
Chapter 2
Fundamental Science and Applications for Biomaterials
Ali S. Ayoub1,2 and Lucian A. Lucia1
1Archer Daniels Midland Company, ADM Research, Chicago, IL, USA
2North Carolina State University, Department of Forest Biomaterials, Raleigh, NC, USA
The fuel of the future is going to come from fruit like that sumac out by the road, or from apples, weeds, sawdust - almost anything. There is fuel in every bit of vegetable matter that can be fermented. There's enough alcohol in one year's yield of an acre of potatoes to drive the machinery necessary to cultivate the fields for a hundred years.
Henry Ford, 1925
2.1 Introduction
The basic construct of biopolymer matrices remains a virtually insurmountable obstacle to the “best laid plans of mice and men” of providing products to compete with petro-based chemicals and associated commodity items. A more robust and precise understanding of the factors that limit a widespread use of lignocellulosic substrates in society is perhaps the most pressing challenge that the emergent bio-economy faces. The goal, therefore, of this chapter is to elucidate the fundamental physico-chemistry of the biomaterials, emphasize their value proposition for supplanting petrochemicals, tackle the challenges of conversion, and ultimately provide a milieu of possibilities for the biomaterials. The reader will be conversant and knowledgeable of the critical issues that surround the field of lignocellulosic intransigence, possible successful strategies to cope with their inertness, and potential pathways for the successful use of lignocellulosics and starch in the new bio-economy.
2.2 What are the Biopolymers that Encompass the Structure and Function of Lignocellulosics?
In the history of energy usage, wood has occupied a particularly noteworthy and prominent role for most of humankind. It was not until about 100 years ago (the early part of the twentieth century) that its hegemony and utility came into serious question principally due to the discovery of a cheap, seemingly inexhaustible, and easily implementable product known as petroleum (literally, “rock oil”). At that time, a famous entrepreneur and industrialist, Mr. Henry T. Ford, opined that it behooved mankind to foster the exploitation and use of natural materials (such as wood) for their use as fuels; for example, he planned to power his Model Ts with ethanol while early diesel engines were run with peanut oil. The ability to use wood, plants, and their by-products for energy and other valuable products was important at that time, and it continues to be even more paramount now with the specter of the scarcity of petroleum reserves looming and the concomitant ever-increasing price of gasoline. However, efficient use of such raw materials demands a keen and in-depth understanding of their constituents and the processes in place to unlock their fuel and material value. A good understanding of these materials is key to their future use.
Wood is a raw material that has served humankind very well over its history. Locked within its macrostructure are three significant polymers (biopolymers) whose utility can rival that of petroleum and its by-products. The principal building blocks of wood (and hence nearly all lignocellulosics) are cellulose, heteropolysaccharides (or “hemicelluloses”), and lignin. These raw polymers are among the most abundant materials in the biosphere. They are only rivaled by chitin, another polysaccharide like cellulose that is found in the exoskeletons of marine and terrestrial life forms, as to their dominance in the material world. The symmetry found in the natural world is extraordinary especially when comparing the exquisite twofold screw structure of cellulose, the most dominant biomaterial on land, to its analogue, chitin, and the most dominant biomaterial in the sea. Figure 2.1 demonstrates this awesome symmetry.
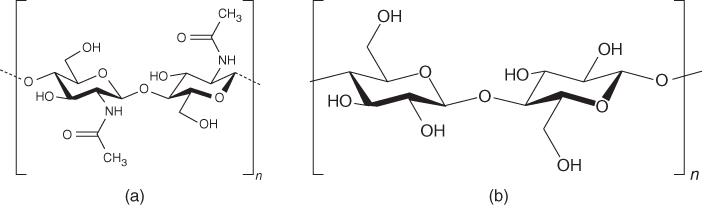
Figure 2.1 The archetypal structures of the most abundant biomaterials on the planet. (a) The repeating unit (N-acetylglucosamine) of the biopolymer chitin. (b) The repeating unit (glucose) of the biopolymer cellulose.
2.2.1 Cellulose
Cellulose, the major constituent of lignocellulosics, occupies up to 50% or more of the overall composition of the matter [1]. In general, it is considered to be the most abundant renewable material on the planet, with annual cellulosic biomass production in the order of approximately 2 teratons (or 2 × 1012 tons). Although it has the same structural motif in each material that it comprises (i.e., the same corkscrew twofold symmetry), its degree of polymerization (DP or “n” shown in Figure 2.1) and crystallinity (degree of packing order as evidenced by X-ray crystallography) can vary widely. Estimates of the DP found in the literature show that it can vary from about n = 300 (in wood) to upward of 10,000 (cotton and bacterial cellulose, BC). It can also show wide trends in the overall crystalline morphology; for example, it can show a relatively low crystallinity in wood, whereas it can be highly crystalline in Valonia [2]. Nevertheless, cellulose tends to be rather nonreactive to chemicals other than cellulose enzymes. It is a linear polymer that is made up of β-1,4-linked d-glucopyranose units.
One of the glucose units within the chain can be described as having a rotation of 180° relative to its neighbor within the chain. The chemical description of a “dimer” or two units is cellobiose unit, which technically could be used to describe the polymeric structural pattern. Classically, a cellulose chain, in as much as its glucose monomer makeup, presents two different terminal hydroxyl groups: the non-reducing end group (NREG) and the reducing end group (REG). The NREG consists of a 4-hydroxyl group (at the 4 carbon), whereas the REG is categorized in organic chemistry as a hemiacetal linkage (or aldehyde hydrate group). Figure 2.2 shows a classical representation of the NREG and REG.
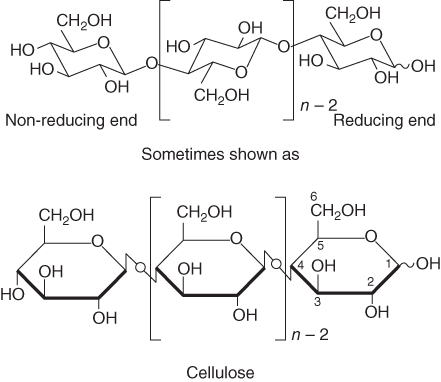
Figure 2.2 A simplified representation of the stereochemical asymmetry present in cellulose: the existence of a non-reducing end group (NREG) versus an opposite reducing end group (REG) give cellulose different terminal chemistries.
Each glucose unit presents three reactive hydroxyl groups (C2, C3, and C6), which expedite the formation of both intra- and interchain hydrogen bonding. The stiffness and compactness of the chain can almost exclusively be attributed to interchain interactions. However, the structural uniformity of cellulose in any plant is lacking; typically, there exist both highly ordered (crystalline) domains and amorphous (low degree of order) domains, which as a rule depend on the raw material and the treatments it has been subjected to. The relative robustness of cellulose is critically dependent on these features.
Cellulose generally provides the main mechanical properties of any lignocellulosics owing to its packing and hydrogen-bonded structure; in general, it provides the overall load-bearing capacity of wood and plants but does not typically complex with the other biopolymers in the lignocellulosic matrix.
Cellulose is synthesized in vascular plants1 in the plasma membrane of the rosette terminal complexes (RTCs), a class of macromolecular protein structures approximately 25 nm in diameter. These structures contain the cellulose synthase enzymes (at least three different sythases are involved that are encoded by CesA2 genes) that are responsible for synthesizing the individual cellulose chains. The RTCs are able to spin a bundle of cellulose chains known as a microfibril into the cell wall. Scheme 2.1 shows a simplified representation of the production of microfibrils leading to cellulose fibers.
