These results were in agreement with Guardiola (2002), who reported that a single COP detected by GC‐MS in single ion‐monitoring mode displayed different LODs and LOQs, probably due to different molecular structures and competitive interactions (Cardenia et al. 2012).
Table 7.8 Analytical parameters of fast and conventional GC‐MS methods.
Source: Reproduced with kind permission WILEY‐VCH Verlag GmbH & Co. KGaA (Cardenia et al. 2012).
| Analyte | Intraday (RSD) | Interday (RSD) | LOD (ng mL−1) | LOQ (ng mL−1) | ||||
| Fast | Conventional | Fast | Conventional | Fast | Conventional | Fast | Conventional | |
| 7α‐HC | 2.36 | 2.37 | 2.36 | 4.38 | 2.18 | 2.82 | 6.93 | 9.40 |
| 7β‐HC | 2.85 | 3.72 | 4.02 | 5.01 | 3.68 | 3.11 | 12.27 | 10.37 |
| β‐CE | 1.35 | 4.01 | 5.79 | 4.34 | 4.97 | 4.19 | 16.57 | 13.97 |
| α‐CE | 3.09 | 0.49 | 4.12 | 2.34 | 5.07 | 5.14 | 16.90 | 17.13 |
| Triol | 3.35 | 4.28 | 5.40 | 4.30 | 2.65 | 2.03 | 8.83 | 6.77 |
| 25‐HC | 4.29 | 2.82 | 3.66 | 3.66 | 2.85 | 2.73 | 9.50 | 9.10 |
| 7‐KC | 1.81 | 4.02 | 4.61 | 4.61 | 3.92 | 4.84 | 13.07 | 16.13 |
RSD, relative standard deviation; LOD, limit of detection; LOQ, limit of quantification.
Souza et al. (2017) developed a novel microwave‐assisted direct saponification method for the simultaneous determination of cholesterol and cholesterol oxides in shrimp. They showed very low detection (≤0.57 μg mL−1) and quantification (≤1.73 μg mL−1) limits, good repeatability (≤10.50% intraday and ≤8.56% interday), and low artifact formation. The LOD and LOQ were, however, lower than reported by Cardenia et al. (2012). The uses of solid phase microextraction and subsequent analysis by GC‐FID can also be very effective for the analysis of cholesterol and its oxidation products (Narvaez‐Rivas et al. 2014).
7.4.1.2 Liquid Chromatography
There are many published HPLC methods for cholesterol determination; however, most were not developed for routine analysis, but rather to separate cholesterol from other unsaponifiable compounds for specific research purposes (Mestre Prates et al. 2006; Daneshfar et al. 2009). Because of the slight polarity caused by the hydroxyl group, either normal‐phase (NP) or reversed‐phase (RP) HPLC can be used for analysis of cholesterol. NP‐HPLC has been used to separate triglycerides, diglycerides, and cholesterol (Fenton 1992). As mentioned previously, the GC system is ineffective for separating cholesterol from tocopherols; however, Katsanidis and Addis (1999) successfully separated and quantified cholesterol, vitamin E, and vitamin E homologues using an NP‐HPLC technique with a 25 cm Zorbax RX‐Sil column (particle size = 5 μm). The compounds were detected with a UV detector at 295 nm for vitamin E and 202 nm for cholesterol. The column was made of ultra‐clean porous silica microparticles. The mobile phase was 99% hexane and 1% isopropanol. Most NP‐HPLC methods use an NP (polar stationary phase) column made of highly pure, porous silica microparticles, such as μPorasil (Kermasha et al. 1994), Zorbax RX‐Sil (Katsanidis and Addis 1999), InertSil ODS‐2 (Sion et al. 2001), Zorbax RX‐Sil (Ponte et al. 2008), or Spherisorb S5W (Costa et al. 2006). Generally, the particle size is 5 μm; however, the column used by Kermasha et al. (1994) was 10 μm. Smaller particle size leads to significantly increased column pressures, but also increases the resolution (theoretical plate number). More polar stationary phases, such as cyanopropylsilica and alcohol‐bonded silica, have been used (Abidi 2001). The mobile phase for NP‐HPLC has primarily been an isocratic phase of 1–3% isopropanol in hexane (Kermasha et al. 1994; Katsanidis and Addis 1999; Abidi 2001; Ponte et al. 2008). In order to use light‐scattering detection and improve peak shape, Sion et al. (2001) used a gradient mobile phase made of 98 : 2 methanol/water (v/v), 30 : 6 : 10 chloroform/methanol/water (v/v/v), and 50 : 50 methanol/chloroform (v/v). A constant flow of 1 mL min−1 or lower has been used in most studies.
Grun and Besseau (2016) developed NP‐ LC coupled to an atmospheric pressure photoionization MS method for the determination of cholesterol and phytosterol oxidation products. They reported the separation of 15 sterol oxidation products, as shown in Figure 7.4. The best chromatographic separation with the highest sensitivity was accomplished by employing a solvent system consisting of hexane and isopropanol on a diol‐bonded silica column. The dopant of choice was chlorobenzene, added post‐column to the solvent stream at 10% of the flow rate. The developed NP‐LC‐APPI‐MS method proved to be a valuable tool for the separation and detection of sterol oxidation products.
Figure 7.4 NP‐LC‐APPI‐MS analysis of cholesterol oxidation products. (1) cholesta‐3,5‐diene, (2) 5‐cholesten‐3‐one, (3) 4‐cholesten‐3‐one, (4) cholesta‐4,6‐dien‐3‐one, (5) cholesterol, (6) 22‐ketocholesterol, (7) 5 α,6α‐epoxycholesterol, (8) 20α‐hydroxycholesterol, (9) 7‐ketocholesterol, (10) 25‐hydroxycholesterol, (11) 22(S)‐hydroxycholesterol, (12) 19‐hydroxycholesterol, (13) 7 α‐hydroxycholesterol, (14) 7 β‐hydroxycholesterol, and (15) 5 α,6β‐dihydroxycholesterol.
Source: Reproduced with kind permission of Elsevier (Grun and Besseau 2016).
Although NP‐HPLC was the first technique used successfully to separate cholesterol in meat such as beef (Abreu et al. 2016), bovine tissues (Katsanidis and Addis 1999), and chicken (Ponte et al. 2008), RP‐HPLC has been preferred because it offers a wider range of column selectivity and separable compounds based on polarity. The RP‐HPLC method uses a nonpolar column or a column with low polarity and a polar mobile phase. The simple addition of some polar solvents, such as methanol, ethanol, or water, can dramatically change mobile phase polarity and alter elution (Abidi 2001). Most RP‐HPLC methods of cholesterol determination use acetonitrile as the polar organic solvent, with the addition of either ethanol (Daneshfar et al. 2009), methanol (Simsek et al. 2009), or isopropanol (Komprda et al. 2003). They have typically employed silica (Abidi 2001) as a 5 μm particle support material covalently bonded with either octadecyl (Salvatori et al. 2008) or octyl (Nakajima et al. 1995; Daneshfar et al. 2009). The RP‐HPLC C‐18 column was found to be superior to the C‐8 column because it has a much stronger retention of cholesterol and other sterols (Fenton 1992). Therefore, a mobile phase that is less polar should be employed for cholesterol separation on a C‐18 column. The HPLC technique, although superior to GC in terms of resolution of multiple unsaponifiable compounds, can be further improved by programmed temperature elution.
Constantinou et al. (2015) reported a baseline separation of seven COPs in several food samples. The separation was achieved with the help of a cyano‐bonded HPLC column and isocratic mobile phase consisting of n‐hexane/2‐propanol/acetone (97 : 1.5 : 1.5, v/v), as shown in Figure 7.5. The LOD and LOQ were in the range 0.15–0.63 and 0.45–1.91 μg mL−1, respectively, for the seven COPs.

Figure 7.5 Optimum HPLC chromatographic separation of seven cholesterol oxidation products (COPs). Baseline separation was achieved by the use of n‐hexane:2‐propanol : acetone (97 : 1.5 : 1.5, v/v) at a flow rate of 1 ml min−1 and a temperature of 25 °C. (1) 20α‐ΟΗ, (2) 22R‐OH, (3) 22S‐OH, (4) 25‐OH, (5) 7‐keto, (6) 7β‐ΟΗ, (7) 7α‐ΟΗ.
Source: Reproduced with kind permission of Springer Nature (Constantinou et al. 2015).
Georgiou et al. (2016) used a novel approach for the fast determination of the most important COPs in several Cypriot foods (halloumi cheese, feta cheese, hiromeri, snail, etc.) using UPLC‐ESI‐MS (Table 7.9). Among them, 7‐keto was detected in all of the samples analysed. This finding confirms that 7‐keto can be used as an appropriate marker of cholesterol oxidation in food systems, especially fried products.
Table 7.9 Levels of cholesterol oxidation products (COPs) detected in Cypriot food products.
Source: Reproduced with kind permission of WILEY‐VCH Verlag GmbH & Co. KGaA (Georgiou et al. 2016).
| Samples | COPs (ng per 250 mg fat) | |||
| 7‐OH | 7‐Keto | 5,6‐EP | Total COPs | |
| Yogurt | 41 ± 1.7 | 57 ± 5.2 | 1314 ± 1.7 | 1412 |
| Feta cheese | 1430 ± 1.8 | 73 ± 2.8 | n.d | 1503 |
| Halloumi cheese | 616 ± 1.1 | 82 ± 3.0 | n.d | 698 |
| Anari cheese | 105 ± 2.4 | 1099 ± 2.7 | n.d | 1204 |
| Snail | 1239 ± 4.7 | 1424 ± 3.0 | n.d | 2663 |
| Hiromeri | 395 ± 2.2 | 155 ± 2.8 | 105 ± 2.7 | 655 |
| Sausage | 444 ± 1.7 | 490 ± 4.7 | n.d | 934 |
| Salami | 972 ± 2.5 | 141 ± 3.7 | 442 ± 3.0 | 1555 |
| Caul fat | 122 ± 1.7 | 331 ± 3.6 | 1406 ± 2.3 | 1859 |
| Bacon (raw) | 1104 ± 1.5 | 158 ± 3.0 | n.d. | 1262 |
| Bacon (fried) | 2166 ± 1.5 | 237 ± 1.6 | 232 ± 2.8 | 2635 |
| Rabbit (fried) | 843 ± 1.8 | 681 ± 1.4 | 202 ± 2.6 | 1726 |
| Rabbit (oven‐cooked) | 2530 ± 0.3 | 938 ± 1.8 | 1594 ± 2.7 | 5062 |
Values are mean with standard deviations (n = 4).
Another rapid and sensitive HPLC‐APCI‐MS/MS method for the determination of COPs in milk powder‐based foods was reported by Gorassini et al. (2017). The method consists in the direct saponification of the sample and purification of oxysterols by reversed‐phase C‐18‐SPE followed by HPLC‐MS/MS analysis. By this procedure, the extraction and enrichment of oxysterols are combined in a unique step, reducing sample manipulation and the possible formation of artifacts. The LOD and LOQ were in the concentration ranges of 2–8 and 8–30 ng g−1, respectively.
These studies suggest that HPLC‐MS provides a very sensitive analytical tool to determine the cholesterol or COP contents in fried foods. However, it should be kept in mind that sample preparation is highly important: Georgiou et al. (2014) showed that it contributes to artifact formation, perhaps altering the chemical composition of foods under study. Further, no method used to date can be considered precise and accurate for routine analysis of COPs in foods.
7.4.2 Analysis of Phytosterol Oxidation
Plant foods also contain sterols called phytosterols, which can be oxidized to form phytosterol oxidation products (POPs). Thermal oxidation of phytosterols occurring in foods encompasses a sequence of reactions resulting in primary (hydroperoxides), secondary (polar: ketones, alcohols, epoxides; unpolar: steradienes, steratrienes), and tertiary (dimers, oligomers, polymers) oxidation products. Due to the analytical capabilities of each lab, the focus has almost exclusively been put on the secondary polar POPs. Accordingly, the term ‘POPs’ here refers to the keto‐, epoxy‐, and hydroxy‐compounds derived from the respective sterols/stanols.
There are a number of analytical methodologies available, based on (i) lipid extraction and saponification or transesterification, (ii) isolation and purification via TLC or solid phase extraction, (iii) derivatization to trimethylsilyl ethers, and (iv) detection via HPLC‐ and GC‐based techniques (Guardiola et al. 2004). GC‐based analysis of POPs is usually not preferred at high temperatures. GC‐FID has been used for phytosterol analysis (Duong et al. 2016). The use of MS detectors has improved the sensitivity of phytosterols in foods (Inchingolo et al. 2014).
These methods have been employed to generate a broad spectrum of analytical data on model systems involving the thermal treatment of phytosterol standards (Oehrl et al. 2001; Soupas et al. 2004; Barriuso et al. 2012). Thermal oxidations under different time and temperature conditions revealed that some of the secondary POPs constitute intermediates, which are further transformed or degraded in the course of the reaction (Rudzińska et al. 2009). Initial attempts to isolate fractions containing dimers, trimers, and tetramers via size‐exclusion chromatography (SEC) have been described, and structures for sterol dimers have been proposed (Lampi et al. 2009; Rudzińska et al. 2010; Sosińska et al. 2013, 2014).
Heating of stigmasterol at 180 °C for 3 hours resulted in a loss of the intact sterol of 61%. Polar, midpolar, and nonpolar oxidation products accounted for 39% of the loss; the formation of dimers and polymers accounted for 30%. This means that there is a gap in the mass balance, leaving 31% of the stigmasterol loss unexplained (Menéndez‐Carreño et al. 2010).
There are data available indicating qualitative and quantitative differences in oxidation profiles between free and esterified phytosterols (Soupas et al. 2004; Lehtonen et al. 2011). Complex mixtures are to be expected, owing to potential oxidations in the sterol as well as in the FA moiety of phytosteryl esters of unsaturated FAs (Lehtonen et al. 2012). Very recent studies describe approaches to the analysis of intact oxidized phytosteryl FA esters via HPLC‐ESI‐MS (Julien‐David et al. 2014).
Sun et al. (2017) used 4'‐carboxy‐substituted rosamine (CSR) as a derivative reagent for the determination of phytosterols. The CSR reacted with the hydroxyl group of phytosterol to form a CSR‐phytosterol complex (Figure 7.6).
Figure 7.6 Derivatization reaction of phytosterols with 4'‐carboxy‐substituted rosamine (CSR).
Source: Reproduced with kind permission of Wiley‐VCH Verlag GmbH & Co. KGaA (Sun et al. 2017).
