
Chapter 1
Silicon and Silicon Carbide Oxidation 1
This chapter is devoted to the physics of silicon and silicon carbide oxidation. We will find in this chapter, an examination of the main techniques of deposition and growth of thin films. The reader will then discover how the laws governing oxidation are established, in particular those concerning silicon oxidation. This remains nowadays the most widespread method in the manufacture of integrated circuits and of MEMS in the broad sense. This chapter has a double purpose. First, it is written to expose in detail the theoretical principles that are particularly interesting for researchers, and secondly, to review a certain number of experimental results, useful in the practice of any process engineer.
1.1. Introduction
The substantial improvement of the electrical and physical characteristics of the SiO2/Si interface leads to an impressive development of integrated circuits. That was made possible by a better understanding, over time, of the way in which silica is manufactured by deposition or growth on silicon.
Although thermal growth is nowadays one of the most frequently used methods, other techniques have also been developed. In section 1.2, we review the main passivation techniques employed today in industry as well as in research laboratories.
In the semiconductor industry, silica can be manufactured by thermal growth from silicon substrates placed at atmospheric pressure in a flux of water vapor, oxygen or a mix of oxidizing gases. Chemical vapor deposition is also usually used to obtain thick passivation films.
In laboratories, more often aiming at the study of physical phenomena governing the production of layers, the techniques used will be based on the physics of the methods (anodic oxidation in an electrolytic environment or by oxygen plasma) or on specific conditions of thermal oxidation (low or high pressure, etc.).
In section 1.3, we detail the silica properties useful for understanding the growth phenomena. We pay particular attention to showing the properties of atomic transport and the solubility of gases in silica. For a more complete review of the physical characteristics of silica, we refer the reader to the works of Bruckner [BRU 70]. For self-scattering and chemical diffusion in glasses, we recommend the works of Frischat [FRI 75].
In section 1.4, we develop the general equations of transport taking place during oxide growth, and the borderline cases arising from various approximate assumptions that can be made on the flux of mobile species in the case of oxidation. Thus, we also could deduct the expressions of the flux of the transported ion species, in the case of anodic oxidation.
In section 1.5, we show how it is possible to give a satisfactory answer to the two following questions: Which are the mobile species in silicon and in silicon carbide oxidation? How do they move? For this purpose, isotopic labeling techniques are also presented, as well as the experimental results.
In section 1.6, we state the transport equations after the simplifying hypotheses. Two cases are considered, where the transported species are either neutral or charged. When they are charged, two cases still emerge. For the films with a thickness lower than 3 nm, where the tunneling currents are established, the Cabrera and Mott theory predicts the growth laws. For the larger thicknesses, equations become very complex.
The classic Deal and Grove theory, describing silicon oxidation is detailed in section 1.7. It is based on the fact that oxidizing species (O2, H2O) are dissolved in silica in interstitial positions and migrate towards the SiO2/Si interface, to form there the new oxide by a chemical reaction assumed to be of the first order. This theory leads to the linear-parabolic growth kinetics, which we largely resort to in industry. They give a good account of the growth kinetics in the case of oxidation under water vapor, and in the case of oxidation under O2, for thicknesses higher than 20–30 nm.
Whereas the Deal and Grove theory assumes that species diffuse through oxide without interacting with it, Breed and Doremus have proposed a theory developed in section 1.8, assuming the interaction of the diffusing species with the silica network. The concentration profiles of the various species that we can expect from it are compared to the experimental results.
In section 1.9, we examine the growth kinetics of film slower than 30 nm and those of very thin films (< 3 nm) obtained by thermal growth, for which the Deal and Grove theory is no longer satisfactory. Several alternative models are stated to explain the initial regime of fast growth.
Finally, in section 1.10, we show how the important oxidation parameters, kP and kL, are affected by the experimental oxidation conditions such as the pressure, temperature and crystalline direction of the substrate and doping.
Several English journals, already published, explain the classical theories linked to the subjacent oxidation mechanisms. Those show the crucial interest for the industry to control and understand silicon oxidation, which must produce increasingly thin functional oxide layers. This chapter relies on the work of Rigo [RIG 86], while updating them by taking into account all the latest breakthroughs of the field.
1.2. Overview of the various oxidation techniques
1.2.1. General information
There are many techniques used to form silica films on silicon. They are included in two categories, according to whether they rely on an oxidation or deposition process.
1.2.1.1. Oxidation process
In oxidation processes, the film growth depends on the injection phenomena taking place at the two interfaces: oxide/gas and oxide/substrate. Those are a priori rather well defined. In all cases, the oxide film itself always plays a fundamental role. The atomic transport, from one interface to another, of at least one component through oxide is necessary to the growth. The oxide composition is determined by the external environment and by the laws of thermodynamics. Oxidation can be carried out in two ways:
— Thermal oxidation: heating a silicon sample at high temperatures under an oxidizing atmosphere, increases the hopping probability of the atoms. Indeed, the hopping probability being proportional to exp(−W/kBT), raising the temperature thus increases the transport of most mobile species. Moreover, fluxes at the interfaces are then increased. At this stage, it is necessary to stress that mobile species can be either electrically charged or neutral.
— Anodic oxidation: the injection and transport of charged species (anions and cations) are caused by an important electrical field (10 MV.cm − 1), which decreases the potential barrier that the ions must overcome. This type of growth can be carried out at room temperature.
1.2.1.2. Deposition process
In deposition processes, the films’ growth depends on phenomena taking place outside the silica film or its surface. The main part of the film already formed does not play any role in the following growth stages (if the film is maintained at a sufficiently low temperature). The composition of the formed film should vary considerably according to the techniques used: evaporation, sputtering of a material or chemical vapor deposition. We will find a comparison of the physical and chemical properties of these films in relation to their formation process in the summary article by Pliskin [PLI 77].
1.2.2. Most frequently used methods in the semiconductor industry
1.2.2.1. Thermal oxidation
Among all the methods employed in the silicon industry, thermal oxidation is the most widely used. It can be done either with dry oxygen (dry oxidation), or with water vapor (wet oxidation).
The intermediate case, i.e. oxidation in an O2+H2O mixture, will not be discussed in this chapter. The two types of thermal oxidation will be developed in distinct sections. However, they are sometimes used sequentially, during the manufacture of integrated circuits (for example, wet oxidation followed by a stage of dry oxidation), to grow thick field oxides.
1.2.2.1.1. Oxidation under dry oxygen
Typically, dry oxidation is carried out in a device schematized in Figure 1.1b. The furnace is a quartz tube heated by the Joule effect. After being cleaned, the silicon wafers are placed in a socket. The latter is slowly introduced in the center of the furnace, in an oxygen flux, so as to avoid thermal shocks. When we use a pure O2 flux, the oxidation is carried out with a PO2 oxygen pressure equal to the atmospheric pressure.
By using a mixture of O2 and of inert gases, we can oxidize with lower partial oxygen pressures.
To form very thin oxides (a few nm), we will use the rapid processing systems (RTF — rapid thermal furnace) principle. They rely on a rapid heating system (halogen lamps), in order to increase the temperature of a silicon wafer from the room temperature to 1,000°C in a fraction of a second. These furnaces allow very short processing times at high temperature, to obtain thin oxides with good electrical properties. A rapid thermal processing system under controlled atmosphere is schematized in Figure 1.1a.
Figure 1.1a. Schematic diagram of a rapid thermal oxidation furnace under a controlled atmosphere (according to Ganem [GAN 92])
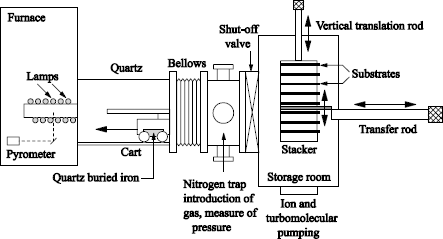
Figure 1.1b. Schematic diagram of a of thermal oxidation furnace (according to Rigo [RIG 86])
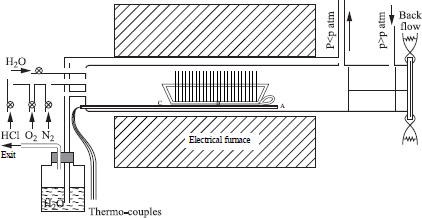
Dry oxidation, although very simple, has some disadvantages:
— the process can become complicated if we need to grow several films of different thicknesses;
— to obtain a Si-SiO2 interface of good quality, the oxide must be annealed (for example, under a N2 flux, after stopping the O2 flux).
A pre-oxidation stage can be necessary (for example, under a flux of chlorinated compounds).
To oxidize under an atmosphere of pure oxygen, we must reduce the contamination sources to a minimum. They were listed by Revesz [REV 79] and are primarily CO2 and H2O. The gas flux always contains traces of contaminants, which are mainly water vapor and hydrocarbons. The latter will react with oxygen in the hot area of the furnace, to form CO2 and H2O. It is thus necessary to burn these hydrocarbons (for example by injecting the gas into a small quartz tube containing quartz wool heated to 900°C) [IRE 78a]. H2O and CO2 are then eliminated by sending gas through zeolites and/or by the means of a low temperature trap.
A third possible contaminant is H2. Above 1,000°C, the hydrogen diffuses through the quartz tube from the outside. Various techniques have already been conceived (in the laboratory), in order to avoid this phenomenon. We then use a tube with double or triple inner surfaces, in between which an inert gas circulates [REV 69, VAN 72].
Lastly, the native oxide and the adsorbed impurities are also contamination sources. The silicon substrate is covered at room temperature with a native oxide layer (thickness ~1 nm) and with adsorbed impurities. This layer can be removed before the oxidation, through 10 minute processing under H2 at 1,200°C [REV 69], followed by a neutral gas annealing.
Another means reported by several authors [KAM 77, HOR 78] would consist of carrying out oxidations with low partial oxygen pressures. This is made possible by mixing O2 with inert gases (N2 or Ar). However, knowing that neutral gases always contain traces of impurity, it is almost impossible to simultaneously obtain dry oxygen and low values of PO2.
1.2.2.1.2. Oxidation under water vapor or wet oxidation
The vapor flux with atmospheric pressure is obtained by heating a balloon containing very high purity water. The entry channel of the vapor into the furnace is itself heated to avoid any condensation. By mixing the vapor with O2, we can also carry out the oxidation under wet oxygen. In order to improve the purity of the vapor, we resort to H2O produced by pyrogenic synthesis [DEA 78]. It is obtained by the reaction of O2 with H2, simultaneously introduced and heated, before they reach the oxidation furnace.
1.2.2.2. Chemical vapor deposition
At the same time as oxidation, there are other methods widely spread in the semiconductor industry, including CVD (chemical vapor deposition), evaporation and the sputtering of targets. We will limit ourselves to the case of the CVD technique.
Chemical vapor deposition can be carried out by pyrolysis of silane SiH4 in the presence of oxygen. The basic equations are then as follows:
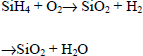
Derivatives of silane such as Si(OR)4 and the oxidizing compounds such as N2O and CO2 can also be used.
Whereas the high partial pressures of silane PSiH4 and the high temperatures (600–1,000°C) support the formation of H2O, low pressures and low temperatures lead to the formation of H2. Silica films are formed by a heterogeneous reaction on the surface. However, in the gas phase, due to nucleation process, a homogeneous cloud of colloidal particles also occurs. This is a significant contamination source. Indeed, some of these particles condense in a white powdery coating on the surfaces at low temperatures, such as the walls of the reactor.
There are several types of CVD techniques at room temperature:
— low temperature with normal pressure (low temperature CVD –LTCVD);
— low pressure (low pressure CVD – LPCVD);
— enhanced plasma (plasma enhanced CVD – PECVD).
1.2.2.2.1. Low temperature with normal pressure (LTCVD)
LTCVD reactors function under normal pressure conditions and at temperatures lower than 500°C. In most commercial facilities, the substrate is heated by the Joule effect, whereas the walls and entry points of gas are cooled to remove heterogeneous nucleation in the gas phase. The complete elimination of the contaminating particles constitutes one of the most serious difficulties of this technique.
1.2.2.2.2. Low pressure (LPCVD)
The pressure can vary between 0.5 and 1 Torr and the temperature between 700 and 800°C. This way, we manage to increase the mass transfer of the gas phase towards the substrate of an order of magnitude. It is the surface reaction that determines the oxide growth rate. Thus, we decrease the effects related to mass transfer the variations. Since the walls of the low pressure reactor are hot, the films deposit there in the same way as on the substrate. The schematic diagram of a LPCVD reactor is presented in Figure 1.2.
Figure 1.2. Schematic diagram of a LPCVD reactor (according to Kern and Rosler [KER 77])

1.2.2.2.3. Plasma enhanced (PECVD)
A glow discharge plasma is generally created with pressures ranging between 0.01 and 1 Torr. This plasma splits up the gas molecules to form very reactive species, in order to carry out a deposition in chemical phase of low temperature films. J.R. Hollahan mentions the silica film deposition for temperatures ranging between 250 and 350°C [HOL 79].
An article by W. Kern and R.S. Rosler [KER 77] reviews the basic principles, the deposition parameters, the advantages and the limitationss of various CVD techniques depositing dielectric passivation films.
1.2.3. Other methods
We mention here other methods used in the laboratory to grow silica films on silicon.
1.2.3.1. Anodic oxidation
An oxide is grown in an electrolytic cell by implementing a potential difference between the silicon wafer, acting as an anode, and a cathode plate immersed in an electrolyte or a gas plasma [DEL 71].
The aqueous solutions containing salt traces are not desirable for silicon oxidation, because they cause its corrosion. The oxide thus formed is porous.
The following non-aqueous solutions have been used to form compact SiO2 films [CRO 71a, DRE 66, SCH 57]:
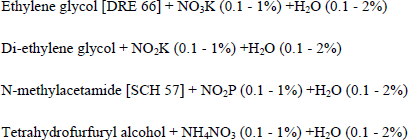
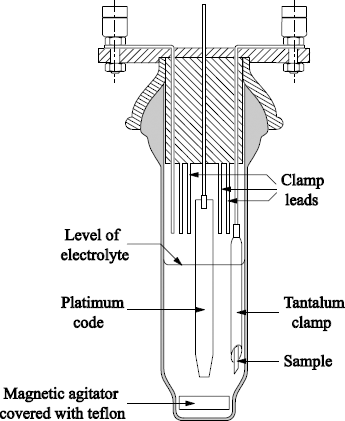
A schematic representation of an anodic oxidation cell is given in Figure 1.3.
Oxidations are performed at a constant current (typically 1 to 10 mA.cm-2). As the oxide thickness grows, the cell voltage increases up to a pre-established value. This value is then maintained constant until the density of the current decreases, to reach a value of about 10 µA.cm-2.
The oxidation process can last from 1 to 10 hours, depending on the desired oxide thickness. With these experimental parameters, we obtain SiO2 films of 0.6 nm per volt. In practice, it is rather easy to carry out oxidations up to 300 V (180 nm of silica). For values of higher voltages, it is necessary to be freed from the corrosion phenomena of impurities, such as fluorine ions [CRO 71b].
Voltages going up to 500 V (i.e. X = 300 nm) can be reached using a bath of dihydroxydiethyl ether containing 6.7 × 10-3 mole/liter of Al(NO3)3, 9H2O.
With this type of oxidation method, the film growth is due to the ionization current, representing unfortunately only 1% of the total current. Indeed, the main part of the current (99%) is an electronic current circulating through the oxide during growth.
Figure 1.3. Anodic oxidation cell (according to Croset et al. [CRO 71a])
We can also dope the oxides during the anodic growth thanks to suitable salts, such as diethylphosphate [SCH 64]. The doped oxides can be used as diffusion sources of controlled composition [SCH 65].
This technique has some advantages:
— oxidations are carried out at room temperature;
— we can produce homogeneous films and repeatable thicknesses even in the case of low thicknesses, of a few nm.
It however involves disadvantages:
— the method is performed in a wet environment;
— the insulating properties of the obtained materials are not as good as those reached by thermal oxidation. They are deteriorated by the presence of water, coming from the external environment [DRE 66, NAN 70], as well as by the possible contamination of the electrolyte by alkaline ions.
1.2.3.2. Oxidation assisted by plasma
In 1963, Miles and Smith [MIL 63] suggested using plasmas to make oxides grow. Since then, this technique was applied to various metals (such as Ta, Nb, Al, Mg, etc.) and to semiconductors (Ge, Si, GaAs, etc.). A review discusses the physical mechanisms implied for this type of oxidation [GOU 81].
In this method, the electrolyte is substituted by a gaseous plasma of oxygen. The oxide surface is bombarded by a large number of charged and neutral species (molecules, atoms, ions, free radical and electrons), whose states of charge and concentrations depend on the various phenomena taking place in the plasma. These will vary with the type of discharge and with the experimental parameters, such as geometry, pressure, etc.
This method is called “plasma oxidation”, when the substrate is electrically floating, with respect to the plasma, or “plasma anodization”, when the substrate is positively biased. During plasma oxidation, no current circulates through oxide, following the example of what occurs during thermal oxidation. On the other hand, in the case of plasma anodization, an electrical current goes through the oxide, as in the case of the electrolytic anodic oxidation. The latter is the only way to obtain important thicknesses. Several types of discharge and geometry can be used.
1.2.3.2.1. Discharge with constant current
This is obtained by applying a continuous voltage (around 1 kV) between the anode and the cathode immersed in a gas at low pressure (10-2 to 1 Torr). The electronic density is of the order of 1010cm-3. A common device is presented in Figure 1.4 [OHA 70].
The highly energetic ions due to the high voltage can produce sputtering of the electrodes. This can then involve a strong contamination of the oxide film by the sputtered cathode material, even if the sample is not facing the electrode [LES 78]. This contamination affects the oxide characteristics, as well as the growth kinetics. The ion bombardment on the substrate can also involve an ablation of the oxide film itself. In order to reduce this effect, the sample must be located as far as possible from the electrodes. Moreover, the electrode must be made of a material with low sputtering rates (Al, Si, etc.).
Copeland and Pappu [COP 71] reported a low growth rate of oxide (100 nm in 4–5 hours) at low temperatures (32–55°C).
Figure 1.4. Typical device of a plasma reactor (according to O’Hanlon [OHA 70])

