
8. How Cellular Information Is Altered
We have discussed some aspects of how cells inherit information and how a chemostat (covered in Chapter 6, “How Cells Grow”) can be used as a tool to select for individual cells with different or augmented metabolism. The mechanism for DNA replication in procaryotes was summarized in Chapter 4, “How Cells Work.” This process is a good example of the exchange of genetic information from one generation to another. However, some individual cells can receive additional genetic information through natural or artificial means. The initial genetic information within a cell may also undergo rearrangements or alterations. In this chapter, we discuss some mechanisms causing alterations in a cell’s genetic information and ways that we can manipulate those mechanisms to improve bioprocesses.
8.1. Evolving Desirable Biochemical Activities Through Mutation and Selection
Although the cell has a well-developed system to prevent errors in DNA replication and an active repair system to correct damage to a DNA molecule, mistakes can still occur. These mistakes are called mutations. Before we discuss mutations, we need to establish the working vocabulary of microbial genetics.
The sum of the genetic construction of an organism constitutes its genotype. The characteristics expressed by a cell constitute its phenotype. The phenotypic response of a culture may change reversibly with alterations in environmental conditions, whereas the genotype is constant irrespective of the environment. A mutation is a genotypic change and is irreversible. A whole culture undergoes a phenotypic response, whereas only a rare individual will undergo a genotypic change. For example, if a culture changes in color from white to green when oxygen levels fall and then changes from green to white upon an increase in dissolved oxygen, the change would be phenotypic. Now consider an alternative experiment where white cells were removed from a culture, and placed on a plate (a small circular dish filled with nutrients solidified with agar) and allowed to grow into separate colonies. If one colony, but not the others, turned green and if cells obtained from the green colony remain green when cultured under the original conditions, it would be evidence for a genotypic change. In this case, the white cells would be the wild type and the green cells the mutants.
Let us consider what mechanisms may lead to genotypic change.
8.1.1. How Mutations Occur
Most mutations occur because of mistakes in DNA synthesis. Some examples are shown in Figure 8.1. One common form is a point mutation. A point mutation results from the change of a single base (e.g., cytosine instead of thymine). Some point mutations are silent mutations because the altered codon still codes for the same amino acid (e.g., UCU and UCA both code for serine). Even if the point mutation causes the substitution of a different amino acid, it may or may not alter protein activity substantially. A change of amino acid near the active site might alter protein activity greatly, whereas the same substitution at another site might have little to no effect.

Figure 8.1. (a) Possible effects of base-pair substitution in a gene encoding a protein. (b) Shifts in the reading frame of mRNA caused by insertions and deletions. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
One type of point mutation that usually has a profound effect is a nonsense mutation, whereby a stop codon is introduced into a DNA sequence (e.g., the CAA codon for glutamine changed to the UAA codon for “stop” in the mRNA derived from the altered DNA). A nonsense mutation results in premature termination of translation and a truncated, incomplete, and usually nonfunctional protein product.
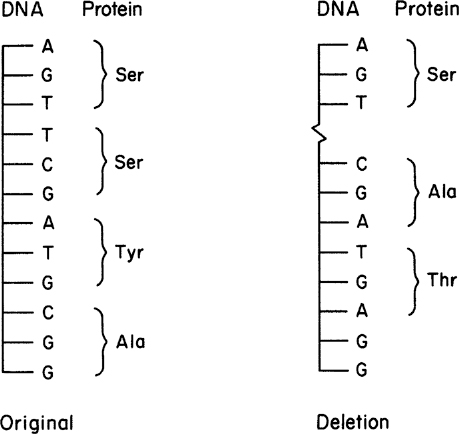
Generally, deletion mutations have profound effects on cellular metabolism. By deleting or adding one or more bases, we can alter the whole composition of a protein, not just a single amino acid. A deletion can shift the reading frame when translating the resulting mRNA. This effect is illustrated in Figure 8.2.
Additions often take place through insertion-sequence (IS) elements. These elements are about 700 to 1400 base pairs in length; there are 37 different IS elements present in the genome of Escherichia coli strain MG1655. These elements can move on the chromosome from essentially any site to another. Often, they will insert in the middle of a gene, totally destroying its function.