Agilent SB C‐18 column with the specification 2.1 × 50 mm, 1.8 μm was used for separation via UHPLC‐MS/MS. With the help of 0.1% formic acid in the mobile phase, better separations were obtained for five phytosterols and diosgenin derivatives using acetonitrile versus methanol (Figure 7.7). The major product ions for all CSR derivatives of phytosterols and diosgenin are m/z 443.1 and 399.0. In this work, the product ion at m/z 443.1 was used for quantitation, and the product ion at m/z 399.0 was employed to confirm the identification.

Figure 7.7 (a) Representative multiple reaction monitoring (MRM) chromatograms of 4'‐carboxy‐substituted rosamine (CSR) derivatives of five phytosterols and diosgenin in corn oil. (b) Product ion spectrum and proposed fragmentation pathways of a CSR‐stigmasterol derivative.
Source: Reproduced with kind permission of Wiley‐VCH Verlag GmbH & Co. KGaA (Sun et al. 2017).
Delgado‐Zamarreño et al. (2016) proposed an LC‐MS method, which was successfully applied to the analysis of free phytosterols in almonds, cashew nuts, hazelnuts, peanuts, tiger nuts, sunflower seeds, and pistachios. Another method was reported by Yuan et al. (2017) for the simultaneous determination of tocopherols and phytosterols in edible oil. The authors determined the content of plant sterols, which ranged from 94.9 mg /100 g in camellia oil to 314.1 mg /100 g in corn oil, 164.1 mg /100 g in soybean oil, and 127.8 mg /100 g in olive oil. Ito et al. (2017b) also reported an HPLC‐FLD method for determining the phytosterol contents of several plant foods. There is a large amount of literature available regarding phytosterol and its oxidation products in foods, suggesting that HPLC with different detection systems offers superior analytical tools for studying the frying of foods.
7.5 Analysis of Sensory Metabolites
Zhang et al. (2015) showed that a palatable aroma, which is mainly attributed to the volatile compounds formed during the frying process, is one of the reasons that fried food is so popular. The two major sources of volatile compounds are the low‐molecular‐weight TAG oxidation decomposition compounds, such as aldehydes, alcohols, ketones, hydrocarbons, and acids, and the Maillard reaction products, such as sulfur‐ and nitrogen‐containing compounds (Martin and Ames 2001). Due to their direct influence on the quality of fried food (both positive and negative), the identification of these volatile compounds in the frying process is useful and vital (Romano et al. 2013; Aladedunye and Matthäus 2014).
Because of their volatility, the determination of volatile compounds during or at the end of the frying process is not easy. However, precisely designed devices for identifying volatile products have been reported to overcome this difficulty. Early isolation of volatiles was mainly achieved using a Likens–Nickerson apparatus modified to perform simultaneous steam distillation and extraction (SDE) with an organic solvent such as hexane or diethyl ether. As a result, the volatiles from deep‐fat‐fried, microwave‐heated, and oven‐baked garlic slices were successfully analysed (Yu et al. 1993). Briefly, samples were mixed with distilled water to be steam‐distilled and were then extracted into 60 mL pentane/ether (1 : 1, v/v) solvent in a modified Likens–Nickerson apparatus for 3 hours. Before the drying treatment of the isolate with anhydrous sodium sulfate, a certain amount of 1‐propyl butyrate stock solution was added as the internal standard. Then, the distillate was concentrated to a minimum volume using a spinning‐band distillation apparatus. Finally, the distillate was concentrated by blowing nitrogen gas to a volume of 0.2 mL for GC‐FID and GC‐MS analysis. A similar sample pretreatment was also performed to monitor the volatile compounds released from used frying oil (Takeoka et al. 1996). Later, the formation of volatiles from the Maillard reaction that occurs between lysine and xylose or glucose and that is influenced by various types of vegetable oils was investigated using the same volatile extraction method (Negroni et al. 2001). This method was also utilized to effectively investigate the flavour of deep‐fried shallots as influenced by various types of frying oil (Chyau and Mau 2001).
Another simultaneous gas‐purging and SE apparatus for the isolation of volatiles was designed to use nitrogen in the transfer of volatiles. With the help of this tool, volatile compounds produced in heated peanut oil (Chung et al. 1994) and menhaden fish oil (Horiuchi et al. 1998) with different amounts of cysteine were precisely measured by GC‐MS. Volatile compounds that were trapped in a Tenax trap prior to GC‐MS were measured in order to study the formation of Strecker reaction aldehydes and pyrazines in a fried potato model system. The results showed that both sugars and amino acids present in the potato were crucial to the development of flavour compounds in the final product (Martin and Ames 2001).
Recently, an extraction apparatus was designed to determine the volatile aldehydes formed when coconut oil, safflower oil, canola oil, and extra virgin olive oil were heated for 6 hours at 180, 210, 240, and 270 °C, as shown in Figure 7.8 (Katragadda et al. 2010). By collecting the fumes using Tedlar bags and later analysing them by GC‐MS, saturated aldehydes such as acetaldehyde, propanal, butanal, pentanal, heptanal, octanal, and nonanal and unsaturated aldehydes such as acrolein, 2‐hexenal, 2‐heptenal, 2‐octenal, 2‐decenal, 2,4‐heptadienal, and 2,4‐decadienal were identified in all frying oils. Some of these volatiles, such as 2‐hexenal and 2,4‐decadienal, endow the oily flavour of fried food; however, some of them, such as acrolein and 2,4‐decadienal, are harmful to consumers. Previously, the same fume‐trapping method and similar aldehyde emissions were reported when canola oil, olive oil, and extra virgin olive oil were heated at 180 and 240 °C in a closed Pyrex Instatherm reaction flask (Fullana et al. 2004).

Figure 7.8 Schematic representation of an experimental system used in a frying simulation.
Source: Reproduced with kind permission of Elsevier Ltd (Katragadda et al. 2010).
Different volatile trapping methods, solvent extraction (SE), SDE, and nitrogen purge and steam distillation (NPSD) were tested for the collection of flavour substances from a traditional Dalmatian smoked ham that had previously undergone a dry‐curing and frying treatment (Jerković et al. 2007). After GC and GC‐MS analysis, it was shown that SE extracted the most isolated volatiles, due to its thorough extraction of medium‐volatile compounds, which were only partially isolated by SDE. NPSD extracted the fewest, because it could only extract headspace volatiles. Volatile compounds formed in a simulated frying process can be thoroughly identified by means of these methods. However, practical food frying occurs in an open environment, in which the volatile compounds are released throughout the high‐temperature treatment process. Therefore, determination of the volatile compounds produced at the end of the frying process captures only a portion of the total volatiles produced.
In addition to the foregoing static and dynamic SE, SDE, and NPSD methods, volatile compounds can also be absorbed by absorption fibres coated with special materials, such as carbowax (CW), polydimethylsiloxane (PDMS), divinyl benzene (DVB), carboxen (CAR), and polyethylene glycol. This isolation method is usually known as headspace solid‐phase micro‐extraction (SPME). SPME is recognized as a rapid and convenient sample preparation tool when compared with the traditional methods (Doleschall et al. 2003), and it has been extensively applied to the study of the volatiles of oils and fats and the evaluation of their stability (Gromadzka and Wardencki 2010; Petersen et al. 2012).
The odour released from palm olein deep fried with frozen, breaded chicken at 182 °C for a total of 136 hours (8 hours per day for 17 consecutive days) was investigated by the SPME‐GC‐MS technique (Osawa et al. 2013). Briefly, 5 g of oil was placed in a glass vial with a Teflon septum and an aluminium cap containing a magnetic stir rod. A 50/30 μm DVB/CAR/PDMS fibre with a length of 2 cm coupled to a holder was inserted into the headspace of the vial and positioned approximately 1 cm above the oil level. A total of 45 minutes was timed for the capture of the volatiles by agitation at 500 rpm. A water bath was used to maintain the samples at 80 °C. Then, the fibre was removed from the vial and immediately desorbed into the injector of the GC equipment. A similar isolation procedure was effectively used to collect the volatile compounds released during the storage of potato chips that had previously been fried in mid oleic sunflower oil (Lee and Pangloli 2013). Recently, a new sampling system based on the use of SPE‐GC‐MS to determine the volatile compounds generated during the heating of sunflower and extra virgin olive oils above their smoking point was developed by Ontanón et al. (2013). The relatively sealed environment and the SPE device guaranteed the comprehensiveness of the volatiles formed during the heating process. The main volatiles formed during food frying can be successfully measured using this method.
It is very important to achieve maximum extraction of volatiles in order to analyse them via GC or GC‐MS. Many factors, such as the variety of fibre coating, the extraction temperature and time, the addition of salt to promote volatilization, and whether the sample is stirred can influence the final extraction result. Furthermore, the time gap between food frying and sample extraction should be considered, because it is easy to lose some volatile compounds during this interval. Therefore, conducting SPME of the oil sample as soon as possible after frying and selecting appropriate SPME conditions are crucial for the analysis of volatiles by SPME‐GC‐MS during or at the end of the food frying process.
The analysis of a single volatile compound or of a single class of volatiles has also been studied (Mubiru et al. 2014). Acrolein is a typical low‐molecular‐weight volatile aldehyde formed when oils and fats undergo frying. Derivatization with 2,4‐dinitrophenylhydrazine (2,4‐DNPH) before SPME treatment was conducted to study the acrolein profile in potato chips that had been fried in soybean, corn, canola, sunflower, or palm oils (Osório and De Lourdes Cardeal 2011). This sample pretreatment promoted the extraction of acrolein and improved the accuracy, sensitivity, and linearity of the SPME‐GC‐MS method in the range of interest. The evolution of oxygenated α,β‐unsaturated aldehydes in extra virgin olive oil, sunflower oil, and virgin linseed oil heated at 190 °C for periods of 8 h day−1 was monitored using the SPME‐GC‐MS method. As a result, 4‐hydroxy‐(E)‐2‐nonenal, (E)‐4,5‐epoxy‐(E)‐2‐decenal, 4‐hydroxy‐(E)‐2‐hexenal, and 4‐oxo‐(E)‐2‐nonenal were first detected using standards or taking into account their retention times, mass spectra, and matching rates with a commercial mass spectrum library or with mass spectra published in previous references (Guillén and Uriarte 2012).
Nitrogen‐containing heterocyclic compounds are typical volatile products that result from the interactions that occur between the constituents of food materials and the components of frying oil during the food frying process. Three methods – GC‐ion trap (IT)/MS, GC‐time‐of‐flight (ToF)/MS, and GC × GC‐ToF/MS – combined with SPME were compared for the critical measurement of substituted pyrazines and related substances in potato chips prepared using different frying techniques (Lojzova et al. 2009). Among these compounds, pyrazines, pyrroles, pyridines, thiazoles, thiazolines, oxazoles, oxazolines, thiophenes, and pyrrolidinones were reported. These volatiles are the typical components of the meat flavour generated through the Maillard reaction. Although some pairs of pyrazine isomers, such as 2‐ethyl‐5(6)‐methylpyrazine (4(5)) + 2,3,5‐trimethylpyrazine (6) and 2‐ethyl‐3,5(6)‐dimethylpyrazine (8(9)) + 2,3‐diethylpyrazine (10) were not fully separated from each other, the use of GC × GC‐ToF/MS offered the best solution because it had the lowest quantification limits for all of the targeted alkyl pyrazines among the three methods. Accordingly, the results shown using a two‐dimensional diagram were more visual and effective than those shown in a one‐dimensional diagram.
The formation and emission of volatile compounds, including the aldehydes and some toxic compounds of oil samples, pure refined olive‐pomace oil (ROPO), and the blended ROPO/ refined coconut oil (RCO) (80–20%), were carried out during deep frying at 180 °C (Ben Hammouda et al. 2017). The volatile profile of both oil samples was evaluated using an optimized HS‐SPME‐GC/MS method, before and after 20, 40, and 60 successive sessions of deep frying. The blended ROPO/RCO revealed fewer formations of unsaturated aldehydes, including toxic ones, such as acrolein, and showed a greater stability against oxidative thermal degradation compared to ROPO pure.
In addition to the GC‐MS analysis, volatile aldehydes were also identified using the HPLC‐ultraviolet visible (UV/VIS) technique. Non‐prefried frozen sliced potatoes were fried in DAG‐rich oil and TAG‐rich oil with different unsaturated degrees of FA moieties in a round‐bottom flask at an average temperature of 170 °C (Katsuta et al. 2008). Quantitative analysis of aldehydes was achieved through an external standard method. The same derivatization pretreatment and HPLC‐DAD‐MS method was successfully used to monitor the carbonyl compounds formed during the thermal oxidation of canola oil treated for different heating times and using different metal ions (Bastos and Pereira 2010). As a result, formaldehyde, acetaldehyde, acrolein, propanal, butanal, hexanal, (E)‐2‐heptenal, and octanal were identified in the emissions of the heating system. 2,4‐Decadienal is a dominant volatile compound mainly formed during the peroxidation of linoleic acid and arachidonic acid. 2,4‐Decadienal was extracted from the used frying oil by a methanol/water solution and redissolved in acetonitrile. The acetonitrile solution was further purified by RP‐HPLC separation. Subsequently, the 2,4‐decadienal was identified by GC‐MS analysis (Andrikopoulos et al. 2004). The DNPH derivatization of aldehydes for HPLC analysis was shown to sensitively and precisely identify the volatile aldehydes formed during the frying process. Wang et al. (2016) reported an LC‐MS‐based method involving derivatization of volatiles formed in heated frying oils and French fries with 2‐hydrazinoquinoline. The authors showed that aldehyde formed in French fries is comparable with that formed in frying oil.
7.6 Analysis of Heterocyclic Amines
Heterocyclic amines (HCAs) are Maillard reaction products with potential mutagenicity and carcinogenicity that are found in fried food. Generally, the analysis of HCAs includes sample pretreatments, such as extraction, enrichment, and purification; and qualitative and quantitative analyses using HPLC‐based methods. SPE and other clean‐up procedures have typically been adapted to improve the sensitivity and chromatographic resolution of the HPLC analysis of HCAs formed in fried meat products (Gibis and Weiss 2012; Keşkekoğlu and Üren 2014). The quantitative analysis of HCAs by SPE and HPLC coupled with photodiode‐array UV and FLD was investigated to evaluate the influence of oil type on the evolution of HCAs in fried beef burgers (Johansson et al. 1995). An ODS 80TM column (250 × 4.6 mm; 5 μm) and an LC‐18‐DB precolumn (20 × 4.6 mm) were combined to perform the separation. Three solvents, 10 mM triethylamine with pH 3.2, 10 mM triethylamine with pH 3.6, and acetonitrile, were selected to constitute the mobile phase in linear gradient mode. Identification and quantitative analysis were achieved by comparing the spectra and a peak area of the spiked HCAs. As a result, the amounts of HCAs in the meat and the pan residue were significantly lower when sunflower oil and margarine were used as the frying media than when butter, liquid margarine, margarine fat phase, or rapeseed oil was used.
