
Chemistry With Controlled Ions
Stefan Willitsch
Department of Chemistry, University of Basel, Klingelbergstrasse 80, 4056 Basel, Switzerland
Contents
- I. Introduction
- II. Cooling and Control of Ions in the Gas Phase
- III. Control of Neutral Molecules in the Gas Phase
- IV. Ion–Molecule Reaction Dynamics: A Brief Outline
- V. Applications and Examples
- VI. Conclusions and Outlook
- Acknowledgments
- References
I. Introduction
Over the past years, the development of experimental methods for the manipulation of atoms and molecules in the gas phase has made impressive progress. The starting point for these developments was the advent of laser cooling of atoms in the 1970s and 1980s which paved the way for the preparation of trapped ensembles of ultracold atoms and atomic ions [1]. These experimental breakthroughs initiated many exciting applications and new research directions in atomic physics, quantum science, and precision measurements [2–4].
Inspired by the successes with atoms, a variety of methods for the cooling of molecules and the preparation of cold molecular samples have been conceived since the mid-1990s. Techniques such as Stark deceleration [5], Zeeman deceleration [6, 7], Rydberg deceleration [6], photoassociation [8, 9], magnetic association [10, 11], velocity selection [12], and optical deceleration [13] enable the preparation of translationally cold molecules with kinetic energies corresponding to a few Kelvin down to hundreds of nanokelvin with exquisite quantum-state selectivity for some of these approaches. These developments have been reviewed extensively in the recent past [5–11, 14–27].
The ability to cool implies a large degree of control over the motion of molecules. Consequently, in recent years a new focus has developed to employ these techniques for the precise control of molecular motions with the objective to precisely control molecular collisions and chemical reactions [17, 28–32]. As opposed to coherent-control schemes which aim at shaping wavefunctions with short laser pulses [33], the approach discussed here concentrates on the precise manipulation of the translational motion, the position in space, the internal quantum state, the energy and the geometric structure of the reaction partners using external electric, magnetic, and optical fields. The ultimate objective of these efforts is to gain a comprehensive control over all dynamic properties of the collision partners, at the level of single molecules, in order to elucidate in detail chemical dynamics and reaction mechanisms as well as to control chemical processes. This approach can be viewed as taking to the extreme the control philosophy traditionally adopted in synthetic chemistry in which reaction conditions are modified by changing the temperature, the pressure, and the reactant concentrations and by using catalytic agents in order to influence the outcome of chemicalreactions.
This chapter focuses on the application of controlled-molecule techniques for precise studies of ion–molecule reactions in the gas phase. Sections II and III give an overview of the most important techniques presently used for the control of the translational motion, internal quantum states, and structural properties of molecular ions and neutral molecules in the gas phase. Section IV gives a brief outline of salient concepts of ion–molecule reaction dynamics which are relevant for the present discussion. In Section V, some illustrative examples are presented in which cold- and controlled-molecule techniques have been used to study the mechanisms and dynamics of ionic reactions.
II. Cooling and Control of Ions in the Gas Phase
A. Ion Trapping
Many state-of-the-art techniques for the control of ions rely on trapping. Among the instruments most widely used for the confinement of ions are linear radiofrequency (RF) traps in which an assembly of electrodes is arranged in a quadrupolar [34–36] or higher-order multipolar [37, 38] configuration. Charged particles are dynamically trapped in the center of these devices by the application of time-varying and static voltages to the electrodes.
The functionality of an RF ion trap is illustrated with the schematic of a typical linear quadrupole trap shown in Fig. 1(a) [24]. The arrangement of the electrodes ensures the generation of a quadrupolar electrostatic potential in the center of the trap. The electrodes are separated into segments to allow the application of time-varying and static voltages. RF voltages ![]() , with
, with ![]() the RF amplitude and
the RF amplitude and ![]() the angular RF frequency, are applied to all segments of each electrode with a phase difference of 180
the angular RF frequency, are applied to all segments of each electrode with a phase difference of 180![]() between adjacent electrodes. The resulting time-varying electric potential ensures a dynamic trapping of the ions in the
between adjacent electrodes. The resulting time-varying electric potential ensures a dynamic trapping of the ions in the ![]() plane perpendicular to the longitudinal trap axis [34, 37]. The application of static voltages to the outermost segments of each electrode enables the confinement of the ions in the longitudinal trap direction
plane perpendicular to the longitudinal trap axis [34, 37]. The application of static voltages to the outermost segments of each electrode enables the confinement of the ions in the longitudinal trap direction ![]() .
.
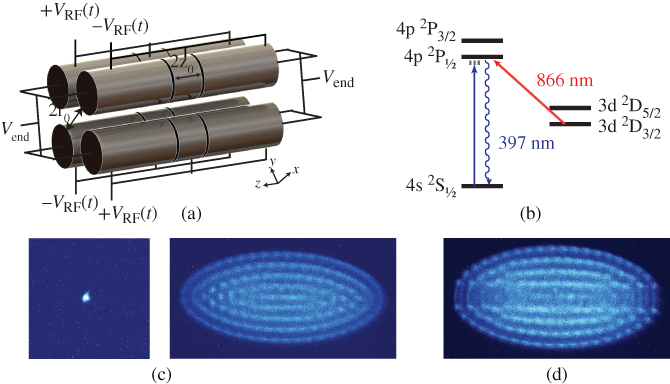
Figure 1 Linear radiofrequency (RF) ion traps and Coulomb crystals. (a) Schematic representation of a linear RF ion trap consisting of four cylindrical electrodes arranged in a quadrupolar configuration. The electrodes are sectioned into segments for the application of RF voltages 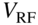 and static voltages
and static voltages 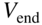 to allow the confinement of charged particles in the center of the trap. (b) Laser-cooling scheme for
to allow the confinement of charged particles in the center of the trap. (b) Laser-cooling scheme for 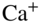 . See text for details. (c) Fluorescence images of Coulomb crystals of laser-cooled
. See text for details. (c) Fluorescence images of Coulomb crystals of laser-cooled  ions. Left: a single
ions. Left: a single 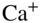 ion, right: a large spheroidal Coulomb crystal consisting of several hundred ions. (d) Bicomponent Coulomb crystal consisting of laser-cooled
ion, right: a large spheroidal Coulomb crystal consisting of several hundred ions. (d) Bicomponent Coulomb crystal consisting of laser-cooled 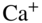 ions and sympathetically cooled
ions and sympathetically cooled  ions. The
ions. The 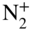 ions form a nonfluorescing string of ions in the center and are only indirectly visible as a dark core of the crystal.
ions form a nonfluorescing string of ions in the center and are only indirectly visible as a dark core of the crystal.
Willitsch, 2012 [24]. Reproduced with permissions of Taylor & Francis.
The classical motion of a single ion in a quadrupole RF trap follows a set of Mathieu equations [34, 35, 39]

where ![]() is any of the Cartesian coordinates
is any of the Cartesian coordinates ![]() . The Mathieu parameters
. The Mathieu parameters ![]() and
and ![]() are defined by1
are defined by1

In Eq. (5.2), ![]() and
and ![]() stand for the mass and the charge of the ion, respectively,
stand for the mass and the charge of the ion, respectively, ![]() is the distance between the endcaps,
is the distance between the endcaps, ![]() is a geometry parameter, and
is a geometry parameter, and ![]() is the distance between two diagonally opposed quadrupole rods as shown in Fig. 1(a). The specific combinations of
is the distance between two diagonally opposed quadrupole rods as shown in Fig. 1(a). The specific combinations of ![]() for which a stableconfinement of ions is feasible are usually represented in terms of stability diagrams [34, 35, 39].
for which a stableconfinement of ions is feasible are usually represented in terms of stability diagrams [34, 35, 39].
The dynamic confinement using RF voltages imprints a fast oscillating motion on the ions termed “micromotion.” The amplitude of the micromotion, and therefore the ion kinetic energy, scales with the magnitude of the RF potential at the position of the ion. In an adiabatic approximation [34, 37], the average kinetic energy stored in the micromotion appears as an effective (pseudo)potential ![]() which governs the slow thermal (secular) motions of the ions in the trap. For quadrupolar traps, the pseudopotential assumes a harmonic form:
which governs the slow thermal (secular) motions of the ions in the trap. For quadrupolar traps, the pseudopotential assumes a harmonic form:
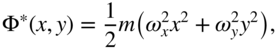
where ![]() and
and ![]() are the secular frequencies for the ion's motion along
are the secular frequencies for the ion's motion along ![]() and
and ![]() , respectively. In a linear trap like the one shown in Fig. 1(a),
, respectively. In a linear trap like the one shown in Fig. 1(a), ![]() holds in the absence of static fields which break the symmetry. Higher-order multipole traps exhibit shallower potentials close to the trap center and therefore offer a reduced micromotion, however, at the expense of a tight confinement of the ions [37].
holds in the absence of static fields which break the symmetry. Higher-order multipole traps exhibit shallower potentials close to the trap center and therefore offer a reduced micromotion, however, at the expense of a tight confinement of the ions [37].
B. Cooling by Collisions with Neutrals
“Traditional” methods for the cooling of ions rely on collisions with neutral gas particles which affect both the internal (vibrational) and external (rotational, translational) degrees of freedom of the ions. Cooling of the internal degrees of freedom manifests itself by a confinement of the populations to the lowest quantum states. Cooling of the translational motion of the ion results in a compression of the velocity distribution of the ion ensemble.
For decades, cooling in adiabatic gas expansions has been a method of choice for preparing internally cold molecules [40] and molecular ions [41, 42]. An adiabatic expansion of a gas from a high-pressure into a low-pressure zone results in the formation of a molecular beam and leads to the cooling of the internal degrees of freedom of the molecules through the conversion of internal energy to kinetic energy by collisions in the expansion region. Rotational temperatures down to about 1 K can be achieved using state-of-the-art pulsed gas nozzles [43] and sufficiently high pressure differentials between the gas reservoir and the expansion chamber maintained at high vacuum. The adiabatic expansion also results in translational cooling by a compression of the velocity distribution in the moving frame of the gas. Thus, the translational energy spread and therefore the temperature is small in the moving frame, butthe molecules are still fast in the laboratory frame. While the translational temperatures in the moving frame are often comparable to the rotational ones, the vibrational temperatures of the molecules can be significantly higher owing to less efficient cooling of the vibrational motion by collisions in the expanding gas.
Another important technique for the cooling of ions relies on their immersion into a cryogenic buffer gas, for example, helium [37, 44]. In this approach, the ions have to be confined to a well-defined region of space using a trap. The translational and internal temperatures achievable with this method are limited by the temperature of the buffer gas which is typically on the order of 10 K.
C. Laser and Sympathetic Cooling: Coulomb Crystallization of Ions
Recently, advanced methods such as laser and sympathetic cooling have been developed which enable to reach ion translational temperatures in the millikelvin regime, that is, more than three orders of magnitudes lower than achievable with the collisional methods discussed earlier. These techniques have been treated in detail elsewhere [1, 16, 20, 24, 45, 46] and are briefly summarized here.
Laser cooling of a particle is based on the removal of kinetic energy by the transfer of momentum imparted during the repeated scattering of photons from a laser beam [1, 45]. Since its introduction in the late 1970s [48, 49], it has become a working-horse technique in atomic physics. It is applicable to systems with a sufficiently simple energy level structure which lends itself to the implementation of closed optical cycling transitions so that the population is not lost during the repeated scattering of photons. As an example, a schematic level scheme illustrating the laser cooling of ![]() ions is shown in Fig. 1(b). In this case, laser cooling is performed on the
ions is shown in Fig. 1(b). In this case, laser cooling is performed on the ![]() transition at 397 nm. The laser frequency is slightly red-detuned from the resonance frequency of the transition to ensure that particles with a velocity component antiparallel to the laser beam preferentially absorb photons and are thus cooled (Doppler laser cooling). For neutral atoms, six laser beams in an optical-molasses configuration, that is, two beams counterpropagating on each coordinate axis, are necessary to achieve the cooling of all translational degrees of freedom. For large ensembles of ions in a trap, cooling with a laser beam along a single direction is in principle sufficient because the Coulomb interaction between the particles couples all translational degrees of freedom.
transition at 397 nm. The laser frequency is slightly red-detuned from the resonance frequency of the transition to ensure that particles with a velocity component antiparallel to the laser beam preferentially absorb photons and are thus cooled (Doppler laser cooling). For neutral atoms, six laser beams in an optical-molasses configuration, that is, two beams counterpropagating on each coordinate axis, are necessary to achieve the cooling of all translational degrees of freedom. For large ensembles of ions in a trap, cooling with a laser beam along a single direction is in principle sufficient because the Coulomb interaction between the particles couples all translational degrees of freedom.
