
CHAPTER 3
Compliance Concerns for Medical Equipment
1. Introduction
Medical equipment is highly regulated by all countries, and held to a higher level of safety than nearly all other equipment on the market. The main reasons for this are that medical equipment may be used on patients who are not able to respond to hazardous conditions or pain; an actual electrical connection between the equipment and patient may exist; patient treatment may be based on the output of the medical device; and certain types of medical equipment function as life support, the failure of which could result in the death of the patient. Understanding these requirements early in the design process will result in lower product development cost, faster certification turnaround, and increased product safety.
2. National and International Requirements
2.1. U.S. Requirements
In the United States, the Center for Devices and Radiological Health (CDRH), a branch of the Food and Drug Administration (FDA), separates medical devices into three categories (Class I, II, or III). This separation depends on the degree of regulation necessary to provide a reasonable assurance of safety and effectiveness. The FDA Federal Food, Drug, and Cosmetic Act requires that all medical devices be “safe and effective,” and recognizes safety standards as a means to support a declaration of conformity. You can find a list of the FDA-recognized consensus standards at www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm.
Class I devices are subject to the following requirements: premarket notification, registration and listing, prohibitions against adulteration and misbranding, and rules for good manufacturing practices. Class II devices additionally need to show that they meet recognized consensus standards. Class III devices have the same requirements of Class II devices, but require premarket approval from the FDA.
The collection of documents that are submitted to the FDA for Class I or Class II devices is called an FDA 510(k). This shows that the above requirements are met and that the medical device is substantially equivalent to a medical device that was either in commercial distribution before May 28, 1976 or has been reclassified as Class I or II. The FDA 510(k) submission is examined by the FDA or an accredited third-party reviewer to determine whether the device is substantially equivalent to the specified predicate device and whether it meets the specified recognized consensus standards. If the device is found to be substantially equivalent, the FDA accepts the claim of substantial equivalency; it can then be legally marketed and sold in the United States.
If the device is not substantially equivalent to a predicate device, due to new technology or differences in intended use, the medical device is considered to be Class III. For a Class III device, the company must present detailed information, such as clinical trial data, statistical data, and compliance evaluation and testing results to the FDA to show that the device is safe and effective. Only the FDA may review Class III submissions; if the FDA finds the information and data adequate, it will grant premarket approval for the device, allowing it to be legally marketed and sold in the United States.
In addition to the legal requirements of the FDA, many “authorities having jurisdiction” (AHJ) and purchasers of medical electrical equipment in the United States require a safety certification mark on the equipment. A safety certification mark is issued by a nationally recognized testing laboratory (NRTL), such as Underwriters Laboratories Inc. (UL), Canadian Standards Administration (CSA), Technical Inspection Association (in English) (TUV), or European Test Laboratory (ETL). The NRTL program is run by the Occupational Safety and Health Administration (OSHA); it recognizes private sector organizations that meet the necessary qualifications specified in the regulations for the program. The NRTL determines that specific products meet consensus-based standards of safety to provide the assurance, required by OSHA, that these products are safe for use in the U.S. workplace.
Underwriters Laboratories is the major product-safety certification organization in North America. Manufacturers of medical equipment submit product samples and information to UL for evaluation to the applicable safety standard(s). Products that meet these requirements are authorized to apply the UL Classification Mark. The applicable standards are UL 60601-1 and International Electrotechnical Commission (IEC) 60601-1 collateral and particular standards, which are discussed later in this chapter.
2.2. European Requirements
All but low-risk, non-measuring, non-sterile medical devices placed on the market in Europe must bear the CE Mark with a notified body’s identification number. This mark specifies that the medical device complies with the requirements of the Medical Device Directive (93/42/EEC). A notified body is a third party designated by European authorities to assess products to directives. The Medical Device Directive is essentially European “law” for the requirements of medical devices. The notified body reviews the evidence of compliance to the Medical Device Directive requirements for safety, performance, suitability for intended use, and risk to patients and operators. Manufacturers can choose from several conformity assessment routes, most involving a quality assurance assessment of the manufacturer’s facilities. Low-risk, non-measuring, non-sterile medical devices also require a CE Mark, but are allowed to “self-declare” compliance to the Medical Device Directive without the involvement of a notified body.
2.3. Other Countries
Health Canada reviews medical devices to assess their safety, effectiveness, and quality before being authorized for sale in Canada. The system is similar to the European requirements, but the classification system is different.
Other countries have similar requirements to the United States and Europe, and have adopted the same family of medical consensus standards, with the addition of their national deviations. Since the detailed requirements specific to each country have been changing in recent years, it is not practical to address all of them in this chapter. The key is demonstration of compliance to the family of recognized standards.