1.2.3.2.2. Hot cathode discharge
The hot cathode emits an electron flux producing a plasma with a higher electron density (1010−1011 cm-3), when the plasma is magnetically confined [GOU 80]. This discharge can be maintained with a low DC voltage (lower than 50 V) and the plasma is thus free from very energetic particles. The main problem is the sputtering of the hot cathode in the oxygen atmosphere. High quality SiO2 films have been produced by this method, with fast growth rates (90 nm in 10 minutes), by setting the substrate at a temperature of 225°C [LIG 70].
1.2.3.2.3. High frequency discharge
This discharge is obtained by applying a RF voltage (a few MHz) [PUL 73, PUL 74] or a microwave (a few GHz) [KRA 67, LIG 65]. The oxygen pressure can vary between 0.01 and 1 Torr. The advantage of this technique is that the electron density can reach 1013cm-3 [LIG 65]. The major disadvantage lies in the fact that the energetic particles can damage the oxide.
By using the microwave discharge, we can obtain strong oxidation rates: 200 nm in 5 minutes [LIG 65] and 600 nm in 2 hours [KRA 67], at a substrate temperature of 300–400°C.
In the case of the RF discharge, we reach 3 nm/min [PUL 73] or 5.2 nm/min [PUL 74], with a substrate temperature around 200°C. Chang et al. [CHA 80] have shown that, by mixing a small quantity of CF4 with O2, we considerably increased growth rates with a contamination of fluorine not exceeding 0.5 atom for 100.
Anodic plasma has the following characteristics (for silicon, as well as for other materials):
— very low efficiency of the ionic current (less than 1%);
— if the total current is constant, the oxidation rate generally decreases as growth proceeds;
— the electron bombardment on the substrate surface plays a crucial role in oxidation. The growth rate is enhanced with the electron temperature, but strongly decreases, if we prevent the electrons from reaching the sample [OHA 73].
This technique offers the following advantages:
— this is a process in a dry environment;
— growth rates are significant at low temperatures.
It also has disadvantages, the main ones being:
— the oxides thus produced have in general high densities of electron traps and thus require annealing, which can be at high temperature (approximately 1,000°C) [LIG 65]. This disadvantage can be mitigated by reducing the bombardment of the energetic ions on the surface of the substrate. That was done [CHA 77, TSU 78] by removing the plasma source of the process chamber, where the high frequency discharge is taking place.
1.2.3.3. Low pressure oxidation under an ultra-high vacuum
Ultra-thin layers (1–10 nm) have a very important advantage, because they illustrate the first stages of silicon oxidation. They are also very important practically in the manufacture of very high integration silicon components. These films are often grown under a low oxygen pressure by a mixture of O2 with an inert gas. However, as mentioned previously, this oxidation mode is very sensitive to water vapor contamination. With a set up under ultra-high vacuum, we can use very low partial oxygen pressures, while minimizing contaminations. Moreover, a very clean silicon surface, free from natural oxide, is easily obtained by heating under an ultra-high vacuum.
This type of technique was described and used by Hopper et al. [HOP 75] and Smith et al. [SMI 82]. The wafers were electrically heated, thanks to a current crossing them from a sample-holder made of platinum or molybdenum. The basic vacuum of the device was tiny until approximately 10-10 Torr. The reaction chamber was then drained three times with pure oxygen containing at maximum 3 ppm of water vapor. The pressure there was then reduced to a value lower than 10-7 Torr. Later, the sample was heated up to 1,200°C – 1,350°C for 5–10 minutes, to remove the superficial oxide and the carbonaceous contaminants. Immediately after this cleaning process, the substrate temperature is reduced to the desired value and the oxygen is introduced from bakeable UHV leakage valves, until we reach the desired pressure.
The adjustable experimental parameters have been examined between the following values: 700 – 950°C, 50 – 1,200 Torr in the case of Hopper et al. [HOP 75] and 890 – 1,130°C, 5.10-5 – 5.10-2 Torr for Smith et al. [SMI 82]. The latter have determined the critical values of oxygen pressure and substrate temperature, for which the oxide growth is made possible. These critical conditions are illustrated in Figure 1.5 [SMI 82].
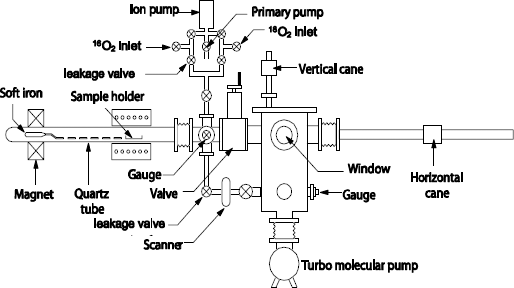
Derrien et al. [DER 82] measured the oxidation kinetics for pressures ranging between 10-4 – 10-2 Torr and 700 – 1,050°C. The silicon surface is thermally cleaned under an ultra-high vacuum at 1,100°C, sometimes leaving very little carbon traces. Figure 1.6 [ROC 84] shows a furnace containing a quartz tube heated by the Joule effect, connected to an ultra-high vacuum room. It enables oxidations in a very wide range of pressures (10-6 - 60 Torr), under an ultra-dry oxygen atmosphere (less than 3 ppm H2O).This oxygen can be strongly enriched in 18O (beyond 99%), to study the oxidation mechanisms. The purity and the labeling of the gas are controlled by a mass spectrometer. However, in this device, the water vapor contamination is always possible, if the gas oxygen reacts with hydrogenated species diffusing through the inner surface of the quartz tube.
Figure 1.5. Diagram representing the critical values of PO2 oxygen pressure and of TS substrate temperature beyond which the oxide growth is feasible. If the experimental data of PO2 and TS are placed in the upper right corner of the graphic, oxidation can take place. If they are located in the lower left corner, the silicon surface is cleaned and oxidation is inhibited (according to Smith and Ghidini [SMI 82])
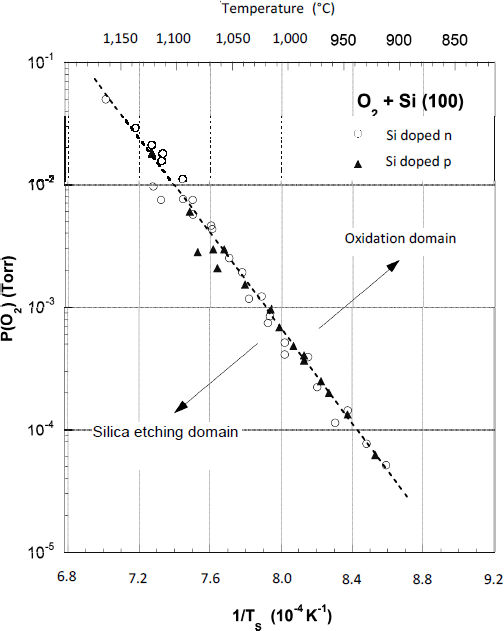
Figure 1.6. Schematic diagram of the ultra-high vacuum furnace for processing under dry gas of 18O2 and 16O2 (according to Rochet et al. [ROC 84])
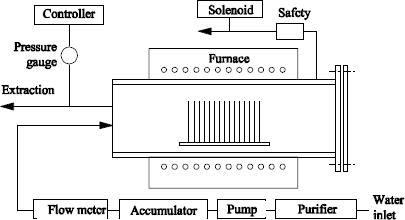
1.2.3.4. High pressure oxidation
Katz and Howells [KAT 79] described a thermal scanning processing system under flowing water vapor at high pressure (see Figure 1.7).The metal walls of the furnace are covered with quartz. Due to their coefficient of thermal expansion, they limit the processing temperature to a maximal value of 750°C. High purity deionized steam is injected into the furnace with a pressure of 20 atm. The pressure regulation is ensured by the opening of a load shedding valve, thanks to a solenoid. The steam flow rate at 20 atm is maintained at 4 liters/min. The oxidation kinetics carried out under such conditions at 725°C were published in [KAT 79]. The latter shows results identical to those obtained at 1,050°C under water vapor at atmospheric pressure.
Thus, 1 µm of oxide is obtained after a 5–hour processing. The oxides carried out at high pressure present higher refractive indexes (1.474 against 1.469), higher densities (2.31 g.cm-3 against 2.21 g.cm-3) and lower etching speed rates in buffered HF solutions (855 Å/min against 922 Å/min), than oxides obtained at 900°C under atmospheric pressure of water vapor. Nevertheless, this type of oxide presents similar electrical characteristics, after being carefully submitted to suitable annealing processing (the measured interface defects are about 3 × 1010 cm-2).
Figure 1.7. Schematic diagram of a high pressure oxidation furnace (according to Katz and Howells [KAT 79])
1.3. Some physical properties of silica
In this section, we present some properties of silica, useful for a better understanding of silicon oxidation and the growth models developed in this chapter.
During silicon oxidation, the oxide growth proceeds because a phenomenon of atomic transport takes place through the film (as shown in the following sections).
Thus, we must consider the transport properties of silica, which should depend in theory on its atomic structure, and thus on its physical characteristics.
1.3.1. The silica structure
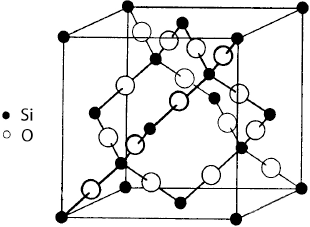
The basic silica structure is represented in Figure 1.8. It is a tetrahedron, with in its center, a silicon atom linked to 4 oxygen atoms located at the corners. Each oxygen atom belongs to 2 adjacent tetrahedrons, while being linked to 2 silicon atoms. The mean value of inter-atomic distances was measured by Mozzi and Warren [MOZ 69] in the fused quartz with a 2.20 g/cm3 density.
They report the following values:
— 1.62 Å for the Si-O bond;
— 2.62 Å for the O-O bond;
— 3.12 Å for the Si-Si bond.
Figure 1.8. Basic structure of SiO2

Table 1.1. Structural characteristics of the most widespread crystalline forms of SiO2 (according to Revesz [REV 70, REV 80])
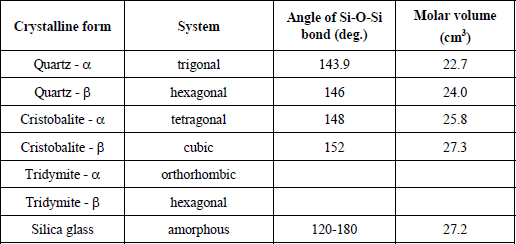
The stability zones of silica and polymorphic transformations are represented in Figure 1.9; and the various structures in Figures 1.10, 1.11 and 1.12.
Figure 1.9. Stability zones of silica at atmospheric pressure and its polymorphic transformations (according to Pascal [PAS 62])
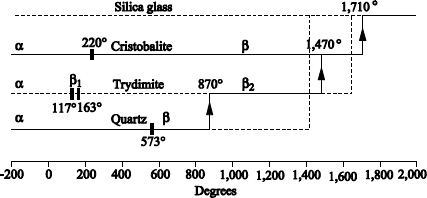
Figure 1.10. Projection in the (001) plane of structures α and β of the quartz (according to Pascal [PAS 62])

Figure 1.11. Structure of tridymite-β (according to Pascal [PAS 62])

Figure 1.12. Structure of cristobalite-β (according to Pascal [NOT 62])
Revesz [REV 80] allocated the wide distribution of measured angles, Φ, for the Si-O-Si bond to the flexibility of the Si-O bond that has three components (covalent-σ, covalent-π and ion (40%)).The contribution of each of its components would vary with Φ without significantly changing the total bonding energy.
Indeed, several configurations of the same energies could coexist (see Table 1.1).
Moreover, it was shown that in the crystalline structures, the π bonding was enhanced along the directions parallel to axis c.
1.3.2. Three useful parameters of silica
The properties of vitreous silica depend on its thermal history and on the impurities it can contain [HET 62] (OH, H, Al, etc.). That could explain the wide range of parameters given in Table 1.2.
Table 1.2. Some important parameters of silica glass (according to Bruckner [BRU 70])
| Refraction index n (at 546 nm) |
Density ρ (g.cm-3) |
Coefficient of thermal expansion (10-6.K-1) |
|---|---|---|
| 1.460 | 2.200–2.206 | 0.5–1.2 |
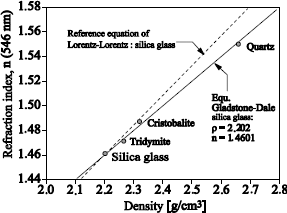
The porosity and the transparency of the structure are two of the most important physical characteristics of silica films. We can quantify them by using mass density values. However, for thin films this is not easily directly measurable, contrary to the refraction index. Two equations connect the density ρ, with the refraction index n, of an environment.
The Gladston-Dale equation:
[1.1] ![]()
The Lorentz-Lorentz equation:
[1.2] ![]()
with k1 and k2, constants where k2 can also be expressed according to the polarizability α and to the molar mass M of silica:
![]()
These equations (see Figure 1.13) are based on the hypothesis that the basic material remains identical in its atomic structure, but with different porosities and optical transparency.
Figure 1.13. Refraction index of silica according to the density for various SiO2 forms (according to Pliskin [PLI 77])
1.3.3. Transport properties in silica
To understand the transport mechanisms taking place during oxidation, we should not only know the SiO2 structure, but also examine the way in which the species diffuse and/or possibly react in amorphous silica. For example, the rare gases diffuse without reacting, whereas hydrogen reacts with its diffusive environment for temperatures higher than 500°C. The species that will be examined are those that we generally encounter during oxidation and annealing processes.
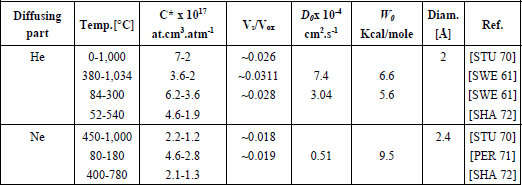
The diffusion of rare gases helps us to define two important concepts: solubility and diffusivity.
1.3.3.1. Diffusion of rare gases in silica
The rare gases (He, Ne, Ar, Xe) diffuse interstitially without reacting with the silica network.
1.3.3.1.1. Solubility
Solubility is the aptitude of a network to dissolve a gas which is immersed there at a given temperature and pressure. It is best described by the C* parameter. The latter is the maximum concentration of gas dissolved in the material, after having reached the thermodynamic equilibrium.
The solubility process can be described by two distinct models: the free volume model, suggested by Doremus [DOR 94] and the complete statistical thermodynamics model, developed by Shackelford and other scientists [SHA 72, SHE 77].
The free volume model relies on the idea that the solid network contains a certain accessible volume Vs, remaining constant with the temperature and the pressure, as long as the structure is not modified. In that case, C* can be expressed as follows:
[1.3] ![]()
with Cg, the atomic concentration per unit of gas volume, and Vox, the volume of the solid.
In the model derived from statistical mechanics, the equilibrium between the gaseous and dissolved states of the same species requires us to know that the Gibbs free energies in the gaseous state Gg equals that in the dissolved state Gs. Thus, C* can be expressed as follows:
[1.4] ![]()
where p is the gas pressure and C0 is the concentration of the solubility sites (2.3 × 1021 and 1.3 × 1021 cm-3 respectively for He and Ne). K(T) is expressed:
[1.5] ![]()
where WSG is the difference between the potential energy that an atom has at rest, on the one hand, in the dissolved state, and on the other hand, in its gaseous environment
(-1.5 kcal/mole and -2.9 kcal/mole respectively for He and Ne). The expression of f(T) depends on the type and on the degree of freedom assumed [STU 70].
1.3.3.1.2. Diffusivity
Diffusivity is the aptitude of a species to diffuse in an environment. This physical characteristic is the best represented by the diffusion coefficient D, given by the Arrhenius equation:
![]()
However, the experimental results show that D0 and W0 would also depend on T, in the range of temperatures considered [PER 71, SHE 77, SWE 61]:
— Sweets [SWE 61] suggests that it could be due to the fact that the considered crystalline forms of silica present strong phase reversal in the range of temperatures [160°C – 280°C].
— Perkins and Begeal [PER 71] expose models derived from the statistical model. They predict that the pre-exponential factor D0 should be independent from the temperature, if the partition function fq corresponding to the displacement in the direction of the diffusion is the same as that of a linear oscillator. If fq is the partition function of a linear translation, then D0 must be proportional to T1/2.
— By taking an interest in the amorphous structure of glass, Shelby and Keeton [SHE 74] suggest that the energy barrier to be crossed to go from one site to another can have several different values with a Gaussian distribution. The adjustment of the experimental results for the helium diffusion (24°C – 100°C) confirms the model, and validates the Doremus hypothesis [DOR 62], predicting a linear dependence in T of D0.
— The activation energy W0, of the molecular diffusion increases with the radius r, of the diffusing molecule (see Table 1.3). This behavior is described by the model suggested by Anderson and Stuart [AND 54]: W0 is the potential energy of elasticity necessary to stretch an orifice of radius rD to the radius r of the diffusing molecule, so that:
[1.6] ![]()
G being the shear modulus. A reasonable agreement is obtained with experience by taking rD = 0.6 Å and G = 1.33 1010Pa [PER 71] as values.
1.3.3.2. Molecular diffusion in silica
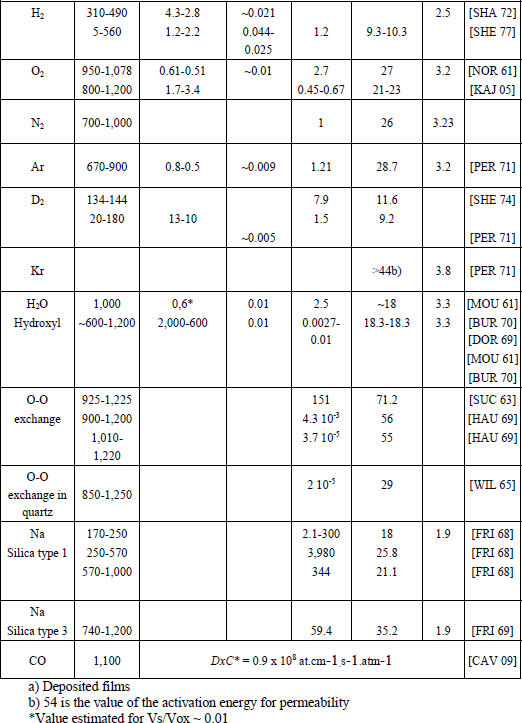
Gas molecules such as H2, O2, H2O, diffuse in a way similar to rare gases.
As we can see in Table 1.3, the activation energy of the molecules diffusion is similar to that of rare gases with the same radius. However, the interactions and chemical reactions with the environment can complicate the observation and extraction of parameters. Moreover, the presence of impurities traces can strongly influence chemical reactions.
Table 1.3. Values of the solubility parameters (C*, Vs/Vox) and of the diffusivity parameters (D0, W0) for various elements. Vs/Vox is calculated according to the hypothesis of free volume
1.3.3.2.1. Hydrogen diffusion
The hydrogen primarily diffuses without reacting up to 500°C [SHE 77, VAN 75]. At higher temperatures, Si-OH groupings are formed and the equilibrium concentration of these hydroxyls varies as the square root of the molecular gas pressure [BEL 62]. Hetherington et al. [BEL 62, HET 64] propose the following reactive mechanism:
[1.7] ![]()
Whereas Van der Steen and Papanikolau [VAN 75] propose the following:
[1.8] ![]()
This hypothesis cannot be checked by measurement of infrared absorption, because SiH groups cannot be detected; their peak of absorption being embedded in the background noise of the network.
1.3.3.2.2. Water diffusion
Water diffuses in silica, while reacting to it [BUR 70, MOU 61]. The concentration of dissolved OH, COH is proportional to the square root of the water vapor pressure PH2O [HET 62, MOU 61]. The equilibrium reaction can be written as:
Several transport mechanisms have been considered:
a — associated diffusion of protons and OH groups [ROB 66];
b — interstitial diffusion of the water molecule. The latter is associated with the more or less quick reaction [1.9] of water with the network [DOR 69];
c — water diffusion in the ambipolar form of hydronium ions (H3O+) and hydroxyl OH-, to form SiOH after the reaction with the network.
The b mechanism is proposed by Doremus [DOR 69]. It seems to better explain the experimental results. The water vapor will react in the network to form SiOH groups. Those can, in their turn, react two to two to release a water molecule, which will be able to diffuse and react in its turn. The diffusion process of the water vapor is complex.
At high temperature (T > 1,000°C), the reaction rate is much more important than the diffusion process. The concentration of SiOH, [OH] of H2O, [H2O], and the effective diffusion coefficient, DOH will be given by the relation:
[1.10] ![]()
D, being the diffusion coefficient of the water molecule, kOH is the equilibrium constant of the reaction given by the relation:
[1.11] ![]()
Experimentally, we find the diffusion coefficient of DOH is proportional to the concentration of hydroxyls groups.
At low temperature (T < 1,000°C), the reaction rate becomes smaller and the hypothesis of a very fast reaction is no longer valid.
The Doremus model seems to be in agreement with the experimental results obtained by isotopic labeling methods, consisting of doing an oxidation under water vapor isotopically enriched (tritium, deuterium [BUR 70, HOL 70], 18O [LIG 60, MIK 81]) with a silica already saturated in natural OH.
The calculation of isotopic profiles is detailed in the reference [RIG 82].They are in harmony with the experimental results.
1.3.3.2.3. Oxygen diffusion
The solubility and diffusivity of the oxygen molecule have been deduced from permeation measurements made by Norton [NOR 61]. Several authors also point out an exchange mechanism of the oxygen, when we submit natural silica to a thermal process under an atmosphere of enriched oxygen in 18O [SUC 63, WIL 65]. Nevertheless, the values of the diffusion coefficients found strongly vary from one experiment to another. They do not help us to get to a conclusion, while also raising the question of the purity of the gases used, notably their level of water vapor.
Is the oxygen exchange an intrinsic phenomenon between the interstitial O2 molecules and the silica network, or is it the result of another transport mechanism lacunar? Several authors tried to answer this question.
D.A. Hutchingson [HUT 54] did not find an exchange with silica for temperatures going up to 1,000°C, and Rochet [ROC 84] showed that there was also no exchange between silica and dry oxygen at 930°C.
Conversely, we know that water vapor exchanges a lot with silica. This makes the following hypothesis plausible: uncontrolled water vapor traces could be responsible for the exchanges observed in the different works [SUC 63, WIL 65].
Thus, molecular oxygen diffusion without reaction in silica seems to be the dominating phenomenon, even if the following reaction has been reported, in the case of silica without hydroxyls groupings:
[1.12] ![]()
If this reaction occurs, it is very difficult to prove, because of the difficulty in obtaining water deprived of oxygen gas.
Silica is a very stable material. In its most reduced form, it can be written SiO2-x. The value of x, measured by UV absorption was found to be equal to 3 × 10-5.
1.4. Equations of atomic transport during oxidation
In the previous sections, we presented most techniques leading to the formation of silica layers on silicon, either by oxidation or deposition. In this section, we will limit ourselves to the various physical aspects of oxidation.
The transport equations presented in this section are valid for any oxidation process; silicon oxidation being an application of the general equations.
During oxidation, the oxide growth can be done only if at least one of the oxide components is transported through the film during growth. The latter will be identified as mobile species. The subjacent principle of this transport is that mobile species move by successive hops from site to site, if they are available to host them.
In a perfect single crystal, each atom occupies a well-defined site. The transport of species through a crystal thus presupposes the existence of defects. They can be interstitial, if the species migrate in an interstitial position or lacunar, if the species are placed in a substitutional position compared to the atoms of the crystal lattice. The interstitial defects can be occupied by atoms, molecules or ions. These species are also called interstitial, following the example of the sites they occupy.
In single crystals, the concept of defects is clearly defined. Indeed, each atomic site has a precise position in the periodic network. The situation is more complex in a vitreous material such as silica, where atoms have a random distribution in the amorphous atomic network.
In this case, the hopping distance can enormously vary according to the position of the atom. We must keep that in mind, when we use simplified models, such as the one described below, from which the transport equations are deduced.
Nevertheless, we will use, independently of the diffusive environment, the expressions of mobile species and of mobile defects, to describe the subjacent transport mechanisms.
1.4.1. Transport equations in the general case
To simplify, we will use a model where transport is carried out in only one dimension of space, with two parallel planes (1) and (2), distant of a and perpendicular to the direction of displacement (a is equal to the distance between two neighboring sites). Let us choose a bench mark, so that the respective positions of the planes are x+a/2 and x−a/2 (see Figure 1.14).
Figure 1.14. Geometry of the 1D transport model