For Doppler laser cooling, the limiting temperature ![]() can be achieved at an optimal detuning from resonance of
can be achieved at an optimal detuning from resonance of ![]() , where
, where ![]() is the natural linewidth of the cooling transition, and is given by [1, 45]
is the natural linewidth of the cooling transition, and is given by [1, 45]

For the laser-cooling scheme of ![]() depicted in Fig. 1(b), one obtains
depicted in Fig. 1(b), one obtains ![]() mK. From the excited state, spontaneous emission can occur back to the ground state or to the
mK. From the excited state, spontaneous emission can occur back to the ground state or to the ![]() level which cannot decay to the ground state by dipole-allowed optical transitions, thus disrupting the laser-cooling cycle. Therefore, another laser beam at 866 nm is required to repump population back to the
level which cannot decay to the ground state by dipole-allowed optical transitions, thus disrupting the laser-cooling cycle. Therefore, another laser beam at 866 nm is required to repump population back to the ![]() state. Suitable systems for laser cooling include alkali atoms [45], alkaline earth ions [24], selected complex atoms such as erbium and dysprosium [50, 51], and a number of molecules exhibiting electronic transitions with near-diagonal Franck–Condon factors [52, 53].
state. Suitable systems for laser cooling include alkali atoms [45], alkaline earth ions [24], selected complex atoms such as erbium and dysprosium [50, 51], and a number of molecules exhibiting electronic transitions with near-diagonal Franck–Condon factors [52, 53].
Laser cooling of ions is typically performed in quadrupole ion traps which afford a tight trapping potential for charged particles (see Fig. 1) (a)). The tight confinement of the ions ensures their concentration in a small volume of space to facilitate overlap with the cooling laser beams and thus efficient cooling. The cooling of the ions under these conditions results in their spatial localization to form an ordered structure referred to as a “Coulomb crystal” [16, 24, 27, 46]. Coulomb crystallization occurs when the ratio of the Coulomb repulsion energy between the ions and their thermal energy, the so-called plasma-coupling parameter, exceeds a value of about 150–170 [54, 55]. Typically, under tight trapping conditions in which the distances between the ions are on the order of 10 ![]() m, this criterion implies cooling of the ions down to tens of millikelvin which can readily be achieved with Doppler laser cooling.
m, this criterion implies cooling of the ions down to tens of millikelvin which can readily be achieved with Doppler laser cooling.
Figure 1(c) shows images of Coulomb crystals obtained by monitoring the fluorescence generated during laser cooling of ![]() ions. As can be seen from the images, individual ions can be observed, and therefore also addressed and manipulated, under these conditions. Consequently, the position of every single particle and by virtue of Eq. (3) its average micromotion energy is well defined 2.
ions. As can be seen from the images, individual ions can be observed, and therefore also addressed and manipulated, under these conditions. Consequently, the position of every single particle and by virtue of Eq. (3) its average micromotion energy is well defined 2.
As laser cooling is only applicable to a limited range of molecules [53], alternative methods resulting in the Coulomb crystallization of molecular ions need to be established. Over the past 15 years, sympathetic cooling has emerged as the method of choice for this purpose. In this approach, laser-cooled atomic ions are simultaneously trapped with molecular ions. The latter are cooled by collisions with the former, leading to the formation of bicomponent Coulomb crystals which contain both species (see Fig. 1(d)). As this method relies on the transfer of momentum through collisions between charged species, it is extremely versatile and has been applied to molecular ions ranging in size from diatomics [57, 58] to small proteins [59]. To optimize the momentum transfer during collisions and thus cooling, the coolant and the cooled species should have a similar mass-to-charge-ratio [60]. In contrast tocollisional cooling with neutral particles described in Section II.C, sympathetic cooling with ions is mediated by collisions dominated by the Coulomb interaction and, therefore, does not appreciably affect the internal degrees of freedom of the molecules [61]. This problem can be solved by combining sympathetic cooling of the translational motion with cryogenic-buffer-gas cooling of the internal degrees of freedom, as has recently been shown in Ref. [62], or alternatively by using state-preparation techniques in combination with sympathetic cooling as detailed in the following section.
D. Selective Preparation of Quantum States
Besides the control of the translational motion, preparation of the internal quantum state of molecular ions is a main prerequisite on the way to controlling chemical reactions. Since the 1980s, (multiphoton) photoionization has been a key method for preparing molecular ions in selected internal states [63]. Various variants of this approach have been implemented. Vibrationally state-selected ions have been produced by resonantly ionizing via specific vibrational states of a suitable Rydberg state of the neutral precursor molecule and capitalizing on diagonal Franck–Condon factors in the ionization step [63–65]. Rotational state selection has been achieved by pulsed-electric-field ionization of high Rydberg states of the neutral precursor molecule converging onto specific rotational states of the ion [66, 67]. Alternatively, ions can be produced in selected rotational states by photoionizing slightly above the lowest rotational ionization thresholds accessible from a well-defined intermediate state by photoionization propensity rules [68]. This approach has recently been applied to the state-selective preparation of ![]() ions in combination with sympathetic cooling of their translational motion to generate Coulomb-crystallized state-selected ions [58, 69]. The specific photoionization sequence used in these experiments is depicted in Fig. 2(a).
ions in combination with sympathetic cooling of their translational motion to generate Coulomb-crystallized state-selected ions [58, 69]. The specific photoionization sequence used in these experiments is depicted in Fig. 2(a).
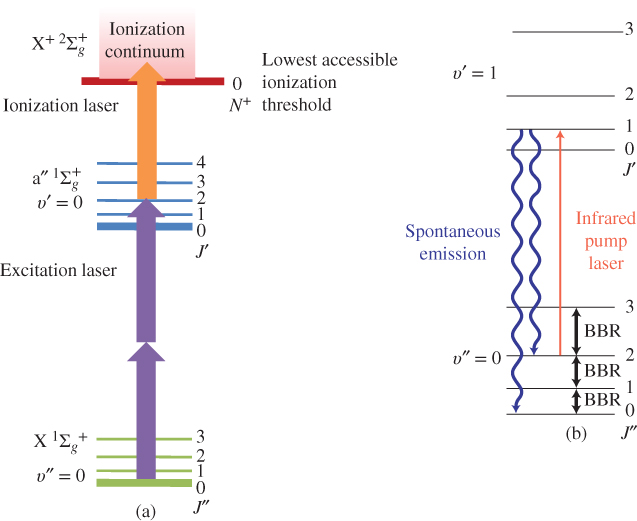
Figure 2 Preparation of molecular ions in selected rotational and vibrational states. (a) [2+1′] multiphoton threshold-photoionization scheme in 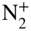 from Ref. [58].
from Ref. [58].  and
and  denote the rotational quantum numbers in the neutral and ionic states, respectively. An initial two-photon excitation step selects a specific rotational level (here with rotational quantum number
denote the rotational quantum numbers in the neutral and ionic states, respectively. An initial two-photon excitation step selects a specific rotational level (here with rotational quantum number 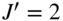 ) in the excited
) in the excited 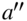 electronic state. Photoionization with an additional photon just above the lowest accessible rotational ionization threshold (corresponding to the cationic rotational ground state
electronic state. Photoionization with an additional photon just above the lowest accessible rotational ionization threshold (corresponding to the cationic rotational ground state 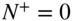 in this case) results in the generation of rotationally state-selected molecular ions. Tong et al., 2010 [58]; Tong et al., 2011 [69]. Reproduced with permissions of American Physical Society. (b) Rotational laser cooling in
in this case) results in the generation of rotationally state-selected molecular ions. Tong et al., 2010 [58]; Tong et al., 2011 [69]. Reproduced with permissions of American Physical Society. (b) Rotational laser cooling in 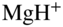 using an infrared laser irradiating the
using an infrared laser irradiating the 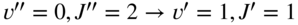 vibrational-rotational transition as implemented in Ref. [70]. The interplay between optical pumping on this transition (thin solid arrow), fluorescence from the upper level back to rotational levels of the ground state (wavy arrows), and population redistribution between the rotational levels of the ground vibrational state by black-body radiation (BBR, thick solid arrows) leads to an accumulation of the population in the absolute ground state
vibrational-rotational transition as implemented in Ref. [70]. The interplay between optical pumping on this transition (thin solid arrow), fluorescence from the upper level back to rotational levels of the ground state (wavy arrows), and population redistribution between the rotational levels of the ground vibrational state by black-body radiation (BBR, thick solid arrows) leads to an accumulation of the population in the absolute ground state 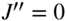 . See text for details.
. See text for details.
Staanum et al., 2010 [70]; Willitsch 2010 [71]. Reproduced with permissions of Macmillan Publishers Limited (Nature.com).
An alternative approach for the internal-state preparation of molecular ions is “rotational laser cooling” [70, 72]. In this scheme, continuous-wave infrared excitation connecting specific rotational levels in the ground vibrational state (vibrational quantum number ![]() ) and excited states (
) and excited states (![]() ) of a molecule is implemented as illustrated with the example of
) of a molecule is implemented as illustrated with the example of ![]() in Fig. 2(b). In this specific example, population is pumped from the lower state (with rotational quantum number
in Fig. 2(b). In this specific example, population is pumped from the lower state (with rotational quantum number ![]() ) to the upper state
) to the upper state ![]() . The excited state can fluoresce back to the
. The excited state can fluoresce back to the ![]() state or the absolute rotational–vibrational ground state
state or the absolute rotational–vibrational ground state ![]() as dictated by electric-dipole selection rules. In addition, the rotational levels in
as dictated by electric-dipole selection rules. In addition, the rotational levels in ![]() are coupled by the ambient black-body radiation (BBR) field which redistributes population between them. Thus, the
are coupled by the ambient black-body radiation (BBR) field which redistributes population between them. Thus, the ![]() level is constantly replenished with population from neighboring levels to be immediately pumped into the excited
level is constantly replenished with population from neighboring levels to be immediately pumped into the excited ![]() state from where it can fluoresce back into
state from where it can fluoresce back into ![]() state. In the long run, this population-redistribution dynamics leads to an accumulation of population in the
state. In the long run, this population-redistribution dynamics leads to an accumulation of population in the ![]() state.
state.
The efficiency of this scheme can be improved by pumping on multiple rotation–vibration transitions simultaneously. In experiments on ![]() with a single infrared pump laser, populations in the order of 40% could be achieved in the
with a single infrared pump laser, populations in the order of 40% could be achieved in the ![]() state after a cooling time of about a minute. This represents a more than 10-fold increase of the population compared to the thermal value at room temperature [70]. In similar experiments in
state after a cooling time of about a minute. This represents a more than 10-fold increase of the population compared to the thermal value at room temperature [70]. In similar experiments in ![]() [72] utilizing two infrared lasers pumping on different rotational–vibrational transitions, ground-state populations close to 80% could be achieved. Recently, broadband laser-cooling techniques have been implemented [73] which utilize repeated femtosecond-laser pulses exciting a manifold of rotational levels simultaneously on a strong electronic transition. The excitation of the rotational ground state is excluded by spectral filtering of the laser pulses leading to an accumulation of population in the ground state through fluorescence from the excited electronic state. With this approach, it has been possible to reach ground-state populations in the order of 95% after only 140 ms of cooling time in
[72] utilizing two infrared lasers pumping on different rotational–vibrational transitions, ground-state populations close to 80% could be achieved. Recently, broadband laser-cooling techniques have been implemented [73] which utilize repeated femtosecond-laser pulses exciting a manifold of rotational levels simultaneously on a strong electronic transition. The excitation of the rotational ground state is excluded by spectral filtering of the laser pulses leading to an accumulation of population in the ground state through fluorescence from the excited electronic state. With this approach, it has been possible to reach ground-state populations in the order of 95% after only 140 ms of cooling time in ![]() .
.
E. Preparation of Specific Molecular Conformations
The shape of a molecule is a key property influencing its chemical reactivity. Conformers, that is, rotational isomers, are the dominant form of isomers of complex molecules. Hence, there is a need for techniques that enable the study of single molecular conformations. For the preparation of specific conformers of ions, several techniques have been developed in the past. Ion-mobility methods achieve the separation of different conformers, or families of conformers, from a mixture based on their geometrical cross sections for collisions while drifting through a buffer gas [74–76]. Alternatively, photoionization of specific conformers of neutral molecules has been successfully applied for the preparation of conformationally pure samples of ions. Here, conformer selection can either be achieved by single-photon excitation to high Rydberg states of a specific conformer followed by mass-analyzed threshold ionization (MATI) to produce the corresponding ion [77] or in resonant multiphoton ionization schemes in which a specific conformer is selected in the first excitation step to a suitable intermediate state [78].