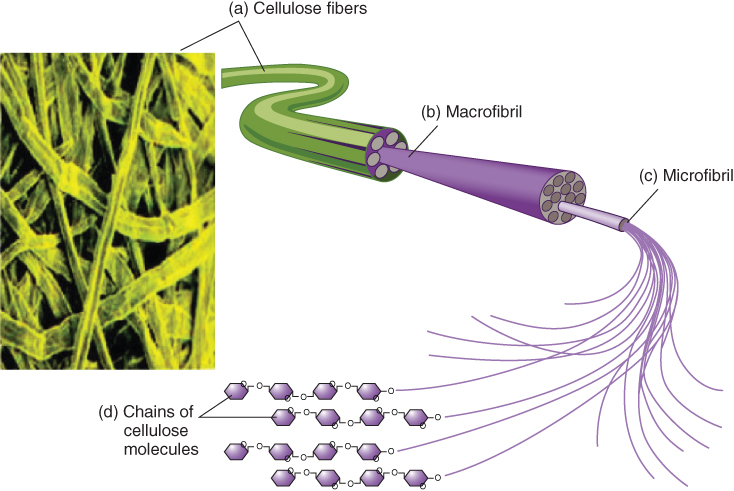
Scheme 2.1 A simplified pictorial summary of the contextual development of cellulose fibers: the evolution of cellulose chains into microfibrils, an elementary unit in cellulose, which contribute to the higher order elementary structures (e.g., the macrofibril is composed of a bundle of microfibrils).
Reprinted with permission from http://alevelnotes.com/Carbohydrate-polymers/65.
Native cellulose whose production is demonstrated in Scheme 2.1 is known from crystallographic data to have two distinct crystalline phases: Iα and Iβ [3]. Recently, exact representations of the principal phases of cellulose (Iα and Iβ) were obtained using atomic-resolution synchrotron and neutron diffraction data. The resulting structure of Iα was a one-chain triclinic unit cell (shown in Figure 2.3 with the principal axes: a, b, and c). The conformation of the glucosidic linkages and hydroxymethyl groups is identical, which is not the case for Iβ. In Iβ, there are two conformationally distinct chains in the monoclinic unit cell (corner and center chains) that requires adjacent glucosyl residues (in the same chain) to be the same. Both triclinic and monoclinic crystal systems derive from the seven crystal systems that are available to describe highly ordered systems.
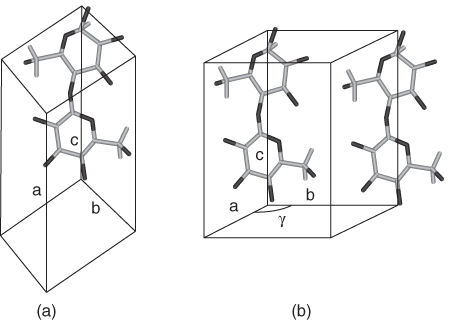
Figure 2.3 A representation of the unit cell mode of chain packing for cellulose. (a) The triclinic unit cell (Iα). (b) The monoclinic unit cell (Iβ).
The triclinic system can be best described as a crystal whose vector descriptors possess unequal length AND are not mutually orthogonal perpendicular to each other as in a classic three-dimensional Cartesian coordinate system. Similarly, a monoclinic system has vectors of unequal length, but they form a rectangular prism with a parallelogram – a base indicating that two vectors are orthogonal, but the third angle, γ, >90°.3
2.2.2 Heteropolysaccharides
Akin to cellulose in form, but not in function, are the heteropolysaccharides (or “hemicelluloses,” a more colloquial, but inexact term for this class of biomaterials) [4, 5]. This class of biomaterials is likely the second most dominant terrestrial material available after cellulose. In general, the amount of hemicelluloses is typically 20–30% of the dry weight of wood. They constitute a variegated class of materials without the chemical precision and homogeneity of cellulose, yet they are naturally produced annually in the order of 60 billion tons. This class of biomaterials is almost mandatorily associated with cellulose in any cellulosic matrix. They act in general to maintain the physical integrity of the cellulosic microfibrils and likely engage in a covalent complex (the lignin–carbohydrate complex, or LCC) with the lignin polymer of the plant cell [6]. They adopt a multiplicity of structural motifs, the most popular form being “xylan,” which is likely the third most abundant biomaterial on the planet [7]. Figure 2.4 shows several representations of xylan, a typical and globally dominant heteropolysaccharide.
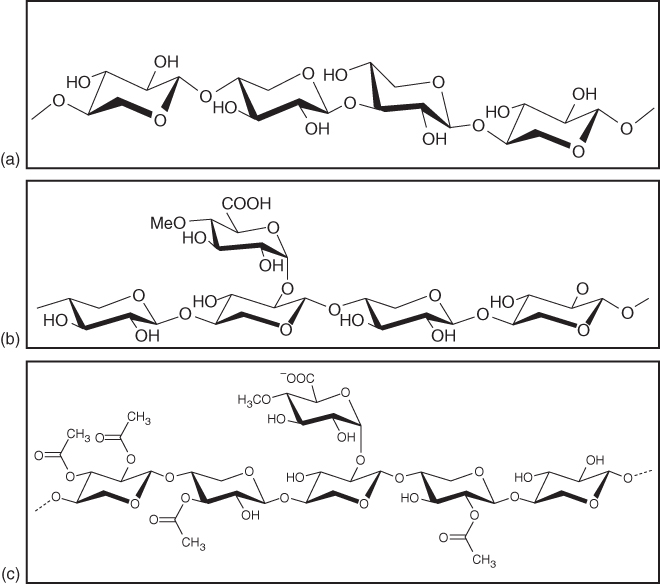
Figure 2.4 Various structural representations of xylan. (a) Simple xylan backbone composed of non-functionalized xylose monomers. (b) Xylan backbone with a pendant 4-O-methylhexenuronic acid residue at the 2-carbon. (c) Representation of a xylan backbone decorated with pendant acetyl groups.
As shown in Figure 2.4, heteropolysaccharides are very much unlike cellulose in a structural sense. Cellulose is crystalline, that is, well organized or packed, of a high molecular weight, and possesses a low polydispersity.4 However, heteropolysaccharides tend to be rather amorphous in their structure (often they are branched as shown in Figure 2.4), of a low molecular weight, and display a low PDI. In fact, the heteropolysaccharides typically do not have just a single repeating unit (such as xylan) but can often contain a multiplicity of monomers. For example, the dominant heteropolysaccharide is xylan, which tends to be fairly localized in angiosperms ( hardwoods), whereas the dominant heteropolysaccharide in gymnosperms (softwoods) is glucomannans. In addition, there are galactoglucomannans, arabinoxylans, and so on. The list is nearly endless in the natural diversity of heteropolysaccharides that are found. Figure 2.5 shows the structure, for example, of galactoglucomannan, a typical branched heteropolysaccharide.
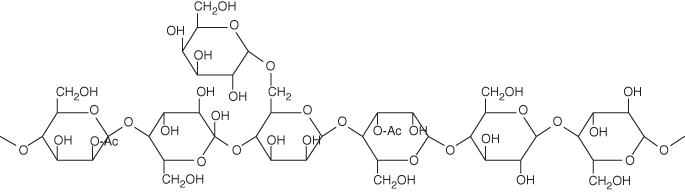
Figure 2.5 Representative structure of the galactoglucomannan heteropolysaccharide. Note that the glucomannan backbone has several Ac (acetyl) groups decorating it, while it possesses a pendant galactose residue on the 6 carbon of a mannose residue.
Given the various physical characteristics stated here for heteropolysaccharides, it is no surprise that they tend to be more susceptible to degradation from both chemical and enzymatic attack. In addition, they can sometimes be more amenable to chemical derivatization and reaction than their cellulose analogue.
2.2.3 Lignin
This last polymer, albeit neglected for many years because of its molecular heterogeneity and seeming lack of tractability, is one of the most important polymers on the planet. It forms the last element of the lignocellulosics that primarily comprise the biomass on this planet. The term “lignin” is in fact taken from the Latin word lignum, which is translated to “wood.” It is found virtually in all biomass especially vascularized plants including herbs and grasses, but it is chiefly located (on a per capita basis) in the cell wall of woody tree species [8, 9]. On a mass basis, up to approximately 30% of all carbon in the biosphere can be attributed to lignin. A representation of its diverse native structure is provided in Figure 2.6 [10]. The structure seemingly lacks the signature monomer associated with polymer structures, but in wood chemistry circles, it is assumed that the monomer is based on a C9 phenylpropanoid residue that will be further elaborated within this section.