Back mutations or reversions are possible. Revertants are cells for which the original wild-type phenotype has been restored. Restoration of a function can occur due to a direct change at the original mutation (e.g., if the original mutation was CAA to UAA, then a second mutation for UAA to CAA restores the original genotype and phenotype). Second-site revertants can occur that restore phenotype (suppressor mutations) but not genotype (e.g., a second deletion mutation that restores the gene to the normal reading frame or a mutation in another gene that restores the wild-type phenotype).
8.1.2. Selecting for Desirable Mutants
Mutants can serve as powerful tools to better understand cell physiology; they are also valuable as industrial organisms because mutation can be used to alter metabolic regulation and cause overproduction of a desired compound. Methods to induce mutations and then select for mutants are important tools for catalyst development in bioprocessing.
Natural (spontaneous) rates of mutation vary greatly from gene to gene (10–3 to 10–9 per cell division), with 10–6 mutations in a gene per cell division being typical. Chemical agents (mutagens) and/or radiation are often used in the laboratory to increase mutation rates. Mutagens are nonspecific and may affect any gene.
The selection of a mutant with desirable properties is no easy task. Mutations are classified as selectable and unselectable. A selectable mutation confers upon the mutant an advantage for growth or survival under a specific set of environmental conditions; thus, the mutant can grow and the wild type will die. An unselectable mutant requires a cell-by-cell examination to find a mutant with the desired characteristics (e.g., green pigment). Even with mutagens, the frequency of mutation is sufficiently low to make prohibitive a brute-force screening effort for most unselectable mutants.
Selection can be direct or indirect. An example of direct selection would be to find a mutant resistant to an antibiotic or toxic compound. A culture fluid containing 108 to 1010 cells/ml is subjected to a mutagenic agent. A few drops of culture fluid are spread evenly on a plate, with the antibiotic incorporated into the solidified medium. Only antibiotic-resistant cells can grow, so any colonies that form must arise from antibiotic-resistant mutants. If one in a million cells has this particular mutation, we would expect to find about 10 to 100 colonies per plate if 0.1 ml of culture fluid was tested.
Indirect selection is used for isolating mutants that are deficient in their capacity to produce a necessary growth factor (e.g., an amino acid or a vitamin). Wild-type E. coli grow on glucose and mineral salts. Auxotrophic mutants would not grow on such a simple medium unless it were supplemented with the growth factor that the cell could no longer make (e.g., a lysine auxotroph has lost the capacity to make lysine, so lysine must be added to the glucose and salts to enable the cell to grow). The wild-type cell that needs no supplements to a minimal medium is called a prototroph. Consider the selection of a rare mutant cell that is auxotrophic for lysine from a population of wild-type cells. This selection cannot be done directly because both cell types would grow in the minimal medium supplemented with lysine.
A method that facilitates selection greatly is called replica plating (see Figure 8.3). A master plate using lysine-supplemented medium will grow both the auxotroph and wild-type cells. Once colonies are well formed on the master plate, an imprint is made on sterile velveteen. The bristles on the velveteen capture some of the cells from each colony. The orientation of the master plate is carefully noted. Then a test plate with minimal medium is pressed against the velveteen; some cells, at the point of each previous colony, then serve to inoculate the test plate at positions identical to those on the master plate. After incubation (approximately 24 hours for E. coli), the test plate is compared to the master plate. Colonies that appear at the same positions on both plates arise from wild-type cells, whereas colonies that exist only on the master plate must arise from the auxotrophic mutants.