III. Control of Neutral Molecules in the Gas Phase
Controlling ion–molecule reactions necessitates control over both reaction partners, that is, the ionic and neutral counterparts in a reaction. Similar to ions, cooling and translational control are in many cases two sides of the same coin also in the case of neutrals. The last 15 years have seen impressive progress in the development of methods to control the translational motion of neutral molecules in the gas phase. These techniques have been reviewed extensively in the recent past; see, for example, Refs [5–7, 17–21, 23, 26, 31, 79], and only a brief summary of the methods most relevant for the present purpose shall be given here.
A. Cooling and Control of the Translational and Internal Molecular Degrees of Freedom
A powerful approach for producing translationally controlled molecules consists in the manipulation of the velocity of pulsed supersonic molecular beams using external electric, magnetic, or optical fields. One of the most widely used methods is Stark deceleration which relies on the force ![]() generated by inhomogeneous electric fields on polar molecules [5, 6, 19, 32, 80]:
generated by inhomogeneous electric fields on polar molecules [5, 6, 19, 32, 80]:
where ![]() is the Stark energy of the molecule in a specific quantum state,
is the Stark energy of the molecule in a specific quantum state, ![]() is the effective dipole moment of the molecule in this state and
is the effective dipole moment of the molecule in this state and ![]() is the strength of the external electric field.
is the strength of the external electric field. ![]() is obtained by solving the relevant Stark problem of the system; see, for example, Refs [5, 81, 82]. Stark deceleration relies on molecules in low-field seeking Stark states, that is, states that increase in energy with increasing electric field, so that a force is exerted which repels the molecules from regions of high field strength. In this scheme, a pulsed molecular beam of polar molecules such as CO,
is obtained by solving the relevant Stark problem of the system; see, for example, Refs [5, 81, 82]. Stark deceleration relies on molecules in low-field seeking Stark states, that is, states that increase in energy with increasing electric field, so that a force is exerted which repels the molecules from regions of high field strength. In this scheme, a pulsed molecular beam of polar molecules such as CO, ![]() , or OH is injected into a linear array of pairs of cylindrical electrodes (Fig. 3(a)). High voltages are applied when a packet of molecules approaches a given pair of electrodes. The Stark forces slightly decelerate the molecules while they have to surmount the potential-energy barrier posed by the electric field (Fig. 3(b)). The fields are switched off before the molecules reach the top of the barrier to prevent their acceleration once they have passed the electrode stage and are applied to the next pair of electrodes to repeat the sequence (Fig. 3(c)). Depending on the timing sequence of the application of the electric fields with respect to the position of the molecules within the electrode array, the molecules can be either decelerated or accelerated enabling the generation of velocity-controlled molecular beams [17]. This scheme can be used to bring the molecules to an effective stand still in the laboratory frame enabling their electric or magnetic trapping [83, 84].
, or OH is injected into a linear array of pairs of cylindrical electrodes (Fig. 3(a)). High voltages are applied when a packet of molecules approaches a given pair of electrodes. The Stark forces slightly decelerate the molecules while they have to surmount the potential-energy barrier posed by the electric field (Fig. 3(b)). The fields are switched off before the molecules reach the top of the barrier to prevent their acceleration once they have passed the electrode stage and are applied to the next pair of electrodes to repeat the sequence (Fig. 3(c)). Depending on the timing sequence of the application of the electric fields with respect to the position of the molecules within the electrode array, the molecules can be either decelerated or accelerated enabling the generation of velocity-controlled molecular beams [17]. This scheme can be used to bring the molecules to an effective stand still in the laboratory frame enabling their electric or magnetic trapping [83, 84].
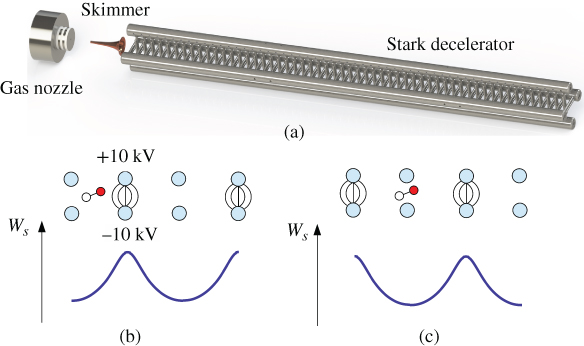
Figure 3 Principle of Stark deceleration. (a) Schematic representation of a Stark deceleration experiment consisting of a gas nozzle for producing a pulsed supersonic molecular beam which is coupled into an array of pairs of cylindrical electrodes. (b) Alternating pairs of electrodes are switched to high voltage creating a potential-energy 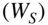 barrier for molecules in low-field seeking Stark states. Thus, the molecules are slowed down when approaching the electrodes. (c) When the molecules approach the top of the barrier, the fields are switched off and applied to the next pair of electrodes. Repeated application of this scheme results in a gradual deceleration of the packet of molecules along the decelerator.
barrier for molecules in low-field seeking Stark states. Thus, the molecules are slowed down when approaching the electrodes. (c) When the molecules approach the top of the barrier, the fields are switched off and applied to the next pair of electrodes. Repeated application of this scheme results in a gradual deceleration of the packet of molecules along the decelerator.
Stark deceleration can only be employed to polar molecules showing a sufficiently strong Stark effect [85] which restricts the chemical diversity of the systems available to this method. The packets of molecules exiting a Stark decelerator possess a very high degree of state purity as the Stark force is very sensitive to the internal state of a molecule [5, 32]. Typically, the voltage switching sequence is optimized for themanipulation of a specific Stark state, and molecules in other states are usually lost from the molecular beam while traversing the electrode array. Recently, the technique has been refined to utilize traveling electric traps generated in a linear array of ring electrodes which afford an improved control over the packet of decelerated molecules compared with conventional Stark deceleration [86].
A variant of this approach is the Stark deceleration of Rydberg atoms and molecules [6, 79, 87]. Because of the large dipole moments and concomitant large Stark forces achievable in Rydberg states, a single electrode stage is often sufficient to bring light molecules to an effective stand still. A chief advantage of this scheme is that it is applicable to polar and nonpolar species alike but requires the preparation of Rydberg states with sufficiently long lifetimes to cover at least the deceleration period (typically milliseconds).
A related approach is Zeeman deceleration which utilizes the magnetic forces produced by inhomogeneous magnetic fields on paramagnetic atoms and molecules [6, 7, 88]. In analogy to Stark deceleration, a pulsed molecular beam is coupled into a linear array of coils in which high currents are rapidly switched to create strong time-varying inhomogeneous magnetic fields used to decelerate (or accelerate) packets of molecules in low-field seeking Zeeman states. Like Stark deceleration, the scheme is highly state specific and produces velocity-controlled molecular beams with a high state purity. Also, it has recently been extended to traveling magnetic traps which yield an improved control over the deceleration process [89, 90].
Another variation on the beam-deceleration theme is optical deceleration which relies on optical dipole forces produced by focused laser beams [13, 91]. The potential energy ![]() of a molecule interacting with a far-off-resonant optical field is given by
of a molecule interacting with a far-off-resonant optical field is given by

where ![]() is the effective polarizability of the molecule,
is the effective polarizability of the molecule, ![]() the intensity of the optical field,
the intensity of the optical field, ![]() the vacuum permittivity, and
the vacuum permittivity, and ![]() the speed of light. With laser radiation that is far red-detuned from all electronic transitions, the particles are high-field seeking and drawn into the focus of the laser beam. Using the intense field of a focused nanosecond pulsed laser such as a Nd:YAG, it is possible to bring a packet of molecules delivered in a pulsed molecular beam to an effective stand still in a single laser shot. The scheme is in principle applicable to virtually any polarizable species.
the speed of light. With laser radiation that is far red-detuned from all electronic transitions, the particles are high-field seeking and drawn into the focus of the laser beam. Using the intense field of a focused nanosecond pulsed laser such as a Nd:YAG, it is possible to bring a packet of molecules delivered in a pulsed molecular beam to an effective stand still in a single laser shot. The scheme is in principle applicable to virtually any polarizable species.
An alternative method to manipulate the velocity distribution of molecules is velocity filtering of an effusive molecular beam. In this approach, the slowest molecules from a thermal distribution are separated from the fast ones using the Stark forces generated in bent electric-multipole guides [12, 92, 93]. A schematic of a setup based on an electric-quadrupole guide is shown in Fig. 4(a). The quadrupolar electric field inside the guide generates Stark forces on polar molecules which drive them toward the center when the molecules are in low-field seeking states. When the molecules approach the bend of the guide, only the slowest ones for which the Stark force exceeds the centrifugal force in the bend are retained, whereas the faster molecules leave the bend and are lost from the guide.
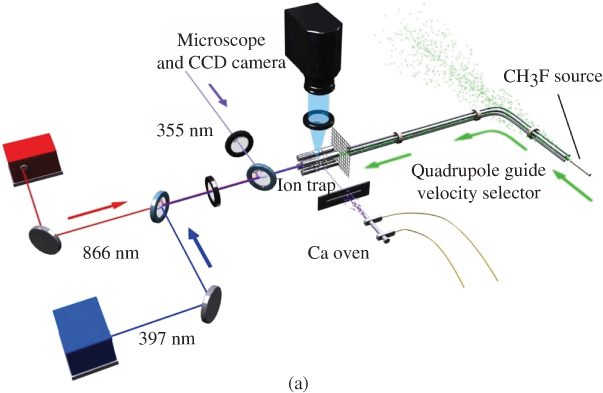
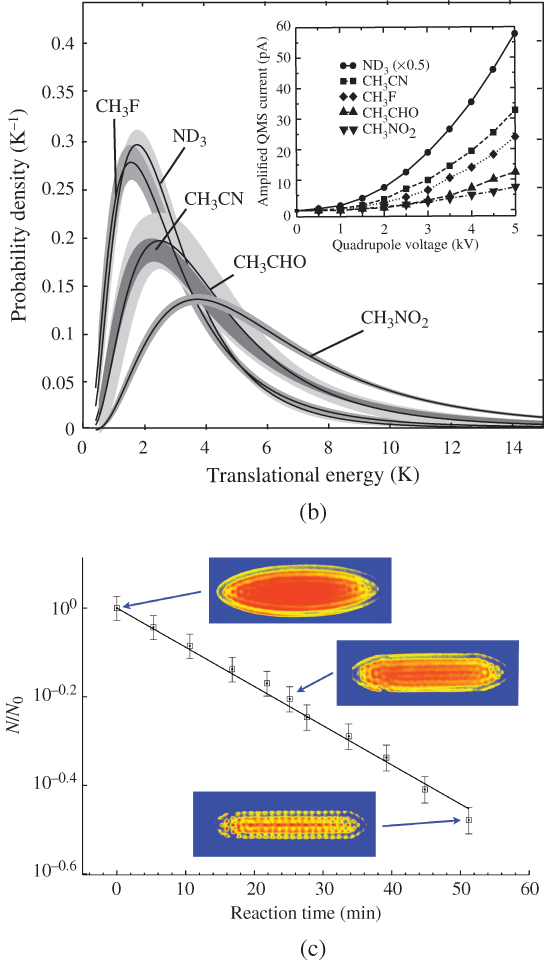
Figure 4 Reactions of cold ions with velocity-selected molecules. (a) Schematic of an experimental setup for studying translationally cold ion–molecule reactions. An electrostatic quadrupole-guide velocity selector produces a continuous beam of translationally cold polar molecules. The molecular beam enters a linear RF ion trap containing Coulomb crystals of cold ions. Laser beams for the cooling and generation of  ions are labeled by their wavelengths 397, 866, and 355 nm. Willitsch et al., 2008 [47]. Reproduced with permissions of American Physical Society. (b) Translational energy distributions of various polar molecules which have been velocity-filtered by the electrostatic guide at a voltage of
ions are labeled by their wavelengths 397, 866, and 355 nm. Willitsch et al., 2008 [47]. Reproduced with permissions of American Physical Society. (b) Translational energy distributions of various polar molecules which have been velocity-filtered by the electrostatic guide at a voltage of 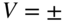 4 kV applied to the quadrupole electrodes. Inset: Averaged signal of polar molecules detected after the velocity filter at different quadrupole voltages. Bell et al., 2009 [92]. Reproduced with permissions of The Royal Society of Chemistry. (c) Rates of chemical reactions between cold
4 kV applied to the quadrupole electrodes. Inset: Averaged signal of polar molecules detected after the velocity filter at different quadrupole voltages. Bell et al., 2009 [92]. Reproduced with permissions of The Royal Society of Chemistry. (c) Rates of chemical reactions between cold 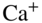 ions and cold
ions and cold  F molecules determined by monitoring the decrease of the volume of the Coulomb crystals, reflecting the decrease of the number
F molecules determined by monitoring the decrease of the volume of the Coulomb crystals, reflecting the decrease of the number 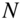 of
of  ions, as a function of the reaction time. The bimolecular rate constant of the reaction is determined from a fit (solid line) of the experimental data to an integrated pseudo-first-order rate law; see text for details.
ions, as a function of the reaction time. The bimolecular rate constant of the reaction is determined from a fit (solid line) of the experimental data to an integrated pseudo-first-order rate law; see text for details.