3. Medical Device Certification
Product certification agencies, such as UL, CSA, TUV, and ETL use recognized consensus standards to evaluate various types of products. These safety standards are documents that define the minimum construction and performance requirements. The base standard that covers medical devices, IEC 60601-1, is accepted by many countries.
The current (second) edition of IEC 60601-1 has two amendments. These amendments contain additions and corrections to the base standard. The standard also has collateral (horizontal) standards, numbered IEC 60601-1-xx, and particular (vertical) standards, numbered IEC 60601-2-xx. The collateral standards include requirements for specific technologies or hazards; they apply to all applicable equipment. Examples are medical systems (IEC 60601-1-1), EMC or electromagnetic compatibility (IEC 60601-1-2), radiation protection in diagnostic x-ray equipment (IEC 60601-1-3), and software (IEC 60601-1-4). The particular standards apply to specific equipment types, such as high-frequency surgical equipment (IEC 60601-2-2), infusion pumps (IEC 60601-2-24), and hospital beds (IEC 60601-2-38).
The UL 60601-1safety standard, published in April 2003, contains the full text of IEC 60601-1 with amendments, and adds U.S. deviations. The U.S. deviations contain national requirements, such as those for the mains circuits, component requirements, lower leakage current limits, enclosure flame ratings, and production line testing. Since these deviations do not conflict with the base standard, equipment that complies with UL 60601-1 also complies with IEC 60601-1. The UL 60601-1 standard is an editorial change to the older UL 2601-1 standard; it does not change any requirements.

Figure 3.1: Examples of standards for compliance of medical devices.
Figure 3.1 illustrates the organization of the collateral and particular IEC 60601 standards. The base standard, national deviations, and applicable collateral and particular standards are used together to evaluate the medical electrical equipment. All the applicable standards use the same clause numbering, which allows cross-referencing of the requirements between these standards.
4. Philosophy of the Standards
The underlying philosophy of the IEC 60601-1 harmonized standards is that equipment must be safe in normal condition (NC) and single fault condition (SFC). To understand the electrical safety requirements, we need to first define a few terms:
• An applied part is any piece(s) of the equipment that can intentionally or unintentionally be brought into contact with the patient.
• Creepage is spacing along a surface (as an ant crawls).
• Clearance is spacing through the air (as a bug flies).
• LOP is a level of protection (level 2 is required by standard).
• Basic insulation (BI) is a spacing or a physical insulation barrier providing 1 LOP.
• Supplemental insulation (SI) is spacing or a physical insulation barrier providing 1 LOP.
• Double insulation (DI) is BI plus SI and provides 2 LOPs.
• Reinforced insulation (RI) is a single spacing or physical barrier that provides 2 LOPs.
• Protective impedance is a component, such as a resistor, that provides 1 LOP.
• Protective earth (PE) is a well-grounded part that provides 1 LOP.
• Class I equipment is defined as using PE as 1 LOP.
• Class II equipment (known as double insulated) is defined as not using PE, 1 LOP.
For electrical safety, the standard requires 2 LOPs against excessive unintentional current, defined as leakage current, passing through the patient or operator. An insulation diagram (also known as an isolation diagram) graphically depicts the 2 LOPs in a device at each part of the circuit.
Figure 3.2 shows the 2 LOPs between the live part (mains) and the patient (1A and 2A), and between the live part and the enclosure (1B and 2B). In the cases of 1A and 2A, the levels of protection are BI and SI, and for 1B and 2B, they are BI and PE.
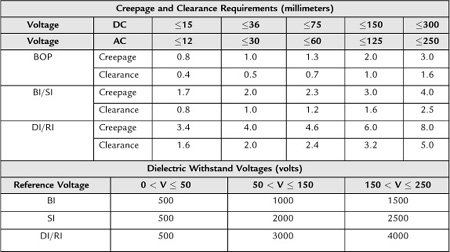
The minimum spacing requirements and dielectric (hipot) requirements for these barriers are specified in the base standard. Table 3.1 shows these requirements from the base standard.
If the insulation does not meet both the dielectric and the spacing requirements, it cannot be considered as a level of protection and can be shorted as a normal condition. Note that BI and SI spacing requirements are the same; however, the SI dielectric values are greater than the BI values above 50 V. To be considered protectively earthed, the grounding path of the equipment must pass 15 amps or 1.5 x-rated current for 5 seconds from the protectively earthed part to the earth connection, 0.1 ohms resistance for equipment with a detachable power supply cord or 0.2 ohms for equipment with a non-detachable power-supply cord.

Figure 3.2: Example of protective layers against leakage current through human patients.
Table 3.1: Minimum spacing requirements and dielectric (hipot) requirements for these barriers of the IEC 60601-1 harmonized standards
To demonstrate that medical equipment is safe in normal and single-fault conditions, the following must be addressed when designing and evaluating the medical equipment. These conditions are specified throughout the standard.