Let us call J the flux, expressed in number of defects cm-2.s-1, due to the transport of a type of defect (or of mobile species). Let us note C(x−a/2) and C(x+a/2), the respective defects concentrations on the level of the planes, expressed in number per volume unit. Between the planes, we assume that there is a potential barrier that should be overcome to carry out a hop from one site towards another. γ12 and γ21 are the probabilities per time unit, of effective hops between plans (1) and (2) in both directions. J12 and J21 are the flux derived from it, and J is the resulting net flux expressed as follows:

[1.13] ![]()
By developing C(x), we obtain:
[1.14] 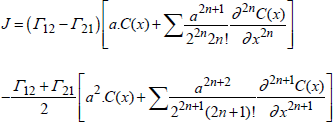
At this stage, it is necessary to make approximations to make the expression of the transport equation more easily usable. We discuss in the next two sections the two most frequently used approximations.
1.4.2. First approximation: C(x) varies slowly with the depth x
If C(x) varies slowly with the depth, we can limit the development to the first term, as follows:
[1.15] ![]()
and the flux is reduced to:
[1.16] ![]()
1.4.2.1. Application to the transport of neutral species in an isotropic environment
In such a situation, we have γ0 = γ12 =γ21. Thus the first term of the flux is cancelled and reduced to:
[1.17] 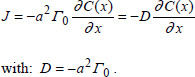
This is in fact the first Fick law with 1D.
1.4.2.2. Application to the transport of species charged under electric field ε(x)
In this case γ12 ≠ γ21. The periodic potential of the network is altered by the electric field, as illustrated in Figure 1.15, for Zqε(x) > 0, with Zq the electric charge of the species.
We will make the following approximations:
— the distance between a minimum and a maximum of the potential barrier is half the hop distance a. The latter is not altered by the electric field ε(x);
— the potential is projected in only 1D in x;
— the fluctuation of the potential barrier, due to the electric field between two minima is worth a.ε(x) and ε(x), is the mean value of the field between x−a/2 and x + a/2.
Figure 1.15. Potential energy of the charged species in presence (----) and without electric field (-)
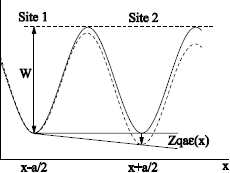
When ε(x) = 0, the potential has a periodicity of a. To move from site to site, the mobile species must cross an energy barrier of a height W. Thus, the hopping probability from site 1 to site 2 is expressed as follows:
[1.18] ![]()
v = v0 exp(ΔS/kBT) is the frequency coefficient. ΔS is the entropy fluctuation, v0 is an oscillation frequency (v0 ≈ 1012s-1), kB the Boltzmann constant (kB = 8.67 × 10-7 eV.K-1) and T the temperature in K.
When ε(x) ≠ 0, the potential energy is modified (dotted line in Figure 1.15). Thus, the potential barrier decreases for a hop from 1 to 2, and increases for a hop from 2 to 1:
[1.19] 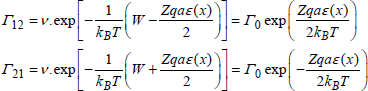
Thus:
![]()
Two limiting cases must be considered:
1) Low field, if |Zqaε(x)/2kBT| << 1, then by developing the hyperbolic functions at the first order:

2) Strong field, if |Zqaε(x)/2kBT| >>1, then the flux can be written:
[1.20b] 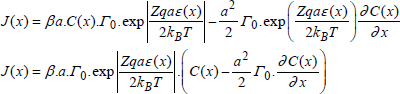
with β = +1 for Zε(x) > 0 and β = -1 for Zε(x) < 0.
If C(x) varies little from one site to another, the flux is written as follows:
1.4.3. Second approximation: ε(x) varies slowly with the depth x
This case was solved by Dignam [DIG 72], who formulated the following hypothesis:
a) the oxide growth is in a stationary state, i.e. the flux J(x) = J is independent of the depth x;
b) the fluctuation of the electric field is sufficiently low to be neglected, thus ε(x) = ε and thus:
[1.22] ![]()
There are two extreme cases:
1) Low field so that:
![]()
Then:
2) Strong field if ![]() then:
then:
[1.24] ![]()
and if, moreover ![]() then:
then:
Let us notice that if equations [1.23] and [1.25] are similar to equations [1.20] and [1.21], they do not answer to the same starting hypothesis and thus cannot be applied to the same domains of variable parameters. For example, the expression J of equation [1.23] remains valid for field values corresponding to equation [1.21].
Dignam [DIG 72] discusses in detail the validity domains of the hypothesis exposed above and indicates the corrections to be made, when they no longer apply (for very strong fields or high concentrations of charged defects). This study goes beyond the account in this chapter.
1.4.4. Applications of the transport equations to thermal and anodic oxidation
The previous hypotheses do not apply in the same manner for these two types of oxidation.
1.4.4.1. Anodic oxidation
In this case, a net current crosses the oxide; the electric field there is intense (ε of about 10 MV.cm-1) and the atoms are transported by means of charged atomic defects. By analogy with an ion current, the charged atomic defects represent the ion species and the net current would be the ion current Ji:
[1.26] ![]()
with Ci, a concentration of charged atomic species.
1.4.4.2. Thermal oxidation
Contrary to what occurs for anodic oxidation, there is no applied external electric field. Consequently, if the charged species take part in the species transport, the sum of all the electric currents (ions and electronic charges) in oxide and at the interfaces must be null. Thus, the ion current Ji must be counterbalanced by the electronic current Je. We will find more details on the transport mechanisms in section 1.5.
At this stage, let us note that the transport equations have been established without knowing the nature of the mobile species, nor by which mean they move. If we want to obtain the laws controlling the growth during thermal or anodic oxidation, we must answer in each case the two following questions: which are the mobile species and how do they move? The answers are given in the following section, with two different experimental approaches.
1.5. Is it possible to identify the transport mechanisms taking place during oxidation?
1.5.1. Identification using isotopic labeling techniques
We present here the principles of isotopic labeling techniques, using a stable 18O oxygen isotope. Silicon isotopes can be the subject of a similar reasoning, if we resort to it.
Silicon oxidation is carried out in at least two stages: a first natural oxygen oxidation (H2O, O2, O3, etc.) upto a thickness x0, then a second oxidation in oxygen strongly enriched in 18O up to a thickness x0+Δx. Various oxidation mechanisms will produce specific concentration profiles of the oxygen isotopes in the oxide film. The measurement of these profiles helps us to determine the oxidation mechanisms implied in the film growth.
Let us consider the mechanisms that could occur.
1.5.1.1. First hypothesis: silicon is the only mobile species
Whatever the implied mechanism, the 18O will be set at the external surface of the oxide, with an abrupt interface 18O/16O (see Figure 1.16a).
1.5.1.2. Second hypothesis: oxygen is the only mobile species
The concentration profile of the 18O depends on the implied process.
Some magnitudes must be defined:
— [O]n: atom concentration in the silica network.
— ![]() : 18O atom concentration in the silica network.
: 18O atom concentration in the silica network.
— ![]() : enrichment in 18O in the silica network.
: enrichment in 18O in the silica network.
— [O]c: O atom concentration in the gas dissolved in silica.
— ![]() : 18O atom concentration in the gas dissolved in silica.
: 18O atom concentration in the gas dissolved in silica.
— ![]() : enrichment in 18O in the gas dissolved in silica.
: enrichment in 18O in the gas dissolved in silica.
— [O]n = 0.44 × 1023 at.cm-3.
— [O]c = S×P, where S is the solubility and P the oxidation pressure. In the case of O2 at 1,100°C, solubility is S ≈ 0.5 × 1023 at.cm−3.atm−1 [KAJ 05].
— [O]c/[O]n ≈ 1 ppm for 1 atm at 1,100°C.
— ![]() : enrichment in 18O of the gas.
: enrichment in 18O of the gas.
1.5.1.2.1. Oxygen diffusion without interaction with the silica network
Oxygen diffuses through the already formed oxide during the first oxidation without reacting with it and reacts with silicon to form SiO2. The 18O atoms are set at the interface oxide/silicon; the 16O/18O interface is abrupt and the enrichment in 18O of the new oxide layer is equal to that of gas (see Figure 1.16b).
1.5.1.2.2. Oxygen diffusion with interaction with the silica network
Oxygen diffuses interstitially through oxide (diffusion coefficient D), using a chemical reaction with it. Oxygen atom exchange occurs between the silica networks formed during the first oxidation and the oxidizing gas. γ is the exchange frequency of O atoms in the molecules of the oxidizing species and in the silica network. At the same time, oxygen reacts with silicon at the Si/SiO2 interface, in order to form a new oxide. Due to the isotopic exchange, the enrichment in 18O at the interface is lower than that of gas, and the amount of 18O is higher than the one corresponding to the thickness increase. The shape of the 18O profile after the second oxidation depends on the oxygen exchange frequency and on the growth rate of the oxide. The complete calculation of the profile is given in the reference [RIG 82], when the oxide growth is negligible. The calculation of various profiles, in the presence of oxide growth, is given in the reference [RIG 88].
We show here the cases where the differential equations are simplified, by considering in particular that the equilibrium is reached, i.e. that the concentration of the dissolved oxidizing species in the film [O]c is independent of the depth x and of the oxidation time t.
According to the exchange frequency γ, the oxygen profiles are as follows (see Figure 1.16c).
High exchange frequency
The exchange frequency γ is important (γt>> 1): isotopic equilibrium is reached (enrichment in 18O is the same in the gaseous phase and in the silica network). The profile of 18O is very close to a complementary error function in the presence, or not, of oxide growth:
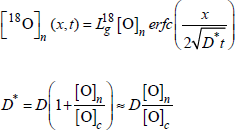
Low exchange frequency
The exchange frequency γ is small (γt << 1): isotopic equilibrium is not reached and the enrichment of the silica network is lower than that of the gaseous phase:
— in the absence of oxide growth, the profiles are as follows:

![]() is the characteristic length of exchange, i.e. the characteristic distance between two events, during which a specific molecule of the oxidizing species exchanges one of its atoms with the silica network;
is the characteristic length of exchange, i.e. the characteristic distance between two events, during which a specific molecule of the oxidizing species exchanges one of its atoms with the silica network;
— in the presence of oxide growth, the general form of the oxide profiles will be:
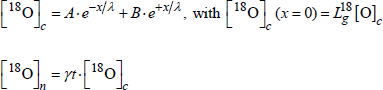
Taking into account an additional boundary condition allows us to solve the equation system.
For the linear regime, this boundary condition is: ![]() .
.
The profiles will thus be:

For the parabolic regime, the boundary condition is:
![]()
and the profiles are as follows:
[1.27] 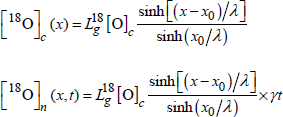
1.5.1.2.3. Oxygen diffusion by a gradual movement of oxygen atoms of the network
The oxygen transport gradually takes place by a movement of oxygen atoms of the silica network. The exchange of oxygen atoms of the gas with those of the silica network occurs only on the external surface of oxide (and not in the volume). This exchange is made possible by the specific configuration of this surface, which looks like a break in the existing volume structure. At the same time, the diffusion of isolated defects present in the oxide involves the diffusion in the oxide volume of the 18O atoms of the surface. We postulate the transfer coefficient between gas and silica to be sufficiently high, so that all the atoms of the surface are completely exchanged in a very short time.
Movement in all directions
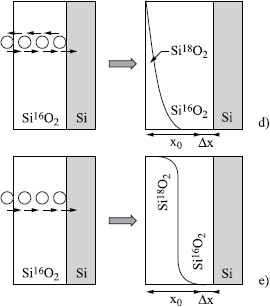
If the defects migrate in all directions, according to a simple diffusion process, there is an isotopic exchange, and the total amount of 18O is higher than the quantity fixed by the growth. In the absence of oxide growth, the profile is a complementary error function (erfc). Without any oxide growth, the calculation [ROC 81] shows that the profile remains close to an erfc function shape, in all cases, for neutral, single or double charged defects (see Figure 1.16d):
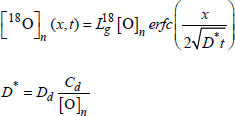
where Cd and Dd are respectively the concentration and the diffusion coefficient of the defects.
Movement in only one direction
If the defects migrate in only one direction (case of a charged defect in an electric field), the 18O atoms move only in the direction of the growth. In this case, no oxygen atom leaves the silica network, and there is no isotopic exchange. The boundary 18O /16O has a light mix, due to the statistics of hops. The quantity of set 18O corresponds to the oxide growth (see Figure 1.16e).
Figures 1.16. Principle of isotopic labeling with 18O applied to put the mechanisms of silicon oxidation in the spotlight. Silicon is first oxidized in 16O2 and then in 18O2. Five types of transport mechanisms are represented, as well as the corresponding 18O profiles. The black circles represent the Si atoms, the white circles, the oxygen atoms. a) Only silicon is mobile; b) oxygen diffuses through the already formed oxide during the first oxidation without reacting with it, and reacts with silicon to form SiO2
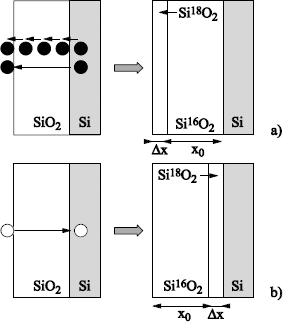
Figure 1.16c. As previously, with c) oxygen diffuses interstitially through oxide by exchanging its oxygen atoms with it, with an exchange frequency γ. The form of the profile depends on the value of γ
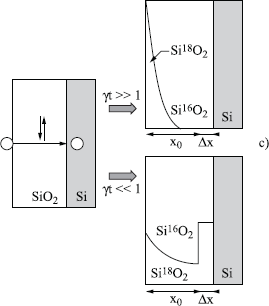
Figures 1.16d and e. As previously, with d) and e) oxygen transport occurs by a gradual movement of oxygen atoms of the silica network, because of the presence of diffusive defects. These defects can migrate in all the directions (d) or in only one direction (e)
1.5.2. Important results for the thermal oxidation of silicon under dry O2
1.5.2.1. Films with thickness higher than 20 nm
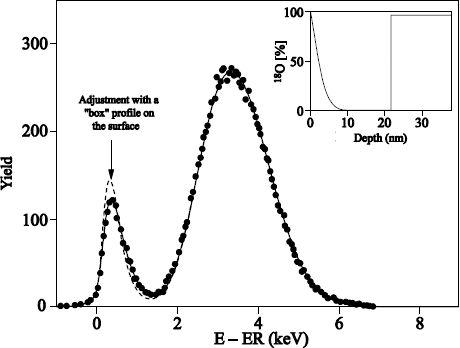
Figure 1.17 shows the typical profile of 18O obtained after sequential 16O2/18O2 oxidations.
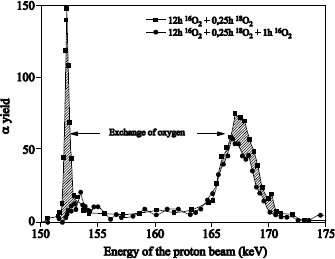
The 18O profile was measured by using the narrow resonance at 151 keV of the 18O (p, α) 15N reaction [BAT 92].
18O oxygen is localized on the external surface of oxide and at the SiO2/Si interface. The 18O profile, close to the external surface of oxide could be adjusted with an erfc profile. The one in proximity to the SiO2/Si interface can be adjusted by a “box” profile, with an enrichment equal to that of the oxidizing gas.
The 18O concentration in the oxide volume was set at 0.2% (concentration in 18O of natural oxygen). The 18O profile at the interface shows that 18O2 oxygen diffuses through oxide, without reacting with it, and reacts with silicon at the SiO2/Si interface. This result validates the transport model suggested by Deal and Grove [DEA 65].
The 18O profile on the surface, which can be adjusted by an erfc function, is then explained by two mechanisms:
— gradual movement of oxygen atoms of the network, induced by the presence of defects in oxide;
— diffusion with exchange between oxygen and the network of an oxygenated species. This will be different from those responsible for fixing 18O at the interface (that is to say enriched 18O steam).
The hypothesis, according to which the water vapor would be responsible for the fixing of 18O on the surface, was rejected by Rochet et al. [ROC 84]. Effectively, the partial pressure of H218 must be up to two orders of magnitude higher than those measured by mass spectrometry.
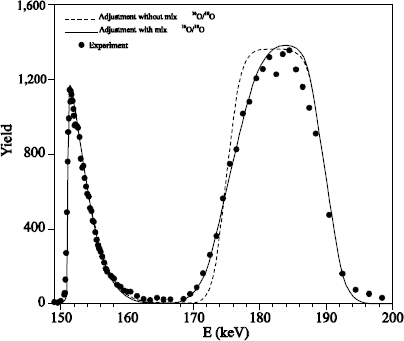
We can conclude that heavy oxygen fixation near the surface is due to anisotopic exchange phenomena and the 18O diffusion, induced by the presence of defects in the oxide. Such a mechanism also produces a light mix at the 18O/16O interface. In Figure 1.17, where oxidation in 18O2 is carried out at 930°C, this mix is not detectable. On the contrary, in Figure 1.18, where oxidation in 18O2 is carried out at 1,100°C, this mix is observable.
Figure 1.17. 151 keV resonance excitation curve for the 18O (p,a)15N reaction obtained on a 20.5 nm Si16O, on reoxidized Si substrate under 100 mbar of 18O2 at 930°C during 5 hours. The 18O profile used to adjust the experimental curve (unbroken line) is also presented: on the external surface, the supposed profile is an erfc function, and is constant close to the SiO2/Si interface. The dotted line represents the adjustment when the erfc function is replaced by a rectangular profile of the same surface and surface concentration
Isotopic labeling experiments have been carried out by Costello and Tressler [COS 84] at 1,000°C and at 1,300°C. At 1,000°C, they are qualitatively in harmony with the results mentioned above. At 1,300°C, all the initial oxygen has been exchanged by 18O; the diffusion mechanism of the defects contributing to considerable oxide growth. Other experiments [HAN 88], carried out between 900 and 1,150°C, are also in qualitative harmony with the previous results.
A study using the pressure of oxidation in 18O2 as the variable parameter was undertaken, in order to try to identify the defect responsible for the oxygen fixing on the surface [TRI 89]. The peroxyl bridge (O-O bond) has then been proposed, as the defect leading to the 18O diffusion in the proximity of the surface.
It is created on the surface according to the reaction:
![]()
Figure 1.18. 151 keV resonance excitation curve for the reaction 18O (p, α)15N obtained on a 103 nm Si16O2 on a reoxidized Si substrate under 200 mbar of 18O2 at 1,100°C during 4 hours. The dotted line corresponds to the adjustment of the experimental curve with a complementary error function on the surface and a profile of type “box” at the interface. The full line corresponds to the adjustment of the experimental curve by taking into account the 18O/16O mix, produced by the presence of defects
The quantity of 18O fixed at the external surface increases when the initial oxide thickness decreases [HAN 88, ROC 84]. A study [TRI 93] could show that the increase in the fixing of 18O on the surface was not related to the increase of the oxidation rate at the interface. Other mechanisms are to be considered, such as for example the modification in the structure of silica during the growth, altering the diffusion coefficient of the defects.
An oxygen exchange at the SiO2/Si interface was highlighted by Akermark et al. [AKE 99], by means of sequential oxidations 16O2/18O2/16O2 at 1,100°C (see Figure 1.19). After the first oxidation, the oxide thickness is 120 nm and the growth regime is parabolic. After the second oxidation, the quantity of 18O set at the interface is equivalent to 1 nm. After the third oxidation in 16O2, a loss in 18O of 0.3 nm is observed at the interface. This result is not predicted by the Deal and Grove model, for which the oxidant concentration at the SiO2/Si interface is null, in the regime.
Figure 1.19. 151 keV resonance excitation curves for the reaction 18O (p, α) 15N obtained after the oxidation sequences 16O2/18O2 and 16O2/18O2/16O2, at 1,100°C and under 210 mbar (from [AKE 99])
1.5.2.2. Films with thicknesses lower than 10 nm
Rochet et al. [ROC 86] have grown films of thickness ≤ 6 nm, under an oxygen pressure of 13.8 mbar and in a temperature range from 600°C to 1,090°C. 18O oxygen is fixed primarily on the surface. At temperatures ranging 810°C – 1,090°C, an exchange phenomenon is observed. The dominant transport mechanism is thus the interstitial transport of oxygen via defects. At 600°C, no exchange is observed, which means that the growth occurs by the movement in only one direction of the oxygen atoms of the network. This mechanism implies the transport of charged defects in the presence of an electric field.
Ganem et al. [GAN 92, GAN 93b] carried out sequential oxidations 16O/18O by rapid thermal oxidation. An oxide film with an initial thickness of 5 nm, reoxidized in 18O2, up to a thickness of 10 nm presents a profile similar to films obtained by conventional thermal oxidation. For a film with an initial thickness of 3 nm, reoxidized in 18O2, up to a thickness of 5 nm, we observe the presence of a pure Si18O2 layer at the interface, but also the fixing of 18O in the oxide volume with a concentration of up to 30%. These results can be explained by the presence of silicon fragments in these very thin oxides. These non-oxidized silicon fragments can be the result of the roughness of the silicon surface, before oxidation and injection of silicon atoms in the SiO2/Si interface, during oxidation. Direct observations of such fragments have been made by electron microscopy in transmission [GIB 89] and by electron diffraction [OCR 86].
In the case of the 3 nm film, the oxidation of these fragments is incomplete and continues during the second oxidation in 18O2. This leads to the fixing of 18O in the oxide volume. In the case of the 5 nm film, the fragments would be completely oxidized and the reoxidation under 18O2 does not cause any more fixing of 18O in the oxide volume. This experimental result could explain the first stages of oxidation, characterized by a regime of rapid growth. Indeed, the oxidation rate at the SiO2/Si interface is proportional to the number of reactive sites. Therefore, as the oxidation rate at the SiO2 interface is proportional to the number of reactive sites, the presence or the formation of silicon fragments or of a roughening of the interface, would involve an increase amongst reactive sites, and thus of the oxidation rate. When these silicon fragments are finally oxidized, the oxidation rate drops to reach its value in steady state.
1.5.3. Important results for wet thermal oxidation
Si16O2 silica layers with thicknesses ranging between 90 and 250 nm have been thermally processed in water vapor strongly enriched in 18O (> 93%), under an atmosphere of 13 Torr and at temperatures from 400 to 900°C [RIG 82].
Regardless of the oxidation durations and temperatures, the fixed quantities of 18O are higher than those corresponding to the increased thickness, revealing a strong isotopic exchange.
At 930°C, the 18O oxygen profiles could be adjusted with an erfc function. From 400°C to 600°C, most 18O profiles could be adjusted with a “cosh” function.
These results indicate that oxygen diffuses interstitially through oxide, while reacting with it.
The equilibrium reaction is as follows:
![]()
1.5.4. Conclusions on the atomic transport mechanisms during silicon thermal oxidation
In all the cases exposed here, oxygen is identified as the mobile species.
For this reason, the choice of oxygen as an isotopic label should be the most suitable.
1.5.5. Experimental results and conclusions on the transport mechanisms during the anodic oxidation of silicon
It was shown that water traces (typically 0.4%) contained in organic solvents used for anodic oxidation were the main oxygen source for the oxide growth [CRO 71a].
In the Croset et al. experiment [CRO 76], described below, it was intentionally added to the solvent, 0.4% of water enriched in 18O (i.e. H218O).
The oxides were initially grown in a bath of dihydroxydiethyl ether enriched in 18O, to reach 150 V, i.e. approximately 90 nm of oxide thickness. These oxides were then reoxidized, but in a solvent containing 0.4% of H216O for voltages from 180 to 300 V (108 to 180 nm).
Figure 1.20 represents the profiles of 18O obtained by SIMS (secondary ion mass spectroscopy) for various phases of the second stage of oxidation. We clearly see that the sequence 18O/16O is preserved, but a mixture 16/18 appears, and a loss of 18O is highlighted.
Figure 1.20. 18O profiles measured by SIMS: a) after oxidation in 18O; b) after a reoxidation in 16O of 20 nm; c) after a reoxidation in 16O of 57 nm; d) after reoxidation in 16O of 80 nm
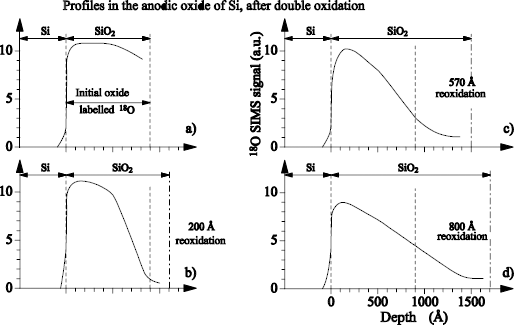
These results show that, during anodic oxidation, the oxygen atoms move in a gradual displacement, in the direction of the electric field, as well as in the opposite direction.
Knowing that at room temperature, only the charged species move, this result requires careful study of the transport mechanisms at an atomic scale. The transport in two directions could be explained in two ways. Either the mobile oxidizing species exist under two states of charge (positive and negative), or they can switch from one state to the other during transport.
1.5.6. Important experimental results from dry SiC thermal oxidation
Silicon carbide is a semiconductor with wide gap, interesting for the high frequencies and high power applications. Its oxidation produces SiO2. Various oxidation models have been proposed [HIJ 09, SON 04], based on the adjustment of the oxide growth kinetics. These models are derivative from the Deal and Grove model for silicon oxidation, and take into account the CO (or CO2) production during the oxidation reaction of SiC under O2. Isotopic labeling has been used for the study of the oxidation mechanisms of SiC (see [VIC 07] for a review of the results obtained by this technique).
Figure 1.21. Excitation curves of the resonance at 151 keV of the 18O (p, α) 15N reaction obtained on an oxide of 109 nm of Si16O2 on a 6H -SiC substrate (oxidation of the carbon face), annealed under 200 mbar of 18O2 at 1,100°C during 2, 4 and 8 hours
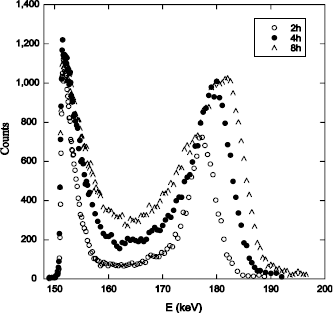
Figure 1.21 represents the excitation curves of the resonance at 151 keV of the 18O (p, α) 15N reaction, obtained after sequential oxidations 16O2/18O2. These curves show that the 18O oxygen is located on the surface and at the interface, as in the case of silicon oxidation, but also in the oxide volume. The interaction of the CO molecule with silica was studied by isotopic labeling [CAV 09].
Results obtained during the 13C18O processing of Si16O2 films on silicon showed that 18O oxygen has been fixed on the surface (gradual transport of oxygen atoms induced by the presence of defects) and in the oxide volume (CO diffusion through oxide with a low exchange frequency). The results of Figure 1.20 could thus be interpreted as the highlighting of CO production, during SiC oxidation.
1.6. Transport equations in the case of thermal oxidation
In section 1.4, we established the transport equations for oxidation in general and we saw how these are applied for thermal and anodic oxidation. In section 1.5, we have shown how it is possible to experimentally identify the species moving during oxidation and by which means they move, for the two types of oxidation. In this section, we will pay more attention to detailing how transport equations are applied in the case of thermal oxidation. The proceedings developed in this section could be applied in the case of silicon oxidation, as well as with other materials.
1.6.1. General information on flux and on growth kinetics
If Ni is the number of “defects” or of mobile species necessary to form a unit oxide volume, then the growth rate is expressed as follows:
[1.28] ![]()
with X, the oxide thickness, t, the oxidation time and |J|, the flux amplitude of mobile species, where the growth occurs (SiO2/Si interface in the case of the Si oxidation). The problem is to determine J, and two scenarios arise:
— mobile species are neutral;
— mobile species are charged.
1.6.2. Flux calculation for neutral mobile species
In this case, the flux is expressed by:
[1.29] ![]()
If the flux is conservative (there is no accumulation of mobile species in oxide), ![]() is constant and could be calculated from the concentrations of the mobile species present at the gas/oxide interface Cs and at the oxide/silicon interface Cin (see following section).
is constant and could be calculated from the concentrations of the mobile species present at the gas/oxide interface Cs and at the oxide/silicon interface Cin (see following section).
1.6.3. Flux calculation for ion mobile species
Even though no external electric field is applied during a thermal growth for reasons of equilibrium, the chemical potential of the substrate and of the dissolved oxygen creates a potential V, through oxide.
1.6.3.1. Case of very thin films (X ≤ 3 nm)
This theory has been developed by Cabrerra and Mott [CAB 49]. When the oxide thickness is lower than 3 nm, the electric field resulting from the potential can be very large. Thus, the Ji flux of “atomic defects” is given by:
![]()
Cabrera and Mott showed that the interfaces (gas/oxide and oxide/substrate) determine the Ji flux. The product a.Ci represents the density of mobile species injected to the interfaces, and γ 0 the hopping probability per unit of time. The potential barrier that the species must cross at the interfaces is a priori different from the one existing within oxide.
If the film is sufficiently thin, an electron flux by tunneling current Je will come to counterbalance the ion flux Ji. If the concentration of charged species is negligible in oxide, we will then be able to neglect the variations of ε(x) with the depth, and we will be able to write:
![]()
X being the obtained oxide thickness, thus:
![]()
and the growth rate is given by:
![]()
Cabrera and Mott assume that the potential V remains constant during oxidation and that it is determined by the balance currents of the interfaces. Thus:
[1.30] ![]()
with ![]() and
and ![]() .
.
By integration, the processing duration t, required to have an oxide thickness X, is:
[1.31] ![]()
This integral can only be calculated digitally [FRO 77]. We will nevertheless be able to give approximations by neglecting, for example, the higher orders in X/X1. In this case:
[1.32] ![]()
and:
[1.33a] ![]()
[1.33b] ![]()
A better approximation has been proposed by Ghez [GHE 73]:
[1.33c] ![]()
1.6.3.2. Case of thick films (X >> 3 nm)
The internal electric field is now low. The atomic transport is thus now to the diffusion and the conduction of mobile species. In stationary state, the electric current must be equal to zero and the transport of the mobile charged species imposes that an electronic current must be set up, to ensure the electrical neutrality of the system, defined by:
where Je(x) is an electron flux (hole or electron) with γ = +1 for the holes and γ = −1 for the electrons. Ji(x) is the flux of the mobile species of index i and of charge Zi (it is an ion flux).
Thus, Ji(x) and Je(x) can be expressed by:
with μi and μe the respective mobilities of ion and electronic species.
In general, the last three equations are difficult to use. However, they can be simplified if we consider that only the ion species move. Thus, the electro-neutrality will be ensured by electronic species of opposite polarity compared to the ion species, and in using the Einstein law:
![]()
We can write that:
[1.37] ![]()
Equation [1.34] becomes:
[1.38] ![]()
Thus, relations [1.35] and [1.36] become:
While combining [1.39] and [1.40], we have:
[1.41] ![]()
To satisfy the electric neutrality, we have:
[1.42] 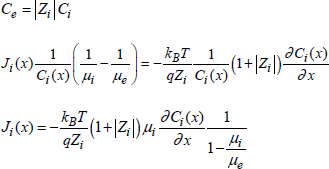
[1.43] 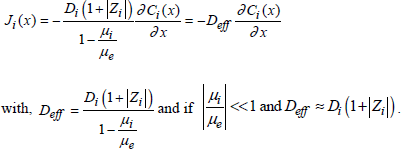
When several species are mobile, equations [1.34], [1.35] and [1.36] become too complex to be solved analytically. This case can be solved when electrons and holes are at the equilibrium and when their mobility is much higher than the ion mobility. The other cases are too complicated and have not been looked at for the silicon thermal oxidation.
1.7. Deal and Grove theory of thermal oxidation
The basic assumption of Deal and Grove is that oxidizing species, dissolved in oxide in interstitial position, migrate up to the SiO2/Si interface, to react there and form the new oxide. During transport, they could also react with the silica network. Thanks to this process, the authors were able to establish the growth kinetics of silica on silicon, largely used nowadays.
This theory is in agreement with the experimental results regarding thermal oxidation under O2 for thicknesses higher than 40 nm, as well as oxidation under water vapor for the whole range of thicknesses.
In order to establish growth kinetics, we must, first of all, determine the flux compared to the above basic assumptions, without taking into account of a possible reaction of the species in silica during their transport. This particular point will be the subject of section 1.8.
1.7.1. Flux calculation
Let us call Fg the flux of oxidizing species in the gaseous phase, F1 the flux at the gas/oxide interface, F2 the flux in oxide, and F3 the flux at the oxide/silicon interface. Figure 1.22 shows the concentrations of species, as well as these flux.
In steady flow, all flux are equal and constant:
[1.44] ![]()
Figure 1.22. Flux and distributions of the oxidizing species according to the Deal and Grove theoretical model
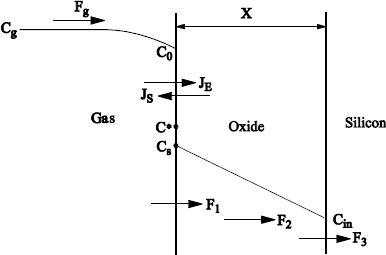
1.7.1.1. Flux in gas Fg and net flux at the gas/oxide interface F1
For a given gas pressure, Pg, we will have a concentration of oxidizing species Cg proportional to Pg (perfect gases hypothesis). Near the interface, a part of the molecules dissolves in oxide, by the way producing a slight drop of the oxidizing species concentration at the proximity of the surface in the gas C0. It will induce a flux, Fg = hg (Cg − C0), where hg is a transfer speed of molecules in gaseous phase.
F1, a net flux at the gas/oxide interface, can be expressed by the difference between the entering and outgoing flux:
[1.45] ![]()
If Cs is the concentration of oxidizing species dissolved in oxide on the surface, and if we make the hypothesis of a first order proportionality between the outgoing flux Js and Cs, we will have:
[1.46] ![]()
When oxidation stops, F1 = 0 and Cs reaches the limiting value C*, which depends on the external pressure of the gas.
With the free volume model, we can write:
where ![]() , is the ratio of the free volumes in the gas and in the oxide of the transported species. C* is of about a few ppm at atmospheric pressure. Expression [1.47] is equivalent to the Oswald law, stipulating that at equilibrium, the concentration of species dissolved in the solid, is proportional to the one in gas. It is also an adaptation of Henry’s law, of the dissolution of a gas in a solid and not in a liquid.
, is the ratio of the free volumes in the gas and in the oxide of the transported species. C* is of about a few ppm at atmospheric pressure. Expression [1.47] is equivalent to the Oswald law, stipulating that at equilibrium, the concentration of species dissolved in the solid, is proportional to the one in gas. It is also an adaptation of Henry’s law, of the dissolution of a gas in a solid and not in a liquid.
NOTE.— In the case of steam oxidation, we must consider that C* is really in relation to the mobile species, and not with the OH clusters, formed in silica during oxidation.
Thus, if F1 = 0 with CS = C*, we have 0 = JE − h Cs and thus:
1.7.1.2. Net flux in oxide, F2
The first Fick law in 1D (one dimension) gives us:
![]()
As the flux is constant at the equilibrium and assuming that D is a constant, we can write that:
if X is the thickness of the formed oxide and Cin, the concentration of oxidizing species present at the oxide/substrate interface.
1.7.1.3. Flux at the oxide/silicon interface, F3
If we consider a first order chemical reaction at the interface (e.g. ![]() , then:
, then:
with kr the reaction constant. By using equations [1.48], [1.49] and [1.50], and by setting that in steady state F1 = F2 = F3 = F, we have:
[1.51] ![]()
As C* is the only variable directly connected to the gas concentration,. it is convenient to express all in functions of C* and:
[1.52] 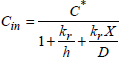
[1.53] 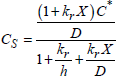
Thus:
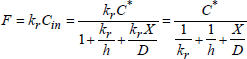
By writing that ![]() in [1.54]:
in [1.54]:
[1.55] 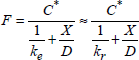
Bearing in mind that, generally, h >> kr, even in the event of non-equilibrium, we can write that CS ≈ C* and ke ≈ kr.
1.7.2. Growth kinetics equations
Let us remember that Ni represents the number of oxidizing species necessary to form a unitary volume of silica (Ni = 2.25 1022 cm-3 for O2 and Ni= 4.5 × 1022cm-3 for H2O). Thus, in agreement with [1.54], we obtain the growth rate:
[1.56] 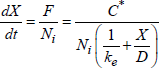
By integration:
[1.57] ![]()
By using the initial conditions that t = 0, X = X0 (X0 being the initial oxide thickness), then:
[1.58] ![]()
which can be written:
with:
![]()
[1.59] will have as solution:
[1.60] ![]()
Let us consider the two limiting cases:
— Short times, low thicknesses when: ![]() .
.
In this case, the equation [1.59] could be approximated by:
[1.61] ![]()
with ![]() , the linear constant giving place to the linear oxidation regime.
, the linear constant giving place to the linear oxidation regime.
— Long times, high thicknesses when: ![]() .
.
In this case, equation [1.59] could be approximated by:
![]()
That is to say:
[1.62] ![]()
with ![]() , the parabolic constant giving place to the oxidation regime in
, the parabolic constant giving place to the oxidation regime in ![]() .
.
In such a situation, the oxidation rate is controlled by the diffusion, but also by the concentration limit C* defined by the gas pressure.
The constant ![]() , represents a critical thickness below which oxidation follows a linear law and beyond which it follows a parabolic law.
, represents a critical thickness below which oxidation follows a linear law and beyond which it follows a parabolic law.
Equation [1.59] can be also written as:
1.7.3. Remarks on the variations of the oxidation constants kP and kL
As seen previously, if we use the free volume model, C* is proportional to 1/T; this dependence will be more complex in a statistical model.
Experimentally, it was shown that C* slightly decreases when T increases. Thus, for a restricted temperature range, we will be able to consider as a first approximation that C* remains constant. And if the hypothesis is made that the thin SiO2 films behave in the same way as thicker silica, we will be able to establish that oxidation constants kP and kL, proportional to C*, will be proportional to the gas pressure of Cg processing.
Moreover, ke ≈ kr, then ![]() , that is to say that kL and kr will have the same activation energies, if we neglect the variations of C* depending on T. In the same way, kP will have the same activation energy as C*, if we neglect the variations of C* depending on T.
, that is to say that kL and kr will have the same activation energies, if we neglect the variations of C* depending on T. In the same way, kP will have the same activation energy as C*, if we neglect the variations of C* depending on T.
In addition, the critical thickness A proportional to D/kr, will be independent of the pressure and will have an activation energy equal to the difference of the one of D(Wo) and the one of kr(Wr). So, when A decreases T increases, if Wr is higher than Wo, as we will see later on.
1.7.4. Determination of the oxidation parameters from experimental results
By deriving equation [1.63], we have:
[1.64] ![]()
which gives a linear dependence of the reverse of the speed:
[1.65] ![]()
Thus, from experimental results of X as a function of t, dt/dX is represented according to X in Figure 1.23.
Figure 1.23. Two curves for dt/dX functions: a) X; and of b) X according to (t+ τ)/X
The slope of this curve will be equal to 2/kP and the intersection with the axis X = 0 will be equal to 1/kL.
Another mean to determine the constants is to notice that equation [1.59] can be written:
[1.66] ![]()
By delineating X according to (t+ τ)/X (Figure 1.23b), we can also determine kP and kL.
t can be given previously by plotting X as a function of t, by extrapolating the curve at the ordinate axis X = 0 (see Figure 1.24).
Figure 1.24. Kinetics of growth: X functions of t curve
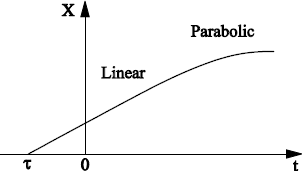
1.7.5. Confrontation of the Deal and Grove theory with experimental results
Nevertheless, the linear parabolic law of the Deal and Grove theory is in good agreement with the experimental results in the following cases:
— in the case of wet oxidation for all thicknesses;
— in the case of dry oxidation under O2 for thicknesses higher than 20–30 nm (for thinner thicknesses, the growth is characterized by a fast growth regime). In Figure 1.25, this restriction is illustrated for an oxidation carried out under O2 at 700°C for variable times [DEA 65]. The value of X0 which must be added in the Deal and Grove equation is equal to 20–30 nm for processing temperatures ranging between 700°C – 1,200°C; the processes being carried out at atmospheric pressure.
Figure 1.25. Silicon oxidation under dry oxygen at 700°C (according to Deal and Grove [DEA 65])
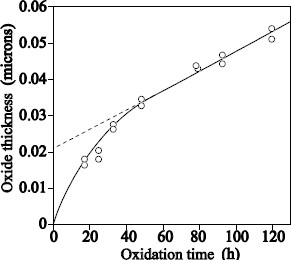
Figures 1.26 [DEA 65, WOL 69] and 1.27 [IRE 78b] show oxidation kinetics. The variations of oxidation constants B, B/A and A as functions of 1/T at atmospheric pressure are represented in Figures 1.28, 1.29 and 1.30 for wet oxidation, as well as for dry oxidation. We will further detail the experimental results. However, we can see that the constants kP and kL are higher for water vapor than in the case of O2.
Figure 1.26. Oxide thickness as a function of the time for various processing temperatures. The full lines represent the results for wet oxidation and the dotted lines, for dry oxidation. Silicon (111) oxidation according to Wolf [WOL 69] based on the data of Deal and Grove [DEA 65]
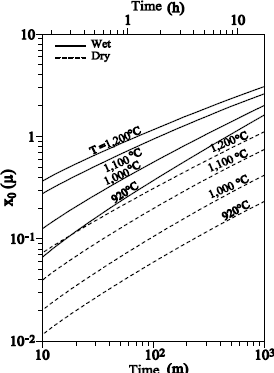
The difference in the case of the parabolic constant can easily be understood by the fact that the diffusivity and solubility of the H2O molecule in silica are higher than those of O2. The respective values of activation energy (16.3 kcal for H2O and 28 kcal for O2) are in harmony with those found in the case of compact silica.
The difference observed for the linear constant can be due to the highest solubility of the water vapor in silica and/or to a more important reactivity of the latter at the SiO2/Si interface. The corresponding value of the activation energy is independent of the environment and is worth 45 kcal, which is very close to the value necessary to break the Si-Si bond, i.e. 42.2 kcal.
Figure 1.27a. Oxide thicknesses obtained by silicon (100) oxidation, little doped (STD), very doped phosphorus (P), very doped boron (B), for various temperatures under dry oxygen at atmospheric pressure (according to Irene and Dong [IRE 78b])
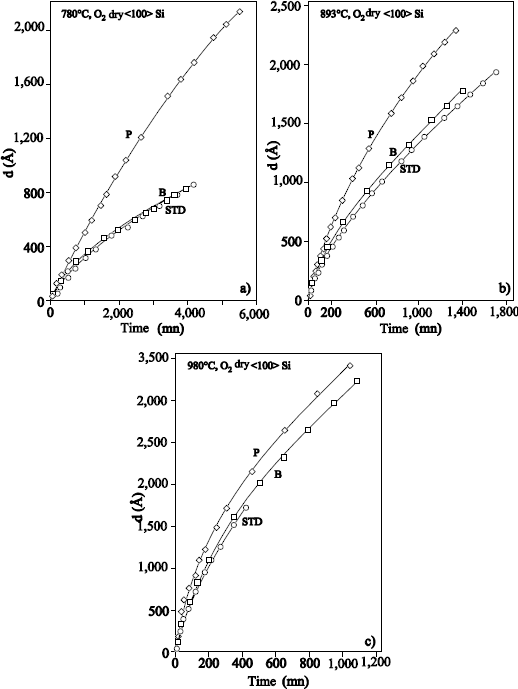
Figure 1.27b. As in Figure 1.27a for other temperatures (according to Irene and Dong [ANGER 78b])