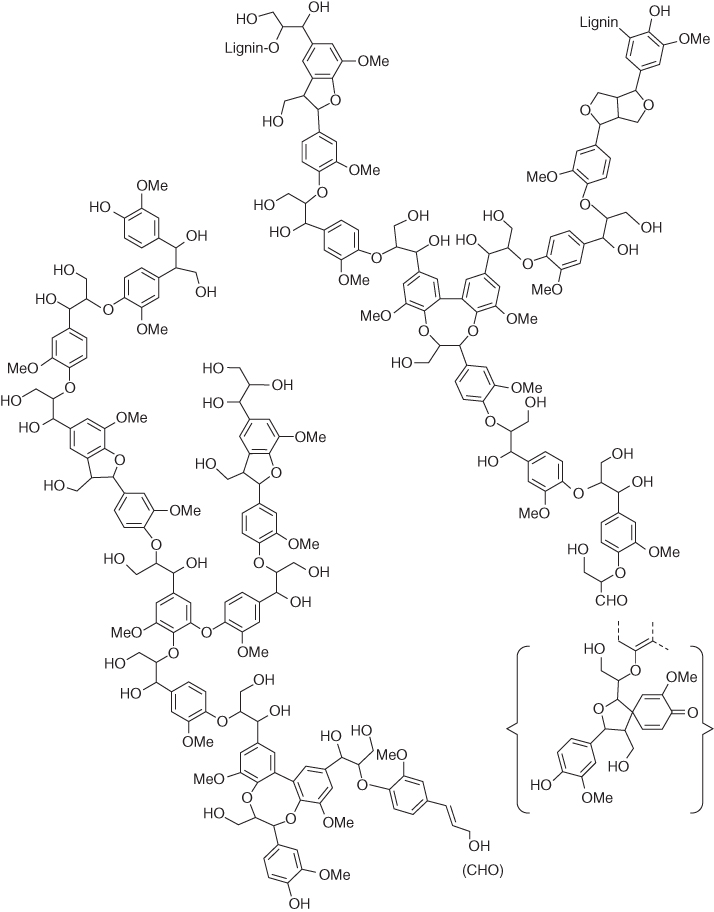
Figure 2.6 A generic representation of the lignin that is believed to be localized within angiosperm wood cells.
The structure elucidated here is speculative at best in nature and represents a best guess attempt because not only is its X-ray structural characterization impossible, but it is extremely divergent in its structure between species and even within the same species. In terms of its role among the natural polymers in woody and plant species, lignin maintains unique and powerful functionality within the bio-system. It is essentially a natural “glue” that helps to keep the polysaccharide bundle intact and whole. The current theory for its particular niche in lignocellulosics is that it covalently complexes with the heteropolysaccharides in a so-called LCC. In addition to its chemical attachment to polysaccharides, it is believed that lignin also acts as a plant's second line of defense (after the bark/extractives) against microbial attack/infestation.
Lignin as shown in Figure 2.6 is a complex aromatic network polymer (but it is three-dimensional) that is polydisperse and contains a number of divergent functionalities and branching points. Although PDI may not be objectively applicable in the case of lignin, lignin nevertheless is a branched network polymer that has its origins in well-characterized lignol subunits. Figure 2.7 shows the principal monomeric units that constitute the biosynthesized lignin.
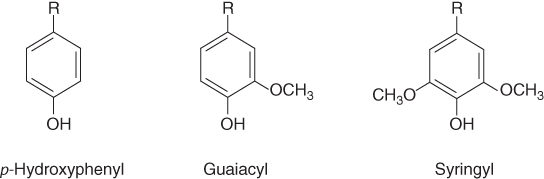
Figure 2.7 The generic lignin monomeric structural motifs that constitute the totality of all of the lignin polymers that are extant in nature.
Each of the structures shown in Figure 2.7 is available in differing amounts in different classes of woods or plants. For example, monocotyledonous grasses tend to have almost exclusively the p-hydroxyphenyl monomer unit, whereas gymnosperms are almost exclusively constructed out of the guaiacyl units. Angiosperms, on the other hand, tend to have some combination of syringyl (principal component) and guaiacyl units.
The lignin extant in nature generally has a support role for the polysaccharide matrix. In woody tissue, it tends to surround the wood cell helping to ensure not only integrity but to maintain proper surface energetics (hydrophobicity) to allow for the uninterrupted circulation of water and nutrients. Figure 2.8 demonstrates the respective localization of lignin within a representative wood cell.

Figure 2.8 A fluorescence micrograph obtained from a cross-section of wood cells. Shown in the micrograph are the major elements of the wood cell starting from the inner lumen (dark, no fluorescence), normal lignified cell wall (second part extending from the inner dark sphere), to the outer middle lamella.
The cell wall is the region that contains the most lignin polymer, but it is highly concentrated between cells, in essence, to ensure good mechanical integrity among the macrofiber bundle [11]. This material has for roughly 130 years been the targeted polymer for deconstructing cellulosic fibers for papermaking. A huge global paper pulp industry has emerged since 1879 when Dahl, a German chemist, used sodium sulfate as a makeup chemical for soda pulping to regenerate sodium hydroxide. Serendipitously, sodium sulfide was formed and unexpectedly gave far better pulping results in terms of rates and pulp quality. The process was termed kraft from the German for “strong” [12].
2.2.4 The Discovery of Cellulose and Lignin
Anselme Payen (1795–1891) was born in Paris to Marc and Jean Payen, a family in which there was great respect for science, law, and chemistry. In fact, young Anselme (at the tender age of 13) began a lifelong love of studying science first with his father, a man whose entrepreneurial spirit led him to establish chemical factories, and then Anselme went off to study chemistry, physics, and mathematics at the École Polytechnique under the tutelage of Louis Nicolas Vauquelin and Michel Eugène Chevreul. Interestingly, after this quasi-internship, he returned to work for his father as the superintendent of a borax refining plant when he was only 23 years old (1818). What was so unique about this situation was that Payen's father had devised a better way of producing borax from boric acid that allowed him better market opportunities. After Payen's father died in 1820, he held sole custody of the family estate. Payen turned his interest to one of his father's factories that refined sugar from beets. Interestingly, he employed activated charcoal to decolorize the sugar, a method that has been in vogue ever since that impressive discovery. In addition, this work helped to expedite the transition, on a world stage, from obtaining sugar from cane to beets. One of his most remarkable efforts and perhaps what he may be best known for is the discovery of diastase (from the Greek for “separate”), an enzyme that converts starch to glucose. He was able to isolate this substance from malt extract in 1833; interestingly, it was the first isolated enzyme, an organic system that demonstrates catalysis, that is, enhancing the rate of reaction without being consumed. His choice of terminology, diastase, led to the tradition of using the suffix -ase in biochemistry for the naming of enzymes. In 1834, Payen began the systematic study of wood from which he discovered a substance from the plant cell walls that could be hydrolyzed (broken down with water) to glucose units (similar to starch). He called the substance “cellulose,” which began another tradition of using “-ose” as the suffix to name carbohydrates. In 1835, he departed from industrial work in favor of a professorship of industrial and agricultural chemistry at the École Centrale des Arts et Manufactures. In 1839, he then accepted a joint appointment at the nearby Conservatoire des Arts, which he held in conjunction with his original appointment until his death in 1871. He was a prolific researcher, publishing over 200 papers and 10 books on topics such as dextrans, sugars, lignin, cellulose, and starch. His seminal paper on cellulose and lignin isolation was published in 1838. These experiments in 1838 revealed that in addition to cellulose, wood contained an oxidizable crusty substance that was later designated as “lignin” in 1857 by Schulze. Payen treated the wood with a concentrated nitric acid solution and later washed the residue with an alkaline solution (sodium hydroxide) to dissolve the crusty substance. Payen noted significant differences between the wood and the crusty substance; he is also credited with the first Klason lignin isolation method whereby he used concentrated sulfuric acid to remove the polysaccharides, which then allowed for the isolation of the “lignin” fraction. Today, the American Chemical Society (ACS) honors Anselme Payen's memory by awarding each year a prize in his name to the scientist who in the opinion of the Cellulose and Renewable Materials Division (CELL) of the ACS has contributed the most to the science, engineering, and technology of cellulose and renewable materials (more information can be found at the ACS site: http://cell.sites.acs.org/anselmepayenaward.htm).