The HCAs formed in beef steak roasted with extra virgin olive oil for 5 minutes at 200 °C were quantitatively and qualitatively investigated by HPLC‐ESI‐MS (Lee et al. 2011). HCAs were extracted by a series of sample pretreatments, which included sample extraction with sodium hydroxide by sonicating and shaking, elution with dichloromethane/toluene (95 : 5, v/v) on a ChemElut‐20 ML column, SPE with ammonium acetate on a polysulfonic acid cartridge, and SPE with methanol on a C‐18 cartridge. A Zorbox RX‐C‐8 column and a mobile phase consisting of ammonium formate buffer (pH 3.7) and acetonitrile in gradient mode were selected to perform the chromatographic separation. By comparing the retention times of the spiked HCAs, 3‐amino‐1,4‐dimethyl‐5H‐pyrido‐[4,3‐b]indole, 3‐amino‐1‐methyl‐5H‐pyrido[4,3‐b]indole, 9H‐pyrido[3,4‐b]indole, 1‐methyl‐9H‐pyrido[3,4‐b]indole, 2‐amino‐9H‐pyrido[2,3‐b]indole, 2‐amino‐3‐methyl‐9H‐pyrido[2,3‐b]indole, 2‐amino‐3,8‐dimethylimidazo[4,5‐f]‐quinoxaline, and 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]‐pyridine were detected in all the cooked beef steaks.
An alternative HPTLC technique coupled with UV/FLD was selected to quantify the HCAs formed in beef patties fried at 230 ± 1 °C after SPE treatment (Jautz et al. 2008). The chamber was initially automatically saturated with a solution of ammonia aqueous solution/ultrapure water (1 : 4, v/v) for 20 minutes. The plate activity was adjusted to 34% relative humidity with a saturated magnesium chloride aqueous solution. The chromatographic separation was performed up to a migration distance of 60 mm with a solution of methanol/chloroform (1 : 9, v/v) at ambient temperature for 30 minutes. Immersion of the layer in the solution of paraffin/n‐hexane (1 : 1, v/v) was adopted to enhance the fluorescence of the analytes. Multi‐wavelength scanning at 262 nm for absorbance measurements and at 313/>340 and 366/>400 nm for FLD was selected to perform the densitometry. The concentration ranges of HCAs obtained by the HPTLC‐ and the HPLC‐based methods were similar, based on highly satisfactory correlations between the two (R2 = 0.8875–0.9751). However, the HPTLC‐based method was faster and less expensive than the HPLC‐based method.
Ultra‐performance LC‐MS/MS was used to determine the HCA contents of roasted beef patties (Zeng et al. 2016). Another method was reported for HCA analysis in hamburger patties, involving microwave‐assisted extraction of HCA and analysis using HPLC (Aeenehvand et al. 2016). It has been observed that HPLC with specific extraction procedures is the best choice for measurement of HCA in fried foods.
7.7 Analysis of Acrylamide
The quantitative analysis of acrylamide (AA) in foodstuffs or fried foods has been extensively studied and reviewed (Oracz et al. 2011; Tekkeli et al. 2012; Matthäus and Haase 2014; Hu et al. 2015). According to previous studies, GC and HPLC coupled with appropriate detectors, such as an electron‐capture detector (ECD) or a DAD and MS or MS/MS, are conventionally used to monitor AA.
The characterization of AA formed in fried foods has been reported mainly in starch‐based foods, such as potato chips, due to the extensive market for these products. Initial AA analysis was unsuccessful when GC‐based methods in starch‐based samples were used, due to the strong gelatinization effect. To overcome this obstacle, the use of GC‐MS in SIM mode after the bromination of AA to form a derivative with high‐mass ions after fragmentation was investigated. After a series of sample preparations, AA in chips was successfully identified and quantified by the GC‐MS method with a DB‐5MS capillary column and monitored ions of m/z 106 and 109 (Gertz and Klostermann 2002). A DB‐23 capillary column with a programmed temperature and selected ions of m/z 74, 71, 58, and 55 was also adopted to investigate the influences on the AA concentration during the deep fat frying of potatoes. In addition, a DB‐17 capillary column and selected ions of m/z 106, 108, 150, and 152 for the brominated products and 120 and 122 for the brominated methacrylamide were investigated and used to monitor the formation and amount of AA in French fries (Romani et al. 2009).
A modified sample pretreatment method was studied, consisting in the addition of 13C3‐labelled AA as the internal standard, defatting with hexane, extraction with a sodium chloride aqueous solution, derivatization with potassium bromate and potassium bromide, and liquid–liquid extraction with ethyl acetate. The final extract was quantitatively measured by the GC‐ECD method with an HP‐INNOWax capillary column (30 m × 0.32 mm × 25 μm, 100% polyethyleneglycol). After further purification of the derivatized extracts with OASIS HLB cartridges, the analytes were confirmed by GC‐MS with an HP5‐MS capillary column and selected ions of m/z 70, 149, and 151 for 2‐bromopropenamide and m/z 110 and 154 for 2‐bromo(13C3)‐propenamide (Zhang et al. 2006). With this method, a low limit of detection of 0.1 μg kg−1 was estimated. According to the quantification and identification results, this method represents a robust and low‐cost alternative for the conventional characterization of AA in fried food.
In addition to GC analysis, HPLC‐UV and HPLC‐MS/MS methods have been conventionally used to monitor AA formed in fried food (Wang et al. 2013; Lim et al. 2014). As with the preparation procedures already described, the general pretreatment process for AA analysis in fried food via HPLC includes defatting, centrifugation, ultrafiltration, and further purification by SPE columns or cartridges. To investigate the influence of oil type on the concentration of AA formed in the model system, as well as in French fries, two SPE columns – an Oasis HLB column and a Bond Elut‐Accucat column – were successively used to perform a complete extraction (Mestdagh et al. 2005). As a result, no significant difference in the formation of AA was found among the nine lipids investigated, although the homemade potato powder mixture containing vegetable oil may significantly have influenced the AA concentration. By using this method, degradation products (such as glycerol, MAGs, and DAGs) of soybean oil, corn oil, soybean fat, and commercial palm fat mixtures (80% palm oil and 20% palm stearin) were confirmed to have no significant influence on the formation of AA during either the frying of French fries or the heating of an artificial mixture (Mestdagh et al. 2007). In addition, the colour of the fried potato strips, represented by L* and a*, was also closely correlated with the concentration of AA (Pedreschi et al. 2006). The effect of frying time and the shape of the potato sample on the formation of AA in deep‐fat‐fried potato cubes was also investigated using this joint HPLC‐MS/MS method (Carrieri et al. 2010). The detection of AA by MS/MS can alternatively be achieved by UV detection at 210 and 225 nm (Wang et al. 2008) and DAD at 226 nm with continuous monitoring of the peak spectra within the range 190–350 nm (Gokmen et al. 2005).
Both GC‐ and HPLC‐based methods can be utilized, as they gave satisfactory qualitative and quantitative analyses of AA with high sensitivity in a food matrix. However, no derivatization treatment is needed in the HPLC‐based method, which makes the sample preparation for AA analysis easier (Zhang et al. 2015).
7.8 Analysis of Tocopherols
Tocopherols and tocotrienols are important antioxidants that are found both in frying medium and in foods. Saini and Keum (2016) reviewed some methods for the extraction, chromatographic separation, and detection of tocopherols in plants or their products. Supercritical fluid extraction is an emerging technique for the extraction of tocols, while RP‐HPLC and MS in negative mode are sensitive tools. Extraction may be required in fried samples, but not in frying oils. Several GC methods have been reported for the determination of tocopherols in edible oils or foods (Schwartz et al. 2008; Mitei et al. 2009). However, due to their stable and nonpolar nature, HPLC is a better choice (Table 7.10). Only two NP‐HPLC methods have been reported since 2011 (Cruz and Casal 2013; Górnaś et al. 2014). The solvent in this method was 1,4‐dioxane and n‐hexane, while FLD and DAD were used as detectors for the analysis of tocopherols in green vegetables and butter.
Table 7.10 Analytical chromatographic methods for the determination of vitamin E in frying medium and foods.
| Sample | Mode of separation | Detection system | Solvent system | Column | References |
| Marine and freshwater fish Species | RP | FLD | Isocratic: ACN‐methanol (50 : 50) | Develosil RPAQUEOUS (150 × 4.6 mm, 5 μm) | Özogul et al. (2011) |
| Grape | RP | FLD | Gradient: ACN and methanol | Develosil RPAQUEOUS (150 × 4.6 mm, 5 μm) | Tangolar et al. (2011) |
| Cereals | RP | FLD | Gradient: 2‐propanol and water | PerfectSil Target ODS‐3 (250 × 4.6 mm, 3 μm) | Irakli et al. (2012) |
| Green vegetables | NP | FLD and DAD | Isocratic: 1,4‐dioxane in n‐hexane (2.5%, v/v) | Supelcosil LC‐SI (7.5 cm × 3 mm, 3 μm) | Cruz and Casal (2013) |
| Tropical fruits | UPC | DAD | Gradient: CO2 and methanol | ACQUITY UPC2 BEH (100 × 3.0 mm, 1.7 μm) | Gong et al. (2014) |
| Butter and seed samples | RP | FLD | Isocratic: methanol‐water (93 : 7, v/v) | Luna PFP (150 × 4.6 mm, 3 μm) | Górnaś et al. (2014, 2015) |
| Butter samples | NP | FLD | Isocratic: n‐hexane with 1,4‐dioxane (96 : 4, v/v) | LiChrosorb Si60 (4.6 × 250 mm, 5 μm) | Górnaś et al. (2014) |
| Vegetables | RP | FLD and DAD | Gradient: methanol‐water (85 : 15) and tert‐methylbutyl‐ ether‐methanol‐water (80 : 18 : 2) | Develosil RP aqueous C‐30 (150 × 3 mm, 3 μm) and Kinetex PFP (150 × 3 mm, 2.6 μm) | Knecht et al. (2015) |
| Rice | RP | DAD and FLD | Gradient: water and methanol | Kinetex PFP (250 × 4.6 mm, 5 μm) | Shammugasamy et al. (2015) |
| Sunflower and olive oils | RP | FLD | Gradient: ACN and methanol | C‐18 (250 × 4.6 mm, 5 μm) | Bakre et al. (2015) |
| Animal feed and food | RP | UV | Gradient: methanol‐water solution (94 : 6, v/v) and methanol | Supelcosil LC‐18 (25 cm × 4.6 mm, 5 μm) | Claeys et al. (2016) |
| Foods | RP | FLD and UV | Isocratic: ACN 100% | Restek Ultra IBD (150 × 3 mm, 3 μm) | Sunarić et al. (2017) |
| Vegetable oils | RP | ESI‐MS | Gradient: methanol‐water‐ammonium hydroxide (99 : 1 : 0.1, v/v/v) and 2‐propanol | Acquity UPLC BEH C‐18 (100 × 2.1 mm, 1.7 μm) | Ansolin et al. (2017) |
| Tomatoes | RP | FLD | Gradient: ACN and methanol | Acquity UPLC BEH C‐18 (50 × 2.1 mm, 1.7 μm) | Figueira et al. (2017) |
RP, reversed phase; NP, normal phase; HPLC, high‐performance liquid chromatography; UPLC, ultra‐pressure liquid chromatography; DAD, diode array detector; ESI, electrospray ionization; MS, mass spectrometry; UV, ultraviolet; FLD, fluorescence detector; ACN, acetonitrile.
RP‐HPLC is a commonly applied technique for the separation of different tocopherols and tocotrienols in food samples. The most widely used detector in HPLC was found to be FLD for marine and freshwater fishes (Özogul et al. 2011), grape seeds (Tangolar et al. 2011), cereals (Irakli et al. 2012), butter samples (Górnaś et al. 2014, 2015), vegetables (Knecht et al. 2015), rice (Shammugasamy et al. 2015), vegetable oils (Bakre et al. 2015), animal feed and food (Claeys et al. 2016), and other foods and fruits (Figueira et al. 2017; Sunarić et al. 2017). A DAD or UV detector (Cruz and Casal 2013; Gong et al. 2014; Knecht et al. 2015; Shammugasamy et al. 2015; Claeys et al. 2016; Sunarić et al. 2017) and an ESI‐MS detector (Ansolin et al. 2017) have also been successfully used. These showed that acetonitrile and methanol were the major solvents used during separation on C‐18 columns. Irakli et al. (2012) demonstrated that column temperatures affect the elution and separation of tocopherols, as shown in Figure 7.9.


Figure 7.9 HPLC separation of T and T3 isomers at different temperatures with PerfectSil Target stationary phase. Mobile phase, isopropanol‐water (75 : 25, v/v); flow rate, 0.3 mL min−1.
Source: Reproduced with kind permission of American Chemical Society (Irakli et al. 2012).
Tocopherol has been identified simultaneously with carotenoids or with phenolic compounds. In green vegetables, tocopherols were extracted with carotenoids and chlorophylls and separated using a C‐18 column and gradient elution of methanol, water, and tertiary methyl butyl ether (MTBE) (Zeb 2017). Similarly, tocopherols have been identified and quantified simultaneously with carotenoids and free and esterified sterols in canola oils (Flakelar et al. 2017) using NP‐HPLC. These results show that HPLC with FLD is the best choice for the analysis of tocopherols during frying.
7.9 Analysis of Polyphenolic Compounds
Phenolic compounds are found in a variety of plant foods. The most widely used means of determining total polyphenol contents is a spectrophotometric method using Folin Ciocaltaeu reagent. The modes of analysis, extraction, and detection have been reviewed by Ignat et al. (2011). Details regarding polyphenols are usually obtained via separation by HPLC and detection with a DAD detector. The RP‐HPLC‐DAD method was developed for the determination of polyphenolic compounds in plant leaves (Zeb 2015b) and was successfully used for the determination of changes in polyphenolic compounds during frying of chapli kebab (Zeb and Haq 2017). For phenolic compounds in the presence or absence of an authentic standard, the use of MS with chromatography is warranted. Oueslati et al. (2017) reported a method using HPLC‐DAD‐ESI‐TOF‐MS for the determination of changes in phenolic compounds during heating or microwave heating. These studies show that phenolic compounds and their variations can be accurately assessed with the help of LC and MS detectors.