Figure 1.28. Effect of the temperature on kP = B (according to Deal and Grove [DEA 65])
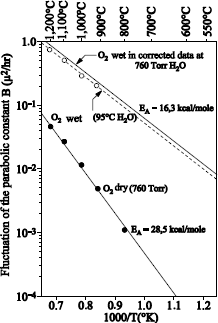
Figure 1.29. Effect of the temperature on kL = B/A (according to Deal and Grove [DEA 65])

Figure 1.30. Effect of the temperature on kP /kL = A (according to Deal and Grove [DEA 65])
1.7.6. Conclusions on the Deal and Grove theory
As seen in section 1.5, the theoretical framework leading to the equation of kinetics [1.59] does not take account of the three following phenomena:
— the exchange of oxygen on the surface;
— the oxygen loss at the interface probably due to the SiO exo-diffusion towards the gaseous phase;
— the initial phase with roughening/existence of fragments and/or presence of an electric field.
If we do not expect a specific incidence on the kinetics from the first phenomenon, on the other hand, the two other phenomena should be taken into account, in order to predict with more accuracy the growth kinetics in case of dry oxidation.
For practical reasons, we will be able, as a first approximation, to use the results obtained by the Deal and Grove theory, and more particularly the constants of this, defined in Figures 1.28, 1.29 and 1.30. However, it is necessary to pay particular attention to the processing conditions, such as the temperature, the partial pressure, the orientation, the doping, etc. This aspect is discussed in section 1.10.
1.8. Theory of thermal oxidation under water vapor of silicon
In section 1.7, the Deal and Grove theory supposes that mobile species diffuse through the oxide without reacting with it. If these species react, is it possible to produce a theory taking account of this reaction and which can satisfactorily explain the experimental results?
SiO2 is not the only chemical species produced during the reaction of the water vapor with silica. Hydrogen is produced at the SiO2/Si interface. Hydrogen and water react with silica and produce SiOH and SiH. The composition of the silica film is altered, which can change its physical and transport properties. Breed and Doremus [BRE 76] proposed a theory, developed below, supposing that H2O is the mobile species.
1.8.1. Concentration profiles expected for H2O
In stationary state, the flux of mobile molecules of H2O is constant and expressed by:
[1.67] ![]()
where ![]() and [H2O] are respectively the diffusion coefficient and the concentration of water molecules.
and [H2O] are respectively the diffusion coefficient and the concentration of water molecules.
If ![]() and [H2O] are the concentrations of the water molecules on the external surface and at the SiO2/Si interface, we have:
and [H2O] are the concentrations of the water molecules on the external surface and at the SiO2/Si interface, we have:
[1.68] ![]()
where X is the silica thickness. By integration, we obtain:
[1.69] ![]()
1.8.2. Concentration profiles expected for the OH groups
If we postulate that the SiOH groups and H2O molecules are in equilibrium according to the reaction:
![]()
we have:
![]()
where ![]() is the equilibrium constant of the previous reaction, we obtain:
is the equilibrium constant of the previous reaction, we obtain:
[1.70] ![]()
If ![]() is negligible, we have:
is negligible, we have:
[1.71] ![]()
1.8.3. Concentration profiles expected for H2
The water molecules react at the SiO2-Si interface, to form SiO2 and H2, according to the reaction:
![]()
The hydrogen formed at the interface diffuse through the oxide. In stationary state, the flux of ![]() of H2 is constant, equal in absolute value to that of the water molecules:
of H2 is constant, equal in absolute value to that of the water molecules:
[1.72] ![]()
where ![]() and [H2] are respectively the diffusion coefficient and the concentration of H2 molecules.
and [H2] are respectively the diffusion coefficient and the concentration of H2 molecules.
If ![]() and
and ![]() are the concentrations of H2 molecules on the external surface and at the SiO2/Si interface, we have:
are the concentrations of H2 molecules on the external surface and at the SiO2/Si interface, we have:
[1.73] ![]()
We thus obtain:
[1.74] ![]()
Since ![]() , we have the following relations:
, we have the following relations:
[1.75] ![]()
If ![]() and
and ![]() are negligible, we have:
are negligible, we have:
[1.76] ![]()
[1.77] ![]()
[1.78] ![]()
1.8.4. Concentration profiles expected for H
H2 reacts with silica. If we assume that the reaction:
[1.79] ![]()
occurs at the equilibrium, we have:
[1.80] ![]()
where [H] is the concentration of SiH groups, and ![]() is the equilibrium constant of the reaction.
is the equilibrium constant of the reaction.
By using the previous expressions of [H2] and [OH ] in parabolic regime, we obtain:
[1.81] 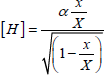
α is given by the expression:
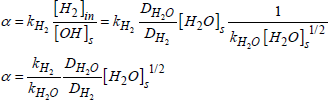
Figure 1.31. Expected concentration profiles of the various species present during the silicon oxidation in water vapor