2.3 Chemical Reactivity of Cellulose, Heteropolysaccharides, and Lignin
A proper understanding of the chemical, biological, and mechanical reactivity of the biopolymers in biomass is crucial to determining the best approaches for their strategic utilization. Throughout history, a basic understanding of lignocellulosics has lagged behind their usage on a societal level. This is not surprising because pragmatism dictates that this should be the case; people will always be practical in approaching the usage of materials before a fundamental survey of the properties of the materials is accomplished. However, advances in bio-fuels and biomaterials cannot be made from a pragmatic perspective because there are too many technical hurdles to overcome and, additionally, the economic barriers to implementation are severe. Therefore, this section attempts to survey the individual reactivities of the biopolymers under scrutiny. The survey examines several different stressors or reactants used on the biopolymers and their responses.
2.3.1 Cellulose Reactivity
One of the complicating factors in the proper understanding of the reactivity of cellulose is its accessibility in addition to its chemical makeup. It is organized elegantly within a lignocellulosic matrix that consists of its interplay with the heteropolysaccharides and the lignin. Today, the reactivity of cellulose is a topic of grave importance because of its ability to supply an ever burgeoning bio-fuels and biomaterials community [13, 14]. As demonstrated earlier, the reactivity of cellulose is a function of accessibility, which is severely hampered by its compact structure. This compact structure is a function of the presence of a very strong hydrogen bonding network that gives rise to highly ordered region.
Critical factors that have been suggested by a number of experts include the number and size of pores in the cellulose structure, the molecular size and type of chemical that is added to the cellulose, the internal surface as controlled by the size of fibrils/aggregates, and the morphology of the cellulose macromolecules. These critical factors can only be addressed by (i) ensuring that the cellulosic pores are sufficiently open to accommodate the chemicals/reagents added, (ii) deconstructing fibrillar aggregates, and (iii) deconstructing the ordered regions to no longer adopt a stiff, compact network structure, that is, the hydrogen bonding network must be disrupted. An optimal chemical interaction, therefore, must consider these latter three criteria; recently, chemical, mechanical, and biological treatments have been tested, with a great push toward the environmentally benign biological treatments.
Chemical treatments of cellulose [15] are employed to enhance the swelling of the cellulose fibers. This swelling not only facilitates the passage of chemical agents, but its primary purpose is to disrupt the hydrogen bonds because of the high osmotic pressure induced by the swelling phenomenon. Thus, the hydrogen-bonded network becomes disrupted, and as a result the compact structure is no longer available leading to a more accessible structure. A pictorial representation of this phenomenon is shown in Figure 2.9.
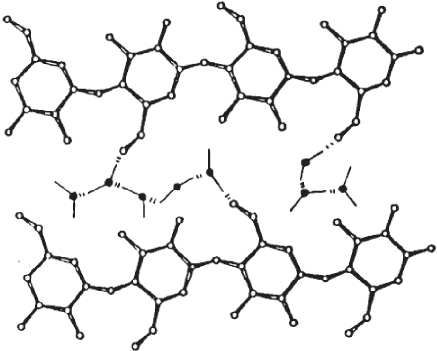
Figure 2.9 Shown is a simple cartoon that illustrates the effect of introducing water molecules within the H-bonded network structure of cellulose.
Reprinted with permission from Westermark, S. “Use of Mercury Porosimetry and Nitrogen Adsorption in Characterisation of the Pore Structure of Mannitol and Microcrystalline Cellulose Powders, Granules and Tablets.” Academic Dissertation, November 2000, University of Helsinki, Helsinki, Finland.
The scheme illustrates the inclusion of water molecules that interfere with the hydrogen bonding between the C6, and the C3 and C2 hydroxyls of chain neighbors and act to therefore swell the cellulosic substrate. As a result of this inclusion of water molecules, the swelled structure loses its ordered nature and in some cases, there is a complete loss of crystallinity, which consequently leads to an increase in the active surface area or exposed hydroxyl groups (which were heretofore buried within the tightly packed cellulosic crystallite). There have been a number of studies within this particular area that have shown such accessibility to solvents including among others the use of sodium hydroxide and its combination with urea. Urea is conjectured to insert within the interchain cellulosic matrix and disrupt the hydrogen bonding paradigm. The purpose of such treatments is to facilitate accessibility for a particular
In addition to chemical treatments, cellulosics can be treated with a mechanical process [16]. Mechanical treatment of the cellulose fibers is used in the pulp and paper industry because of its capacity to enhance fiber–fiber bonding, to cut or make the fibers stronger, and to produce changes on the cellulose structure. For instance, strong bonds among fibers give the printing paper strong and smooth properties. When the pulp is subjected to mechanical treatment, the interfibrillar bonds, which are mainly located in the primary wall and in the outer lamella of the S1 layer of the cell wall, are disrupted. This effect leads to an increase in the reactive surface area of the fibers, improving the accessibility of the cellulose. In several studies, mechanical treatment has been used in combination with other treatments.
Moreover, enzymatic treatments can be used as well [17]. Enzymes have broad industrial applications. They have been used in the detergent, food, and pharmaceutical sectors. Enzymes have also been studied in the pulp and paper industry, and they are currently used for several applications, including deinking and as bleaching agents. The effect of enzymatic treatments on cellulose reactivity has also been investigated. It has been reported that enzymatic treatments, especially that of cellulases on dissolving pulps, hold a great potential for increasing cellulose reactivity.
One of the enzymes is the cellulases: monocomponent endoglucanases. Cellulases are enzymes that hydrolyze the 1,4-β-d-glucosidic bonds of the cellulose chain. There are three major groups of cellulases: endoglucanases, cellobiohydrolases or exoglucanases, and glucosidases. These enzymes can act alone or together on the cellulose chain or together. When they act together, a synergistic phenomenon is often generated, resulting in an efficient degradation of the cellulose structure.
Endoglucanases are enzymes that randomly cleave the amorphous sites of the cellulose creating shorter chains (oligosaccharides) and, therefore, new chain ends. Cellobiohydrolases or exoglucanases attack the reducing and non-reducing ends of the cellulose chains, generating mainly glucose or cellobiose units. This type of cellulose can also act on microcrystalline cellulose by a peeling mechanism. Glucosidases act on cellobiose generating glucose units. It has been suggested that there are three primary parameters affecting the degree of enzymatic hydrolysis: the crystallinity, the specific surface area, and the degree of polymerization of the cellulose.
Most cellulases consist of two domains. The first is a catalytic domain, which is responsible for the hydrolysis of the cellulose chain. The catalytic domain of endoglucanases is “cleft shaped.” Exoglucanase, on the other hand, has a “tunnel-shaped” catalytic domain structure. The second is a cellulose-binding domain (CBD), which helps the enzyme to bind to the cellulose chain bringing the catalytic domain close to the substrate. An interdomain linker serves as a connection between the two domains.
2.3.1.1 Reactivity Measurements
Several methods have been developed to measure cellulose reactivity, including iodine sorption water retention value of pulps, swelling water coefficient, and viscose filter value [18–20].
2.3.1.2 Dissolving-Grade Pulps
For regenerated cellulose manufacturing, it is generally known that the raw material is required to have a high cellulose content (over 90%) and low levels of hemicellulose, lignin, extractives, and minerals [21–23]. Today, the raw materials used for the production of regenerated cellulose are dissolving-grade pulps and, to a lesser extent, cotton linters. Dissolving-grade pulps are produced mainly by two different processes: the sulfite process and the prehydrolysis kraft process. Other pulping processes have been investigated for the production of these pulps, including organosolv pulping. This process is based on the use of organic solvents; however, the expense of solvent recovery is the biggest drawback of this process. Dissolving-grade pulps are costlier than kraft pulps. This can be attributed to several factors, such as wood costs (the production of these pulps has a lower yield since hemicelluloses are dissolved and washed away); capital costs (because the yield is low, more equipments may be needed to have a high production); chemical costs; production rates (lower than for paper-grade pulps); and the inventories and storage space (the pulps are produced for specific customers with certain requirements, which implies a high control of the inventory). As a consequence, the viability of converting paper-grade pulps into dissolving-grade pulps arises.