Normal conditions (likely to occur)
• Reverse polarity of supply mains
• Failure of insulation less than basic (operational)
Single-fault conditions (could occur)
• Interruption of protective earth
• Interruption of one supply conductor
• Mains voltage on floating (F-type) applied part(s)
• Mains voltage on communication ports
• Failure of electrical components, one at a time
• Failure of mechanical parts, one at a time
• Failure of temperature limiting devices, one at a time
• Shorting of basic or supplemental insulation
• Overload of mains supply transformers
• Interruption and short circuit of motor capacitors
• Locking of moving parts
• Impairment of cooling (fans, vents blocked)
Not evaluated (unlikely to occur)
• Total breakdown of double or reinforced insulation
• Loss of protective earth on permanently installed equipment
• More than one single-fault condition at a time
• Failure of a UL-recognized optocoupler barrier
• Failure of a UL-recognized Y1 capacitor, acting as a barrier
5. Evaluation Process
5.1. Preliminary Evaluation
The process of evaluating medical equipment for compliance to the requirements of standards includes not only the equipment itself, but the user’s manual, markings, software (if it mitigates a hazard), biocompatibility of applied parts, and electromagnetic compatibility (EMC). Before submitting equipment for evaluation, the following information should be developed. This is typically done as a preliminary evaluation.
1. Ensure that the equipment fits the scope of the standard.
2. Review whether the equipment fits the scope of any collateral or particular standards.
3. List of all equipment functions and accessories that can be used with it.
4. Check if the equipment can connect to any other equipment (computer, printer, etc.).
• Any non-medical equipment connected must have IEC certification (evaluated to the applicable IEC standard) or be part of the medical equipment evaluation and meet the medical standard.
• Computers and other IT equipment are considered to have ground-referenced 50 V in normal condition, mains in single-fault condition on their data ports.
5. Create an insulation diagram (graphic illustration of the required electrical barriers).
6. Determine the equipment classifications from the standard.
7. Document all components that cross electrical barriers, per the insulation diagram.
8. Verify spacings (creepage and clearance), per the insulation diagram.
9. Examine enclosure openings.
• IEC test finger (access to live parts).
• IEC test pin (access to live parts).
• Requirement for a tool to access any live parts.
10. Determine potential mechanical hazards, pinch points, and sharp edges.
11. Determine potential hazards under normal use and foreseeable misuse.
12. Document components that must meet nationally recognized standards.
• Primary circuit components, up to mains transformer(s).
• Lithium batteries (also requires reverse charge protection circuitry).
• CRTs >5 inches.
• Printed wiring boards with >15 W available.
• Wiring/tubing with >15 W available.
• Optical isolators acting as barrier per insulation diagram.
• Conductive coating process.
13. Verify that component ratings meet the equipment’s ratings.
14. List enclosure materials.
• UL 94 flame-rating requirements for polymeric enclosures if there is >15 W available power in the enclosure.
• V-2 minimum for mobile, portable equipment.
• V-0 minimum for fixed or stationary equipment.
15. Verify mains fuse requirements (in the equipment or external power supply).
• Class I: line and neutral required.
• Class II with functional earth: line and neutral required.
• Class II: line only required.
• Permanently installed equipment: line only required.
16. Verify that protective earth conductors are green with yellow stripe.
17. Verify that wires are secured from hazardous movements.
18. Verify equipment-marking requirements (labels on the equipment).
19. Verify requirements for the user manual (accompanying documents).
20. Take photos of the medical equipment, outside and inside of enclosure.
5.2. Testing
Once you have completed the preliminary evaluation, the safety evaluation of the equipment can continue. One or more samples are required, depending on the equipment type and time requirements. Multiple samples of device components may be needed to perform destructive tests, such as transformers, relays, plastic enclosures, and motors. Once the test lab receives the samples, it can perform the required testing, including electrical, mechanical, temperature, and abnormal condition, as well as essential performance requirements (typically specified in the particular standards section). If software is required for mitigating fire, shock, or mechanical hazards, or if it is specified as required in an applicable particular standard, the IEC 60601-1-4 standard is used to evaluate the software design process and implementation. Electromagnetic compatibility (EMC) testing per collateral standard (IEC 60601-1-2), and the review of biocompatibility documentation are conducted. After the evaluation and testing, the critical component table is developed. Any components that may affect compliance with the requirements of the applicable standard(s) or could have an effect on the testing results are considered critical components.
5.3. Compliance Reports
The documentation developed as a result of a safety evaluation depends on the manufacturer’s requirements. The common types of documentation are a letter report, an informative test report, and a certified body report.