Willitsch et al., 2008 [16]. Reproduced with permissions of The Royal Society of Chemistry.
This approach is capable of generating continuous beams of molecules with compressed velocity distributions corresponding to translational temperatures of a few Kelvin (Fig. 4(b)). Like Stark deceleration, it is only applicable to polar species. Starting from a room-temperature source and a corresponding thermal distribution of rotational populations, typically a wide range of Stark states are filtered by the guide. As a consequence, the velocity-filtered beam of molecules is translationally cold, but rotationally warm [92–94]. This problem can be mitigated by feeding the guide with molecules from a cryogenic source which establishes a cooling of the internal degrees of freedom by collisions with a cryogenic buffer gas prior to velocity selection [95, 96].
B. Conformer Selection
The selective study of individual conformers of complex neutral molecules in the gas phase is a challenging problem from an experimental perspective. In many cases, several conformers are populated at room temperature which may interconvert into one another over low energy barriers. Recent years have seen impressive progress in the spectroscopic characterization of specific molecular conformations [97–99] and in the characterization of conformer-specific photodissociation dynamics [77, 78]. A common feature of these approaches is the adiabatic cooling of the molecules in a supersonic expansion in order to ensure the confinement of the populations to the energetically lowest conformers, thus reducing the conformational complexity, and to prevent their interconversion by thermal motion. Zwier and coworkers also achieved the “synthesis” and subsequent cooling of energetically excited molecular conformations by optical pumping on infrared transitions promoting the interconversion between conformers in the high-pressure zone of a supersonic expansion [100, 101].
A method for the spatial separation of individual conformers in a molecular beam has recently been developed by Küpper, Stapelfeldt, Meijer, and coworkers [32, 102]. The separation is based on different mass-to-dipole moment ratios of different conformers. In this approach, a molecular beam containing a mixture of conformers is coupled into an electrostatic deflector consisting of two high-voltage electrodes creating a strong inhomogeneous electric field, see Fig. 8(a) for an illustration. Conformers with different dipole moments experience different Stark forces in the inhomogeneous field which deflect them perpendicularly to the beam axis. As a consequence, the conformers spatially separate and the molecular beam splits up into conformational components. The successful application of this scheme is subject to several prerequisites required to achieve a clean spatial separation of the conformers. First, the target molecule should not exhibit too many conformers which are populated in the molecular beam. Second, these conformers also need to exhibit sufficiently different ratios of their mass to their effective dipole moments to enable a clean separation. Thus far, this method has successfully been applied to medium-sized organic molecules like 3-aminophenol, 3-fluorophenol, and indole-water clusters [32].
IV. Ion–Molecule Reaction Dynamics: A Brief Outline
Many ion–molecule reactions are effectively barrierless and are therefore governed by long-range intermolecular forces. In these cases, the reaction rates are determined by the efficiency of the capture of the reaction partners at long range. The long-range ion–molecule interaction potential ![]() , where
, where ![]() is the separation of the center of masses of the two particles, is usually expressed in terms of a power series [103–107]. For ion–neutral interactions, this series can be formulated in atomic units as
is the separation of the center of masses of the two particles, is usually expressed in terms of a power series [103–107]. For ion–neutral interactions, this series can be formulated in atomic units as

where ![]() is the charge number of the ion,
is the charge number of the ion, ![]() is the permanent dipole moment of the neutral molecule,
is the permanent dipole moment of the neutral molecule, ![]() is the angle between
is the angle between ![]() and
and ![]() ,
, ![]() is the quadrupole moment of the neutral molecule,
is the quadrupole moment of the neutral molecule, ![]() is the angle between the axis of
is the angle between the axis of ![]() and
and ![]() , and
, and ![]() is the isotropic polarizability of the neutral molecule. The terms in the order given in Eq. (7) correspond to the interaction of the charge of the ion with the permanent dipole moment, with the permanent quadrupole moment and with the induced dipole moment of the neutral molecule. The last term which scales with
is the isotropic polarizability of the neutral molecule. The terms in the order given in Eq. (7) correspond to the interaction of the charge of the ion with the permanent dipole moment, with the permanent quadrupole moment and with the induced dipole moment of the neutral molecule. The last term which scales with ![]() is often called the Langevin interaction and is the leading term for an ion interacting with a structureless neutral particle, for example, an atom in an
is often called the Langevin interaction and is the leading term for an ion interacting with a structureless neutral particle, for example, an atom in an ![]() state.
state.
A long-range potential of the form of Eq. (7) is generally attractive and barrierless. The only barrier that plays a role is the centrifugal barrier which arises from nonzero collisional angular momentum ![]() . The centrifugal potential is given by
. The centrifugal potential is given by ![]() , where
, where ![]() is the reduced mass of the collision, and needs to be added to the interaction potential. Classically, the collisional angular momentum is given by
is the reduced mass of the collision, and needs to be added to the interaction potential. Classically, the collisional angular momentum is given by ![]() , where
, where ![]() is the collision velocity and
is the collision velocity and ![]() is the impact parameter. As ion–neutral interaction potentials are long range according to Eq. (7), most collisions occur at large impact parameters and therefore with large angular momentum.
is the impact parameter. As ion–neutral interaction potentials are long range according to Eq. (7), most collisions occur at large impact parameters and therefore with large angular momentum.
The classical capture rate is given by all collisions that have enough kinetic energy to surmount the centrifugal barrier and lead to inward-spiraling trajectories that bring the particles close enough to react [108, 109]. For a charge-induced dipole interaction, this leads to the well-known Langevin rate constant ![]() which is independent of the collision energy. More advanced capturemodels such as adiabatic capture theory [107, 110, 111] or the statistical adiabatic channel model [105, 106, 112] also take into account the effects of the quantum rotation of the neutral molecule while preserving a classical treatment of the translational motion. For very low collision energies at which individual collisional partial waves are resolved, the relative motion of the collision partners cannot be treated classically anymore and quantum capture treatments have been formulated (see, e.g., Refs [113–115]).
which is independent of the collision energy. More advanced capturemodels such as adiabatic capture theory [107, 110, 111] or the statistical adiabatic channel model [105, 106, 112] also take into account the effects of the quantum rotation of the neutral molecule while preserving a classical treatment of the translational motion. For very low collision energies at which individual collisional partial waves are resolved, the relative motion of the collision partners cannot be treated classically anymore and quantum capture treatments have been formulated (see, e.g., Refs [113–115]).
V. Applications and Examples
A. Translationally Cold Ion–Molecule Collisions
The combination of techniques for the control of the velocities of ions and neutrals discussed in Sections II and III, respectively, recently enabled the implementation of reaction experiments with controlled relative velocities of the reaction partners down to the “cold regime” defined as ![]() K. At low collision energies, the reaction dynamics often becomes very sensitive to subtleties of the underlying potential-energy surface which opens up new possibilities for studies of reaction mechanisms and intermolecular interactions. The following examples may serve as an illustration.
K. At low collision energies, the reaction dynamics often becomes very sensitive to subtleties of the underlying potential-energy surface which opens up new possibilities for studies of reaction mechanisms and intermolecular interactions. The following examples may serve as an illustration.
In Ref. [47], the combination of an electrostatic quadrupole-guide velocity selector with an ion trap was introduced for studies of reactions between translationally cold neutrals and Coulomb-crystallized ions. A schematic of the setup is displayed in Fig. 4(a). With this approach, collision energies corresponding to ![]() 5–100 K could be reached in ion–molecule collisions by tuning the velocity distribution of the neutrals exiting the velocity filter and/or varying the size and shape of the Coulomb crystals in the trap [92].
5–100 K could be reached in ion–molecule collisions by tuning the velocity distribution of the neutrals exiting the velocity filter and/or varying the size and shape of the Coulomb crystals in the trap [92].
First experiments focused on reactions of laser-cooled ![]() ions with
ions with ![]() F,
F, ![]() F
F![]() , and
, and ![]() Cl [47, 116]. The rates of these reactions were determined from monitoring the decrease of the number
Cl [47, 116]. The rates of these reactions were determined from monitoring the decrease of the number ![]() of
of ![]() ions in the Coulomb crystals as a function of the reaction time
ions in the Coulomb crystals as a function of the reaction time ![]() (Fig. 4(c)). Because of the excess of neutral molecules in comparison to the ions, the reactions showed pseudo-first-order kinetics according to the integrated rate law
(Fig. 4(c)). Because of the excess of neutral molecules in comparison to the ions, the reactions showed pseudo-first-order kinetics according to the integrated rate law ![]() , where
, where ![]() is the bimolecular rate constant and
is the bimolecular rate constant and ![]() the number density of neutral molecules in the velocity-filtered beam. From a fit of the experimental data to the integrated rate law (solid line in Fig. 4(c)), the rate constant
the number density of neutral molecules in the velocity-filtered beam. From a fit of the experimental data to the integrated rate law (solid line in Fig. 4(c)), the rate constant ![]() could be determined with the known the neutral molecule density
could be determined with the known the neutral molecule density ![]() .
.
Reactions of this type are initiated by an attack of the metal ion on a halogenatom on the neutral co-reactant leading to halogen abstraction from the molecule [117]. The energy profile along the reaction coordinate is reminiscent of an ![]() 2 reaction [118] exhibiting a central energy barrier corresponding to the transition state of the halogen abstraction step and minima in the entrance and exit channels of the reaction corresponding to ionic complexes [117]. Depending on the substitution pattern of the neutral reaction partner, the barrier maxima are located slightly above or below the energy of the reactants. These constitute dynamical bottlenecks which slow down the reaction on the ground-state potential-energy surface [116]. As the
2 reaction [118] exhibiting a central energy barrier corresponding to the transition state of the halogen abstraction step and minima in the entrance and exit channels of the reaction corresponding to ionic complexes [117]. Depending on the substitution pattern of the neutral reaction partner, the barrier maxima are located slightly above or below the energy of the reactants. These constitute dynamical bottlenecks which slow down the reaction on the ground-state potential-energy surface [116]. As the ![]() ions are constantly laser-cooled and therefore electronically excited during the experiment, a fraction of the reactive collisions occurs with ions in the excited
ions are constantly laser-cooled and therefore electronically excited during the experiment, a fraction of the reactive collisions occurs with ions in the excited ![]() or
or ![]() states. In these cases, the reaction is initiated on an excited surface with rates close to the capture limit implying effectively barrierless processes. In cases such as
states. In these cases, the reaction is initiated on an excited surface with rates close to the capture limit implying effectively barrierless processes. In cases such as ![]() +CH
+CH![]() F
F![]() which exhibit high barriers on the lowest potential-energy surface, the reaction rate observed in the experiment was completely dominated by the excited-state processes and the effective reaction rate could be tuned by varying the populations in the excited states [116].
which exhibit high barriers on the lowest potential-energy surface, the reaction rate observed in the experiment was completely dominated by the excited-state processes and the effective reaction rate could be tuned by varying the populations in the excited states [116].
Even lower collision energies could be reached by immersing laser or sympathetically cooled ions into clouds of ultracold atoms [119–125]. An example of such a “hybrid”-trapping experiment is shown in Fig. 5(a) in which a linear RF ion trap has been combined with a magneto-optical trap (MOT) for ultracold neutral atoms, in this case ![]() Rb [126–128]. A MOT relies on a combination of six cooling-laser beams in optical-molasses configuration and a quadrupolar magnetic field generated by two coils in anti-Helmholtz configuration to cool and confine atoms in its center. See, for example, Refs [1, 45, 129] for a detailed discussion of the working principle of a MOT. Apart from MOTs, [119, 120, 124, 125, 127, 130, 131], ion traps have also been combined with magnetic or optical dipole traps which allow the preparation of even colder samples of atoms down to temperatures of hundreds of nanokelvin [132–134].