2.3.1.3 Converting Paper-Grade Pulps into Dissolving-Grade Pulps
In recent years, several studies have used different methods to examine the feasibility of modifying paper-grade pulps for further use as dissolving-grade pulps [24, 25]. These studies have focused mainly on the optimal removal of hemicelluloses because in the production of viscose, hemicelluloses can affect the viscose filterability, the xanthation of cellulose, and the strength of the end product. Several methods have been reported for the removal of hemicelluloses, including treatments with alkaline extraction, nitren and cuen extraction, and a combination of pretreatments using xylanases and alkaline extraction. However, little attention has been paid to the effect of changes in the accessibility and reactivity of the cellulose after these treatments.
2.3.2 Hemicellulose Reactivity
Several methods have been used to extract hemicellulose from woody tissues [26–28]. Those methods include dilute-acid pretreatments, alkaline extraction, alkaline peroxide extraction, liquid hot-water extraction, steam treatment, microwave treatment, and ionic liquid extraction.
Dilute-acid pretreatment, often involving about 0.5–1% sulfuric acid, is a useful procedure for hemicellulose isolation. By using this method, the majority of the original amount of hemicellulose from the poplar tree (hardwood) can be recovered as dissolved sugar. However, in the course of such treatment, a high amount of the hemicellulose monomers are degraded, leading to the generation of by-products. On the other hand, extraction of hemicellulose from sugarcane bagasse by using hot-water extraction at a temperature of 150–170 °C recovers almost 90% of the hemicellulose as dissolved sugar, but with relatively less monomer degradation than that of the dilute-acid method. Because hot-water treatment cleaves some of the acetate groups from hemicellulose, the pH decreases. The reduced pH results in additional generation of acetic acid, leading to a phenomenon called “autohydrolysis.” It has been shown that autohydrolysis can be promoted by irradiating wood with microwaves in water.
Due to severe treatments caused by acidic and partly applied heated extraction processes, hemicellulose degrades by losing its chain length, and consequently, exhibits high polydispersity. Steam and microwave treatments are combined with chemicals to dissolve the hemicellulose; however, due to the complexity of those methods, they have been applied mainly on a small scale for hemicellulose extraction. In steam treatment, ester bonds in the hemicelluloses will be cleaved and then the wood is treated with steam, resulting in the formation of acetic acid. This will lower the pH and, thus, induce autohydrolysis of the glycosidic bonds in the hemicelluloses. Such a process will generate a low-molecular-weight, water-soluble hemicellulose. From this method, one can get a low percentage yield of hemicellulose with significant contamination of dissolved cellulose and lignin and degradation products of the hemicellulose.
An ionic liquid/cosolvent extraction system has been used by Froschauer and coworkers to separate hemicellulose and cellulose from wood pulp. In this study, hemicellulose-rich birch kraft pulp was selectively separated, with high levels of purity, into pure cellulose and hemicellulose fractions by using mixtures of cosolvents (water, ethanol, or acetone) and the cellulose-dissolving ionic liquid 1-ethyl-3-methylimidazolium acetate (EMIM OAc) with reaction conditions of 60 °C for 3 h under stirring. This process was used to generate dissolving pulp that met the manufacture of revived cellulose products and cellulose derivatives because of its extraordinary cellulose content, which was assumed to be more than 90%, as well as the product's high brightness and uniform molecular-weight distribution.
Alkaline extraction is well studied and is known as a strong and efficient method for hemicellulose extraction. Indeed, alkaline treatment can cleave the ester linkage between ferulic acid of lignin and the glucan and arabinan residues of hemicellulose in the cell wall, thus releasing oligo- and polymeric hemicellulose rather than sugars, as obtained from dilute-acid extraction or hot-water extraction. The extraction of hemicelluloses from various biomasses is expected to be closely related to the amount of LCCs. The LCCs entail covalent linkages between lignin and carbohydrates, mainly hemicelluloses. The major types of LCCs include phenyl glycoside, benzyl ether, and benzyl ester types of linkages. Different biomasses have different frequencies of LCC, for example, the amounts of ether and ester LCC linkages in pine and aspen cellulolytic enzyme lignin (CEL) have been reported to be about 2.2–2.5 and 0.3–0.6 per 100 monomeric lignin units, respectively.
The decision on which method to use for hemicellulose extraction is highly dependent on the final application of the recovered hemicellulose. For example, if the extracted hemicellulose is targeted for the production of bio-ethanol, then the monomeric form of sugars is required, and thus, it may be preferred to use dilute acid or hot water. Some other applications require a high-molecular-weight polymer (blended plastics, hydrogels, and others). For those applications, it may be extremely important to use the alkaline method for the isolation of hemicellulose from the biomass.
2.3.2.1 Structural Characterization of Hemicellulose
Great interest in converting lignocellulosic biomass into valuable, green fuels and chemicals has challenged researchers to develop methods for determining the structure, accurate chemical composition, quantity, and potential uses of hemicellulose in the lignocellulosic biomass [26]. Several analytical methods have been used to characterize hemicellulose: high-performance liquid chromatography (HPLC), gas chromatography (GC), size-exclusion chromatography (SEC), gas chromatography mass spectrometry (GC-MS), high-performance anion-exchange pulse amperometric detection (HPAE-PAD) chromatography, 1D and 2D NMR spectrometry, and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS.
2.3.3 Lignin Reactivity
Many new techniques were developed to isolate lignin formulations to study [29–33]. Generally, three approaches were taken toward investigating lignin structure: degradation reactions, biosynthetic work, and spectroscopy studies. In the most common approach, lignin formulations were subjected to various degradation reactions yielding identifiable products that gave useful structural information. Many new analytical techniques were developed during these times that were highly useful for the lignin chemists. In the final approach, model compounds were synthesized from postulated lignin precursors to produce new products for study. This was a time in which much work was applied to developing new methods to isolate lignin. In 1936, Bailey at the University of Washington showed by microdissection that the middle lamella was composed mainly (72%) of lignin rather than pectin. In 1935, Van Beckum and Ritter at the US Forest Products Laboratory (USFPL) removed lignin from plant tissues with hypochlorite followed by NaOH. The material that remained termed holocellulose consisted of the total carbohydrate mass present in the plant tissues. In 1939, Brauns reported that neutral solvent extraction of woody tissue and subsequent purification created a few percent of what he named native lignin or Braun's lignin. Presently, many investigators view this material as a mixture of lower molecular weight lignins and/or lignans. This work reflected the continuing search by some classical organic chemists for a lignin that could be extracted simply by use of solvents without chemical reaction. In 1947, Richie and Purves at McGill University oxidized wood at pH 4 with aqueous 5% sodium periodate. The periodate lignin preparation thus obtained was 86–96% Klason lignin, insoluble in organic solvents even at boiling temperature but completely soluble under conditions of sulfite pulping. A periodate lignin from spruce closely duplicated the behavior of spruce lignin in situ toward many degradative procedures.
2.4 Composite as a Unique Application for Renewable Materials
Plastic composite processors worldwide are becoming increasingly aware that environmentally sustainable products have become mainstream, and it can no longer be considered only a niche market that can be ignored [34–37]. Moreover, in the light of the recent Paris climate agreement in 2015, development of environmentally sustainable new technologies and materials is of growing importance; state and local governments are mandating it; and now, even the largest retailers are building it into the foundation of their marketing strategies. The development of renewable/sustainable materials is perceived by the industry as a hedge against the prospect that traditional plastics will be much more costly in the future due to dramatically higher petroleum prices. The sustainability movement is further seen as a positive development for plastic processors since it will drive further innovation and a new generation of materials with properties more comparable to commodity plastics. For instance, packaging and containers constitute a nearly $500 billion global market. Plastic container sales alone account for $130 billion worldwide.