7.10 Analysis of Other Minor Compounds
In addition to the previously described GC and HPLC analyses of the common products formed during oil heating and food frying, products present in low concentrations have also been reported. These products, such as α‐tocopherol oxides and polycyclic aromatic hydrocarbons (PAHs), directly or indirectly influence the final quality of the frying oil and fried food. Those compounds, such as trans fatty acids (TFAs), that do not have a significant effect on the final food will not be discussed here. By using specialized sample pretreatment and an appropriate detector, the HPLC‐based method is commonly used to conduct qualitative and quantitative analyses of minor compounds of interest.
The typical variation of lipid microcomponents involves changes in antioxidants during the frying process (Hwang et al. 2013; Aladedunye et al. 2017). The frying process causes the number of antioxidants, and thus the corresponding antioxidative activity, to decrease, which reduces the ability of the system to prevent the deterioration of oil and to inhibit the formation of products with potential health risks. These changes can be attributed to reactions involving the antioxidants and to the loss of the antioxidative configurations during the frying process. Oxidation can also take place in natural antioxidants. The oxidation products of α‐tocopherol formed in a model triolein system with 0.35% α‐tocopherol heated at 240 °C for 90 minutes were analysed by NP‐HPLC‐FLD with a silica column and an isocratic mobile phase consisting of 99.5% hexane and 0.5% isopropyl alcohol at 1.5 mL min−1. The excitation and emission wavelengths of FLD were set at 230 and 292 nm, respectively. The quantification and identification of the oxidative products were achieved with the help of pure standards and the aforementioned synthetic α‐tocopherol oxidation products. As a result, α‐tocopherol quinone, 4a,5‐epoxy‐α‐tocopherol quinone, and 7,8‐epoxy‐α‐tocopherol quinone were confirmed to be present during the heating process (Verleyen et al. 2001). In addition, α‐tocopherol quinone (1.4–7.7%) and epoxy‐α‐tocopherol quinone (4.3–34.8%) were quantitatively monitored in the model system containing a triolein and tripalmitin mixture with 0.1% α‐tocopherol heated at 150–250 °C over a certain heating period using this method. Moreover, the amounts of the oxidation products of α‐tocopherol were changeable during the heating process, and the fate of the α‐tocopherol itself depended on the heating time and temperature.
As a class of toxicants, PAHs are ubiquitous in the environment and in human bodies (Ramesh et al. 2004). A number of analyses of PAHs during the heating of vegetable oils or the frying process have been reported in recent years (Sjaastad et al. 2010). As with the analysis method used for HCAs, alkaline hydrolysis, clean‐up or purification by SPE or column chromatography, and HPLC‐UV/FLD are usually involved in the preparation and analysis of PAHs formed in fried meat (Rivera et al. 1996). A purified extract was separated by a Hypersil Green PAH column (250 × 4.6 mm; 5 μm) connected with a guard column (10 × 4 mm; 5 μm) and an isocratic mobile phase (84% acetonitrile and 16% water). Through the addition of PAH standards, and using the external calibration curve method, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]anthracene, benzo[a]pyrene, dibenzo[a,h]anthracene, and benzo[g,h,i]perylene were quantitatively characterized in fried pork meat and gravy (Janoszka 2011). Naphthalene, acenaphthylene, fluorine + acenapthene, phenanthrene, anthracene, fluoranthene, and pyrene, benzo(a)anthracene + chrysene, benzo(k)fluoranthene, benzo‐(b)fluoranthene, B(a)P, dibenzo(a,h)anthracene, indino(1,2,3‐cd)pyrene, and benzo(g,h,i)perylene were also quantitatively analysed in repeatedly boiled sunflower oil using this method (Srivastava et al. 2010). In addition, the concentrations and particle size distributions of 19 major PAHs in the oil mist emitted by grilled pork, trout, beef, prawns, and corn were also studied by GC‐MS with a special HAC collection apparatus (Saito et al. 2014).
Other compounds, such as 3‐monochloropropane‐1,2‐diol (3‐MCPD) and 2‐monochloropropane‐1,3‐diol (2‐MCPD), are also present as contaminants in foods. Xu et al. (2013) reported a GC‐MS method for the determination of 3‐MCPD. Similarly, Karl et al. (2016) developed GC‐MS method for the determination of 3‐MCPD in fishes. The 3‐MCPD was found to be affected by salt concentration during frying (Wong et al. 2017).
7.11 Key Concepts
- Several analytical methods for TAG determination have been reported recently, which show that RP‐HPLC provides high sensitivity.
- Oxidized TAGs have been studied using normal‐ and reversed‐phase TLC and HPLC, while limited studies are available using GC‐MS.
- The best technique for the analysis of oxidized TAGs is RP‐LC coupled with MS.
- FA oxidation products can be determined with GC‐MS, which has superior sensitivity.
- Better extraction methods such as SPE and derivatization can give more sensitive results for oxygenated FAs.
- Cholesterol and phytosterols have largely been analysed using GC‐based techniques; HPLC coupled to MS is also a good option.
- Secondary metabolites such as volatile products are usually collected and isolated using specific extraction techniques and analysed using GC.
- GC coupled to a variety of detectors, including MS, provides highly sensitive and accurate results.
- Heterocyclic aromatic amines (HAAs) have been analysed using RP‐HPLC with DAD or MS detection.
- GC‐ and HPLC‐based methods can be utilized, as they give satisfactory qualitative and quantitative analyses of AA with high sensitivity in a food matrix.
- Tocopherols have been determined using HPLC with FLD detection, which provides superior sensitivity.
- Polyphenols can be analysed using RP‐HPLC, which gives better sensitivity on MS detection.
- Several minor compounds can be identified and quantified using GC‐ or HPLC‐based techniques.
References
- Abdallah, M., Vergara‐Barberán, M., Lerma‐García, M.J. et al. (2016). Use of triacylglycerol profiles established by HPLC‐UV and ELSD to predict cultivar and maturity of Tunisian olive oils. European Food Research and Technology 242: 1607–1619.
- Abidi, S.L. (2001). Chromatographic analysis of plant sterols in foods and vegetable oils. Journal of Chromatography A 935: 173–201.
- Abreu, F.F., Souza, A.C., Teixeira, S.A. et al. (2016). Elucidating the role of oxidative stress in the therapeutic effect of rutin on experimental acute pancreatitis. Free Radical Research 50: 1350–1360.
- Adewuyi, A. and Oderinde, R.A. (2012). Analysis of the lipids and molecular speciation of the triacylglycerol of the oils of Luffa cylindrica and Adenopus breviflorus. CyTA Journal of Food 10: 313–320.
- Aeenehvand, S., Toudehrousta, Z., Kamankesh, M. et al. (2016). Evaluation and application of microwave‐assisted extraction and dispersive liquid–liquid microextraction followed by high‐performance liquid chromatography for the determination of polar heterocyclic aromatic amines in hamburger patties. Food Chemistry 190: 429–435.
- Akasaka, K. and Ohrui, H. (2000). Development of phosphine reagents for the high‐performance liquid chromatographic‐fluorometric determination of lipid hydroperoxides. Journal of Chromatography A 881: 159–170.
- Akasaka, K., Ijichi, S., Watanabe, K. et al. (1992). High‐performance liquid chromatography and post‐column derivatization with diphenyl‐1‐pyrenylphosphine for fluorimetric determination of triacylglycerol hydroperoxides. Journal of Chromatography 596: 197–202.
- Akasaka, K., Ohrui, H., Meguro, H., and Tamura, M. (1993). Determination of triacylglycerol and cholesterol ester hydroperoxides in human plasma by high‐performance liquid chromatography with fluorometric postcolumn detection. Journal of Chromatography 617: 205–211.
- Aladedunye, F. and Matthäus, B. (2014). Phenolic extracts from Sorbus aucuparia (L.) and Malus baccata (L.) berries: antioxidant activity and performance in rapeseed oil during frying and storage. Food Chemistry 159: 273–281.
- Aladedunye, F., Przybylski, R., and Matthaus, B. (2017). Performance of antioxidative compounds under frying conditions: a review. Critical Reviews in Food Science and Nutrition 57: 1539–1561.
- Andrikopoulos, N.K., Chiou, A., Mylona, A. et al. (2004). Monitoring of 2,4‐decadienal in oils and fats used for frying in restaurants in Athens, Greece. European Journal of Lipid Science and Technology 106: 671–679.
- Ansolin, M., de Souza, P.T., de Almeida Meirelles, A.J., and Batista, E.A.C. (2017). Tocopherols and tocotrienols: an adapted methodology by UHPLC/MS without sample pretreatment steps. Food Analytical Methods 10: 2165–2174.
- Araujo, F.B., Barbosa, D.S., Hsin, C.Y. et al. (1995). Evaluation of oxidative stress in patients with hyperlipidemia. Atherosclerosis 117: 61–71.
- Armstrong, D. (2009). Lipidomics: Methods and Protocols. New York, NY: Humana Press.
- Baiocchi, C., Medana, C., Dal Bello, F. et al. (2015). Analysis of regioisomers of polyunsaturated triacylglycerols in marine matrices by HPLC/HRMS. Food Chemistry 166: 551–560.
- Bakre, S.M., Gadmale, D.K., Toche, R.B., and Gaikwad, V.B. (2015). Rapid determination of alpha tocopherol in olive oil adulterated with sunflower oil by reversed phase high‐performance liquid chromatography. Journal of Food Science and Technology 52: 3093–3098.
- Bansal, G., Zhou, W., Barlow, P.J. et al. (2010). Review of rapid tests available for measuring the quality changes in frying oils and comparison with standard methods. Critical Reviews in Food Science and Nutrition 50: 503–514.
- Barriuso, B., Otaegui‐Arrazola, A., Menéndez‐Carreño, M. et al. (2012). Sterols heating: degradation and formation of their ring‐structure polar oxidation products. Food Chemistry 135: 706–712.
- Bastos, L.C.S. and Pereira, P.A.D.P. (2010). Influence of heating time and metal ions on the amount of free fatty acids and formation rates of selected carbonyl compounds during the thermal oxidation of canola oil. Journal of Agricultural and Food Chemistry 58: 12777–12783.
- Bauer‐Plank, C. and Steenhorst‐Slikkerveer, L. (2000). Analysis of triacylglyceride hydroperoxides in vegetable oils by nonaqueous reversed‐phase high‐performance liquid chromatography with ultraviolet detection. Journal of the American Oil Chemists' Society 77: 477–482.
- Beccaria, M., Sullini, G., Cacciola, F. et al. (2014). High performance characterization of triacylglycerols in milk and milk‐related samples by liquid chromatography and mass spectrometry. Journal of Chromatography A 1360: 172–187.
- Beccaria, M., Costa, R., Sullini, G. et al. (2015). Determination of the triacylglycerol fraction in fish oil by comprehensive liquid chromatography techniques with the support of gas chromatography and mass spectrometry data. Analytical and Bioanalytical Chemistry 407: 5211–5225.
- Beermann, C., Winterling, N., Green, A. et al. (2007). Comparison of the structures of triacylglycerols from native and transgenic medium‐chain fatty acid‐enriched rape seed oil by liquid chromatography‐‐atmospheric pressure chemical ionization ion‐trap mass spectrometry (LC‐APCI‐ITMS). Lipids 42: 383–394.
- Ben Arfa, K., de Person, M., Hmida, D. et al. (2017). UHPLC‐APCI‐MS profiling of triacylglycerols in vegetable oils – application to the analysis of four North African sesame seed varieties. Food Analytical Methods 10: 2827–2838.
- Ben Hammouda, I., Freitas, F., Ammar, S. et al. (2017). Comparison and characterization of volatile compounds as markers of oils stability during frying by HS–SPME‐GC/MS and chemometric analysis. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences 1068–1069: 322–334.
- Berdeaux, O., Márquez‐Ruiz, G., and Dobarganes, M. (1999). Characterization, quantitation and evolution of monoepoxy compounds formed in model systems of fatty acid methyl esters and monoacid triglycerides heated at high temperature. Grasas y Aceites 50: 53–59.
- Bhuiyan, M., Tucker, D., and Watson, K. (2013). Determination and differentiation of triacylglycerol molecular species in Antarctic and non‐Antarctic yeasts by atmospheric pressure‐chemical ionization‐mass spectrometry. Journal of Microbiological Methods 94: 249–256.
- Biedermann‐Brem, S., Biedermann, M., Fankhauser‐Noti, A. et al. (2007). Determination of epoxidized soybean oil (ESBO) in oily foods by GC‐FID or GC‐MS analysis of the methyl diepoxy linolate. European Food Research and Technology 224: 309–314.
- Blanksby, S.J. and Mitchell, T.W. (2010). Advances in mass spectrometry for lipidomics. Annual Review of Analytical Chemistry 3: 433–465.
- Bohac, C.E., Rhee, K.S., Cross, H.R., and Ono, K. (1988). Assessment of methodologies for calorimetric cholesterol assay of meats. Journal of Food Science 53: 1642–1644.
- Bou Khalil, M., Hou, W., Zhou, H. et al. (2010). Lipidomics era: accomplishments and challenges. Mass Spectrometry Reviews 29: 877–929.
- Bragagnolo, N. (2009). Cholesterol and cholesterol oxides in meat and meat products. In: Handbook of Muscle Food Analysis (ed. L.M.L. Nollet and F. Toldra). Boca Raton, FL: CRC Press.
- Brühl, L., Weisshaar, R., and Matthäus, B. (2016). Epoxy fatty acids in used frying fats and oils, edible oils and chocolate and their formation in oils during heating. European Journal of Lipid Science and Technology 118: 425–434.
- Byrdwell, W.C. and Neff, W.E. (1999). Non‐volatile products of triolein produced at frying temperatures characterized using liquid chromatography with online mass spectrometric detection. Journal of Chromatography A 852: 417–432.
- Byrdwell, W.C. and Neff, W.E. (2001). Autoxidation products of normal and genetically modified canola oil varieties determined using liquid chromatography with mass spectrometric detection. Journal of Chromatography A 905: 85–102.
- Byrdwell, W.C. and Neff, W.E. (2002). Dual parallel electrospray ionization and atmospheric pressure chemical ionization mass spectrometry (MS), MS/MS and MS/MS/MS for the analysis of triacylglycerols and triacylglycerol oxidation products. Rapid Communications in Mass Spectrometry 16: 300–319.
- Byrdwell, W.C. and Neff, W.E. (2004). Electrospray ionization MS of high M.W. TAG oligomers. Journal of the American Oil Chemists' Society 81: 13–26.
- Caldironi, H.A. and Manes, M.E. (2006). Proximate composition, fatty acids and cholesterol content of meat cuts from tegu lizard Tupinambis merianae. Journal of Food Composition and Analysis 19: 711–714.