A letter report is a list of applicable standards used, and a summary of the evaluation, stating whether or not the device complies with the specified standards. An informative test report completely documents all the requirements in a standard (N/A, pass, or fail), the test record, an insulation diagram, illustrations, equipment markings, and other applicable information. It is the preferred document for technical files; some hospitals and clinics request it before equipment purchases.
A UL report is an informative test report, with the addition of authorizing the manufacturer to apply the UL/C-UL (United States/Canada) Mark to products covered in the report. UL conducts quarterly audits using this report to verify that the equipment bearing the UL/C-UL Mark is the same as the equipment that was tested.
A certified body (CB) report is an informative test report, but contains a certificate from the issuer, who is required to be a member of the IECEE CB scheme. A CB report is used to obtain third-party certification marks (UL, CSA, TUV, VDE, SEMKO (a Swedish testing and certification institute), etc.) in various countries without repeating all equipment testing. The informative test report or CB report is very important to have for the equipment’s technical files, as they serve as an international “passport” for the device.
5.4. Common Noncompliances
When medical equipment is submitted for evaluation, designers and manufacturers are expected to be aware that they would need to meet the applicable standards. While this is not always the case, most medical devices require limited changes to meet the standards.
The first and most critical type of noncompliance typically seen is the choice of a power supply. There are a great number of medical power supplies available that are UL recognized to the UL 60601-1 medical standard. Despite this, many devices are submitted with ITE (information technology equipment) power supplies. These power supplies are evaluated to the IEC 60950 standard for ITE equipment, which is different than the medical standard, and will not provide adequate isolation from the mains (wall outlet voltage).
The next type of noncompliance typically seen in markings and accompanying documents, is less critical, but more common. All of the IEC 60601 standards have very specific requirements for markings and inclusions in the accompanying documents. Since most companies have separate departments that create these documents, they are often not aware of the requirements.
Another common noncompliance is inadequate spacing. When designing medical equipment, it is important to be aware that there are minimum spacing requirements for electrical barriers. Inadequate spacings on circuit boards are another typical noncompliance. An example of this is the required barrier between circuitry and accessible parts of the device. This means that there are required spacings between any circuit board, motor housing, connector pins, and so on, and any accessible part of the device. These required spacings are based on the insulation diagram generated in the preliminary evaluation.
Another common noncompliance is misapplied flammability. For equipment with plastic enclosures, there will also be flammability requirements for any plastic that is acting as part of the enclosure (creating a fire enclosure). The plastic chosen for the enclosure must be at least a UL-recognized V-0 flame rating for fixed equipment, or at least a UL-recognized V-2 flame rating for all other types of equipment.
The last common mistake relates to indicator lights. Red indicator lights can only be used for a warning and yellow for caution. Keep this in mind when selecting LEDs for indicator lights.
These common noncompliances can be easily avoided with knowledge of the applicable standards, and they are the major reasons that preliminary investigations of medical equipment are best done in the early design phase.
6. Conclusion
Medical equipment is highly regulated and is required to show compliance to applicable safety standards. Understanding these requirements, as well as the certification and regulatory process in the design phase of the equipment will result in cost reductions in equipment development, faster certification turnaround, and increased product safety.
Bibliography
European Committee for Electrotechnical Standardization (CENELEC). Medical electrical equipment—Part 1, general requirements for safety, EN 60601-1. Brussels: European Committee for Electrotechnical Standardization (CENELEC); 1991.
International Electrotechnical Commission. IEC standards database. Available at: www.iec.ch/searchpub/cur_fut.htm.
International Electrotechnical Commission. Medical electrical equipment—Part 1, general requirements for safety, IEC 60601-1. Geneva: IEC; 1988.
Medical Equipment Compliance Associates. Evaluation package for IEC/UL/CSA/EN 60601-1, Oak Creek, WI: Medical Equipment Compliance Associates, LLC; 2007. Available at:60601-1.com/documents.htm.
Medical Equipment Compliance Associates. Medical equipment compliance: Design and evaluation to the ‘601’ standards. Seminar. Oak Creek, WI: Medical Equipment Compliance Associates, LLC; 2008.
UK Department of Health. Medical device directive: Council Directive 93/42/EEC, of June 1993. London: Department of Health; 1993.
Underwriters Laboratories Inc. Medical electrical equipment—Part 1, General requirements for safety, UL 60601-1. Northbrook, IL: Underwriters Laboratories Inc.; 2006.
Underwriters Laboratories Inc. UL StandardsInfoNet, catalog of UL standards for safety. Available at: ulstandardsinfonet.ul.com/.
U.S. Department of Health and Human Services. Implementation of third party programs under the FDA Modernization Act of 1997. Final guidance for staff, industry, and third parties. Washington, DC: U.S. Department of Health and Human Services; 2001.