The combination of lignocellulose and starch would mean a further step ahead in the utilization of bio-based materials for challenging applications such as Styrofoam-like foams, plastics, and packaging made from petroleum resources.
After an extensive literature review on this topic, we have concluded that there is a great need for systematic and accurate mapping of product structure characteristics.
2.4.1 Rationale and Significance
Plastics industries manufacture has a wide variety of materials, which plastics are produced or derived from major non renewable energy sources such as crude oil, natural gas and coal. Nearly 6% of the world's crude oil production is used for making approximately 245 million metric tons of plastics globally on an annual basis. These are used to meet both the requirements of cheap mass production and of highly specific applications. The worldwide economy dependence on petroleum-based plastics is not sustainable, based on the extremely volatile oil and energy situation, coupled with major changes in supply and demand patterns. The fluctuation in costs is a challenge but in addition, the limited feedstock availability is tightening and impacting supply and demand worldwide, and putting the industry under tremendous pressure.
Moreover, plastics now make up a significant part of a typical municipal solid waste (MSW) stream, and represent the fastest growing component. A huge 44 billion pounds of plastics enter United States' MSW stream each year, equivalent to ½ pound per day per person. In the United States, plastics on average account for 10% by weight of MSW, more than metals (8%) and glass (6%). The cost of waste management is now a matter of great public concern. There is more carbohydrate on earth than all other organic material combined. Polysaccharides are the most abundant type of carbohydrate and make up approximately 75% of all organic matter. The use of biodegradable, starch- and wood-based products as proposed here would be a significant improvement to the economy, environment, and society. Furthermore, starch-cellulosic fibers are viable feedstocks for enzymatic conversion to ethanol or are high heat value material suitable for combustion, alternatives to landfilling.
Many industry announcements regarding new and innovative plastic products occur on an ever more frequent basis. Coca-Cola recently announced that they will begin utilizing polyethylene terephthalate (PET) bottles containing 30% renewable content from sugarcane-derived ethylene glycol. They also announced plans to convert all their plastic packaging to the new material by 2020. Heinz will use the same material to make 120 million bottles for their ketchup products this year. PepsiCo claims to have developed the world's first totally bio-based PET bottle. It is made from biomass including switchgrass, pine bark, and corn husks. Pilot-scale production began in 2012. Other interesting new materials entering the market include a new family of resins (Panacea) containing 10–40% finely ground soy-based protein and an injection mold-grade cellulose-based resin. The cellulose-based resin is being used to make the first biodegradable tubes for toothpaste.
Much of the focus on renewable and sustainable plastics involves the use of starch either as a feedstock or as a component for industrial products. Industrial products in the United States that utilize starch have grown from 13 million metric tons (MMT) in 1975 to over 160 MMT today. Starch is inexpensive, widely available, and one of the most abundant biomass products in nature. It is produced in many different plant organs including roots, leaves, seeds, and stems. However, the hydrophilic nature of starch and its tendency to embrittle with age do not make it suitable as a replacement for plastics – hence, the importance of adding lignocellulose to the starch matrix system.
2.4.2 Starch-Based Materials
The development and production of biodegradable starch-based materials have been spurred by oil shortages and the growing interest in easing the environmental burden of petrochemically derived polymers. Starch is one of the most studied and promising raw materials for the production of biodegradable plastics, which is a natural renewable carbohydrate polymer obtained from a great variety of crops (Figure 2.10). Starch is low cost material in comparison to most synthetic plastics and is readily available. Starch has been investigated widely for the potential manufacture of products such as water-soluble pouches for detergents and insecticides, flushable liners and bags, and medical delivery systems and devices. Native starch commonly exists in a granular structure, which can be processed into thermoplastic starch (TPS) under the action of high temperature and shear by melt extrusion. Unfortunately, the properties of TPSs are not satisfactory for some applications such as packaging materials and composites engineering products.
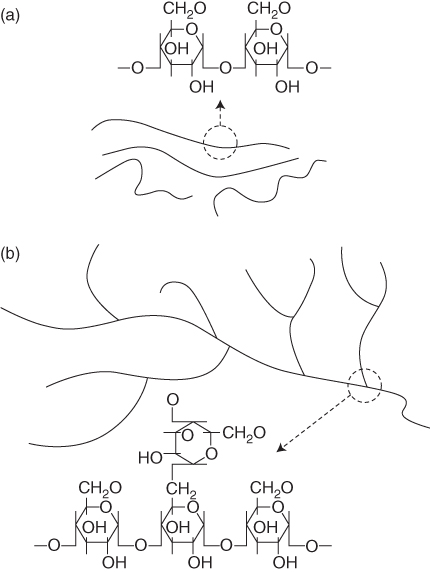
Figure 2.10 Chemical structures and physical schematic representation of (a) amylose starch and (b) amylopectin starch.
One of the unique characteristics of starch-based polymers is their processing properties, which are much more complex than conventional polymers. The processing of starch-based polymers involves multiple reactions, for example, water diffusion, granule expansion, gelatinization, decomposition, melting, and crystallization (Figure 2.11). Among the various phase transitions, gelatinization is particularly important because it is closely related to others, and it is the basis for the conversion of starch to a thermoplastic. Furthermore, the decomposition temperature of starch is higher than its melting temperature before gelatinization. Without physical force (shear stress), the process of gelatinization depends mainly on water content and temperature conditions. A previous study has shown that shear stress can result in the fragmentation of starch granules during extrusion. Indeed, both the mechanical and thermal energies are transferred to starch dough during extrusion in molten medium. The main objectives of most starch-processing techniques are melting and mixing, which are adjusted to minimize chain degradation.
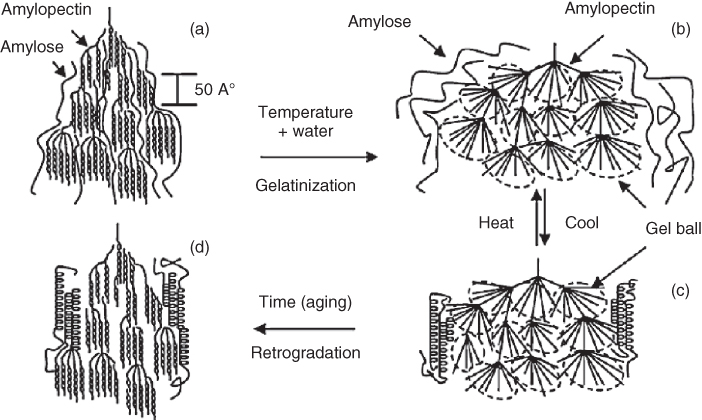
Figure 2.11 Schematic representation of the phase transitions of starch during thermal processing and aging.
Starch use in papermaking dates back to the invention of paper 2000 years ago, when it was applied to obtain a stronger and smoother writing surface. Starch contributes to paper manufacturing because it serves as a binding agent that can enhance the mechanical properties of paper and improve paper manufacturing by increasing paper pulp retention on the paper machine. Starch was also chemically modified into cationic forms to further improve interconnections between fibers, increasing thus the paper strength. Non-covalent binding of starch and cellulosic fiber in biocomposites were created using a polymer suspension either by, for example, drying or hot-pressing.
2.4.3 Starch-Based Plastics
Plastic ranks as the second most used packaging material in the United States. In contrast to paper, only 7% of plastic generated as waste is recycled. This explains why more plastics ultimately end up in landfills than paper or any other packaging material. China, one of the world's largest plastic producers, noted that the largest source of its marine pollution was from discharging wastewater to sea. In Europe alone, an estimated 2–3 million tons of plastics is used each year in agricultural applications. Polyethylene films are used extensively to increase yields, extend growing seasons, reduce the usage of pesticides and herbicides, and help conserve water. Films made of starch blends were some of the first films containing renewable content tested as agricultural mulch.