- Caldwell, J.D., Cooke, B.S., and Greer, M.K. (2011). High performance liquid chromatography–size exclusion chromatography for rapid analysis of total polar compounds in used frying oils. Journal of the American Oil Chemists' Society 88: 1669–1674.
- Cardenia, V., Rodriguez‐Estrada, M.T., Baldacci, E. et al. (2012). Analysis of cholesterol oxidation products by fast gas chromatography/mass spectrometry. Journal of Separation Science 35: 424–430.
- Carrieri, G., Anese, M., Quarta, B. et al. (2010). Evaluation of acrylamide formation in potatoes during deep‐frying: the effect of operation and configuration. Journal of Food Engineering 98: 141–149.
- Chao, P., Chao, C., Lin, F., and Huang, C. (2001). Oxidized frying oil up‐regulates hepatic acyl‐CoA oxidase and cytochrome P450 4 A1 genes in rats and activates PPARα. Journal of Nutrition 131: 3166–3174.
- Cheung, M., Young, A.B., and Harrison, A.G. (1994). O(−) and OH(−) chemical ionization of some fatty acid methyl esters and triacylglycerols. Journal of the American Society for Mass Spectrometry 5: 553–557.
- Choi, J., Zhang, W., Gu, X. et al. (2011). Lysophosphatidylcholine is generated by spontaneous deacylation of oxidized phospholipids. Chemical Research in Toxicology 24: 111–118.
- Christensen, T.C. and Holmer, G. (1996). Lipid oxidation determination in butter and dairy spreads by HPLC. Journal of Food Science 61: 486–489.
- Chung, T.Y., Eiserich, J.P., and Shibamoto, T. (1994). Volatile compounds produced from peanut oil heated with different amounts of cysteine. Journal of Agricultural and Food Chemistry 42: 1743–1746.
- Chyau, C.‐C. and Mau, J.‐L. (2001). Effects of various oils on volatile compounds of deep‐fried shallot flavouring. Food Chemistry 74: 41–46.
- Claessens, H.A. and van Straten, M.A. (2004). Review on the chemical and thermal stability of stationary phases for reversed‐phase liquid chromatography. Journal of Chromatography A 1060: 23–41.
- Claeys, E., Vossen, E., and De Smet, S. (2016). Determination of α‐tocopherol by reversed‐phase HPLC in feed and animal‐derived foods without saponification. Journal of the Science of Food and Agriculture 96: 522–529.
- Clark, P.K. and Snyder, H.E. (1991). Hydroperoxide formation in soybean seeds during storage. Journal of the American Oil Chemists Society 68: 346–347.
- Constantinou, M.S., Georgiou, C.A., and Kapnissi‐Christodoulou, C.P. (2015). Development of a reliable analytical protocol for the isolation of cholesterol oxidation products – a comparison of different lipid extraction and saponification methods. Food Analytical Methods 8: 1499–1507.
- Correia, A.C., Dubreucq, E., Ferreira‐Dias, S., and Lecomte, J. (2015). Rapid quantification of polar compounds in thermo‐oxidized oils by HPTLC‐densitometry. European Journal of Lipid Science and Technology 117: 311–319.
- Costa, P., Roseiro, L.C., Partidário, A. et al. (2006). Influence of slaughter season and sex on fatty acid composition, cholesterol and α‐tocopherol contents on different muscles of Barrosã‐PDO veal. Meat Science 72: 130–139.
- Cruz, R. and Casal, S. (2013). Validation of a fast and accurate chromatographic method for detailed quantification of vitamin E in green leafy vegetables. Food Chemistry 141: 1175–1180.
- Daneshfar, A., Khezeli, T., and Lotfi, H.J. (2009). Determination of cholesterol in food samples using dispersive liquid–liquid microextraction followed by HPLC–UV. Journal of Chromatography, B: Analytical Technologies in the Biomedical and Life Sciences 877: 456–460.
- Davis, T.A., Gao, L., Yin, H. et al. (2006). In vivo and in vitro lipid peroxidation of arachidonate esters: the effect of fish oil omega‐3 lipids on product distribution. Journal of the American Chemical Society 128: 14897–14904.
- Delgado‐Zamarreño, M.M., Fernández‐Prieto, C., Bustamante‐Rangel, M., and Pérez‐Martín, L. (2016). Determination of tocopherols and sitosterols in seeds and nuts by QuEChERS‐liquid chromatography. Food Chemistry 192: 825–830.
- Dinh, T.T.N., Thompson, L.D., Galyean, M.L. et al. (2011). Cholesterol content and methods for cholesterol determination in meat and poultry. Comprehensive Reviews in Food Science and Food Safety 10: 269–289.
- Dobarganes, C. and Márquez‐Ruiz, G. (2003). Oxidized fats in foods. Current Opinion in Clinical Nutrition and Metabolic Care 6: 157–163.
- Dobarganes, M., Velasco, J., and Dieffenbacher, A. (2000). Determination of polar compounds, polymerized and oxidized triacylglycerols, and diacylglycerols in oils and fats: results of collaborative studies and the standardized method (technical report). Pure and Applied Chemistry 72: 1563–1575.
- Doleschall, F., Recseg, K., Kemény, Z., and Kővári, K. (2003). Comparison of differently coated SPME fibres applied for monitoring volatile substances in vegetable oils. European Journal of Lipid Science and Technology 105: 333–338.
- Dorsey, J.G. and Dill, K.A. (1989). The molecular mechanism of retention in reversed‐phase liquid chromatography. Chemical Reviews 89: 331–346.
- Dugo, P., Kumm, T., Lo Presti, M. et al. (2005). Determination of triacylglycerols in donkey milk by using high performance liquid chromatography coupled with atmospheric pressure chemical ionization mass spectrometry. Journal of Separation Science 28: 1023–1030.
- Dugo, P., Beccaria, M., Fawzy, N. et al. (2012). Mass spectrometric elucidation of triacylglycerol content of Brevoortia tyrannus (menhaden) oil using non‐aqueous reversed‐phase liquid chromatography under ultra high pressure conditions. Journal of Chromatography A 1259: 227–236.
- Duong, S., Strobel, N., Buddhadasa, S. et al. (2016). Rapid measurement of phytosterols in fortified food using gas chromatography with flame ionization detection. Food Chemistry 211: 570–576.
- Ekroos, K., Janis, M., Tarasov, K. et al. (2010). Lipidomics: a tool for studies of atherosclerosis. Current Atherosclerosis Reports 12: 273–281.
- Endo, Y., Ohta, A., Kido, H. et al. (2011). Determination of triacylglycerol composition in vegetable oils using high‐performance liquid chromatography: a collaborative study. Journal of Oleo Science 60: 451–456.
- Fang, L., Harkewicz, R., Hartvigsen, K. et al. (2010). Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high cholesterol diet: macrophage binding and activation. Journal of Biological Chemistry 285: 32343–32351.
- Fankhauser‐Noti, A., Fiselier, K., Biedermann‐Brem, S., and Grob, K. (2005). Epoxidized soy bean oil migrating from the gaskets of lids into food packed in glass jars ‐ Analysis by on‐line liquid chromatography‐gas chromatography. Journal of Chromatography A 1082: 214–219.
- Fankhauser‐Noti, A., Fiselier, K., Biedermann‐Brem, S., and Grob, K. (2006). Assessment of epoxidized soy bean oil (ESBO) migrating into foods: comparison with ESBO‐like epoxy fatty acids in our normal diet. Food and Chemical Toxicology 44: 1279–1286.
- Fasciotti, M. and Pereira Netto, A.D. (2010). Optimization and application of methods of triacylglycerol evaluation for characterization of olive oil adulteration by soybean oil with HPLC‐APCI‐MS‐MS. Talanta 81: 1116–1125.
- Fenton, M. (1992). Chromatographic separation of cholesterol in foods. Journal of Chromatography A 624: 369–388.
- Figueira, J.A., Pereira, J.A.M., and Câmara, J.S. (2017). Quantification of δ‐, γ‐ and α‐tocopherol in tomatoes using an improved liquid‐dispersive solid‐phase extraction combined with ultrahigh pressure liquid chromatography. Food Analytical Methods 10: 2507–2517.
- Flakelar, C.L., Prenzler, P.D., Luckett, D.J. et al. (2017). A rapid method for the simultaneous quantification of the major tocopherols, carotenoids, free and esterified sterols in canola (Brassica napus) oil using normal phase liquid chromatography. Food Chemistry 214: 147–155.
- Frankel, E.N. (1984). Chemistry of free radical and singlet oxidation of lipids. Progress in Lipid Research 23: 197–221.
- Frankel, E.N., Neff, W.E., and Miyashita, K. (1990). Autoxidation of polyunsaturated triacylglycerols. II. Trilinolenoylglycerol. Lipids 25: 40–47.
- Frankel, E.N., Selke, E., Neff, W.E., and Miyashita, K. (1992). Autoxidation of polyunsaturated triacylglycerols. IV. Volatile decomposition products from triacylglycerols containing linoleate and linolenate. Lipids 27: 442–446.
- Fullana, A., Carbonell‐Barrachina, Á.A., and Sidhu, S. (2004). Volatile aldehyde emissions from heated cooking oils. Journal of the Science of Food and Agriculture 84: 2015–2021.
- Gao, B., Luo, Y., Lu, W. et al. (2017). Triacylglycerol compositions of sunflower, corn and soybean oils examined with supercritical CO2 ultra‐performance convergence chromatography combined with quadrupole time‐of‐flight mass spectrometry. Food Chemistry 218: 569–574.
- Georgiou, C.A., Constantinou, M.S., and Kapnissi‐Christodoulou, C.P. (2014). Sample preparation: a critical step in the analysis of cholesterol oxidation products. Food Chemistry 145: 918–926.
- Georgiou, C.A., Constantinou, M.S., Andreou, R. et al. (2016). Novel approach to fast determination of cholesterol oxidation products in Cypriot foodstuffs using ultra‐performance liquid chromatography‐tandem mass spectrometry. Electrophoresis 37: 1101–1108.
- Gertz, C. and Klostermann, S. (2002). Analysis of acrylamide and mechanisms of its formation in deep‐fried products. European Journal of Lipid Science and Technology 104: 762–771.
- Gibis, M. and Weiss, J. (2012). Antioxidant capacity and inhibitory effect of grape seed and rosemary extract in marinades on the formation of heterocyclic amines in fried beef patties. Food Chemistry 134: 766–774.
- Giuffrida, F., Destaillats, F., Skibsted, L.H., and Dionisi, F. (2004). Structural analysis of hydroperoxy‐ and epoxy‐triacylglycerols by liquid chromatography mass spectrometry. Chemistry and Physics of Lipids 131: 41–49.
- Gladovič, N., Zupančič‐Kralj, L., and Plavec, J. (1997). Determination of primary oxidation products of linoleic acid and triacylglycerols. Journal of Chromatography A 767: 63–68.
- Goicoechea, E. and Guillen, M.D. (2010). Analysis of hydroperoxides, aldehydes and epoxides by 1H nuclear magnetic resonance in sunflower oil oxidized at 70 and 100 degrees C. Journal of Agricultural and Food Chemistry 58: 6234–6245.
- Gokmen, V., Senyuva, H.Z., Acar, J., and Sarioglu, K. (2005). Determination of acrylamide in potato chips and crisps by high‐performance liquid chromatography. Journal of Chromatography A 1088: 193–199.
- Gong, X., Qi, N., Wang, X. et al. (2014). A new method for determination of α‐tocopherol in tropical fruits by ultra performance convergence chromatography with diode array detector. Food Analytical Methods 7: 1572–1576.
- Gonzalez‐Covarrubias, V. (2013). Lipidomics in longevity and healthy aging. Biogerontology 14: 663–672.
- Gorassini, A., Verardo, G., Fregolent, S.C., and Bortolomeazzi, R. (2017). Rapid determination of cholesterol oxidation products in milk powder based products by reversed phase SPE and HPLC‐APCI‐MS/MS. Food Chemistry 230: 604–610.
- Górnaś, P., Siger, A., Czubinski, J. et al. (2014). An alternative RP‐HPLC method for the separation and determination of tocopherol and tocotrienol homologues as butter authenticity markers: a comparative study between two European countries. European Journal of Lipid Science and Technology 116: 895–903.
- Górnaś, P., Soliven, A., and Segliņa, D. (2015). Seed oils recovered from industrial fruit by‐products are a rich source of tocopherols and tocotrienols: rapid separation of α/β/γ/δ homologues by RP‐HPLC/FLD. European Journal of Lipid Science and Technology 117: 773–777.
- Gotoh, N., Matsumoto, Y., Nagai, T. et al. (2011). Actual ratios of triacylglycerol positional isomers consisting of saturated and highly unsaturated fatty acids in fishes and marine mammals. Food Chemistry 127: 467–472.
- Gromadzka, J. and Wardencki, W. (2010). Static headspace sampling and solid‐phase microextraction for assessment of edible oils stability. Chromatographia 71: 81–86.
- Gross, R.W., Jenkins, C.M., Yang, J. et al. (2005). Functional lipidomics: the roles of specialized lipids and lipid‐protein interactions in modulating neuronal function. Prostaglandins & Other Lipid Mediators 77: 52–64.
- Grun, C.H. and Besseau, S. (2016). Normal‐phase liquid chromatography‐atmospheric‐pressure photoionization‐mass spectrometry analysis of cholesterol and phytosterol oxidation products. Journal of Chromatography A 1439: 74–81.
- Guardiola, F. (2002). Cholesterol and Phytosterol Oxidation Products: Analysis, Occurrence, and Biological Effects. Urbana, IL: The American Oil Chemists Society, AOCS Press.
- Guardiola, F., Bou, R., Boatella, J., and Codony, R. (2004). Analysis of sterol oxidation products in foods. Journal of AOAC International 87: 441–466.
- Guillén, M.D. and Uriarte, P.S. (2012). Aldehydes contained in edible oils of a very different nature after prolonged heating at frying temperature: presence of toxic oxygenated α,β unsaturated aldehydes. Food Chemistry 131: 915–926.
- Hachey, D.L., Patterson, B.W., Reeds, P.J., and Elsas, L.J. (1991). Isotopic determination of organic keto acid pentafluorobenzyl esters in biological fluids by negative chemical ionization gas chromatography/mass spectrometry. Analytical Chemistry 63: 919–923.
- Harrison, K.A., Davies, S.S., Marathe, G.K. et al. (2000). Analysis of oxidized glycerophosphocholine lipids using electrospray ionization mass spectrometry and microderivatization techniques. Journal of Mass Spectrometry 35: 224–236.