Rb [126–128]. A MOT relies on a combination of six cooling-laser beams in optical-molasses configuration and a quadrupolar magnetic field generated by two coils in anti-Helmholtz configuration to cool and confine atoms in its center. See, for example, Refs [1, 45, 129] for a detailed discussion of the working principle of a MOT. Apart from MOTs, [119, 120, 124, 125, 127, 130, 131], ion traps have also been combined with magnetic or optical dipole traps which allow the preparation of even colder samples of atoms down to temperatures of hundreds of nanokelvin [132–134].

Figure 5 Cold ion–atom reactions. (a) Schematic of an ion-atom hybrid-trapping experiment consisting of a linear RF trap for the laser and sympathetic cooling of ions integrated into a magneto-optical trap (MOT) for the laser cooling and trapping of neutral atoms, in the present case 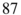 Rb. The MOT consists of two coils situated above and below the ion trap which are operated in an anti-Helmholtz configuration to create a quadrupolar magnetic field. In combination with six laser beams at 780 nm arranged in an optical-molasses configuration, the atoms are laser-cooled and confined to the center of the trap. The circular inset shows a fluorescence image of two laser-cooled
Rb. The MOT consists of two coils situated above and below the ion trap which are operated in an anti-Helmholtz configuration to create a quadrupolar magnetic field. In combination with six laser beams at 780 nm arranged in an optical-molasses configuration, the atoms are laser-cooled and confined to the center of the trap. The circular inset shows a fluorescence image of two laser-cooled 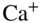 ions immersed into a cloud of trapped ultracold Rb atoms. Hall et al., 2013 [126]. Reproduced with permissions of Taylor & Francis. (b) Rate constant
ions immersed into a cloud of trapped ultracold Rb atoms. Hall et al., 2013 [126]. Reproduced with permissions of Taylor & Francis. (b) Rate constant 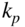 as a function of the collision energy for reactions of
as a function of the collision energy for reactions of 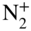 ions with Rb atoms in the excited
ions with Rb atoms in the excited 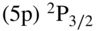 state predicted by classical capture theory taking into account the charge-induced dipole (CID, Langevin, lower trace) and charge-induced dipole plus charge–quadrupole (CQ) interactions (upper trace). The inset shows a comparison of the predicted CID + CQ capture rate constants with experimental data. See text for details. Adapted from Ref. [121].
state predicted by classical capture theory taking into account the charge-induced dipole (CID, Langevin, lower trace) and charge-induced dipole plus charge–quadrupole (CQ) interactions (upper trace). The inset shows a comparison of the predicted CID + CQ capture rate constants with experimental data. See text for details. Adapted from Ref. [121].
As the neutral atoms are usually kept at temperatures ![]() mK while the ions are held at
mK while the ions are held at ![]() mK in the majority of these experiments, the collision energies are limited by the kinetic energies of the ions [134, 135]. The majority of the studies performed so far focused on studying sympathetic cooling of the ions by the atoms [125, 130, 132, 133, 136] and chemical processes between the cold ions and the atoms [124, 126, 127, 137–140]. See Refs [121–123] for reviews of recent studies.
mK in the majority of these experiments, the collision energies are limited by the kinetic energies of the ions [134, 135]. The majority of the studies performed so far focused on studying sympathetic cooling of the ions by the atoms [125, 130, 132, 133, 136] and chemical processes between the cold ions and the atoms [124, 126, 127, 137–140]. See Refs [121–123] for reviews of recent studies.
An example demonstrating the potential of these type of cold-reaction studies for the elucidation of chemical dynamics can be found in Ref. [128] in which charge-exchange reactions between translationally cold ![]() ions and ultracold Rb atoms were studied in the hybrid-trapping setup depicted in Fig. 5(a). The
ions and ultracold Rb atoms were studied in the hybrid-trapping setup depicted in Fig. 5(a). The ![]() ions were sympathetically cooled into the center of a
ions were sympathetically cooled into the center of a ![]() Coulomb crystal forming strings of ions located on the central symmetry axis of the trap, as shown in Fig. 1(c), where the micromotion energy of the ions is minimized. In this way, collision energies as low as
Coulomb crystal forming strings of ions located on the central symmetry axis of the trap, as shown in Fig. 1(c), where the micromotion energy of the ions is minimized. In this way, collision energies as low as ![]() 20 mK could be reached. As the Rb atoms were constantly laser-cooled during the experiment, collisions occurred with atoms in the ground
20 mK could be reached. As the Rb atoms were constantly laser-cooled during the experiment, collisions occurred with atoms in the ground ![]() as well as the excited
as well as the excited ![]() state. It was found that the rate constant for collisions with Rb in the excited state exceeded the relevant Langevin rate by a factor
state. It was found that the rate constant for collisions with Rb in the excited state exceeded the relevant Langevin rate by a factor ![]() in the collision energy interval between 20 and 60 mK (Fig. 5(b)). This finding could be rationalized in terms of a capture-limited process which is dominated by a long-range interaction between the charge of the ion and the permanent quadrupole moment of the Rb atom in its
in the collision energy interval between 20 and 60 mK (Fig. 5(b)). This finding could be rationalized in terms of a capture-limited process which is dominated by a long-range interaction between the charge of the ion and the permanent quadrupole moment of the Rb atom in its ![]() state. In contrast to the Langevin rate constant which is independent of energy (Section IV), the rate constant arising from charge–quadrupole capture increases with decreasing energy so that it dominates the effective reaction rate in the cold regime (Fig. 5(b)). At collision energies of a few hundred Kelvin however, the effective rate constant is governed by the Langevin contribution and the effect of the charge–quadrupole interaction does not noticeably influence the reaction kinetics. These findings illustrate the value of controlled experiments at low collision energies for elucidating subtle details of intermolecular interactions which would otherwise be obscured in experiments at room temperature.
state. In contrast to the Langevin rate constant which is independent of energy (Section IV), the rate constant arising from charge–quadrupole capture increases with decreasing energy so that it dominates the effective reaction rate in the cold regime (Fig. 5(b)). At collision energies of a few hundred Kelvin however, the effective rate constant is governed by the Langevin contribution and the effect of the charge–quadrupole interaction does not noticeably influence the reaction kinetics. These findings illustrate the value of controlled experiments at low collision energies for elucidating subtle details of intermolecular interactions which would otherwise be obscured in experiments at room temperature.
B. Single-Ion Chemistry
One of the most intriguing aspects of the Coulomb-crystallization technique is the capability to perform experiments on single ions. In a chemical context, this offers the possibility to investigate chemical processes on the single-particle level, thus reaching an ultimate limit of sensitivity in chemical reaction studies.
Pioneering experiments in this domain were performed by Drewsen and coworkers who devised methods for single-ion mass spectrometry [141], studying reactions on the single-ion level [142] and recycling the products of single-ion reactions in order to carry out repeated reactions with a single particle [143].
An example of a single-ion reaction study is shown in Fig. 6. In this experiment which was performed in the hybrid-trap setup shown in Fig. 5(a), reactions of laser-cooled ![]() ions with ultracold Rb atoms were studied. Fig. 6(a) shows a Coulomb crystal of two laser-cooled
ions with ultracold Rb atoms were studied. Fig. 6(a) shows a Coulomb crystal of two laser-cooled ![]() ions prepared before the reaction experiment. The
ions prepared before the reaction experiment. The ![]() ions are then immersed into a cloud of ultracold Rb atoms until one
ions are then immersed into a cloud of ultracold Rb atoms until one ![]() ion has reacted away. After reaction, the Rb cloud is removed and the product ion is sympathetically cooled by the remaining
ion has reacted away. After reaction, the Rb cloud is removed and the product ion is sympathetically cooled by the remaining ![]() ion forming a two-ion bicomponent crystal (Fig. 6(b)). The product ion, the position of which is indicated by the circle in Fig. 6(b), is not laser-cooled and therefore does not fluoresce, but its presence manifests itself by dislocating the remaining
ion forming a two-ion bicomponent crystal (Fig. 6(b)). The product ion, the position of which is indicated by the circle in Fig. 6(b), is not laser-cooled and therefore does not fluoresce, but its presence manifests itself by dislocating the remaining ![]() ion from the trap center (schematically indicated by an X in Fig. 6(b)) via the mutual Coulomb repulsion. In the experiment shown, the product ion has been identified as
ion from the trap center (schematically indicated by an X in Fig. 6(b)) via the mutual Coulomb repulsion. In the experiment shown, the product ion has been identified as ![]() using resonant-excitation mass spectrometry [141].
using resonant-excitation mass spectrometry [141].

Figure 6 Single-ion reaction experiments. (a) A Coulomb crystal of two laser-cooled 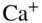 ions prepared in the hybrid-trapping experiment shown in Fig. 5 (a). (b) The same crystal after exposure to a cloud of ultracold Rb atoms. One of the
ions prepared in the hybrid-trapping experiment shown in Fig. 5 (a). (b) The same crystal after exposure to a cloud of ultracold Rb atoms. One of the  ions has undergone a charge-transfer reaction and has been replaced by a nonfluorescing
ions has undergone a charge-transfer reaction and has been replaced by a nonfluorescing 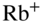 ion (position indicated by the circle). The presence of the product ion manifests itself by the displacement of the remaining
ion (position indicated by the circle). The presence of the product ion manifests itself by the displacement of the remaining  ion from the trap center (labeled with an X) by the Coulomb repulsion between the ions. (c) Histogram of the time-to-reaction of
ion from the trap center (labeled with an X) by the Coulomb repulsion between the ions. (c) Histogram of the time-to-reaction of 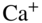 ions with Rb obtained from 49 single-ion reaction experiments. The solid line shows a fit to a pseudo-first-order rate law.
ions with Rb obtained from 49 single-ion reaction experiments. The solid line shows a fit to a pseudo-first-order rate law.
Hall et al., 2013 [126]. Reproduced with permissions of Taylor & Francis.
Rate constants for single-ion reactions can be determined by repeatedly measuring the time-to-reaction of single ions [126, 143]. A histogram of the number of ions ![]() reacting within a given time
reacting within a given time ![]() determined from 49 experiments on single
determined from 49 experiments on single ![]() is shown in Fig. 6(c) [126]. The histogram was fitted to an exponential decay curve (red line) corresponding to a pseudo-first-order rate law (discussed earlier) from which the bimolecular rate constant
is shown in Fig. 6(c) [126]. The histogram was fitted to an exponential decay curve (red line) corresponding to a pseudo-first-order rate law (discussed earlier) from which the bimolecular rate constant ![]() was determined [126].
was determined [126].
C. Energy- and State-Controlled Reactions
The ability to precisely control the translational and internal energies of reaction partners enables accurate studies of the energy balance of chemical processes. A proof-of-principle experiment combining translational-energy- and quantum-state preparation of the reactants with state-resolved detection of the products was reported on the symmetric charge-exchange reaction ![]() +
+ ![]()
![]() +
+ ![]() in Ref. [144]. In this work,
in Ref. [144]. In this work, ![]() ions were produced state-selectively by threshold photoionization of
ions were produced state-selectively by threshold photoionization of ![]() from a molecular beam and sympathetically cooled into a Coulomb crystal as described in Section II.D. The state-selected, translationally cold ions were then reacted with neutral
from a molecular beam and sympathetically cooled into a Coulomb crystal as described in Section II.D. The state-selected, translationally cold ions were then reacted with neutral ![]() molecules from the same molecular beam (Fig. 7(a)). Owing to the cooling in the adiabatic expansion, the neutral
molecules from the same molecular beam (Fig. 7(a)). Owing to the cooling in the adiabatic expansion, the neutral ![]() molecules exhibited rotational temperatures
molecules exhibited rotational temperatures ![]() =10 K corresponding to 50%, 25%, and 22% of the population in the
=10 K corresponding to 50%, 25%, and 22% of the population in the ![]() , and 2 rotational levels and a mean beam velocity of 787 ms
, and 2 rotational levels and a mean beam velocity of 787 ms![]() . The
. The ![]() ions were all produced in their rotational–vibrational ground state forming a string of ions at the center of a
ions were all produced in their rotational–vibrational ground state forming a string of ions at the center of a ![]() Coulomb crystal where they exhibited an average kinetic energy
Coulomb crystal where they exhibited an average kinetic energy ![]() mK. In this way, collisions between the ions and neutrals were investigated at a well-defined collision energy of
mK. In this way, collisions between the ions and neutrals were investigated at a well-defined collision energy of ![]() eV with reaction partners predominantly in their lowest rotational states. The rotational populations of the product
eV with reaction partners predominantly in their lowest rotational states. The rotational populations of the product ![]() ions were probed by a laser-induced charge-transfer (LICT) technique [145]. Using another laser beam around 787 nm, individual rotational-state populations were pumped to vibrationally excited states of the electronic ground state via excitation to the first excited electronic state of
ions were probed by a laser-induced charge-transfer (LICT) technique [145]. Using another laser beam around 787 nm, individual rotational-state populations were pumped to vibrationally excited states of the electronic ground state via excitation to the first excited electronic state of ![]() . From these vibrationally excited levels, the
. From these vibrationally excited levels, the ![]() ions could undergo the vibrationally selective charge-transfer reaction
ions could undergo the vibrationally selective charge-transfer reaction ![]() + Ar
+ Ar ![]()
![]() +
+ ![]() which is energetically suppressed in the vibrational ground state
which is energetically suppressed in the vibrational ground state ![]() . The occurrence of LICT manifested itself in the disappearance of the
. The occurrence of LICT manifested itself in the disappearance of the ![]() ions from the center of the crystal.
ions from the center of the crystal.