Packaging and containers make up the largest sector (29.5%) of plastic waste in MSW. Plastic packaging has become an integral part of the global marketplace. Today, packaging design has begun integrating sustainability as never before, in part, because sustainability itself has become a marketing angle. Retailers are now realizing that customers respond positively to products marketed in more sustainable or green packaging.
The interest in utilizing starch as a replacement for plastics started in the 1970s and intensified in the 1980s right along with the dramatic growth in the use of plastics worldwide and the concerns about the effects of plastics on the environment (Figure 2.12)
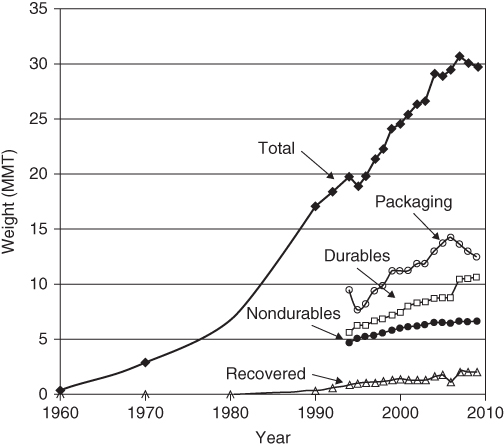
Figure 2.12 Growth of the plastics worldwide.
But starch by itself is a poor substitute for petroleum plastics due to its moisture sensitivity and inferior mechanical properties. However, numerous strategies have been tested resulting in commercialized technologies for incorporating starch in plastics. A survey of the more common approaches has been included in this section as follows:
2.4.3.1 Novamont
One of the early innovators in developing TPS blends for commercial production is the Italian company, Novamont. Founded in 1989, Novamont has developed four different classes of materials all based on blends of TPS and synthetic polymers. The film-grade product is based on blends of TPS and polycaprolactone. This grade degrades in a composting environment in about 1 month. The injection molding grade is a blend of TPS and cellulose derivatives. It degrades in about 4 months and is a rigid material that may replace polystyrene (PS). A foaming grade is also available that contains more than 85% TPS. The foam product is used as a replacement for PS foam for loose-fill packaging. The current production at Novamont has exceeded 60,000 MT.
Novamont continues to develop their technology. In 1997, Novamont purchased the Warner-Lambert patents for TPS/polymer blends. They later acquired (2004) the technology from Eastman Chemical to produce the polyester (Estar Bio). This polyester is now being produced using oil from a non-food crop and is being blended with TPS. The bio-based polyester has enabled Novamont to increase the renewable polymer content of their resins to about 50%. They also intend to commercialize “nanostarch” particles for use in film grades of Mater-Bi. The nanostarch allows for higher renewable content in films while maintaining good strength and clarity.
2.4.3.2 Cereplast
Cereplast is located in El Segundo, CA. It was founded in 1996 and began marketing the Novamont product in North America. In 2000, Cereplast began developing their own TPS technology and started marketing Cereplast products in 2001. Cereplast recently opened a 36,000 MT facility in Indiana.
2.4.3.3 Ecobras
BASF, the world's leading chemical company, has entered the starch blend market. BASF produces the biodegradable polyester, Ecoflex (polybutyrate adipate terephthalate, PBAT). In 2007, BASF aligned itself with Corn Products International and began selling a starch/PBAT blend for the Latin American market. The blend contains about 50% corn starch and is designed for making films although it can be injection molded as well.
2.4.3.4 Biotec
Biotec GmbH in Germany has a capacity of about 11,000 MT. Its products include pure TPS and various blends of starch and copolyesters. It has six commercial formulations for injection molding, rigid and flexible extrusion, and foams. Many of the finished products include biodegradable carrier bags, bin liners, and refuse bags. Most sales are in Europe.
This results in a large number of uses and applications for compostable packaging, short-term consumable articles and special products. Examples from the various production fields are as follows:
| Blown films | Flat films | Injection molding |
| Sack, bags Trash bags Mulsh foils Hygiene products Diaper films Air bubble films Multiple layer films Protective clothing Gloves Double rib bags Labels Barrier ribbons |
Trays for food and non-food articles Flower pots Freezer products and packaging Cups Pharmaceutical packaging |
Disposable cutlery Cans, containers Performed pieces CD trays Gold Trees Cemetery articles Golf tees Toys |
2.4.3.5 Plantic
Plantic Technologies was incorporated in Victoria, Australia, but is located today in Melbourne, Australia. Plantic acquired technology in 2001 for making high-amylose corn starch TPS plastic sheets and trays. They found that TPS sheets could be thermoformed into trays and used to package fatty foods or products with a water activity of 35–70%. The business started making trays for a candy company. Plantic entered the global market in 2004 and recently developed multilayered polymer films with a starch film core that has improved moisture resistance, gas barrier properties, and physical properties. Plantic has announced joint ventures with several companies in recent years including DuPont for making cosmetic and food packaging and Bemis Co., Inc., Neenah, WI, to develop blown film for dry-goods packaging.
2.4.3.6 Biolice
Biolice was developed by Limagrain, a leader in the European agricultural sector. Biolice is a TPS made from cereal flour that is blended with biodegradable polyesters. Biolice is a rigid packaging material that can be thermoformed into single-use items like drink trays and cups. Films can also be made from the resin for agricultural mulch and carrier bags. The product is being marketed in France and is completely biodegradable.
2.4.3.7 KTM Industries
KTM Industries is a company located in Lansing, MI; now it is called Green Cell Foam. The company uses an extrusion process similar to that used to make PS foam sheets. The process involves extruding TPS through an annular die to form a foam tube. The tube is sliced and opened flat to form sheets of starch foam that can then be used for packaging operations. The foam sheets can be cut and glued to form padding for specific packaging applications. The company also makes colored loose-fill products for children craft projects. Other companies using TPS for making starch-based loose-fill products include StarchTech, Inc. and National Starch with its Eco-Foam product.
2.4.3.8 Cerestech, Inc
Cerestech, Inc. was incorporated in 2001 in Montreal, Canada. The company produces blends of TPS and commodity thermoplastics in a one-step extrusion process. The process involves preparing starch/glycerol/water blends of approximately 48%, 32%, and 20%, respectively. The starch preparation is fed into a twin-screw extruder to form a TPS melt. A second single-screw extruder is attached to the twin-screw extruder in a perpendicular position to allow a thermoplastic polyolefin such as high-density polyethylene to be melted and injected directly into the TPS melt. The melt blend is compounded further using high shear to form a blend of the two incompatible resins. Although the polyolefin and TPS form an incompatible blend, the domains of the respective polymers range from several micrometers to less than 1 µm. Blends containing up to 50% starch have been produced with excellent mechanical properties and moisture resistance. The carbon footprint of these blends is significantly reduced compared to the neat polymer due to the starch content. A family of blends (Cereloy) based on starch and various polyolefin resins is being developed. Cerestech has granted a worldwide license to Teknor Apex to produce the blends. The blends are being sold at a similar or lower price than the neat polymer.
2.4.3.9 Teknor Apex
Teknor Apex is also marketing blends of starch and recycled PP and PE to further improve the environmental profile of its products.
We can conclude from the examples cited here that starch as biomaterials is poised to establish an even stronger role in the manufacture of sustainable plastics and other bioproducts largely because it is abundant, renewable, and inexpensive. The cost and availability of starch may improve even further in the future if lignocellulose materials get involved in the process. The proposal herein will boost the strategies for improving the properties of starch-based plastics such as blending starch with other polymers as cellulose materials or others, using starch in composite materials. The prospects for biomaterials in the packaging sector continue to become brighter as the market for sustainable plastics drives further innovation and development in our era.
2.5 Question for Further Consideration
- 1. What are the challenges for sustainable bio-fuel production?
- 2. Could the lignocellulose replace the potential applications of starch new business opportunities?
- 3. What could be the right technology to transform the biomaterials to potential products?
References
- 1 I. Siro and D. Plackett. Microfibrillated cellulose and new nanocomposite materials: a review. Cellulose, 17, 3, 459–494 (2010).