- Hindra, F. and Baik, O.D. (2006). Kinetics of quality changes during food frying. Critical Reviews in Food Science and Nutrition 46: 239–258.
- Horiuchi, M., Umano, K., and Shibamoto, T. (1998). Analysis of volatile compounds formed from fish oil heated with cysteine and trimethylamine oxide. Journal of Agricultural and Food Chemistry 46: 5232–5237.
- Hu, Q., Xu, X., Fu, Y., and Li, Y. (2015). Rapid methods for detecting acrylamide in thermally processed foods: a review. Food Control 56: 135–146.
- Hui, S.P. (2014). Analyses of lipid hydroperoxides and physiologically active lipids by mass spectrometry. Rinsho Byori 62: 283–290.
- Hwang, H.‐S., Winkler‐Moser, J.K., Bakota, E.L. et al. (2013). Antioxidant activity of sesamol in soybean oil under frying conditions. Journal of the American Oil Chemists' Society 90: 659–666.
- Hyötyläinen, T., Kallio, M., Lehtonen, M. et al. (2004). Comprehensive two‐dimensional gas chromatography in the analysis of dietary fatty acids. Journal of Separation Science 27: 459–467.
- Idrus, S.I.S., Latiff, A.A., and Ismail, M.N. (2017). Determination of triacylglycerols in food by high‐performance liquid chromatography. Instrumentation Science & Technology 45: 577–591.
- Ignat, I., Volf, I., and Popa, V.I. (2011). A critical review of methods for characterisation of polyphenolic compounds in fruits and vegetables. Food Chemistry 126: 1821–1835.
- Ikeda, K., Oike, Y., Shimizu, T., and Taguchi, R. (2009). Global analysis of triacylglycerols including oxidized molecular species by reverse‐phase high resolution LC/ESI‐QTOF MS/MS. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences 877: 2639–2647.
- Inchingolo, R., Cardenia, V., and Rodriguez‐Estrada, M.T. (2014). Analysis of phytosterols and phytostanols in enriched dairy products by fast gas chromatography with mass spectrometry. Journal of Separation Science 37: 2911–2919.
- Irakli, M.N., Samanidou, V.F., and Papadoyannis, I.N. (2012). Optimization and validation of the reversed‐phase high‐performance liquid chromatography with fluorescence detection method for the separation of tocopherol and tocotrienol isomers in cereals, employing a novel sorbent material. Journal of Agricultural and Food Chemistry 60: 2076–2082.
- Ito, M., Ishimaru, M., Shibata, T. et al. (2017b). High‐performance liquid chromatography with fluorescence detection for simultaneous analysis of phytosterols (stigmasterol, β‐sitosterol, campesterol, ergosterol, and fucosterol) and cholesterol in plant foods. Food Analytical Methods 10: 2692–2699.
- Ito, J., Shimizu, N., Kobayashi, E. et al. (2017a). A novel chiral stationary phase LC‐MS/MS method to evaluate oxidation mechanisms of edible oils. Scientific Reports 7: 10026.
- Janoszka, B. (2011). HPLC‐fluorescence analysis of polycyclic aromatic hydrocarbons (PAHs) in pork meat and its gravy fried without additives and in the presence of onion and garlic. Food Chemistry 126: 1344–1353.
- Jautz, U., Gibis, M., and Morlock, G.E. (2008). Quantification of heterocyclic aromatic amines in fried meat by HPTLC/UV‐FLD and HPLC/UV‐FLD: a comparison of two methods. Journal of Agricultural and Food Chemistry 56: 4311–4319.
- Jenske, R. and Vetter, W. (2008). Gas chromatography/electron‐capture negative ion mass spectrometry for the quantitative determination of 2‐and 3‐hydroxy fatty acids in bovine milk fat. Journal of Agricultural and Food Chemistry 56: 5500–5505.
- Jerković, I., Mastelić, J., and Tartaglia, S. (2007). A study of volatile flavour substances in Dalmatian traditional smoked ham: impact of dry‐curing and frying. Food Chemistry 104: 1030–1039.
- Jiang, Z., Fenton, M., and Sim, J.S. (1991). Comparison of four different methods for egg cholesterol determination. Poultry Science 70: 1015–1019.
- Johansson, M.A., Fredholm, L., Bjerne, I., and Jagerstad, M. (1995). Influence of frying fat on the formation of heterocyclic amines in fried beefburgers and pan residues. Food and Chemical Toxicology 33: 993–1004.
- Julien‐David, D., Zhao, M., Geoffroy, P. et al. (2014). Analysis of sitosteryl oleate esters in phytosterols esters enriched foods by HPLC–ESI‐MS2. Steroids 84: 84–91.
- Karimi, S., Wawire, M., and Mathooko, F.M. (2017). Impact of frying practices and frying conditions on the quality and safety of frying oils used by street vendors and restaurants in Nairobi, Kenya. Journal of Food Composition and Analysis 62: 239–244.
- Karl, H., Merkle, S., Kuhlmann, J., and Fritsche, J. (2016). Development of analytical methods for the determination of free and ester bound 2‐, 3‐MCPD, and esterified glycidol in fishery products. European Journal of Lipid Science and Technology 118: 406–417.
- Katragadda, H.R., Fullana, A., Sidhu, S., and Carbonell‐Barrachina, Á.A. (2010). Emissions of volatile aldehydes from heated cooking oils. Food Chemistry 120: 59–65.
- Katsanidis, E. and Addis, P.B. (1999). Novel HPLC analysis of tocopherols, tocotrienols, and cholesterol in tissue. Free Radical Biology and Medicine 27: 1137–1140.
- Katsuta, I., Shimizu, M., Yamaguchi, T., and Nakajima, Y. (2008). Emission of volatile aldehydes from DAG‐rich and TAG‐rich oils with different degrees of unsaturation during deep‐frying. Journal of the American Oil Chemists' Society 85: 513–519.
- Kermasha, S., Kubow, S., and Goetghebeur, M. (1994). Comparative high‐performance liquid chromatographie analyses of cholesterol and its oxidation products using diode‐array ultraviolet and laser light‐scattering detection. Journal of Chromatography A 685: 229–235.
- Keşkekoğlu, H. and Üren, A. (2014). Inhibitory effects of pomegranate seed extract on the formation of heterocyclic aromatic amines in beef and chicken meatballs after cooking by four different methods. Meat Science 96: 1446–1451.
- Kinter, M. (1995). Analytical technologies for lipid oxidation products analysis. Journal of Chromatography B: Biomedical Applications 671: 223–236.
- Kıralan, S., Yorulmaz, A., Şimşek, A., and Tekin, A. (2015). Classification of Turkish hazelnut oils based on their triacylglycerol structures by chemometric analysis. European Food Research and Technology 240: 679–688.
- Knecht, K., Sandfuchs, K., Kulling, S.E., and Bunzel, D. (2015). Tocopherol and tocotrienol analysis in raw and cooked vegetables: a validated method with emphasis on sample preparation. Food Chemistry 169: 20–27.
- Komprda, T., Zelenka, J., Fajmonová, E. et al. (2003). Cholesterol content in meat of some poultry and fish species as influenced by live weight and total lipid content. Journal of Agricultural and Food Chemistry 51: 7692–7697.
- Kuksis, A., Suomela, J.P., Tarvainen, M., and Kallio, H. (2009a). Lipidomic analysis of glycerolipid and cholesteryl ester autooxidation products. Molecular Biotechnology 42: 224–268.
- Kuksis, A., Suomela, J.P., Tarvainen, M., and Kallio, H. (2009b). Use of lipidomics for analyzing glycerolipid and cholesteryl ester oxidation by gas chromatography, HPLC, and on‐line MS. Methods in Molecular Biology 580: 39–91.
- Lampi, A.‐M., Kemmo, S., Mäkelä, A. et al. (2009). Distribution of monomeric, dimeric and polymeric products of stigmasterol during thermo‐oxidation. European Journal of Lipid Science and Technology 111: 1027–1034.
- La Nasa, J., Degano, I., Brandolini, L. et al. (2018). A novel HPLC‐ESI‐Q‐ToF approach for the determination of fatty acids and acylglycerols in food samples. Analytica Chimica Acta 1013: 98–109.
- Lee, J.H. and Pangloli, P. (2013). Volatile compounds and storage stability of potato chips fried in mid‐oleic sunflower oil. International Journal of Food Properties 16: 563–573.
- Lee, J., Dong, A., Jung, K., and Shin, H.‐S. (2011). Influence of extra virgin olive oil on the formation of heterocyclic amines in roasted beef steak. Food Science and Biotechnology 20: 159–165.
- Lehtonen, M., Kemmo, S., Lampi, A.‐M., and Piironen, V. (2011). Effects of esterification on the formation and decomposition of steryl hydroperoxides. European Food Research and Technology 232: 255–264.
- Lehtonen, M., Lampi, A.‐M., Riuttamäki, M.‐A., and Piironen, V. (2012). Oxidation reactions of steryl esters in a saturated lipid matrix. Food Chemistry 134: 2030–2039.
- Lerma‐García, M.J., Lusardi, R., Chiavaro, E. et al. (2011). Use of triacylglycerol profiles established by high performance liquid chromatography with ultraviolet–visible detection to predict the botanical origin of vegetable oils. Journal of Chromatography A 1218: 7521–7527.
- Leskinen, H.M., Suomela, J.P., and Kallio, H.P. (2010). Quantification of triacylglycerol regioisomers by ultra‐high‐performance liquid chromatography and ammonia negative ion atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Communications in Mass Spectrometry 24: 1–5.
- Lim, P.K., Jinap, S., Sanny, M. et al. (2014). The influence of deep frying using various vegetable oils on acrylamide formation in sweet potato (Ipomoea batatas L. Lam) chips. Journal of Food Science 79: T115–T121.
- Lin, J.T. and Chen, G.Q. (2010). Acylglycerols containing trihydroxy fatty acids in castor oil and the regiospecific quantification of triacylglycerols. Journal of the American Oil Chemists' Society 87: 1371–1379.
- Lísa, M. and Holčapek, M. (2013). Characterization of triacylglycerol enantiomers using chiral HPLC/APCI‐MS and synthesis of enantiomeric triacylglycerols. Analytical Chemistry 85: 1852–1859.
- Lojzova, L., Riddellova, K., Hajslova, J. et al. (2009). Alternative GC‐MS approaches in the analysis of substituted pyrazines and other volatile aromatic compounds formed during Maillard reaction in potato chips. Analytica Chimica Acta 641: 101–109.
- Mäkinen, M., Piironen, V., and Hopia, A. (1996). Postcolumn chemiluminescence, ultraviolet and evaporative light‐scattering detectors in high‐performance liquid chromatographic determination of triacylglycerol oxidation products. Journal of Chromatography A 734: 221–229.
- Marmesat, S., Velasco, J., and Dobarganes, M.C. (2008). Quantitative determination of epoxy acids, keto acids and hydroxy acids formed in fats and oils at frying temperatures. Journal of Chromatography A 1211: 129–134.
- Márquez‐Ruiz, G., Perez‐Camino, M.C., and Dobarganes, M.C. (1993). Evaluation of hydrolysis and absorption of thermally oxidized olive oil in non‐absorbed lipids in the rat. Annals of Nutrition & Metabolism 37: 121–128.
- Márquez‐Ruiz, G., Tasioula‐Margari, M., and Dobarganes, M.C. (1995). Quantitation and distribution of altered fatty acids in frying fats. Journal of the American Oil Chemists' Society 72: 1171–1176.
- Márquez‐Ruiz, G., Rodríguez‐Pino, V., and de la Fuente, M.A. (2011). Determination of 10‐hydroxystearic, 10‐ketostearic, 8‐hydroxypalmitic, and 8‐ketopalmitic acids in milk fat by solid‐phase extraction plus gas chromatography‐mass spectrometry. Journal of Dairy Science 94: 4810–4819.
- Martin, F.L. and Ames, J.M. (2001). Formation of Strecker aldehydes and pyrazines in a fried potato model system. Journal of Agricultural and Food Chemistry 49: 3885–3892.
- Martínez‐Yusta, A. and Guillen, M.D. (2014a). Deep‐frying food in extra virgin olive oil: a study by (1)H nuclear magnetic resonance of the influence of food nature on the evolving composition of the frying medium. Food Chemistry 150: 429–437.
- Martínez‐Yusta, A. and Guillén, M.D. (2014b). A study by 1H nuclear magnetic resonance of the influence on the frying medium composition of some soybean oil‐food combinations in deep‐frying. Food Research International 55: 347–355.
- Matthäus, B. and Haase, N.U. (2014). Acrylamide – still a matter of concern for fried potato food? European Journal of Lipid Science and Technology 116: 675–687.
- Melton, S.L., Jafar, S., Sykes, D., and Trigiano, M.K. (1994). Review of stability measurements for frying oils and fried food flavor. Journal of the American Oil Chemists' Society 71: 1301–1308.
- Menéndez‐Carreño, M., Ansorena, D., Astiasarán, I. et al. (2010). Determination of non‐polar and mid‐polar monomeric oxidation products of stigmasterol during thermo‐oxidation. Food Chemistry 122: 277–284.
- Mestdagh, F.J., De Meulenaer, B., Van Poucke, C. et al. (2005). Influence of oil type on the amounts of acrylamide generated in a model system and in French fries. Journal of Agricultural and Food Chemistry 53: 6170–6174.
- Mestdagh, F., Maertens, J., De Wilde, T. et al. (2007). Chemical pre‐treatments of potato products: mechanisms of acrylamide mitigation and effects on the sensorial quality. Communications in Agricultural and Applied Biological Sciences 72: 9–12.
- Mestre Prates, J.A., Gonçalves Quaresma, M.A., Branquinho Bessa, R.J. et al. (2006). Simultaneous HPLC quantification of total cholesterol, tocopherols and β‐carotene in Barrosã‐PDO veal. Food Chemistry 94: 469–477.
- Mitei, Y., Ngila, J., Yeboah, S. et al. (2009). Profiling of phytosterols, tocopherols and tocotrienols in selected seed oils from Botswana by GC‐MS and HPLC. Journal of the American Oil Chemists' Society 86: 617–625.
- Miyashita, K., Frankel, E.N., Neff, W.E., and Awl, R.A. (1990). Autoxidation of polyunsaturated triacylglycerols. III. Synthetic triacylglycerols containing linoleate and linolenate. Lipids 25: 48–53.
- Miyazawa, T., Fujimoto, K., and Oikawa, S. (1990). Determination of lipid hydroperoxides in low density lipoprotein from human plasma using high performance liquid chromatography with chemiluminescence detection. Biomedical Chromatography 4: 131–134.