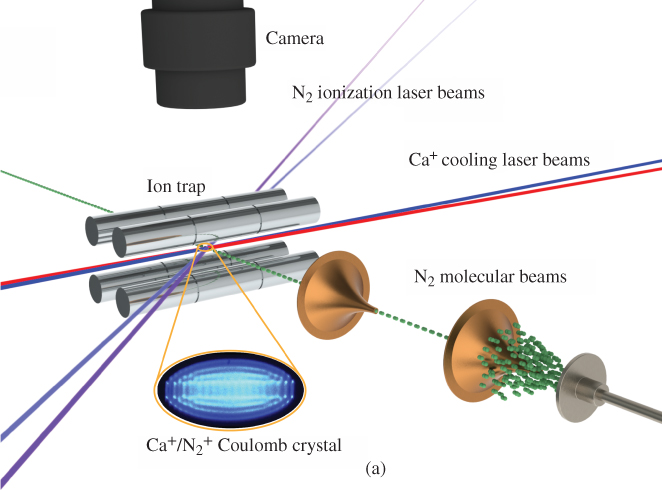
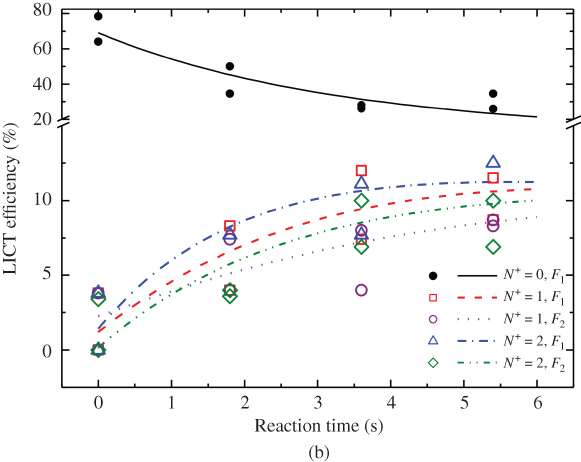
Figure 7 (a) Collision- and state-controlled ion–molecule reactions. (a) Schematic of the experimental setup.  ions were prepared in their rotational–vibrational ground state using the threshold-photoionization scheme shown in Fig. 2(a) and were sympathetically cooled into a Coulomb crystal of laser-cooled
ions were prepared in their rotational–vibrational ground state using the threshold-photoionization scheme shown in Fig. 2(a) and were sympathetically cooled into a Coulomb crystal of laser-cooled  ions (inset). The state-selected
ions (inset). The state-selected  ions were exposed to rotationally cold neutral
ions were exposed to rotationally cold neutral 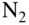 molecules introduced into the ion trap via a doubly skimmed supersonic molecular beam. (b) Dynamics of the populations in different spin-rotational states
molecules introduced into the ion trap via a doubly skimmed supersonic molecular beam. (b) Dynamics of the populations in different spin-rotational states  of sympathetically cooled
of sympathetically cooled 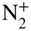 ions as a function of the time of exposure to the molecular beam of neutral
ions as a function of the time of exposure to the molecular beam of neutral  molecules. The populations in the different states have been probed by laser-induced charge transfer (LICT) with Ar atoms. See text for details.
molecules. The populations in the different states have been probed by laser-induced charge transfer (LICT) with Ar atoms. See text for details.
Tong et al., 2012 [144]. Reproduced with permissions of Elsevier.
In the experiment, the state populations of ![]() were monitored as a function of the exposure time to the molecular beam (Fig. 7(b)). It can be seen that the LICT efficiency (which is proportional to the population in the addressed rotational level) decreases as a function of reaction time in the originally prepared rotational ground state
were monitored as a function of the exposure time to the molecular beam (Fig. 7(b)). It can be seen that the LICT efficiency (which is proportional to the population in the addressed rotational level) decreases as a function of reaction time in the originally prepared rotational ground state ![]() but increases in excited levels
but increases in excited levels ![]() .
.
The ![]() ions in the Coulomb crystal were constantly exchanged by charge-transfer reactions with
ions in the Coulomb crystal were constantly exchanged by charge-transfer reactions with ![]() . The experimental results show that the charge transfer is not rotationally adiabatic and may produce ionic products in excited rotational states. With the help of reactive molecular dynamics simulations, this observation could be rationalized by the formation of a long-lived ion–molecule collision complex. Part of the initial collision energy is converted into rotational energy of the products after breakup of the complex. Thus, the increase in rotational energy of the product ions is a signature of translation-to-rotation energy transfer over the course of the reaction. The observation of these subtle energetic effects was only possible through the fine control over the collision and internal energies of the reaction partners in thisexperiment.
. The experimental results show that the charge transfer is not rotationally adiabatic and may produce ionic products in excited rotational states. With the help of reactive molecular dynamics simulations, this observation could be rationalized by the formation of a long-lived ion–molecule collision complex. Part of the initial collision energy is converted into rotational energy of the products after breakup of the complex. Thus, the increase in rotational energy of the product ions is a signature of translation-to-rotation energy transfer over the course of the reaction. The observation of these subtle energetic effects was only possible through the fine control over the collision and internal energies of the reaction partners in thisexperiment.
D. Conformationally Controlled Ion–Molecule Reactions
In the past, studying conformational effects in chemical reaction dynamics has been notoriously difficult because of the challenges involved in the isolation of individual conformations in the gas phase; see Section III.B. Besides matrix-isolation [146] and photodissociation [77, 78] studies, there are only few reports of conformationally resolved bimolecular reactions in the gas phase, for example, Ref. [147]. A new approach to unravel conformational effects in ion–molecule reactions has recently been presented by Chang et al. [148, 149]. It is based on the combination of an electrostatic conformer deflector for neutral molecules (Section B) with a Coulomb crystal experiment as depicted in Fig. 8(a).

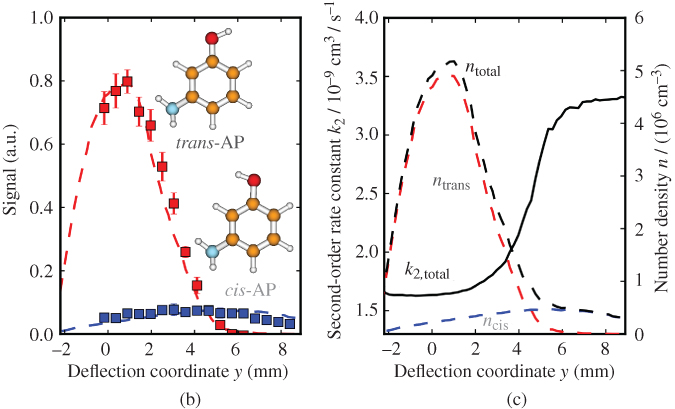
Figure 8 Conformationally controlled ion–molecule reactions. (a) Experimental setup consisting of an electrostatic deflector for conformers with different dipole moments entrained in a supersonic molecular beam. The inset on the left-hand side shows the electric field  generated by the deflector electrodes. The spatially separated conformers (represented by the spheres) are directed at a tightly localized reaction target consisting of an ion Coulomb crystal in a linear RF trap. The inset on the right-hand side shows a Coulomb crystal of
generated by the deflector electrodes. The spatially separated conformers (represented by the spheres) are directed at a tightly localized reaction target consisting of an ion Coulomb crystal in a linear RF trap. The inset on the right-hand side shows a Coulomb crystal of 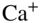 ions before and after reaction with conformers of 3-aminophenol. By tilting the molecular beam assembly, reaction rates can be determined as a function of the coordinate of deflection for the individual conformers enabling to extract conformer-specific rate constants. (b) Density profiles of the deflected cis- and trans 3-aminophenol (AP) conformers as a function of the deflection coordinate at a deflector voltage of
ions before and after reaction with conformers of 3-aminophenol. By tilting the molecular beam assembly, reaction rates can be determined as a function of the coordinate of deflection for the individual conformers enabling to extract conformer-specific rate constants. (b) Density profiles of the deflected cis- and trans 3-aminophenol (AP) conformers as a function of the deflection coordinate at a deflector voltage of  kV. (c) Bimolecular rate constant
kV. (c) Bimolecular rate constant 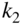 for reactions of cis/trans-AP with laser-cooled
for reactions of cis/trans-AP with laser-cooled  ions as a function of the deflection coordinate.
ions as a function of the deflection coordinate.  denotes the number density of the relevant conformers in the beam. The total rate constant is about a factor of 2 larger at deflection coordinates at which the cis conformer prevails, indicating a higher reactivity for the cis-species. See text for details.
denotes the number density of the relevant conformers in the beam. The total rate constant is about a factor of 2 larger at deflection coordinates at which the cis conformer prevails, indicating a higher reactivity for the cis-species. See text for details.
Chang et al., 2013 [148]. Reproduced with permissions of American Association for the Advancement of Science.
Neutral conformers, as exemplified by the cis- and trans-isomers of 3-aminophenol (AP) (Fig. 8(b)), are spatially separated in the inhomogeneous electric field of the deflector (a typical field distribution is shown in the inset in Fig. 8(a)) so that the molecular beam segregates into conformational components. Deflection profiles, that is, density distributions for the two conformers as a function of the extent of deflection, are shown in Fig. 8(b)). The relative intensities of the signals reflect the populations of the two conformers in the supersonic expansion. cis-AP has a three times larger dipole moment than trans-AP (2.33 and 0.77 D, respectively) so that the cis conformer is more strongly deflected than the trans conformer. At the particular deflector voltage ![]() =7.5 kV of the experiment shown in Fig. 8(b)), the two conformers are not completely separated at the detection point, but regions in which either the trans or the cis conformers are dominant can be found at the smallest and largest deflection coordinates in Fig. 8(b), respectively.
=7.5 kV of the experiment shown in Fig. 8(b)), the two conformers are not completely separated at the detection point, but regions in which either the trans or the cis conformers are dominant can be found at the smallest and largest deflection coordinates in Fig. 8(b), respectively.
For measurements of conformationally dependent reaction rates, the molecular beam was directed at a Coulomb crystal of ![]() ions. Mass spectrometric evidence [149] suggests that
ions. Mass spectrometric evidence [149] suggests that ![]() ions react with the aminophenol conformers predominantly in the
ions react with the aminophenol conformers predominantly in the ![]() excited state by abstraction of one of the functional groups yielding
excited state by abstraction of one of the functional groups yielding ![]() or
or ![]() and the corresponding neutral radicals. The conformation dependence of these processes is probed by tilting the molecular beam apparatus so as to direct only a selected region of the deflected beam at the Coulomb crystal. In this framework, the Coulomb crystal acts as tightly localized reaction target to provide a high spatial and therefore conformational resolution in reactions with the deflected conformers. By spatially scanning the deflected beam across the Coulomb crystal, the reaction rate constant as a function of the deflection coordinate and thus as a function of the conformational composition of the beam was measured (Fig. 8(c)). One can see that the effective bimolecular reaction rate constant
and the corresponding neutral radicals. The conformation dependence of these processes is probed by tilting the molecular beam apparatus so as to direct only a selected region of the deflected beam at the Coulomb crystal. In this framework, the Coulomb crystal acts as tightly localized reaction target to provide a high spatial and therefore conformational resolution in reactions with the deflected conformers. By spatially scanning the deflected beam across the Coulomb crystal, the reaction rate constant as a function of the deflection coordinate and thus as a function of the conformational composition of the beam was measured (Fig. 8(c)). One can see that the effective bimolecular reaction rate constant ![]() increases with deflection and is approximately twice as large at large deflection coordinates, where the cis conformer dominates, compared with small deflection coordinates, where the trans conformer prevails.
increases with deflection and is approximately twice as large at large deflection coordinates, where the cis conformer dominates, compared with small deflection coordinates, where the trans conformer prevails.