1.8.5. Comparison of the expected and the experimental profiles
Beckmann and Harrick [BEC 71] have determined the [OH] and [SiH] concentrations using infrared analysis techniques. It was not possible to distinguish SiOH from H2O, but it is reasonable to think that the H2O concentration is negligible. The experimental profiles obtained are always in good agreement with the theory. The SiH group’s concentration highly increases close to the interface, in agreement with the theory.
However, an increase in the concentration of the SiH groups is observed close to the external surface, contrary to the theory.
Burkard [BUR 67] grew oxides under water vapor enriched in tritium, and measured, by radioactive analysis, the total concentration of the hydrogenated groups [HT].
If we neglect [H2O] and [H2]:
[1.82] 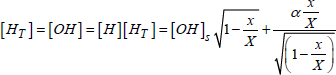
A minimum must be observed for [HT] at the following abscissa:
![]()
We observe a good harmony between the form of the observed profile and the theory.
1.8.6. Wolters theory
Wolters [WOL 80] has developed a theory, according to which the diffusing species are H3O+ and OH-.This theory also tallies with the experimental results of Burkard [BUR 67].
1.9. Kinetics of growth in O2 for oxide films < 30 nm
1.9.1. Introduction
The Deal and Grove theory models the growth kinetics of oxide only beyond the X0 value of the model (30 nm in the range of temperatures 700°C – 1,200°C, at atmospheric pressure). We present here alternative theories able to report a regime of faster initial growth, observed for films < 30 nm (see Figure 1.32).
Figure 1.32. Inverse of the growth rate from the thickness, for oxidations carried out under 1 atm of O2 (according to [HOP 75])
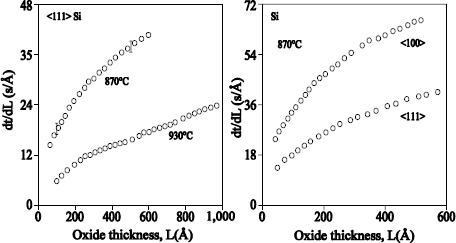
The growth of thin films is sometimes carried out at pressures lower than those of thicker films, or in inert gas/O2 mixtures. It makes it difficult to compare between their kinetics and those of thicker films. Indeed, pressure affects the kinetics.
At low pressure, oxidation is slower on the face (111), than on the face (100). At higher pressure, oxidation is faster on the face (111), than on the face (100), the transition taking place around 10-1 atm [KAM 77, RAI 80]. In the case where oxygen is diluted in an inert gas, the water vapor pressure can become considerable compared to the partial O2 pressure.
Moreover, the surface quality of the substrate (native oxide, impurities) influences the kinetics [DEL 87]. This influence will be greater, as the film thickness is low.
First, we expose below some thin film growth kinetics, which can be useful for the reader.
Figure 1.33. Growth kinetics, Si (100), under O2 atmosphere [CHA 91]
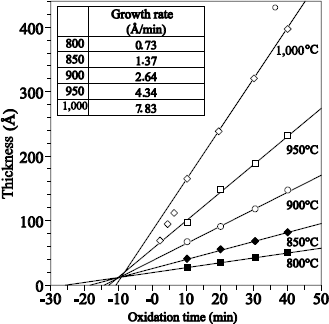
Figure 1.34. Growth kinetics, Si (100), under O2 atmosphere [MAS 85]
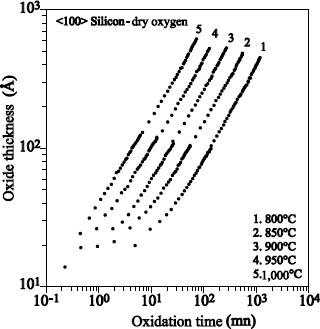
Figure 1.35. Growth kinetics, Si (100), under O2 atmosphere [IRE 78]
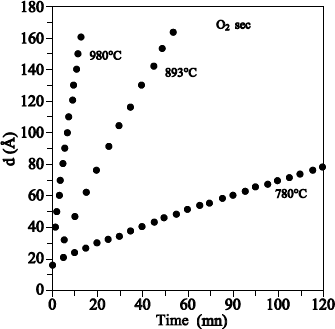
Figure 1.36. Growth kinetics, Si (111), under O2 atmosphere [HOP 75]
Figure 1.37. Growth kinetics, Si (100), under O2 atmosphere, by rapid thermal oxidation [FUK 92]
Figure 1.38. Growth kinetics, Si (100), oxidation in a O2/N2 mixture for various partial O2 pressures [KAM 77]
Figure 1.39. Growth kinetics, oxidation in a O2/N2 mixture, for various partial O2 pressures [VAN 72]
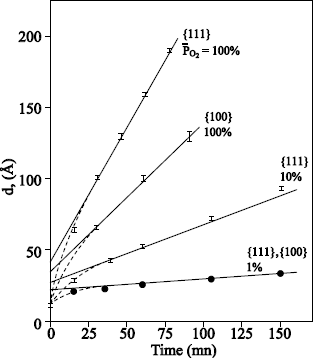
Figure 1.40. Growth kinetics, Si (100), for ultra-thin oxide films [KRZ 07]
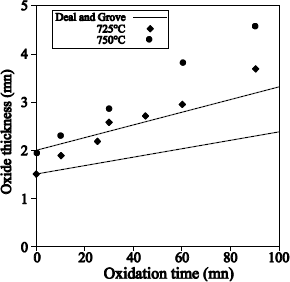
Figure 1.41. Growth kinetics, Si (100), for ultra-thin oxide films, at low temperatures [FEH 72]
Figure 1.42. Growth kinetics, Si (111), for ultra-thin oxide films, at low O2 pressures [DER 82]

1.9.2. Oxidation models of thin films
During the 1970s and 1980s, a very significant effort was made, in order to develop a silicon oxidation model, able to report the initial growth process. For a review of the existing models at the time, we can refer to Massoud [MAS 85] and Irene [IRE 88]. We mention here some of the models published during this period.
Hopper et al. [HOP 75], then Han and Helms [HAN 87], suggested that two oxidation mechanisms took place in parallel. However, isotopic labeling experiments [ROC 84] showed that the contribution of a second mechanism to the growth is negligible beyond 5 nm.
Ghez and Van der Meulen [GHE 72] have put forward the idea that three reactions were in competition at the interface. Blanc [BLA 78] simplified this model with two reactions. But none of these two models satisfactorily adjust the kinetics.
Fargeix et al. [FAR 83a, FAR 83b] and Tiller [TIL 83] suggested the existence of a blocking layer close to the interface, where the diffusion coefficient is lower. One of the potential explanations is a compressive stress located near the SiO2/Si interface [FAR 83b]. With this model, we can correctly adjust the kinetics of growth, but we cannot predict the variations of the oxidation rate with the pressure. More recently, Watanabe et al. [WAT 07] also proposed a model based on the existence of a strained layer of about 1 nm. There again, the model does not predict the variations of oxidation rate with pressure.
In the Massoud model [MAS 85], the oxidation rate in the regime of thin films, under many variable experimental conditions (direction of the substrate, doping, partial oxygen pressure), can be empirically expressed by:
![]()
The first term of the expression corresponds to the Deal and Grove equation. The two exponential terms represent the increase of growth rate in the regime of thin films. The first exponential term has a characteristic length of about 1 nm. It becomes negligible for thicknesses higher than 5 nm. The second exponential term has a characteristic length of about 7 nm and becomes negligible for thicknesses higher than 25 nm. To explain all their experimental results and the fact that the two characteristic lengths are independent of the direction of the substrate, of the doping, of the partial oxygen pressure, Massoud et al. suggested that the increase of the growth rate in the regime of thin films is due to an excess of reactive sites at the interface. This model leads to the correct adjustment of many kinetics, for thicknesses higher than 5 nm, and predicts rather correctly the effects of the pressure on oxidation kinetics.
Kageshima et al. [KAG 99] have rewritten the Deal and Grove transport equations, by adding three transport methods: i) interstitial silicon atoms are created at the interface; ii) the interstitials diffuse in the oxide layer; iii) the interstitials are absorbed in oxide to form Si-O-Si. An additional hypothesis is that the emission of Si at the interface decreases with the interstitial density at the interface. Their equation of dX/dt as afunction of X has the same expression as that of Massoud, for C1=0, if the Si emission rate is much greater than the rate of absorption in oxide. In other words, to a certain extent, their theory can explain the empirical equation of Massoud et al. This model, commonly called the “Si interfacial emission model”, is currently used for the kinetics adjustment. It provides a sub-linear fluctuation of the oxidation rate with the pressure (m = 0.5, slightly different from the values experimentally observed: (m = 0.6–0.8). With this model we cannot correctly adjust kinetics for films grown at sub-atmospheric pressure.
The physical phenomena at the origin of this fast initial regime are not given (strained layer at the interface, silicon emission, etc.). No model has unanimous support, nor is able to universally adjust the kinetics obtained under various experimental conditions. The Massoud empirical model, with C1= 0, is very largely used in simulation software, for its efficiency and its simple analytical form. The model of silicon interfacial emission is the most frequently used for the kinetics adjustment, because it is currently the most satisfactory non-empirical model, most oxidations being carried out at atmospheric pressure.
1.9.3. Case of ultra-thin films (< 5 nm)
We saw in section 1.5, that the gradual movement of oxygen atoms of the network, related to the presence of defects in the oxide, contributed to the growth. This contribution is more important when the film is thin and when the O2 pressure is low [ROC 84, TRI 89].
For these ultra-thin films, Fehlner [FEH 72] obtained a satisfactory adjustment by assuming a Cabrera-Mott transport. Derrien and Commandré [DER 82] adjusted their experimental results by assuming a constant electric field. Ganem et al. proposed the oxidation of Si fragments present in oxide [GAN 93b] and De Almeida et al. [DEA 00] rewrote the transport equations without the hypothesis of a stationary state and an abrupt interface. To this day, there is no valid model for ultra-thin films that is able to fill the gaps of the models of Massoud and of the model of interfacial emission.
1.9.4. On line simulator
Process Lab: Oxidation [CAO 06] is an online simulator of the Deal and Grove and/or Massoud models, available at: http://www.nanohub.org/.
1.9.5. Kinetics and models of SiC oxidation
Figure 1.43. Oxidation kinetics of the logotype 4H-SiC under an O2 atmosphere: a) face C; b) face a; c) face Si (from [SON 04]). The lines correspond to an adjustment according to the Deal and Grove model
Figure 1.44. Oxidation kinetics of the logotype 4H of SiC at 1,150°C, under atmospheres of O2 between 0.25 atm and 4 atm: a) face C; b) face a; c) face Si (from [RAY 08]). The lines correspond to an adjustment according to the Deal and Grove model
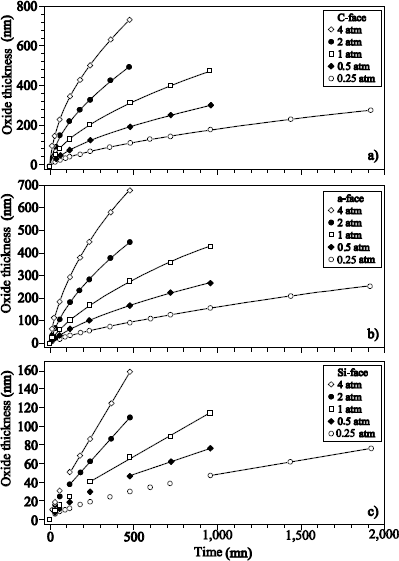
As seen in section 1.5, the oxidation of silicon carbide (SiC) produces SiO2. Various oxidation models have been proposed [HIJ 09, SON 04], based on the adjustment of growth kinetics of the oxide. First of all, we present a set of kinetics of the logotype 4H, for three directions: (000–1) face C, (0001) face Si and (11–20) face a, carried out at 1 atm of O2, for temperatures from 950°C to 1,150°C, and at 1,150°C for O2 pressures between 0.25 and 4 atm.
We now present two models able to report SiC oxidation mechanisms.
To adjust their kinetics, Song et al. proposed a Deal and Grove model [SON 04], corrected in [RAY 08], taking into account the exodiffusion of the products of the oxidation reaction:
![]()
By following the Deal and Grove approach, they obtained modified expressions of B/A and B:
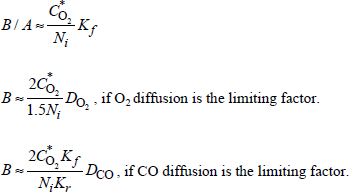
where Kf and Kr are respectively the constants of the direct and opposite reaction.
At 1,150°C, B and B/A are proportional to the O2 pressure, for p = 0.25 − 2 atm [RAY 08]. Moreover, the values of B are similar on faces a and C, suggesting that the limiting factor is the O2 diffusion through oxide.
Yamamoto et al. [RAY 08, YAM 07] have used the empirical equation of Massoud [MAS 85] to fit a large number of oxidation kinetics of the Si and C faces of the 4H polytype of SiC.
In the case of silicon oxidation, the empirical equation by Massoud can be justified by the model of Si interfacial emission. It was thus natural to adapt this model to the SiC oxidation. Hijikata et al. [HIJ 09] proposed the “model of carbon and silicon emission”. This model helped them to correctly adjust curves of oxide growth rate, according to the oxide thickness, for the faces Si and C of the polytype 4H of carbide. However, demonstration of the validity of the model is difficult because of the large number of adjustable parameters that it contains.
1.10. Variations of the oxidation coefficients with experimental conditions
This section is intended for the process engineers, who must predict the oxides thicknesses once the process conditions are established. To be able to use the Deal and Grove relationship, it is necessary to know the oxidation coefficients, kP and kL as precisely as possible. The theory predicts how they vary with certain experimental parameters. The effective experimental conditions can give coefficients different from the theory. Moreover, some other parameters, such as crystalline direction or substrate doping, are not taken into account in the theory.
1.10.1. Role of the pressure
The pressure role is examined, by comparing experimental data under similar conditions.
1.10.1.1. Wet oxidation
In this case, kP and kL vary linearly with PH2O, for all the temperature ranges and pressures examined (50–760 Torr for 1,000°C < T< 1,200°C [DEA 65], 1–20 atm for 800°C < T < 1,000°C [RAZ 81]). This dependence is well predicted by the theory.
1.10.1.2. Dry oxidation
In this case, kP still varies linearly with PO2 pressure, for various conditions (50–760 Torr for T = 1,200°C [DEA 65], 15–760 Torr for 1,100°C [KAM 77], 130–1,200 Torr for 870°C < T < 930°C [HOP 75], 1–20 atm for 800°C < T < 1,000°C [LIE 82]). This result corresponds to the theory.
Contrary to kP, kL varies as ![]() and m takes various values:
and m takes various values:
m = 1 for P=−760 Torr and T = 1,200°C [DEA 65];
0.7 < m < 0.8 for P=−20 atm and 800°C < T < 1,000°C [LIE 82];
m = 0.6 for P=−760 Torr and 1,100°C [KAM 77].
For films thinner than 30nm, we obtain:
![]() , with m varying between 0.59 and 0.83 when 700°C < T < 1,000°C for substrates (111), and 0.54 and 0.66 when 850°C < T < 1,000°C for substrates (100) [GHE 72].
, with m varying between 0.59 and 0.83 when 700°C < T < 1,000°C for substrates (111), and 0.54 and 0.66 when 850°C < T < 1,000°C for substrates (100) [GHE 72].
As seen previously:
![]()
This leads us to an interfacial reaction constant, ![]() , which probably results from the reactive mechanisms occurring at the SiO2 interface, the latter having various configurations.
, which probably results from the reactive mechanisms occurring at the SiO2 interface, the latter having various configurations.
1.10.2. Role of the temperature
The role of temperature is examined by comparison of various work completed for identical pressures.
1.10.2.1. Wet oxidation
The Arrhenius diagrams of the coefficients kP and kL are represented in Figure 1.45 [RAZ 81] for processes under various pressures of pyrogenic steam. For kP the activation energy, corresponding to temperatures higher than 900°C, is 18 kcal/mole (0.78 eV). It reaches 27 kcal/mole (1.17 eV) for lower temperatures.
Figure 1.45. Parabolic and linear coefficent for oxidations under water vapor of silicon (100) and (111) for various temperatures and pressures (from Razouk [RAZ 81])
For kL, the change of activation energy is less obvious and is around 40 kcal/mole.
The change of activation energy could be due to a change of silica structure, which would begin its viscous transition, as Eernisse suggests [EER 79], for temperatures ranging between 900 and 950°C.
1.10.2.2. Dry oxidation
The Arrhenius diagrams of kP and kL, measured by Lie, Razouk and Deal are given in Figure 1.46. They correspond to oxidations at atmospheric pressure for temperatures ranging from 750 to 1,150°C. STD represents samples of average resistivity 2 Ω cm, B and P, bore and phosphorus doped samples of low resistivity (0.001 Ω .cm).
For higher pressures, we will refer to Figure 1.47.
The variations of the activation energies of the coefficients are enumerated and compared in Table 1.4.
The activation energy of kP changes around 900°C as it is in the case of wet oxidation.
Above 900°C, its value is around 1.3 eV and, below, around 2.2 eV. It also refers to a change of oxide structure.
Figure 1.46. Linear (Å/min) and parabolic (Å2/min) coefficients for oxidations under dry O2 of silicon (100) at atmospheric pressure, for various temperatures (from Lie [LIE 82])
Figure 1.47. Parabolic and linear coefficients for oxidations under dry O2 of silicon (111) for various temperatures and pressures (from Lie [LIE 82])
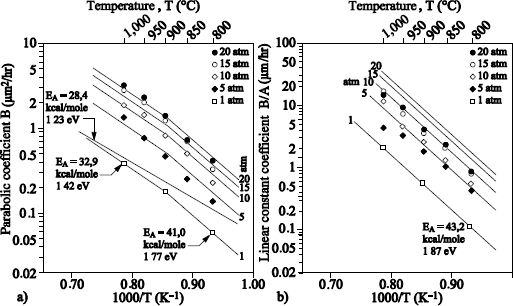
Table 1.4. Compared activation energies for dry oxidation under O2 (from [IRE 78b])
Fargeix and Ghibaudo [FAR 84] point out that the activation energy of kP, measured by the authors, is in fact only an apparent energy Wap, which can take values much higher than the fundamental activation energy Wo of kP. This can be understood, if we express:
![]()
Thus, a variation of 0.1 eV of the value of Wo can explain variations of about 1 eV for Wap. 0.1 eV is compatible with a fluctuation of the oxide diffusivity, because of the unreleased stresses at low-temperature. In this case, we can express the diffusion coefficient by:
![]()
where σ represents the stress, ΔV the consecutive variation of volume, and D0 the diffusion coefficient in unstressed material.
For the kL variations, Lie [LIE 82] reports values of activation energy ranging between 43.3 and 43.7 kcal/mole, for temperatures ranging between 800°C and 1,000°C, in agreement with the energy necessary to break the Si-Si bond (42.2 kcal/mole).
Irene and Dong [IRE 78b] report activation energies of 2.2 eV for temperatures higher than 980°C. However, below 900°C, these values would be 1.5 eV, which corresponds to the energy necessary to break the O-O bond.
1.10.3. Role of the crystal orientation
The oxidation rate also depends on the crystal orientation of the substrate. The oxidation rate is higher for the (111) orientation than for the (100) orientation.
1.10.3.1. Wet oxidation
For this type of oxidation, whereas the parabolic constant would not depend on the direction of the substrate, the linear constant would be strongly altered. Thus, from Figure 1.45, we find:
![]()
Ligenza [LIG 61] explained why the orientation of the substrate modifies the number of reactive sites, which remains much lower than the density of the Si-Si bond on the surface and would be related to the steric hindrance of the water molecule. Thus, by a geometrical calculation, he deducted that the ratio of the densities of available sites would be 1.73 between a (111) and a (100) surface.
1.10.3.2. Dry oxidation
In Figure 1.48, we can clearly see the influence of the crystalline orientation of the substrate on the oxidation rate. From these results, we can express the following relation: kP (111) > kP (110) > kP (100). It is valid for any type of oxidation. Nevertheless, for a temperature higher than 996°C, the parabolic constant no longer depends on the orientation.
Figure 1.48. Oxidation kinetics for various directions and temperatures. The symbol D refers to oxidations under dry atmosphere, and the symbol W refer to a wet atmosphere (from Irene [IRE 74])
Geometrical considerations [LIG 61], lead to the following dependence: kP (111) ≥ kP (110) > kP (100). But this calculation relies on the steric hindrance of the water molecule and not on oxygen. In practice, we will find it at high pressure, as in Table 1.5, kP (110) > kP (111) > kP (100).
At very low pressure, the oxide thickness follows the relation: X (110) > X (100) > kP(110) (see Figure 1.49).
Figure 1.49. Oxidation kinetics under dry oxygen for various crystalline orientations (On the left) PO2 = 20 atm and 800°C < T < 1,000°C (from Lie [LIE 82]); and (on the right), PO2 = 10−3 − 1 atm and T =1,100°C (from Kamigaki and Itoh [KAM 77])
Table 1.5. Values of the oxidation coefficients for T = 1,000°C under O2 atmospheric pressure (from Kamigaki and Itoh [KAM 77])