- Miyazawa, T., Kunika, H., Fujimoto, K. et al. (1995). Chemiluminescence detection of mono‐, bis‐, and tris‐hydroperoxy triacylglycerols present in vegetable oils. Lipids 30: 1001–1006.
- Morales, A., Dobarganes, C., Márquez‐Ruiz, G., and Velasco, J. (2010). Quantitation of hydroperoxy‐, keto‐ and hydroxy‐dienes during oxidation of FAMEs from high‐linoleic and high‐oleic sunflower oils. Journal of the American Oil Chemists' Society 87: 1271–1279.
- Morales, A., Marmesat, S., Dobarganes, M.C. et al. (2012a). Evaporative light scattering detector in normal‐phase high‐performance liquid chromatography determination of FAME oxidation products. Journal of Chromatography A 1254: 62–70.
- Morales, A., Marmesat, S., Dobarganes, M.C. et al. (2012b). Quantitative analysis of hydroperoxy‐, keto‐ and hydroxy‐dienes in refined vegetable oils. Journal of Chromatography A 1229: 190–197.
- Morales, A., Marmesat, S., Ruiz‐Mendez, V. et al. (2014). Formation of oxidation products in edible oils analyzed as FAME derivatives by HPLC‐UV‐ELSD. Food Research International 62: 1080–1086.
- Mubiru, E., Shrestha, K., Papastergiadis, A., and De Meulenaer, B. (2013). Improved gas chromatography‐flame ionization detector analytical method for the analysis of epoxy fatty acids. Journal of Chromatography A 1318: 217–225.
- Mubiru, E., Shrestha, K., Papastergiadis, A., and De Meulenaer, B. (2014). Development and validation of a gas chromatography–flame ionization detection method for the determination of epoxy fatty acids in food matrices. Journal of Agricultural and Food Chemistry 62: 2982–2988.
- Nakajima, M., Yamato, S., Wakabayashi, H., and Shimada, K. (1995). High‐performance liquid chromatographic determination of cholesterol and cholestanol in human serum by precolumn derivatization with 2‐[2‐(isocyanate)ethyl]‐3‐methyl‐1, 4‐naphthoquinone combined with platinum catalyst reduction and electrochemical detection. Biological & Pharmaceutical Bulletin 18: 1762–1764.
- Narvaez‐Rivas, M., Pham, A.J., Schilling, M.W., and Leon‐Camacho, M. (2014). A new SPE/GC‐fid method for the determination of cholesterol oxidation products. Application to subcutaneous fat from Iberian dry‐cured ham. Talanta 122: 58–62.
- Neff, W.E. and Byrdwell, W.C. (1998). Characterization of model triacylglycerol (triolein, trilinolein and trilinolenin) autoxidation products via high‐performance liquid chromatography coupled with atmospheric pressure chemical ionization mass spectrometry. Journal of Chromatography A 818: 169–186.
- Neff, W.E., Frankel, E.N., and Miyashita, K. (1990). Autoxidation of polyunsaturated triacylglycerols. I. Trilinoleoylglycerol. Lipids 25: 33–39.
- Negroni, M., D'agostin, A., and Arnoldi, A. (2001). Effects of olive, canola, and sunflower oils on the formation of volatiles from the Maillard reaction of lysine with xylose and glucose. Journal of Agricultural and Food Chemistry 49: 439–445.
- Noguchi, N., Numano, R., Kaneda, H., and Niki, E. (1998). Oxidation of lipids in low density lipoprotein particles. Free Radical Research 29: 43–52.
- Oehrl, L.L., Hansen, A.P., Rohrer, C.A. et al. (2001). Oxidation of phytosterols in a test food system. Journal of the American Oil Chemists' Society 78: 1073–1078.
- Oette, K. (1965). Identification of some lipid peroxides by thin‐layer chromatography. Journal of Lipid Research 6: 449–454.
- Ohshima, T., Hopia, A., German, J.B., and Frankel, E.N. (1996). Determination of hydroperoxides and structures by high‐performance liquid chromatography with post‐column detection with diphenyl‐1‐pyrenylphosphine. Lipids 31: 1091–1096.
- Ontanón, I., Culleré, L., Zapata, J. et al. (2013). Application of a new sampling device for determination of volatile compounds released during heating olive and sunflower oil: sensory evaluation of those identified compounds. European Food Research and Technology 236: 1031–1040.
- Oracz, J., Nebesny, E., and Zyzelewicz, D. (2011). New trends in quantification of acrylamide in food products. Talanta 86: 23–34.
- Osawa, C.C., Gonçalves, L.A.G., and Da Silva, M.A.A.P. (2013). Odor significance of the volatiles formed during deep‐frying with palm olein. Journal of the American Oil Chemists' Society 90: 183–189.
- Osório, V.M. and de Lourdes Cardeal, Z. (2011). Determination of acrolein in french fries by solid‐phase microextraction gas chromatography and mass spectrometry. Journal of Chromatography A 1218: 3332–3336.
- Oueslati, I., Taamalli, A., Loubiri, A. et al. (2017). Assessment of conventional and microwave heating effects on the variation of the bioactive compounds of Chétoui VOO using HPLC‐DAD‐ESI‐TOF‐MS. Arabian Journal of Chemistry https://doi.org/10.1016/j.arabjc.2017.08.011.
- Özogul, F., Özogul, Y., and Kuley, E. (2011). Simple extraction and rapid HPLC method for tocopherol analysis in marine and fresh‐water fish species. Food Science and Technology Research 17: 595–598.
- Pagliuca, G., Bozzi, C., Gallo, F.R. et al. (2018). Triacylglycerol ‘hand‐shape profile’ of Argan oil. Rapid and simple UHPLC‐PDA‐ESI‐TOF/MS and HPTLC methods to detect counterfeit Argan oil and Argan‐oil‐based products. Journal of Pharmaceutical and Biomedical Analysis 150: 121–131.
- Paul, S. and Mittal, G.S. (1997). Regulating the use of degraded oil/fat in deep‐fat/oil food frying. Critical Reviews in Food Science and Nutrition 37: 635–662.
- Pedreschi, F., Kaack, K., and Granby, K. (2006). Acrylamide content and color development in fried potato strips. Food Research International 39: 40–46.
- Peers, K.E. and Coxon, D.T. (1983). Controlled synthesis of monohydroperoxides by α‐tocopherol inhibited autoxidation of polyunsaturated lipids. Chemistry and Physics of Lipids 32: 49–56.
- Perkins, E.G. and Anfinsen, J.R. (1971). Characterization of the nonvolatile compounds formed during the thermal oxidation of triolein. Journal of the American Oil Chemists' Society 48: 556–562.
- Petersen, K.D., Kleeberg, K.K., Jahreis, G., and Fritsche, J. (2012). Assessment of the oxidative stability of conventional and high‐oleic sunflower oil by means of solid‐phase microextraction‐gas chromatography. International Journal of Food Sciences and Nutrition 63: 160–169.
- Picariello, G., Romano, R., and Addeo, F. (2010). Nitrocellulose film substrate minimizes fragmentation in matrix‐assisted laser desorption ionization time‐of‐flight mass spectrometry analysis of triacylglycerols. Analytical Chemistry 82: 5783–5791.
- Ponte, P.I.P., Alves, S.P., Bessa, R.J.B. et al. (2008). Influence of pasture intake on the fatty acid composition, and cholesterol, tocopherols, and tocotrienols content in meat from free‐range broilers. Poultry Science 87: 80–88.
- Rafferty, J.L., Zhang, L., Siepmann, J.I., and Schure, M.R. (2007). Retention mechanism in reversed‐phase liquid chromatography: a molecular perspective. Analytical Chemistry 79: 6551–6558.
- Rambaldi, D.C., Reschiglian, P., Zattoni, A., and Johann, C. (2009). Enzymatic determination of cholesterol and triglycerides in serum lipoprotein profiles by asymmetrical flow field‐flow fractionation with on‐line, dual detection. Analytica Chimica Acta 654: 64–70.
- Ramesh, A., Walker, S.A., Hood, D.B. et al. (2004). Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. International Journal of Toxicology 23: 301–333.
- Ravi Kiran, C., Sasidharan, I., Soban Kumar, D.R., and Sundaresan, A. (2015). Influence of natural and synthetic antioxidants on the degradation of Soybean oil at frying temperature. Journal of Food Science and Technology 52: 5370–5375.
- Řezanka, T., Matoulková, D., Kolouchová, I. et al. (2013). Brewer's yeast as a new source of palmitoleic acid – analysis of triacylglycerols by LC–MS. Journal of the American Oil Chemists' Society 90: 1327–1342.
- Rivera, L., Curto, M.J., Pais, P. et al. (1996). Solid‐phase extraction for the selective isolation of polycyclic aromatic hydrocarbons, azaarenes and heterocyclic aromatic amines in charcoal‐grilled meat. Journal of Chromatography A 731: 85–94.
- Romani, S., Bacchiocca, M., Rocculi, P., and Dalla Rosa, M. (2009). Influence of frying conditions on acrylamide content and other quality characteristics of French fries. Journal of Food Composition and Analysis 22: 582–588.
- Romano, R., Giordano, A., Le Grottaglie, L. et al. (2013). Volatile compounds in intermittent frying by gas chromatography and nuclear magnetic resonance. European Journal of Lipid Science and Technology 115: 764–773.
- Rudzińska, M., Przybylski, R., and Wąsowicz, E. (2009). Products formed during thermo‐oxidative degradation of phytosterols. Journal of the American Oil Chemists' Society 86: 651–662.
- Rudzińska, M., Przybylski, R., Zhao, Y.Y., and Curtis, J.M. (2010). Sitosterol thermo‐oxidative degradation leads to the formation of dimers, trimers and oligomers: a study using combined size exclusion chromatography/mass spectrometry. Lipids 45: 549–558.
- Ruiz‐Gutierrez, V. and Barron, L.J. (1995). Methods for the analysis of triacylglycerols. Journal of Chromatography B: Biomedical Applications 671: 133–168.
- Saini, R.K. and Keum, Y.‐S. (2016). Tocopherols and tocotrienols in plants and their products: a review on methods of extraction, chromatographic separation, and detection. Food Research International 82: 59–70.
- Saito, E., Tanaka, N., Miyazaki, A., and Tsuzaki, M. (2014). Concentration and particle size distribution of polycyclic aromatic hydrocarbons formed by thermal cooking. Food Chemistry 153: 285–291.
- Salvatori, G., Filetti, F., Di Cesare, C. et al. (2008). Lipid composition of meat and backfat from Casertana purebred and crossbred pigs reared outdoors. Meat Science 80: 623–631.
- Sánchez‐Muniz, F.J., Cuesta, C., and Garrido‐Polonio, C. (1993). Sunflower oil used for frying: combination of column, gas and high‐performance size‐exclusion chromatography for its evaluation. Journal of the American Oil Chemists' Society 70: 235–240.
- Schröter, J., Griesinger, H., Reu, E. et al. (2016). Unexpected products of the hypochlorous acid‐induced oxidation of oleic acid: a study using high performance thin‐layer chromatography electrospray ionization mass spectrometry. Journal of Chromatography A 1439: 89–96.
- Schwartz, H., Ollilainen, V., Piironen, V., and Lampi, A.‐M. (2008). Tocopherol, tocotrienol and plant sterol contents of vegetable oils and industrial fats. Journal of Food Composition and Analysis 21: 152–161.
- Shammugasamy, B., Ramakrishnan, Y., Manan, F., and Muhammad, K. (2015). Rapid reversed‐phase chromatographic method for determination of eight vitamin E isomers and γ‐oryzanols in rice bran and rice bran oil. Food Analytical Methods 8: 649–655.
- Simas, R.C., Catharino, R.R., Cunha, I.B.S. et al. (2010). Instantaneous characterization of vegetable oils via TAG and FFA profiles by easy ambient sonic‐spray ionization mass spectrometry. Analyst 135: 738–744.
- Simsek, U.G., Cerci, I.H., Dalkilic, B. et al. (2009). Impact of stocking density and feeding regimen on broilers: chicken meat composition, fatty acids, and serum cholesterol levels. Journal of Applied Poultry Research 18: 514–520.
- Sion, B., Grizard, G., and Boucher, D. (2001). Quantitative analysis of desmosterol, cholesterol and cholesterol sulfate in semen by high‐performance liquid chromatography. Journal of Chromatography A 935: 259–265.
- Sjaastad, A.K., Jorgensen, R.B., and Svendsen, K. (2010). Exposure to polycyclic aromatic hydrocarbons (PAHs), mutagenic aldehydes and particulate matter during pan frying of beefsteak. Occupational and Environmental Medicine 67: 228–232.
- Sjövall, O., Kuksis, A., Marai, L., and Myher, J.J. (1997). Elution factors of synthetic oxotriacylglycerols as an aid in identification of peroxidized natural triacylglycerols by reverse‐phase high‐performance liquid chromatography with electrospray mass spectrometry. Lipids 32: 1211–1218.
- Sjövall, O., Kuksis, A., and Kallio, H. (2001a). Analysis of molecular species of peroxide adducts of triacylglycerols following treatment of corn oil with tert‐butyl hydroperoxide. Lipids 36: 1347–1356.
- Sjövall, O., Kuksis, A., and Kallio, H. (2001b). Reversed‐phase high‐performance liquid chromatographic separation of tert.‐butyl hydroperoxide oxidation products of unsaturated triacylglycerols. Journal of Chromatography A 905: 119–132.
- Sjövall, O., Kuksis, A., and Kallio, H. (2002). Formation of triacylglycerol core aldehydes during rapid oxidation of corn and sunflower oils with tert‐butyl hydroperoxide/Fe2+. Lipids 37: 81–94.
- Sjövall, O., Kuksis, A., and Kallio, H. (2003). Tentative identification and quantification of TAG core aldehydes as dinitrophenylhydrazones in autoxidized sunflowerseed oil using reversed‐phase HPLC with electrospray ionization MS. Lipids 38: 1179–1190.
- Skipski, V.P., Good, J.J., Barclay, M., and Reggio, R.B. (1968). Quantitative analysis of simple lipid classes by thin‐layer chromatography. Biochimica et Biophysica Acta 152: 10–19.
- Solaesa, Á.G., Bucio, S.L., Sanz, M.T. et al. (2014). Characterization of triacylglycerol composition of fish oils by using chromatographic techniques. Journal of Oleo Science 63: 449–460.
- Sosińska, E., Przybylski, R., Hazendonk, P. et al. (2013). Characterisation of non‐polar dimers formed during thermo‐oxidative degradation of β‐sitosterol. Food Chemistry 139: 464–474.