- 2 J. Blackwell, P. D. Vasko, and J. L. Koenig. Infrared and Raman Spectra of the cellulose from the cell wall of Valonia ventricosa. Journal of Applied Physics, 41, 11, 4375–4379 (1970).
- 3 A. French. Idealized powder diffraction patterns for cellulose polymorphs. Cellulose, 21, 2, 885–896 (2014).
- 4 A. Esker, U. Becker, S. Jamin, S. Beppu, S. Renneckar, and W. Glasser. Self-assembly behavior of some co- and heteropolysaccharides related to hemicelluloses. In: Hemicelluloses: Science and Technology, Chap 14, pp 198–219, ACS Symposium Series, 864 (2003).
- 5 D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz, and J. A. Dumesic. Integrated conversion of hemicellulose and cellulose form lignocellulosic biomass. Energy & Environmental Science, 6, 76–80 (2013).
- 6 M. Lawoko, G. Henriksson, and G. Gellerstedt. Structural differences between the lignin–carbohydrate complexes present in wood and in chemical pulps. Biomacromolecules, 6, 6, 3467–3473 (2005).
- 7 A. Ebringerova and T. Heinze. Naturally occurring xylans structures, isolation procedures and properties. Macromolecular Rapid Communication, 21, 542–556 (2000).
- 8 A. Duval and M. Lawoko. A review on lignin-based polymeric, micro- and nano-structured materials. Reactive and Functional Polymers, 85, 78–96 (2014).
- 9 M. Norgren and H. Edlund. Lignin: Recent advances and emerging applications. Current Opinion in Colloid and Interface Science, 19, 5, 409–416 (2014).
- 10 S. Laurichesse and L. Averous. Chemical modification of lignins: Towards biobased polymers. Progress in Polymer Science, 39, 7, 1266–1290 (2014).
- 11 H. P. S. A. Khalil, M. S. Alwani, and A. K. M. Omar. Chemical composition, anatomy, lignin distribution, and cell wall structure of Malaysian plant waste fiber. BioResources, 1, 2, 220–232 (2006).
- 12 F. S. Chakar and A. J. Ragauskas. Review of current and future softwood kraft lignin process chemistry. Industrial Crops and Products, 20, 2, 131–141 (2004).
- 13 E. Quintana, C. Valls, A. G. Barneto, T. Vidal, J. Ariza, and M. B. Roncero. Studying the effects of laccase treatment in a softwood dissolving pulp: Cellulose reactivity and crystallinity. Carbohydrate Polymers, 119, 2015, 53–61 (2015).
- 14 D. Klemm, B. Heublein, and A. Bohn. Cellulose: Fascinating biopolymer and sustainable raw material. Angewandte Chemie, 44, 22, 3358–3393 (2005).
- 15 M. M. Kabir, H. Wang, K. T. Lau, and F. Cardona. Chemical treatments on plant-based natural fiber reinforced polymer composites: An overview. Composites Part B: Engineering, 43, 7, 2883–2892 (2012).
- 16 C. Tian, L. Zheng, Q. Miao, C. Cao, and Y. Ni. Improving the reactivity of kraft-based dissolving pulp for viscose rayon production by mechanical treatments. Cellulose, 21, 5, 3647–3654 (2014).
- 17 F. M. Gama, J. A. Teixeira, and M. Mota. Cellulose morphology and enzymatic reactivity: A modified solute exclusion technique. Biotechnology and Bioengineering, 43, 5, 381–387 (1994).
- 18 A.-C. Engstrom, M. Ek, and G. Henriksson. Improved accessibility and reactivity of dissolving pulp for the viscose process: Pretreatment with monocomponent endoglucanase. Biomacromolecules, 7, 6, 2027–2031 (2006).
- 19 E. S. Welf, R. A. Venditti, M. A. Hubbe, and J. Pawlak. The effects of heating without water removal and drying on the swelling as measured by water retention value and degradation as measured by intrinsic viscosity of cellulose papermaking fibers. Progress in Paper Recycling, 14, 3, 1–9 (2005).
- 20 U. Weise, T. Maloney, and H. Paulapuro. Quantification of water in different states of interaction with wood pulp fibers. Cellulose, 3, 1, 189–202 (1996).
- 21 H. Sixta, M. Iakovlev, L. Testova, A. Roselli, M. Hummel, M. Borrega, A. van-Heiningen, C. Froschauer, and H. Schottenberger. Novel concepts of dissolving pulp production. Cellulose, 20, 4, 1547–1561 (2013).
- 22 H. Wang, B. Pang, K. Wu, F. Kong, B. Li, and X. Mu. Two stages of treatments for upgrading bleached softwood paper grade pulp to dissolving pulp for viscose production. Biochemical Engineering Journal, 82, 183–187 (2014).
- 23 L. Testova, M. Borrega, L. K. Tolonen, P. A. Penttila, R. Serimaa, P. T. Larsson, and H. Sixta. Dissolving-grade birch pulps produced under various prehydrolysis intensities: quality, structure and applications. Cellulose, 21, 3, 2007–2021 (2014).
- 24 D. Ibarra, V. Kopcke, P. T. Larsson, A.-S. Jaaskelainen, and M. Ek. Combination of alkaline and enzymatic treatments as a process for upgrading sisal paper-grade pulp to dissolving-grade pulp. Bioresource Technology, 101, 19, 7416–7423 (2010).
- 25 J Shen, Z. Song, X. Qian, and W. Liu. Modification of papermaking grade fillers: A brief review. BioResources, 4, 3, 1190–1209 (2009).
- 26 W. Farhat, R. Venditti, M. Hubbe, M. Taha, F. Becquart, and A. Ayoub. A review of water resistant hemicellulose based materials: processing and applications. ChemSusChem, 10, 2, 305–323 (2017).
- 27 M. Marinova, E. Mateos-Espejel, N. Jemaa, and J. Paris. Addressing the increased energy demand of a kraft mill biorefinery: The hemicellulose extraction case. Chemical Engineering Research and Design, 87, 9, 1269–1275 (2009).
- 28 H. Y. Celebioglu, D. Cekmecelioglu, M. Dervisoglu, and T. Kahyaoglu. Effect of extraction conditions on hemicellulose yields and optimization for industrial processes. International Journal of Food Science and Technology, 47, 12, 2597–2605 (2012).
- 29 A. J. Bailey, Lignin in Douglas fir: composition of the middle lamella. Industrial & Engineering Chemistry Analytical Edition, 8, 1, 52–55 (1936).
- 30 W. G. Van Beckum and G. J. Titter. Paper Trade Journal, 105, 18, 127 (1937).
- 31 C. Laaksometsa, E. Axelsson, T. Berntsson, and A. Lundstrom. Energy savings combined with lignin extraction for production increase: case study at a eucalyptus mill in Portugal. Clean Technologies and Environmental Policy, 11, 77–82 (2009)
- 32 X.-F. Sun, R. Cang, P. Fowler, and M. S. Baird. Extraction and characterization of original lignin and hemicelluloses from wheat straw. Journal of Agricultural and Food Chemistry, 53, 4, 860–870 (2005)
- 33 M. Schwanninger and B. Hinterstoisser. Klason lignin: Modifications to improve the precision of the standardized determination. Holzforschung, 56, 2, 161–166 (2005).
- 34 A. Dufresne, D. Dupeyre, and M. R. Vignon. Cellulose microfibrils from potato tuber cells: processing and characterization of starch–cellulose microfibril composites. Journal of Applied Polymer Science, 76, 14, 2080–2092 (2000).
- 35 L. Averous, C. Fringant, and L. Moro. Plasticized starch–cellulose interactions in polysaccharide composites. Polymer, 42, 15, 6565–6572 (2001).
- 36 A. A. S. Curvelo, A. J. F. de Carvalho, and J. A. M. Agnelli. Thermoplastics starch-cellulosic fibers composites: preliminary results. Carbohydrate Polymers, 45, 2, 183–188 (2001).
- 37 X. Ma, J. Yu, and J. F. Kennedy. Studies on the properties of natural fibers-reinforced thermoplastic starch composites. Carbohydrate Polymers, 62, 1, 19–24 (2005).