1.10.4. Role of doping
Experimentally, we observe that highly doped substrates oxidize faster than less doped substrates. To explain that phenomenon, Ho and Plummer [HO 79a, HO 79b] propose a model based on the increase of reactive sites at the SiO2/Si interface. These would be point defects, such as vacancies or dangling bonds (Pb centers), whose concentration would increase for high doping, where the Fermi level moves towards the conduction-band (n-doping) or towards the valence band (p-doping).
Figure 1.50. kL(n)/kL (i) as a function of the concentration of active dopants on a substrate (111), for various temperatures [HO 79b]
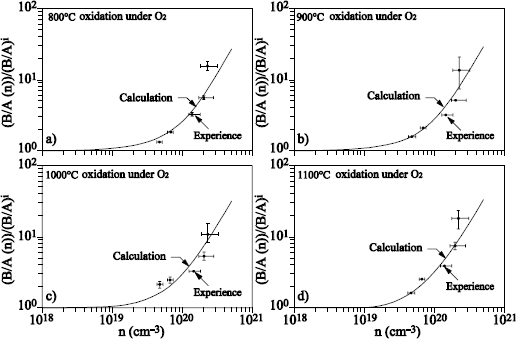
Figures 1.50 and 1.51 present the ratio of linear coefficients of growth kinetics for doped silicon (n-doping and p-doping) to linear coefficients ofgrowth kinetics for intrinsic silicon. The theory and the experiments are in agreement.
Figure 1.51. kL (p)/kL (i) as a function of the concentration of active dopants (As) on a substrate (111) for various temperatures [HO 79b]