The large absolute values of the rate constants (![]() cm
cm![]() s
s![]() ,
, ![]() ) suggests that the reaction kinetics is capture limited and governed by the properties of the long-range interaction between the reaction partners. An analysis within the framework of adiabatic capture theory [111] quantitatively confirmed the different values for the rate constants of the two conformers. It showed that the differences of the rate constants can be rationalized in terms of the different ion–dipole interactions exhibited by the two conformers: the cis conformer with the larger dipole has a stronger and therefore longer-range interaction with an ion. This translates in to a larger capture impact parameter and therefore larger rate constant for this species.
) suggests that the reaction kinetics is capture limited and governed by the properties of the long-range interaction between the reaction partners. An analysis within the framework of adiabatic capture theory [111] quantitatively confirmed the different values for the rate constants of the two conformers. It showed that the differences of the rate constants can be rationalized in terms of the different ion–dipole interactions exhibited by the two conformers: the cis conformer with the larger dipole has a stronger and therefore longer-range interaction with an ion. This translates in to a larger capture impact parameter and therefore larger rate constant for this species.
VI. Conclusions and Outlook
Recent years have seen impressive progress in the control of ions in the gas phase. Molecular ions can be trapped and localized in space with precise control over their kinetic energies and rotational–vibrational quantum states. In combination with techniques for the control of neutral molecules, these methods have paved the way for new studies of ion–molecule reactions which unravel exquisite details of their mechanisms, kinetics and dynamics.
The field is still in its infancy and promises a range of exciting developments for the future. A fascinating prospect is the study of the role of the hyperfine structure in chemical reactions. Recently, evidence has been found for the important role of the reactant hyperfine state in cold ion–neutral collisions [138]. For this purpose, it is highly desirable to develop hyperfine-state preparation techniques for cold molecular ions [150] as a prerequisite for studies of hyperfine effects in cold reactions. Another intriguing prospect is the application of state-control techniques developed in the realm of quantum logic to molecular ions [151, 152] which may ultimately also benefit chemical reaction studies. So far, most experiments have focused on small molecular systems, usually diatomics. The extension of the techniques discussed here to ever larger molecules and the study of polyatomic processes will be a prime objective for the coming years. In this context, improved capabilities for the analysis of the reaction products will be mandatory. For this purpose, several groups have recently developed ion traps coupled to time-of-flight mass spectrometers [153, 154]. Techniques for probing product state distributions, such as discussed in Section V.C, will also have to be refined and extended to a wider range of systems to gain a more comprehensive understanding of ion–molecule reactions dynamics.
Acknowledgments
This work is supported by the Swiss National Science Foundation (grant nrs BSCGI0_157874 and 200021_156182).
References
- 1. Metcalf, H.J. and van der Straten, P. (1999) Laser Cooling and Trapping, Springer, New York.
- 2. Cornell, E.A. and Wieman, C.E. (2002) Rev. Mod. Phys., 74, 875.
- 3. Wineland, D.J. (2013) Rev. Mod. Phys., 85, 1103.
- 4. Haroche, S. (2013) Rev. Mod. Phys., 85, 1083.
- 5. van de Meerakker, S.Y.T., Bethlem, H.L., Vanhaecke, N., and Meijer, G. (2012) Chem. Rev., 112, 4828.
- 6. Hogan, S.D., Motsch, M., and Merkt, F. (2011) Phys. Chem. Chem. Phys., 13, 18 705.
- 7. Narevicius, E. and Raizen, M.G. (2012) Chem. Rev., 112, 4879.
- 8. Jones, K.M., Tiesinga, E., Lett, P.D., and Julienne, P.S. (2006) Rev. Mod. Phys., 78, 483.
- 9. Ulmanis, J., Deiglmayr, J., Repp, M., Wester, R., and Weidemüller, M. (2012) Chem. Rev., 112, 4890.
- 10. Köhler, T., Gorál, K., and Julienne, P.S. (2006) Rev. Mod. Phys., 78, 1311.
- 11. Chin, C., Grimm, R., Julienne, P., and Tiesinga, E. (2010) Rev. Mod. Phys., 82, 1225.
- 12. Rangwala, S.A., Junglen, T., Rieger, T., Pinkse, P.W.H., and Rempe, G. (2003) Phys. Rev. A, 67, 043 406.
- 13. Fulton, R., Bishop, A.I., and Barker, P.F. (2004) Phys. Rev. Lett., 93, 243 004.
- 14. Bethlem, H.L. and Meijer, G. (2003) Int. Rev. Phys. Chem., 22, 73.
- 15. Hutson, J.M. and Soldán, P. (2006) Int. Rev. Phys. Chem., 25, 497.
- 16. Willitsch, S., Bell, M.T., Gingell, A.D., and Softley, T.P. (2008) Phys. Chem. Chem. Phys., 10, 7200.
- 17. van de Meerakker, S.Y.T., Bethlem, H.L., and Meijer, G. (2008) Nat. Phys., 4, 595.
- 18. Krems, R.V., Stwalley, W.C., and Friedrich, B. (eds) (2009) Cold Molecules: Theory, Experiment, Applications, CRC Press, Boca Raton, FL.
- 19. Schnell, M. and Meijer, G. (2009) Angew. Chem. Int. Ed., 48, 6010.
- 20. Bell, M.T. and Softley, T.P. (2009) Mol. Phys., 107, 99.
- 21. Carr, L.D., DeMille, D., Krems, R.V., and Ye, J. (2009) New J. Phys., 11, 055 049.
- 22. Dulieu, O. and Gabbanini, C. (2009) Rep. Prog. Phys., 72, 086 401.
- 23. Hutzler, N.R., Lu, H.I., and Doyle, J.M. (2012) Chem. Rev., 112, 4803.
- 24. Willitsch, S. (2012) Int. Rev. Phys. Chem., 31, 175.
- 25. Stuhl, B.K., Hummon, M.T., and Ye, J. (2014) Annu. Rev. Phys. Chem., 65, 501.
- 26. Jankunas, J. and Osterwalder, A. (2015) Annu. Rev. Phys. Chem., 66, 241.
- 27. Heazlewood, B. and Softley, T.P. (2015) Annu. Rev. Phys. Chem., 66, 475.
- 28. Krems, R.V. (2005) Int. Rev. Phys. Chem., 24, 99.
- 29. Krems, R.V. (2008) Phys. Chem. Chem. Phys., 10, 4079.
- 30. Quéméner, G. and Julienne, P.S. (2012) Chem. Rev., 112, 4949.
- 31. Lemeshko, M., Krems, R.V., Doyle, J.M., and Kais, S. (2013) Mol. Phys., 111, 1648.
- 32. Chang, Y.P., Horke, D.A., Trippel, S., and Küpper, J. (2015) Int. Rev. Phys. Chem., 34, 557.
- 33. Shapiro, M. and Brumer, P. (2003) Principles of the Quantum Control of Molecular Processes, Wiley-Interscience, Hoboken, NJ.
- 34. Major, F.G., Gheorghe, V.N., and Werth, G. (2005) Charged Particle Traps, Springer, Berlin and Heidelberg.
- 35. March, R.E. and Todd, J.F. (2005) Quadrupole Ion Trap Mass Spectrometry, 2nd edn, John Wiley & Sons, Inc., Hoboken, NJ.
- 36. Leibfried, D., Blatt, R., Monroe, C., and Wineland, D. (2003) Rev. Mod. Phys., 75, 281.
- 37. Gerlich, D. (1992) Adv. Chem. Phys., 82, 1.
- 38. Wester, R. (2009) J. Phys. B, 42, 154 001.
- 39. Drewsen, M. and Brøner, A. (2000) Phys. Rev. A, 62, 045 401.
- 40. Scoles, G. (ed.) (1988) Atomic and Molecular Beam Methods, Oxford University Press, Oxford.
- 41. Smith, M.A. (1998) Int. Rev. Phys. Chem., 17, 35.
- 42. Rowe, B.R., Canosa, A., and Le Page, V. (1995) Int. J. Mass Spectrom. Ion Processes, 149/150, 573.
- 43. Even, U. (2015) Eur. Phys. J. Tech. Instrum., 2, 17.
- 44. Gerlich, D. (1995) Ion–neutral collisions in a 22-pole trap at very-low energies. Phys. Scr., T59, 256.
- 45. Foot, C.F. (2005) Atomic Physics, Oxford University Press, Oxford.
- 46. Drewsen, M., Jensen, I., Lindballe, J., Nissen, N., Martinussen, R., Mortensen, A., Staanum, P., and Voigt, D. (2003) Int. J. Mass Spectrom., 229, 83.
- 47. Willitsch, S., Bell, M.T., Gingell, A.D., Procter, S.R., and Softley, T.P. (2008) Phys. Rev. Lett., 100, 043 203.
- 48. Wineland, D.J., Drullinger, R.E., and Walls, F.L. (1978) Phys. Rev. Lett., 40, 1639.
- 49. Neuhauser, W., Hohenstatt, M., Toschek, P., and Dehmelt, H. (1978) Phys. Rev. Lett., 41, 233.
- 50. McClelland, J.J. and Hanssen, J.L. (2006) Phys. Rev. Lett., 96, 143 005.
- 51. Lu, M., Youn, S.H., and Lev, B.L. (2010) Phys. Rev. Lett., 104, 063 001.
- 52. Shuman, E.S., Barry, J.F., and DeMille, D. (2010) Nature, 467, 820.
- 53. Isaev, T.A. and Berger, R. (2016) Phys. Rev. Lett., 116, 063 006.
- 54. Pollock, E.L. and Hansen, J.P. (1973) Phys. Rev. A, 8, 3110.
- 55. Farouki, R.T. and Hamaguchi, S. (1993) Phys. Rev. E, 47, 4330.
- 56. Zhang, C.B., Offenberg, D., Roth, B., Wilson, M.A., and Schiller, S. (2007) Phys. Rev. A, 76, 012 719.
- 57. Mølhave, K. and Drewsen, M. (2000) Phys. Rev. A, 62, 011 401.
- 58. Tong, X., Winney, A.H., and Willitsch, S. (2010) Phys. Rev. Lett., 105, 143 001.
- 59. Offenberg, D., Zhang, C.B., Wellers, Ch., Roth, B., and Schiller, S. (2008) Phys. Rev. A, 78, 061 401.
- 60. Baba, T. and Waki, I. (2002) Appl. Phys. B, 74, 375.
- 61. Bertelsen, A., Jorgensen, S., and Drewsen, M. (2006) J. Phys. B: At. Mol. Opt. Phys., 39, L83.
- 62. Hansen, A.K., Versolato, O.O., Klosowski, L., Kristensen, S.B., Gingell, A., Schwarz, M., Windberger, A., Ullrich, J., Lopez-Urrutia, J.R.C., and Drewsen, M. (2014) Nature, 508, 76–79.
- 63. Anderson, S.L. (1992) Adv. Chem. Phys., 82, 177.
- 64. Morrison, R.J.S., Conaway, W.E., Ebata, T., and Zare, R.N. (1986) J. Chem. Phys., 84, 5527.
- 65. Sage, A.D., Oliver, T.A.A., Dixon, R.N., and Ashfold, M.N.R. (2010) Mol. Phys., 108, 945.
- 66. Mackenzie, S.R. and Softley, T.P. (1994) J. Chem. Phys., 101, 10 609.
- 67. Green, R.J., Qian, J., Kim, H.T., and Anderson, S.L. (2000) J. Chem. Phys., 113, 3002.
- 68. Willitsch, S. and Merkt, F. (2005) Int. J. Mass Spectrom., 245, 14.
- 69. Tong, X., Wild, D., and Willitsch, S. (2011) Phys. Rev. A, 83, 023 415.
- 70. Staanum, P.F., Højbjerre, K., Skyt, P.S., Hansen, A.K., and Drewsen, M. (2010) Nat. Phys., 6, 271.
- 71. Willitsch, S. (2010) Nat. Phys., 6, 240.
- 72. Schneider, T., Roth, B., Duncker, H., Ernsting, I., and Schiller, S. (2010) Nat. Phys., 6, 275.
- 73. Lien, C.Y., Seck, C.M., Lin, Y.W., Nguyen, J.H.V., Tabor, D.A., and Odom, B.C. (2014) Nat. Commun., 5, 4783.
- 74. Shvartsburg, A.A. (2008) Differential Ion Mobility Spectrometry, CRC Press, Boca Raton, FL.
- 75. Bohrer, B.C., Merenbloom, S.I., Koeniger, S.L., Hilderbrand, A.E., and Clemmer, D.E. (2008) Annu. Rev. Anal. Chem., 1, 293.