- Sosińska, E., Przybylski, R., Aladedunye, F., and Hazendonk, P. (2014). Spectroscopic characterisation of dimeric oxidation products of phytosterols. Food Chemistry 151: 404–414.
- Soupas, L., Juntunen, L., Lampi, A.‐M., and Piironen, V. (2004). Effects of sterol structure, temperature, and lipid medium on phytosterol oxidation. Journal of Agricultural and Food Chemistry 52: 6485–6491.
- Souza, H.A.L., Mariutti, L.R.B., and Bragagnolo, N. (2017). Microwave assisted direct saponification for the simultaneous determination of cholesterol and cholesterol oxides in shrimp. Journal of Steroid Biochemistry and Molecular Biology 169: 88–95.
- Srivastava, S., Singh, M., George, J. et al. (2010). Genotoxic and carcinogenic risks associated with the consumption of repeatedly boiled sunflower oil. Journal of Agricultural and Food Chemistry 58: 11179–11186.
- Stan, H.J. and Scheutwinkel‐Reich, M. (1980). Detection of hydroxy fatty acids in biological samples using capillary gas chromatography in combination with positive and negative chemical ionization mass spectrometry. Lipids 15: 1044–1050.
- Steenhorst‐Slikkerveer, L., Louter, A., Janssen, H.G., and Bauer‐Plank, C. (2000). Analysis of nonvolatile lipid oxidation products in vegetable oils by normal‐phase high‐performance liquid chromatography with mass spectrometric detection. Journal of the American Oil Chemists' Society 77: 837–845.
- Stolyhwo, A. and Privett, O.S. (1973). Studies on the analysis of lipid classes by gradient elution adsorption chromatography. Journal of Chromatographic Science 11: 20–25.
- Sugino, K. (1999). Simultaneous determination of different classes of lipid hydroperoxides by high‐performance liquid chromatography with post column detection by a ferrous/xylenol orange reagent. Bioscience, Biotechnology, and Biochemistry 63: 773–775.
- Sun, J., Zhao, X.‐E., Dang, J. et al. (2017). Rapid and sensitive determination of phytosterols in functional foods and medicinal herbs by using UHPLC–MS/MS with microwave‐assisted derivatization combined with dual ultrasound‐assisted dispersive liquid–liquid microextraction. Journal of Separation Science 40: 725–732.
- Sunarić, S., Lalić, J., and Spasić, A. (2017). Simultaneous determination of alpha‐tocopherol and alpha‐tocopheryl acetate in dairy products, plant milks and health supplements by using SPE and HPLC method. Food Analytical Methods 10: 3886–3901.
- Suomela, J.P., Ahotupa, M., Sjövall, O. et al. (2004a). Diet and lipoprotein oxidation: analysis of oxidized triacylglycerols in pig lipoproteins. Lipids 39: 639–647.
- Suomela, J.P., Ahotupa, M., Sjövall, O. et al. (2004b). New approach to the analysis of oxidized triacylglycerols in lipoproteins. Lipids 39: 507–512.
- Suomela, J.P., Ahotupa, M., and Kallio, H. (2005). Triacylglycerol oxidation in pig lipoproteins after a diet rich in oxidized sunflower seed oil. Lipids 40: 437–444.
- Suomela, J.P., Leskinen, H., and Kallio, H. (2011). Analysis of isomeric forms of oxidized triacylglycerols using ultra‐high‐performance liquid chromatography and tandem mass spectrometry. Journal of Agricultural and Food Chemistry 59: 8095–8100.
- Takeoka, G., Perrino, C., and Buttery, R. (1996). Volatile constituents of used frying oils. Journal of Agricultural and Food Chemistry 44: 654–660.
- Tamba Sompila, A.W.G., Héron, S., Hmida, D., and Tchapla, A. (2017). Fast non‐aqueous reversed‐phase liquid chromatography separation of triacylglycerol regioisomers with isocratic mobile phase. Application to different oils and fats. Journal of Chromatography, B: Analytical Technologies in the Biomedical and Life Sciences 1041–1042: 151–157.
- Tangolar, S.G., Özogul, F., Tangolar, S., and Yağmur, C. (2011). Tocopherol content in fifteen grape varieties obtained using a rapid HPLC method. Journal of Food Composition and Analysis 24: 481–486.
- Tarvainen, M., Suomela, J.P., Kuksis, A., and Kallio, H. (2010). Liquid chromatography‐light scattering detector‐mass spectrometric analysis of digested oxidized rapeseed oil. Lipids 45: 1061–1079.
- Tarvainen, M., Nuora, A., Quirin, K.‐W. et al. (2015). Effects of CO2 plant extracts on triacylglycerol oxidation in Atlantic salmon during cooking and storage. Food Chemistry 173: 1011–1021.
- Tekkeli, S.E., Önal, C., and Önal, A. (2012). A review of current methods for the determination of acrylamide in food products. Food Analytical Methods 5: 29–39.
- Terao, J., Shibata, S.S., and Matsushita, S. (1988). Selective quantification of arachidonic acid hydroperoxides and their hydroxy derivatives in reverse‐phase high performance liquid chromatography. Analytical Biochemistry 169: 415–423.
- Tonks, D.B. (2006). The estimation of cholesterol in serum: a classification and critical review of methods. Clinical Biochemistry 39: 446–463.
- Ubhayasekera, S.J.K.A., Verleyen, T., and Dutta, P.C. (2004). Evaluation of GC and GC‐MS methods for the analysis of cholesterol oxidation products. Food Chemistry 84: 149–157.
- Ulberth, F. and Reich, H. (1992). Gas chromatographic determination of cholesterol in processed foods. Food Chemistry 43: 387–391.
- Vaclavik, L., Cajka, T., Hrbek, V., and Hajslova, J. (2009). Ambient mass spectrometry employing direct analysis in real time (DART) ion source for olive oil quality and authenticity assessment. Analytica Chimica Acta 645: 56–63.
- Vaclavik, L., Belkova, B., Reblova, Z. et al. (2013). Rapid monitoring of heat‐accelerated reactions in vegetable oils using direct analysis in real time ionization coupled with high resolution mass spectrometry. Food Chemistry 138: 2312–2320.
- Velasco, J., Berdeaux, O., Márquez‐Ruiz, G., and Dobarganes, M. (2002). Sensitive and accurate quantitation of monoepoxy fatty acids in thermoxidized oils by gas–liquid chromatography. Journal of Chromatography A 982: 145–152.
- Velasco, J., Marmesat, S., Bordeaux, O. et al. (2004). Formation and evolution of monoepoxy fatty acids in thermoxidized olive and sunflower oils and quantitation in used frying oils from restaurants and fried‐food outlets. Journal of Agricultural and Food Chemistry 52: 4438–4443.
- Velasco, J., Marmesat, S., Berdeaux, O. et al. (2005). Quantitation of short‐chain glycerol‐bound compounds in thermoxidized and used frying oils. A monitoring study during thermoxidation of olive and sunflower oils. Journal of Agricultural and Food Chemistry 53: 4006–4011.
- Verleyen, T., Verhe, R., Huyghebaert, A. et al. (2001). Identification of α‐tocopherol oxidation products in triolein at elevated temperatures. Journal of Agricultural and Food Chemistry 49: 1508–1511.
- Viinanen, E. and Hopia, A. (1994). Reversed‐phase high‐performance liquid chromatographic analysis of triacylglycerol autoxidation products with ultraviolet and evaporative light‐scattering detectors. Journal of the American Oil Chemists' Society 71: 537–539.
- Wang, H., Lee, A.W.M., Shuang, S., and Choi, M.M.F. (2008). SPE/HPLC/UV studies on acrylamide in deep‐fried flour‐based indigenous Chinese foods. Microchemical Journal 89: 90–97.
- Wang, H., Feng, F., Guo, Y. et al. (2013). HPLC‐UV quantitative analysis of acrylamide in baked and deep‐fried Chinese foods. Journal of Food Composition and Analysis 31: 7–11.
- Wang, L., Csallany, A.S., Kerr, B.J. et al. (2016). Kinetics of forming aldehydes in frying oils and their distribution in French fries revealed by LC‐MS‐based chemometrics. Journal of Agricultural and Food Chemistry 64: 3881–3889.
- Warner, K., Neff, W.E., Byrdwell, W.C., and Gardner, H.W. (2001). Effect of oleic and linoleic acids on the production of deep‐fried odor in heated triolein and trilinolein. Journal of Agricultural and Food Chemistry 49: 899–905.
- Wijesundera, C., Ceccato, C., Watkins, P. et al. (2008). Docosahexaenoic acid is more stable to oxidation when located at the sn‐2 position of triacylglycerol compared to sn‐1(3). Journal of the American Oil Chemists' Society 85: 543–548.
- Wilson, R. and Lyall, K. (2002). Simultaneous determination by GC‐MS of epoxy and hydroxy FA as their methoxy derivatives. Lipids 37: 917–924.
- Wilson, R., Smith, R., Wilson, P. et al. (1997). Quantitative gas chromatography–mass spectrometry isomer‐specific measurement of hydroxy fatty acids in biological samples and food as a marker of lipid peroxidation. Analytical Biochemistry 248: 76–85.
- Wong, Y.H., Muhamad, H., Abas, F. et al. (2017). Effects of temperature and NaCl on the formation of 3‐MCPD esters and glycidyl esters in refined, bleached and deodorized palm olein during deep‐fat frying of potato chips. Food Chemistry 219: 126–130.
- Xia, W. and Budge, S.M. (2017). Techniques for the analysis of minor lipid oxidation products derived from triacylglycerols: epoxides, alcohols, and ketones. Comprehensive Reviews in Food Science and Food Safety 16: 735–758.
- Xia, W., Budge, S., and Lumsden, M.D. (2016). 1H‐NMR characterization of epoxides derived from polyunsaturated fatty acids. Journal of the American Oil Chemists' Society 93: 467–478. https://doi.org/10.1007/s11746‐016‐2800‐2.
- Xu, X.M., He, H.L., Zhu, Y. et al. (2013). Simultaneous determination of 3‐monochloropropane‐1,2‐diol and acrylamide in food by gas chromatography‐triple quadrupole mass spectrometry with coupled column separation. Analytica Chimica Acta 760: 93–99.
- Yamada, K., Terao, J., and Matsushita, S. (1987). Electrochemical detection of phospholipid hydroperoxides in reverse‐phase high performance liquid chromatography. Lipids 22: 125–128.
- Yen, T.Y., Inbaraj, B.S., Chien, J.T., and Chen, B.H. (2010). Gas chromatography–mass spectrometry determination of conjugated linoleic acids and cholesterol oxides and their stability in a model system. Analytical Biochemistry 400: 130–138.
- Yu, T.H., Wu, C.M., and Ho, C.T. (1993). Volatile compounds of deep‐oil fried, microwave‐heated and oven‐baked garlic slices. Journal of Agricultural and Food Chemistry 41: 800–805.
- Yuan, C., Xie, Y., Jin, R. et al. (2017). Simultaneous analysis of tocopherols, phytosterols, and squalene in vegetable oils by high‐performance liquid chromatography. Food Analytical Methods 10: 3716–3722.
- Zeb, A. (2012). Triacylglycerols composition, oxidation and oxidation compounds in camellia oil using liquid chromatography‐mass spectrometry. Chemistry and Physics of Lipids 165: 608–614.
- Zeb, A. (2015a). Chemistry and liquid chromatography methods for the analyses of primary oxidation products of triacylglycerols. Free Radical Research 49: 549–564.
- Zeb, A. (2015b). A reversed phase HPLC‐DAD method for the determination of phenolic compounds in plant leaves. Analytical Methods 7: 7753–7757.
- Zeb, A. (2017). A simple, sensitive HPLC‐DAD method for simultaneous determination of carotenoids, chlorophylls and α‐tocopherol in leafy vegetables. Journal of Food Measurement and Characterization 11: 979–986.
- Zeb, A. and Haq, I. (2017). Polyphenolic composition, lipid peroxidation and antioxidant properties of chapli kebab during repeated frying process. Journal of Food Measurement and Characterization 12: 555–563. https://doi.org/10.1007/s11694‐017‐9667‐2.
- Zeb, A. and Mehmood, A. (2012). Effects of oxidized vanaspati ghee on the serum lipids profile and radical scavenging activity of the in vitro lipids of liver, brain and muscles. Turkish Journal of Biochemistry 37: 417–423.
- Zeb, A. and Murkovic, M. (2010a). Analysis of triacylglycerols in refined edible oils by isocratic HPLC‐ESI‐MS. European Journal of Lipid Science and Technology 112: 844–851.
- Zeb, A. and Murkovic, M. (2010b). Characterization of the effects of beta‐carotene on the thermal oxidation of triacylglycerols using HPLC‐ESI‐MS. European Journal of Lipid Science and Technology 112: 1218–1228.
- Zeb, A. and Murkovic, M. (2010c). Thin‐layer chromatographic analysis of carotenoids in plant and animal samples. JPC Journal of Planar Chromatography Modern TLC 23: 94–103.
- Zeb, A. and Murkovic, M. (2011). Carotenoids and triacylglycerols interactions during thermal oxidation of refined olive oil. Food Chemistry 127: 1584–1593.
- Zeb, A. and Murkovic, M. (2013a). Determination of thermal oxidation and oxidation products of β‐carotene in corn oil triacylglycerols. Food Research International 50: 534–544.
- Zeb, A. and Murkovic, M. (2013b). Pro‐oxidant effects of β‐carotene during thermal oxidation of edible oils. Journal of the American Oil Chemists Society 90: 881–889.
- Zeng, M., Li, Y., He, Z. et al. (2016). Effect of phenolic compounds from spices consumed in China on heterocyclic amine profiles in roast beef patties by UPLC–MS/MS and multivariate analysis. Meat Science 116: 50–57.
- Zhang, Q., Qin, W., Li, M. et al. (2015). Application of chromatographic techniques in the detection and identification of constituents formed during food frying: a review. Comprehensive Reviews in Food Science and Food Safety 14: 601–633.
- Zhang, X., Eigendorf, G., Stebbing, D.W. et al. (2002). Degradation of trilinolein by laccase enzymes. Archives of Biochemistry and Biophysics 405: 44–54.
- Zhang, Y., Dong, Y., Ren, Y., and Zhang, Y. (2006). Rapid determination of acrylamide contaminant in conventional fried foods by gas chromatography with electron capture detector. Journal of Chromatography A 1116: 209–216.
- Zommick, D.H., Kumar, G.N., Knowles, L.O., and Knowles, N.R. (2013). Translucent tissue defect in potato (Solanum tuberosum L.) tubers is associated with oxidative stress accompanying an accelerated aging phenotype. Planta 238: 1125–1145.