1.11. Conclusion
Because of the many applications of silica in many domains, in particular in the electronics of integrated circuits, silicon oxidation, and more recently silicon carbide oxidation has been studied a lot in laboratories around the entire world. Still, nowadays, with the problems arising from the very high integration of transistors in circuits and in nanotechnologies, many researchers are trying to improve the understanding of the first stages of silica growth, to optimize the functional characteristics. We hope that this chapter will be a useful contribution to the development of new technologies.
1.12. Bibliography
[AKE 99] AKERMARK T., GOSSET L.G., GANEM J.-J., TRIMAILLE I., RIGO S., “Loss of oxygen at the Si-SiO2 interface during dry oxidation of silicon”, Journal of the Electrochemical Society, vol. 146, p. 3389–3392, 1999.
[AND 54] ANDERSON O.L., STUART D.A., “Calculation of activation energy of ion conductivity in silica glasses by classical methods”, Journal of the American Ceramic Society, vol. 37, p. 573–580, 1954.
[BAT 92] BATTISTIG G., AMSEL G., D’ARTEMARE E., VICKRIDGEI., “A very narrow resonance in 18O(p, α)15N near 150 keV: Application to isotopic labeling. II. High resolution depth profiling of 18O”, Nuclear Instruments and Methods in Physics Research Section B, vol. 66, p. 1–10, 1992.
[BEC 71] BECKMANN K.H., HARRICK N.J., “Hydrides and hydroxyls in thin silicon dioxide films”, Journal of the Electrochemical Society, vol. 118, p. 614–619, 1971.
[BEL 62] BELL T., HETHERINGTON G., JACK K.H., “Water in vitreous silica. Part 2. Some aspects of hydrogen-water-silica equilibria”, Physics and Chemistry of Glasses, vol. 3, p. 141–146, 1962.
[BLA 78] BLANC J., “A revised model for the oxidation of Si by oxygen”, Applied Physics Letters, vol. 33, p. 424–426, 1978.
[BRE 76] BREED D.J., DOREMUS R.H., “Hydrogen profiles in water-oxidized silicon”, Journal of Physical Chemistry, vol. 80, p. 2471–2473, 1976.
[BRU 70] BRUCKNER R., “Properties and structure of vitreous silica. I”, Journal of Non-Crystalline Solids, vol. 5, p. 123–175, 1970.
[BUR 67] BURKARD P.J., “Tracer evaluation of hydrogen in steam-grown SiO2 films”, Journal of the Electrochemical Society, vol. 114, p. 196–201, 1967.
[BUR 70] BURN I., ROBERTS J.P., “Influence of hydroxyl content on the diffusion of water in silica glass”, Physics and Chemistry of Glasses, vol. 11, p. 106–114, 1970.
[CAB 49] CaBA$ZERA N., MOTT N.F., “Theory of the oxidation of metals”, Reports on Progress in Physics, vol. 12, p. 163–184, 1949.
[CAO 06] CAO S.V., LIU Y., GRIFFIN P., Process Lab: Oxidation, DOI: 10254/nanohub-r1879.1, 2006.
[CAV 09] CAVEA$ZLIN C.D., TRiMAIA$ZLE I., GANEM J.-J., D’ANGELO M., VICKRIDGE I., PONGRACZ A., BATTISTIG G., “An 18O study of the interaction between carbon monoxide and dry thermal SiO2 at 1 100°C”, Journal of Applied Physics, vol. 105, p. 033501–033501–7, 2009.
[CHA 77] CHANG R.P.H., “Multipurpose plasma reactor for materials research and processing”, Journal of Vacuum Science and Technology, vol. 14, p. 278–280, 1977.
[CHA 80] CHANG R.P.H., CHANG C.C., DARACK S., “Fluorine-enhanced plasma growth of native layers on silicon”, Applied Physics Letters, vol. 36, p. 999–1002, 1980.
[CHA 91] CHAO T.S., LEE C.L., LEI T.F., “Measurement of ultra-thin (<100 Å) oxide films by multiple-angle incident ellipsometry”, Journal of the Electrochemical Society, vol. 138, p. 1756–1761, 1991.
[COP 71] COPELAND M.A., PAPPU R., “Comparative study of plasma anodization of silicon in a column of a DC glow discharge”, Applied Physics Letters, vol. 19, p. 199–201, 1971.
[COS 84] COSTELLO J.A., TRESSLER R.E., “Isotope labeling studies of the oxidation of silicon at 1000° and 1300°C”, Journal of the Electrochemical Society, vol. 131, p. 1944–1947, 1984.
[CRO 71a] CROSET M., PETREANU E., SAMUEL D., AMSEL G., NADAI J.P., “An O18 study of the source of oxygen in the anodic oxidation of silicon and tantalum in some organic solvents”, Journal of the Electrochemical Social, vol. 118, p. 717–727, 1971.
[CRO 71b] CROSET M., DIEUMEGARD D., Proceedings of the Electrochemical Society Meeting, p. 224, Cleveland, United States, October 1971.
[CRO 76] CROSET M., DIEUMEGARD D., “Quantitative secondary ion mass spectrometry analysis of oxygen isotopes and other light elements in silicon oxide films”, Corrosion Science, vol. 16, p. 703–715, 1976.
[DEA 00] DE ALMEIDA R. M. C., GONCALVES S., BAUMVOL I. J. R., STEDILE F. C., “Dynamics of thermal growth of silicon oxide films on Si”, Physical Review B, vol. 61, p. 12992–12999, 2000.
[DEA 65] DEAL B.E., GROVE A.S., “General relationship for the thermal oxidation of silicon”, Journal of Applied Physics, vol. 36 p. 3770–3778, 1965.
[DEA 78] DEAL B.E., “Thermal oxidation kinetics of silicon in pyrogenic H2O and 5% HCl/H2O mixtures”, Journal of the Electrochemical Society, vol. 125, p. 576–579, 1978.
[DEL 87] DELARIOS J.M., HELMS C.R., KAO D.B., DEAL B.E., “Effect of silicon surface cleaning procedures on oxidation kinetics and surface chemistry”, Applied Surface Science, vol. 30, p. 17–24, 1987.
[DEL 71] DELL’OCA C.J.,PULFREY D.L., YOUNG L., “Anodic oxide films”, in H.A. FRANCOMBE, R.W. HOFFMANN (eds), Physics of Thin Films, vol. 6, Academic Press, New York, p. 1, 1971.
[DER 82] DERRIEN J., COMMANDRE M., “SiO2 ultra-thin film growth kinetics as investigated by surface techniques”, Surface Science, vol. 118, p. 32–46, 1982.
[DIG 72] DIGNAM M.J., “Mechanisms of ion transport through oxide films”, in J.W. Diggle (ed.), Oxides and oxide films, volume 1, Marcel Dekker, New York, p. 92–286, 1972.
[DOR 62] DOREMUS R.H., in J.D. MACKENZIE (ed.), Modern Aspects of the Vitreous State, vol. 2, Butterworths, London, p. 12, 1962.
[DOR 69] DOREMUS R.H., in J.W. MITCHELL, R.C. DEVRIES, R.W. ROBERTS, P. CANNON (eds), Reactivity of Solids, John Wiley and Sons, New York, p. 667, 1969.
[DOR 94] DOREMUS R.H, Glass Science, (2nd edition), John Wiley and Sons, New York, 1994.
[DRE 66] DREINER R., “A-C Properties of Anodic Oxide Films on Silicon”, Journal of the Electrochemical Society, vol. 113, p. 1210–1215, 1966.
[DRU 63] DRURY R., ROBERTS J.P., “Diffusion in silica glass following reaction with tritiated water vapour”, Physics and Chemistry of Glasses, vol. 4, p. 79–90, 1963.
[EER 77] EERNISSE E.P., “Viscous flow of thermal SiO2”, Applied Physical Letters, vol. 30, p. 290–293, 1977.
[EER 79] EERNISSE E.P., “Stress in thermal SiO2 during growth”, Applied Physics Letters, vol. 35, p. 8–10, 1979.
[FAR 83a] FARGEIX A., GHIBAUDO G., KAMARINOS G., “A revised analysis of dry oxidation of silicon”, Journal of Applied Physics, vol. 54, p. 2878–2880, 1983.
[FAR 83b] FARGEIX A., GHIBAUDO G., “Dry oxidation of silicon: A new model of growth including relaxation of stress by viscous flow”, Journal of Applied Physics, vol. 54, 7153–7158, 1983.
[FAR 84] FARGEIX A., GHIBAUDO G., “Role of stress on the parabolic kinetic constant for dry silicon oxidation”, Journal of Applied Physics, vol. 56, p. 589–591, 1984.
[FEH 72] FEHLNER F.P., “Formation of ultra-thin oxide films on silicon”, Journal of the Electrochemical Society, vol. 119, p. 1723–1727, 1972.
[FRI 68] FRISCHAT G.H., “Sodium diffusion in SiO2 glass”, Journal of the American Ceramic Society, vol. 51, p. 528–530, 1968.
[FRI 69] FRISCHAT G.H., “Evidence for calcium and aluminum diffusion in SiO2 glass”, Journal of the American Ceramic Society, vol. 52, p. 625–625, 1969.
[FRI 75] FRISCHAT G.H., “Ion diffusion in oxide glasses”, in Y. ADDA, A.D. LECLAIRE, L.M. SLIFKIN, F.H. WÖHLBIER (eds), Diffusion and Defect Monograph Series, 3/4, Trans Tech Publications, Aedermannsdorf, 1975.
[FRO 77] FROMHOLD A.T. JR., “Single carrier steady-state theory for formation of anodic films under conditions of high space charge in very large electric fields”, Journal of the Electrochemial Society, vol. 124, p. 538–549, 1977.
[FUK 92] FUKUDA H., YASUDA M., IWABUCHI T., “Kinetics of rapid thermal oxidation of silicon”, Japanese Journal of Applied Physics, vol. 31, p. 3436–3439, 1992.
[GAN 92] GANEM J.-J., Mécanismes de croissance de diélectriques par nitruration et par oxydation thermique rapide, PHD Thesis, University of Paris 7, 1992.
[GAN 93a] GANEM J.-J., BATTISTIG G., RIGO S., TRIMAILLE I., “A study of the initial stages of the oxidation of silicon using 18O2 and RTP”, Applied Surface Science, vol. 65–66, p. 647–653, 1993.
[GAN 93b] GANEM J.-J., RIGO S., TRIMAILLE I., “Modellization of the silicon rapid thermal oxidation in the initial stages according to the silicon fragments model”, Microelectronic Engineering, vol. 22, p. 35–38, 1993.
[GHE 72] GHEZ R., VAN DER MEULEN YJ., “Kinetics of thermal growth of ultra-thin layers of SiO2 on silicon”, Journal of the Electrochemical Society, vol. 119, p. 1100–1106, 1972.
[GHE 73] GHEZ R., “On the Mott-Cabrera oxidation rate equation and the inverse-logarithmic law”, Journal of Chemical Physics, vol. 58, p. 1838–1843, 1973.
[GIB 89] GIBSON J.M., LANZEROTTI M.Y., “Observation of interfacial atomic steps during silicon oxidation”, Nature, vol. 340, p. 128–131, 1989.
[GOU 80] GOURRIER S., MICEA A., BACAL M., “Oxidation of GaAs in an oxygen multipole plasma”, Thin Solid Films, vol. 65, p. 315–330, 1980.
[GOU 81] GOURRIER S., BACAL M., “Review of oxide formation in a plasma”, Plasma Chemistry and Plasma Processing, vol. 1, p. 217–232, 1981.
[HAN 87] HAN C.J., HELMS C.R., “Parallel oxidation mechanism for si oxidation in dry O2”, Journal of the Electrochemical Society, vol. 134, p. 1297–1302, 1987.
[HAN 88] HAN C.J., HELMS C.R., “18O tracer study of si oxidation in dry O2 using SIMS”, Journal of the Electrochemical Society, vol. 135, p. 1824–1832, 1988.
[HAU 62] HAUL R., DUEMBGEN G., “Oxygen mobility in TiO2, SiO2 and fused quartz by heterogeneous isotopic exchange”, Zeitschrift für Elektrochemie, vol. 66, p. 636–641, 1962.
[HET 62] HETHERINGTON G., JACK K.H., “Water in vitreous silica. Part 1. Influence of water content on the properties of vitreous silica”, Physics and Chemistry of Glasses, vol. 3, p. 129–133, 1962.
[HET 64] HETHERINGTON G., JACK K.H., “The oxidation of vitreous silica”, Physics and Chemistry of Glasses, vol. 5, p. 147–149, 1964.
[HIJ 09] HIJIKATA Y., YAGUCHI H., YOSHIDA S., “A kinetic model of silicon carbide oxidation based on the interfacial silicon and carbon emission phenomenon”, Applied Physics Express, vol. 2, 021203–1–021203–3, 2009.
[HO 79a] HO C.P., PLUMMER J.D., “Si/SiO2 interface oxidation kinetics: a physical model for the influence of high substrate doping levels”, Journal of the Electrochemical Society, vol. 126, p. 1516–1522, 1979.
[HO 79b] HO C.P., PLUMMER J.D., “Si/SiO2 interface oxidation kinetics: a physical model for the influence of high substrate doping levels”, Journal of the Electrochemical Society, vol. 126, p. 1523–1530, 1979.
[HOL 70] HOLMBERG G.L., KUPER A.B., MIRALDI F.D., “Water contamination in thermal oxide on silicon”, Journal of the Electrochemical Society, vol. 117, p. 677–682, 1970.
[HOL 79] HOLLAHAN J.R., “Deposition of plasma silicon oxide thin films in a production planar reactor”, Journal of the Electrochemical Society, vol. 126, p.930–934, 1979.
[HOP 75] HOPPER M.A., CLARKE R.A., YOUNG L., “Thermal oxidation of silicon”, Journal of the Electrochemical Society, vol. 122, p. 1216–1222, 1975.
[HOR 78] HORIUCHI M., KAMIGAKI Y., HAGIWARA T., “Formation and properties of thin tunnelable SiO2 films using a vaporized O2 source at liquid N2 temperature”, Journal of the Electrochemical Society, vol. 125, p. 766–771, 1978.
[HUT 54] HUTCHISON D.A., “Isotopic exchange of oxygen in the systems water-silica and oxygen-silica”, Journal of Chemical Physics, vol. 22, p. 758–759, 1954.
[IRE 74] IRENE E.A., “The effects of trace amounts of water on the thermal oxidation of silicon in oxygen”, Journal of the Electrochemical Society, vol. 121, p. 1613–1616, 1974.
[IRE 76] IRENE E.A., VAN DER MEULEN Y.J., “Silicon oxidation studies: analysis of SiO2 film growth data”, Journal of the Electrochemical Society, vol. 123, p. 1380–1384, 1976.
[IRE 78a] IRENE E.A., “Silicon oxidation studies: some aspects of the initial oxidation regime”, Journal of the Electrochemical Society, vol. 125, p. 1708–1714, 1978.
[IRE 78b] IRENE E.A., DONG D.W., “Silicon oxidation studies: the oxidation of heavily B- and P-doped single crystal silicon”, Journal of the Electrochemical Society, vol. 125, p. 1146–1151, 1978.
[IRE 88] IRENE E.A., “Models for the oxidation of silicon”, CRC Critical Reviews in Solid State and Materials Sciences, vol. 14, p. 175–223, 1988.
[KAG 99] KAGESHIMA H., SHIRAISHI K., UEMATSU M., “Universal theory of Si oxidation rate and importance of interfacial Si emission”, Japanese Journal of Applied Physics, vol. 38, p. L971-L974, 1999.
[KAJ 05] KAJIHARA K., KAMIOKA H., HIRANO M., MIURA T., SKUJA L., HOSONO H., “Interstitial oxygen molecules in amorphous SiO2. III. Measurements of dissolution kinetics, diffusion coefficient, and solubility by infrared photoluminescence”, Journal of Applied Physics, vol. 98, p. 013529–013529–7, 2005.
[KAM 77] KAMIGAKI Y., ITOH Y., “Thermal oxidation of silicon in various oxygen partial pressures diluted by nitrogen”, Journal of Applied Physics, vol. 48, p. 2891–2896, 1977.
[KAT 79] KATZ L.E., HOWELLS B.F. JR, “Low temperature, high pressure steam oxidation of silicon”, Journal of the Electrochemical Society, vol. 126, p. 1822–1824, 1979.
[KER 77] KERN W., ROSLER R.S., “Advances in deposition processes for passivation films”, Journal of Vacuum Science and Technology, vol. 14, p. 1082–1099, 1977.
[KRA 67] KRAITCHAMN J., “Silicon oxide films grown in a microwave discharge”, Journal of Applied Physics, vol. 38, p. 4323–4330, 1967.
[KRZ 07] KRZEMINSKI C., LARRIEU G., PENAUD J., LAMPIN E., DUBOIS E., “Silicon dry oxidation kinetics at low temperature in the nanometric range: Modeling and experiment”, Journal of Applied Physics, vol. 101, p. 064908–064908–8, 2007.
[LES 78] LESLIE J.D., KEITH V., KNORR K., “Auger spectroscopic evidence that plasma anodization involves mass transfer from the cathode to the anode”, Journal of the Electrochemical Society, vol. 125, p. 44–51, 1978.
[LIE 82] LIE L.N., RAZOUK R.R., DEAL B.E., “High pressure oxidation of silicon in dry oxygen”, Journal of the Electrochemical Society, vol. 129, p. 2828–2834, 1982.
[LIG 60] LIGENZA J.R., SPITZER W.G., “The mechanisms for silicon oxidation in steam and oxygen”, Journal of Physicsand Chemistry of Solids, vol. 14, p. 131–136, 1960.
[LIG 61] LIGENZA J.R., “Effect of crystal orientation on oxidation rates of silicon in high pressure steam”, Journal of Physical Chemistry, vol. 65, p. 2011–2014, 1961.
[LIG 65] LIGENZA J.R., “Silicon oxidation in an oxygen plasma excited by microwaves”, Journal of Applied Physics, vol. 36, p. 2703–2707, 1965.
[LIG 70] LIGENZA J.R., KUHN M., Solid State Technology, vol. 13, p. 33, 1970.
[MAS 85] MASSOUD H.Z., PLUMMER J.D., IRENE E.A., “Thermal oxidation of silicon in dry oxygen: growth-rate enhancement in the thin regime”, Journal of the Electrochemical Society, vol. 132, p. 2685–2700, 1985.
[MIK 81] MIKKELSEN J.C., “Oxidant transport during steam oxidation of silicon”, Applied Physics Letters, vol. 39, p. 903–905, 1981.
[MIL 63] MILES J.L., SMITH P.H., “The formation of metal oxide films using gaseous and solid electrolytes”, Journal of the Electrochemical Society, vol. 110, p. 1240–1245, 1963.
[MOU 61] MOULSON A.J., ROBERTS J.P., “Water in silica glass”, Transactions of the Faraday Society articles, vol. 57, p. 1208–1216, 1961.
[MOZ 69] MOZZI R.L., WARREN B.E., “The structure of vitreous silica”, Journal of Applied Crystallography, vol. 2, p. 164–172, 1969.
[NAN 70] NANNONI R., MUSSELIN M.J., “Anodic oxidation of silicon effects of water on oxide properties”, Thin Solid Films, vol. 6, p. 397–406, 1970.
[NOR 61] NORTON F.J., “Permeation of gaseous oxygen through vitreous silica”, Nature, vol. 91, p. 701–701, 1961.
[OHA 70] O’HANLON J.F., “Plasma anodization of metals and semiconductors”, Journal of Vacuum Science and Technology, vol. 7, p. 330–338, 1970.
[OHA 73] O’HANLON J.F., SAMPOGNA M., “A new technique for high speed anodization in a DC oxygen glow discharge”, Journal of Vacuum Science and Technology, vol. 10, p. 450–452, 1973.
[PAS 62] PASCAL P., Nouveau traité de Chimie Minerale, t. 8, Editions Masson, Paris, 1962.
[PER 71] PERKINS W.G., BEGEAL D.R., “Diffusion and permeation of He, Ne, Ar, Kr, and D2 through silicon oxide thin films”, Journal of Chemical Physics, vol. 54, p. 1683–1694, 1971.
[PFE 80] PFEFFER R., OHRING M.G., in G. LUCOVSKY, S.T. PANTELIDES, F.L. GALEENER (eds), The Physics of MOS Insulators, Pergamon Press, New York, p. 162–166, 1980.
[PFE 81] PFEFFER R., OHRING M., “Network oxygen exchange during water diffusion in SiO2”, Journal of Applied Physics, vol. 52, p. 777–784, 1981.
[PLI 77] PLISKIN W.A., “Comparison of properties of dielectric films deposited by various methods”, Journal of Vacuum Science and Technology, vol. 14, p. 1064–1081, 1977.
[PUL 73] PULFREY D.L., HATHORN F.G.M., YOUNG L., “The anodization of Si in an RF plasma”, Journal of the Electrochemical Society, vol. 120, p. 1529–1535, 1973.
[PUL 74] PULFREY D.L., RECHE J.J.H., “Preparation and properties of plasma anodized silicon dioxide films”, Solid-State Electronics, vol. 17, p. 627–628, 1974.
[RAI 80] RAIDER S.I., FORGET L.E., “Reversal of relative oxidation rates of <111> and <100> oriented silicon substrates at low oxygen partial pressures”, Journal of the Electrochemical Society, vol. 127, p. 1783–1787, 1980.
[RAY 08] RAY E.A., ROZEN J., DHAR S., FELDMAN L.C., WILLIAMS J.R., “Pressure dependence of SiO2 growth kinetics and electrical properties on SiC”, Journal of Applied Physics, vol. 103, p. 023522–1–023522–7, 2008.
[RAZ 81] RAZOUK R.R., LIE L.N., DEAL B.E., “Kinetics of high pressure oxidation of silicon in pyrogenic steam”, Journal of the Electrochemical Society, vol. 128, p. 2214–2220, 1981.
[REV 69] REVESZ A.G., EVANS R.J., in J.W. MITCHELL, R.C. DE VRIES, R.W. ROBERTS, P. CANNOn (eds), Reactivity of Solids, John Wiley and Sons, New York, p. 425–454, 1969.
[REV 70] REVESZ A.G., “A new model for defects in non-crystalline silicon dioxide”, Journal of Non-Crystalline Solids, vol. 4, p. 347–356, 1970.
[REV 79] REVESZ A.G., “The role of hydrogen in SiO2 films on silicon”, Journal of the Electrochemical Society, vol. 126, p. 122–130, 1979.
[REV 80] REVESZ A.G., “The defect structure of vitreous SiO2 films on silicon. I. Structure of vitreous SiO2 and the nature of the SiO2 bond”, Physica Status Solidi (a), vol. 57, p. 235–243, 1980.
[RIG 80] RIGO S., ROCHET F., STRABONI A., AGIUS B., in G. LUCOVSKY, S.T. PANTELIDES, F.L. GALEENER (eds), The Physics of MOS Insulators, Pergamon Press, New York, p. 167–188, 1980.
[RIG 82] RIGO S., ROCHET F., AGIUS B., STRABONI A., “An 18O study of cooperative diffusion and chemical reaction during thermal treatments of silica films in water vapor”, Journal of the Electrochemical Society, vol. 129, p. 867–876, 1982.
[RIG 86] RIGO S., “Silica films on silicon” in G. BARBOTTIN, A. VAPAILLE (eds), Instabilities in Silicon Devices: Silicon Passivation and Related Instabilities, p. 5–100, Elsevier, Amsterdam, 1986.
[RIG 88] RIGO S., “Si oxidation mechanisms as studied by oxygen tracer methods”, in C.R. HELMS, B.E. DEAL (eds), The Physics and Chemistry of SiO2 and the Si-SiO2 Interface, p. 75–84, Plenum Press, New York, 1988.
[ROB 64] ROBERTS G.J., ROBERTS J.P., “Influence of thermal history on the solubility and diffusion of water in silica glass”, Physics and Chemistry of Glasses, vol. 5, p. 26–31, 1964.
[ROB 66] ROBERTS G.J., ROBERTS J.P., “An oxygen tracer investigation of the diffusion of water in silica glass”, Physics and Chemistry of Glasses, vol. 7, p. 82–88, 1966.
[ROC 81] ROCHET F., Etude par traçage isotopique à l’oxygène 18 des mécanismes de transport atomique dans les couches minces d’oxyde de silicium et d’oxyde de tantale sur silicium, Postgraduate Thesis, Pierre and Marie Curie University, 1981.
[ROC 84] ROCHET F., AGIUS B., RIGO S., “An 18O study of the oxidation mechanism of silicon in dry oxygen”, Journal of the Electrochemical Society, vol. 131, p. 914–923, 1984.
[ROC 86] ROCHET F., RIGO S., FROMENT M., D’ANTERROCHES C., MAILLOT C., ROULET H., DUFOUR G., “The thermal oxidation of silicon the special case of the growth of very thin films”, Advances in Physics, vol. 35, p. 237–274, 1986.
[SCH 57] SCHMIDT P.F., MICHEL W., “Anodic formation of oxide films on silicon”, Journal of the Electrochemical Society, vol. 104, p. 230–236, 1957.
[SCH 64] SCHMIDT P.F., OWEN A.E., “Anodic oxide films for device fabrication in silicon”, Journal of the Electrochemical Society, vol. 111, p. 682–688, 1964.
[SCH 65] SCHMIDT P.F., O’KEEFE TW., OROSHNIK J., OWEN A.E., “Doped anodic oxide films for device fabrication in silicon. II. Diffusion sources of controlled composition, and diffusion results”, Journal of the Electrochemical Society, vol. 112, p. 800–807, 1965.
[SHA 72] SHACKELFORD J.F., STUDT R.L., FULRATH R.M., “Solubility of gases in glass. II. He, Ne, and H2 in fused silica”, Journal of Applied Physics, vol. 43, p. 1619–1626, 1972.
[SHE 74] SHELBY J.E., KEETON S.C., “Temperature dependence of gas diffusion in glass”, Journal of Applied Physics, vol. 45, p. 1458–1460, 1974.
[SHE 77] SHELBY J.E., “Molecular diffusion and solubility of hydrogen isotopes in vitreous silica”, Journal of Applied Physics, vol. 48, p. 3387–3394, 1977.
[SMI 82] SMITH F.W., GHIDINI G., “Reaction of oxygen with Si(111) and (100): critical conditions for the growth of SiO2”, Journal of the Electrochemical Society, vol. 129, p. 1300–1306, 1982.
[SON 04] SONG Y., DHAR S., FELDMAN L.C., CHUNG G., WILLIAMS J.R., “Modified Deal Grove model for the thermal oxidation of silicon carbide”, Journal of Applied Physics, vol. 95, p. 4953–4957, 2004.
[SPI 61] SPITZER W.G., LIGENZA J.R., “Oxygen exchange between silica and high pressure steam”, Journal of Physics and Chemistry of Solids, vol. 17, p. 196–202, 1961.
[STU 70] STUDT P.L., SHACKELFORD J.F., FULRATH R.M., “Solubility of gases in glass-a monatomic model”, Journal of Applied Physics, vol. 41, p. 2777–2780, 1970.
[SUC 63] SUCOV E.W., “Diffusion of oxygen in vitreous silica”, Journal of the American Ceramic Society, vol. 46, p. 14–20, 1963.
[SWE 61] SWEETS D.E., LEE E.W., FRANK R.C., “Diffusion coefficients of helium in fused quartz”, Journal of Chemical Physics, vol. 34, p. 17–22, 1961.
[TIL 83] TILLER W.A., “On the kinetics of the thermal oxidation of silicon”, Journal of the Electrochemical Society, vol. 130, p. 501–506, 1983.
[TRI 89] TRIMAILLE I., RIGO S., “Use of 18O isotopic labelling to study thermal dry oxidation of silicon as a function of temperature and pressure”, Applied Surface Science, vol. 39, p. 65–80, 1989.
[TRI 93] TRIMAILLE I., RAIDER S.I., GANEM J.J., RIGO S., PENEBRE N.A., “Use of 18O labelling to study growth mechanisms in dry oxidation of silicon”, in C.R. Helms, B.E. Deal (eds), The Physics and Chemistry of SiO2 and the Si-SiO2 Interface 2, Plenum Press, New York, p. 7–14, 1993.
[TSU 78] TSUCHIMOTO T., “Plasma stream transport method (2) Use of charge exchange plasma source”, Journal of Vacuum Science and Technology, vol. 15, p. 1730–1733, 1978.
[VAN 72] VAN DER MEULEN Y.J., “Kinetics of thermal growth of ultra-thin layers of SiO2 on silicon”, Journal of the Electrochemical Society, vol. 119, p.530–534, 1972.
[VAN 75] VAN DER STEEN G.H.A.M., PAPANIKOLAU E., “Introduction and removal of hydroxyl groups in vitreous silica - 2. Chemical and physical solubility of hydrogen in vitreous silica”, Philips Research Report, vol. 30, p. 192–205, 1975.
[VIC 07] VICKRIDGE I., GANEM J., HOSHINO Y., TRIMAILLE I., “Growth of SiO2 on SiC by dry thermal oxidation: mechanisms”, Journal of Physics D., vol. 40, p. 6254–6263, 2007.
[VON 72] VON DER MEULEN Y.J., “Kinetics of thermal growth of ultra-thin layers of SiO2 on silicon”, Journal of the Electrochemical Society, vol. 119, p. 530–534, 1972.
[WAT 07] WATANABE T., OHDOMARI I., “A kinetic equation for thermal oxidation of silicon replacing the Deal–Grove equation”, Journal of the Electrochemical Society, vol. 154, G270-G276, 2007.
[WIL 65] WiA$ZLIAMS E.L., “Diffusion of oxygen in fused silica”, Journal of the American Ceramic Society, vol. 48, p. 190–194, 1965.
[WOL 69] WOLF H.F., Silicon Semiconductor Data, Pergamon Press, New York, 1969.
[WOL 80] WOLTERS D.R., “On the oxidation kinetics of silicon: the role of water”, Journal of the Electrochemical Society, vol. 127, p. 2072–2082, 1980.
[YAM 07] YAMAMOTO T., HIJIKATAO Y., YAGUCHI H., YOSHIDA S., “Growth rate enhancement of (0001)-face silicon–carbide oxidation in thin oxide regime”, Japanese Journal of Applied Physics, vol. 46, p. L770-L772, 2007.
[YAM 08] YAMAMOTO T., HIJIKATAO Y., YAGUCHI H., YOSHIDA S., “Oxide growth rate enhancement of silicon carbide (0001) Si-faces in thin oxide regime”, Japanese Journal of Applied Physics, vol. 47, p. 7803–7806, 2008.
1Chapter written by Jean-Jacques GANEM and Isabelle TRIMAILLE.