- 76. Papadopoulos, G., Svendsen, A., Boyarkin, O.V., and Rizzo, T.G. (2011) Faraday Discuss., 150, 243.
- 77. Park, S.T., Kim, S.Y., and Kim, M.S. (2002) Nature, 415, 306.
- 78. Kim, M.H., Shen, L., Tao, H., Martinez, T.J., and Suits, A.G. (2007) Science, 315, 1561.
- 79. Hogan, S.D. (2016) Eur. Phys. J. Tech. Instrum., 3, 2.
- 80. Bethlem, H.L., Berden, G., and Meijer, G. (1999) Phys. Rev. Lett., 83, 1558.
- 81. Kroto, H.W. (1975) Molecular Rotation Spectra, John Wiley & Sons, Ltd., London.
- 82. Brown, J.M. and Carrington, A. (2003) Rotational Spectroscopy of Diatomic Molecules, Cambridge University Press, Cambridge.
- 83. van de Meerakker, S.Y.T., Vanhaecke, N., van der Loo, M.P.J., Groenenboom, G.C., and Meijer, G. (2005) Phys. Rev. Lett., 95, 013 003.
- 84. Sawyer, B.C., Lev, B.L., Hudson, E.R., Stuhl, B.K., Lara, M., Bohn, J.L., and Ye, J. (2007) Phys. Rev. Lett., 98, 253 002.
- 85. Bethlem, H.L., Crompvoets, F.M.H., Jongma, R.T., van de Meerakker, S.Y.T., and Meijer, G. (2002) Phys. Rev. A, 65, 053 416.
- 86. Osterwalder, A., Meek, S.A., Hammer, G., Haak, H., and Meijer, G. (2010) Phys. Rev. A, 81, 051 401.
- 87. Vliegen, E., Wörner, H.J., Softley, T.P., and Merkt, F. (2004) Phys. Rev. Lett., 92, 033 005.
- 88. Vanhaecke, N., Meier, U., Andrist, M., Meier, B.H., and Merkt, F. (2007) Phys. Rev. A, 75, 031 402.
- 89. Trimeche, A., Bera, M.N., Cromiéres, J.P., Robert, J., and Vanhaecke, N. (2011) Eur. Phys. J. D, 65, 263.
- 90. Akerman, N., Karpov, M., David, L., Lavert-Ofir, E., Narevicius, J., and Narevicius, E. (2015) New J. Phys., 17, 065 015.
- 91. Fulton, R., Bishop, A.I., Shneider, M.N., and Barker, P.F. (2006) J. Phys. B: At. Mol. Opt. Phys., 39, S1097.
- 92. Bell, M.T., Gingell, A.D., Oldham, J., Softley, T.P., and Willitsch, S. (2009) Faraday Discuss., 142, 73.
- 93. Bertsche, B. and Osterwalder, A. (2010) Phys. Rev. A, 82, 033 418.
- 94. Motsch, M., Schenk, M., van Buuren, L.D., Zeppenfeld, M., Pinkse, P.W.H., and Rempe, G. (2007) Phys. Rev. A, 76, 061 402.
- 95. van Buren, L.D., Sommer, C., Motsch, M., Pohle, S., Schenk, M., Bayerl, J., Pinkse, P.W.H., and Rempe, G. (2009) Phys. Rev. Lett., 102, 033 001.
- 96. Twyman, K.S., Bell, M.T., Heazlewood, B.R., and Softley, T.P. (2014) J. Chem. Phys., 141, 024 308.
- 97. Simons, J.P., Jockusch, R.A., Carcabal, P., Hünig, I., Kroemer, R.T., Macleod, N.A., and Snoek, L.C. (2005) Int. Rev. Phys. Chem., 24, 489.
- 98. de Vries, M.S. and Hobza, P. (2007) Annu. Rev. Phys. Chem., 58, 585.
- 99. Rizzo, T.R., Stearns, J.A., and Boyarkin, O.V. (2009) Int. Rev. Phys. Chem., 28, 481.
- 100. Dian, B.C., Longarte, A., and Zwier, T.S. (2002) Science, 296, 2369.
- 101. Dian, B.C., Clarkson, J.R., and Zwier, T.S. (2004) Science, 303, 1169.
- 102. Filsinger, F., Küpper, J., Meijer, G., Hansen, J.L., Maurer, J., Nielsen, J.H., Holmegaard, L., and Stapelfeldt, H. (2009) Angew. Chem. Int. Ed., 48, 6900.
- 103. Hirschfelder, J.O. and Meath, W.J. (1967) Adv. Chem. Phys., 12, 3.
- 104. Stone, A. (2013) The Theory of Intermolecular Forces, 2nd edn, Oxford University Press, Oxford.
- 105. Maergoiz, A.I., Nikitin, E.E., Troe, J., and Ushakov, V.G. (1996) J. Chem. Phys., 105, 6263.
- 106. Maergoiz, A.I., Nikitin, E.E., Troe, J., and Ushakov, V.G. (1996) J. Chem. Phys., 105, 6270.
- 107. Clary, D.C. (1985) Mol. Phys., 54, 605.
- 108. Gioumousis, G. and Stevenson, D.P. (1958) J. Chem. Phys., 29, 294.
- 109. Levine, R.D. (2005) Molecular Reaction Dynamics, Cambridge University Press, Cambridge.
- 110. Clary, D.C. (1995) Chem. Phys. Lett., 232, 267.
- 111. Stoecklin, T., Clary, D.C., and Palma, A. (1992) J. Chem. Soc., Faraday Trans., 88, 901.
- 112. Troe, J. (1987) J. Chem. Phys., 87, 2773.
- 113. Dashevskaya, E.I., Maergoiz, A.I., Troe, J., Litvin, I., and Nikitin, E.E. (2003) J. Chem. Phys., 118, 7313.
- 114. Gao, B. (2010) Phys. Rev. Lett., 104, 213 201.
- 115. Gao, B. (2011) Phys. Rev. A, 83, 062 712.
- 116. Gingell, A.D., Bell, M.T., Oldham, J.M., Softley, T.P., and Harvey, J.M. (2010) J. Chem. Phys., 133, 194 302.
- 117. Harvey, J.N., Schröder, D., Koch, W., Danovich, D., Shaik, S., and Schwarz, H. (1997) Chem. Phys. Lett., 273, 164.
- 118. Mikosch, J., Trippel, S., Eichhorn, C., Otto, R., Lourderaj, U., Zhang, J.X., Hase, W.L., Weidemüller, M., and Wester, R. (2008) Science, 319, 183.
- 119. Smith, W.W., Makarov, O.P., and Lin, J. (2005) J. Mod. Opt., 52, 2253.
- 120. Grier, A.T., Cetina, M., Oručević, F., and Vuletić, V. (2009) Phys. Rev. Lett., 102, 223 201.
- 121. Willitsch, S. (2015) In Proceedings of the International School of Physics “Enrico Fermi”, vol. 189, p. 255.
- 122. Sias, C. and Köhl, M. (2014) In Quantum Gas Experiments, World Scientific Publishing, Singapore, p. 267.
- 123. Härter, A. and Hecker Denschlag, J. (2014) Contemp. Phys., 55, 33.
- 124. Rellergert, W.G., Sullivan, S.T., Kotochigova, S., Petrov, A., Chen, K., Schowalter, S.J., and Hudson, E.R. (2011) Phys. Rev. Lett., 107, 243 201.
- 125. Ravi, K., Lee, S., Sharma, A., Werth, G., and Rangwala, S.A. (2012) Nat. Commun., 3, 1126.
- 126. Hall, F.H.J., Eberle, P., Hegi, G., Raoult, M., Aymar, M., Dulieu, O., and Willitsch, S. (2013) Mol. Phys., 111, 2020.
- 127. Hall, F.H.J., Aymar, M., Bouloufa-Maafa, N., Dulieu, O., and Willitsch, S. (2011) Phys. Rev. Lett., 107, 243 202.
- 128. Hall, F.H.J. and Willitsch, S. (2012) Phys. Rev. Lett., 109, 233 202.
- 129. Raab, E.L., Prentiss, M., Cable, A., Chu, S., and Pritchard, D.E. (1987) Phys. Rev. Lett., 59, 2631.
- 130. Sivarajah, I., Goodman, D.S., Wells, J.E., Narducci, F.A., and Smith, W.W. (2012) Phys. Rev. A, 86, 063 419.
- 131. Haze, S., Hata, S., Fujinaga, M., and Mukaiyama, T. (2013) Phys. Rev. A, 87, 052 715.
- 132. Zipkes, C., Palzer, S., Sias, C., and Köhl, M. (2010) Nature, 464, 388.
- 133. Schmid, S., Härter, A., and Hecker Denschlag, J. (2010) Phys. Rev. Lett., 105, 133 202.
- 134. Meir, Z., Sikorsky, T., Ben-Shlomi, R., Akerman, N., Dallal, Y., and Ozeri, R. (2016) Phys. Rev. Lett., 117, 243 401.
- 135. Cetina, M., Grier, A.T., and Vuletić, V. (2012) Phys. Rev. Lett., 109, 253 201.
- 136. Rellergert, W.G., Sullivan, S.T., Schowalter, S.J., Kotochigova, S., Chen, K., and Hudson, E.R. (2013) Nature, 495, 490.
- 137. Hall, F.H.J., Aymar, M., Raoult, M., Dulieu, O., and Willitsch, S. (2013) Mol. Phys., 111, 1683.
- 138. Ratschbacher, L., Zipkes, C., Sias, C., and Köhl, M. (2012) Nat. Phys., 8, 649.
- 139. Härter, A., Krükow, A., Dei
 , M., Drews, B., Tiemann, E., and Hecker Denschlag, J. (2013) Nat. Phys., 9, 512.
, M., Drews, B., Tiemann, E., and Hecker Denschlag, J. (2013) Nat. Phys., 9, 512. - 140. Haze, S., Saito, R., Fujinaga, M., and Mukaiyama, T. (2015) Phys. Rev. A, 91, 032 709.
- 141. Drewsen, M., Mortensen, A., Martinussen, R., Staanum, P., and Sorensen, J.L. (2004) Phys. Rev. Lett., 93, 243 201.
- 142. Staanum, P.F., Højbjerre, K., Wester, R., and Drewsen, M. (2008) Phys. Rev. Lett., 100, 243 003.
- 143. Hansen, A.K., Sørensen, M.A., Staanum, P.F., and Drewsen, M. (2012) Angew. Chem. Int. Ed., 51, 7960.
- 144. Tong, X., Nagy, T., Yosa Reyes, J., Germann, M., Meuwly, M., and Willitsch, S. (2012) Chem. Phys. Lett., 547, 1.
- 145. Schlemmer, S., Kuhn, T., Lescop, E., and Gerlich, D. (1999) Int. J. Mass Spectrom., 185–187, 589.
- 146. Khriachtchev, L., Domanskaya, A., Marushkevich, K., Räsänen, M., Grigorenko, B., Ermilov, A., Andrijchenko, N., and Nemukhin, A. (2009) J. Phys. Chem. A, 113, 8143.
- 147. Taatjes, C.A., Welz, O., Eskola, A.J., Savee, J.D., Scheer, A.M., Shallcross, D.E., Rotavera, B., Lee, E.P.F., Dyke, J.M., Mok, D.K.W., Osborn, D.L., and Percival, C.J. (2013) Science, 340, 177.
- 148. Chang, Y.P., Dlugolecki, K., Küpper, J., Rösch, D., Wild, D., and Willitsch, S. (2013) Science, 342, 98.
- 149. Rösch, D., Willitsch, S., Chang, Y.P., and Küpper, J. (2014) J. Chem. Phys., 140, 124 202.
- 150. Bressel, U., Borodin, A., Shen, J., Hansen, M., Ernsting, I., and Schiller, S. (2012) Phys. Rev. Lett., 108, 183 003.
- 151. Mur-Petit, J., Pérez-Ríos, J., Campos-Martínez, J., Hernández, M.I., Willitsch, S., and García-Ripoll, J.J. (2012) Phys. Rev. A, 85, 022 308.
- 152. Wolf, F., Wan, Y., Heip, J.C., Gebert, F., Shi, C., and Schmidt, P.O. (2016) Nature, 530, 457.
- 153. Schneider, C., Schowalter, S.J., Chen, K., Sullivan, S.T., and Hudson, E.R. (2014) Phys. Rev. Appl., 2, 034 013.
- 154. Meyer, K.A.E., Pollum, L.L., Petralia, L.S., Tauschinsky, A., Rennick, C.J., Softley, T.P., and Heazlewood, B.R. (2015) J. Phys. Chem. A, 119, 12 449.