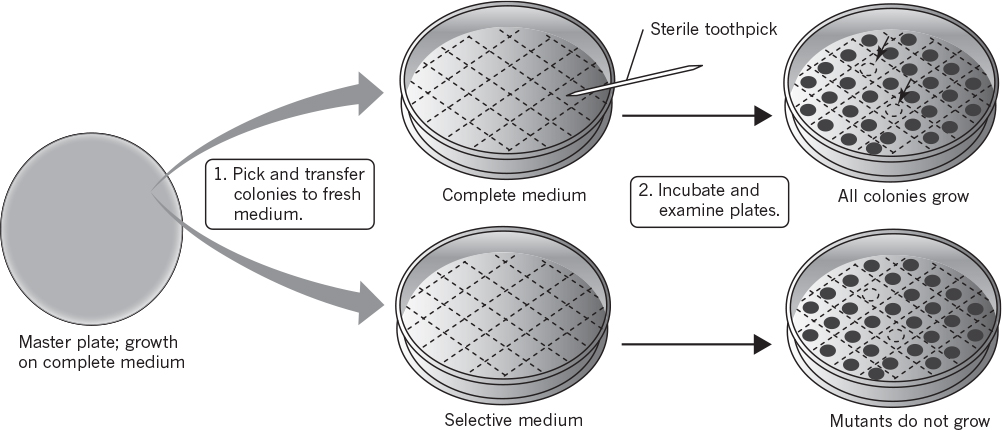
Figure 8.3. Screening for nutritional auxotrophs. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
Another class of mutants is called conditional mutants. Mutations that would normally be lethal to the cell could not be detected by methods we have described so far. However, mutated proteins are often more temperature sensitive than normal proteins. Thus, temperature sensitivity can often be used to select for conditionally lethal mutations. For example, the mutant may be unable to grow at the normal growth temperature (e.g., 37°C for E. coli) but will grow satisfactorily at a lower temperature (e.g., 25°C). Table 8.1 summarizes a variety of mutants and how they may be detected.

Mutation and selection have been used to tremendous advantage to probe the basic features of cell physiology and regulation. They also have been the mainstay of industrial programs for the improvement of production strains. For example, mutation and selection strategies have been primarily responsible for increasing the yield of penicillin from 0.001 g/l in 1939 to current values of about 50 g/l of fermentation broth.
8.2. Natural Mechanisms for Gene Transfer and Rearrangement
Bacteria can gain and express wholly different biochemical capabilities (e.g., the ability to degrade an antibiotic or detoxify a hazardous chemical in their environment) literally overnight. These alterations cannot be explained through inheritance and small evolutionary changes in the chromosome. Rather, they arise from gene transfer from one organism to another and/or large rearrangements in chromosomal DNA. In this section, we discuss genetic recombination, gene transfer, and genetic rearrangements—all mechanisms that can be exploited to genetically engineer cells (see Table 8.1).
8.2.1. Genetic Recombination
Genetic recombination is a process that brings genetic elements from two different genomes into one unit, resulting in new genotypes in the absence of mutations. Genetic recombination in procaryotes is a rare event but sufficiently frequent to be important ecologically as well as industrially. The three main mechanisms for gene transfer are transformation, transduction, and conjugation. Transformation is a process in which free DNA is taken up by a cell. Transduction is a process in which DNA is transferred by a bacteriophage, and conjugation is DNA transfer between intact cells that are in direct contact with one another.
Once donor DNA is inside a cell, the mechanism for recombination is essentially independent of how the donor DNA was inserted. Figure 8.4 summarizes the molecular-level events in general recombination. The donor DNA must be homologous, or nearly so, to a segment of DNA on the recipient DNA.† Under the right conditions, cellular enzymes cut out the homologous section of recipient DNA, allow insertion of the donor DNA, and then ligate or join the ends of the donor DNA to the recipient DNA. Note that pieces of donor DNA that a cell recognizes as foreign are usually degraded by enzymes called restriction endonucleases (these enzymes are essential in genetic engineering) and cut a DNA molecule at a particular predetermined nucleotide sequence. A cell marks its own DNA (e.g., through methylation of certain purine or pyrimidine bases) to distinguish it from foreign DNA. These modifications block the action of a cell’s own restriction endonucleases on its own DNA. Under natural conditions, gene transfer is effective only if the donor DNA is from the same or closely related species.
† Illegitimate recombination between nonhomologous regions of DNA is possible, but rare. See later discussion on transposons.

Figure 8.4. A simplified version of homologous recombination. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
Let us now consider some details of how donor DNA can enter a cell.
8.2.2. Transformation
Transformation is the genetic alteration of a cell resulting from the direct uptake and incorporation of exogenous genetic material (e.g., DNA) from its surroundings through the cell membrane(s). Transformation is a naturally occurring process in some bacteria, but it cannot be performed by all genera of bacteria. Even within transformable genera, only certain strains are transformable (competent). Competent cells have a much higher capacity for binding DNA to the cell surface than do noncompetent cells. Competency can depend on the physiological state of the cell (current and previous growth conditions). Even in a competent population, not all cells are transformable. Typically, about 0.1% to 1.0% are transformable.
Transformation can also be implemented by artificial means in other cells. For example, E. coli cells are not normally competent, but their importance to microbial genetics has led to the development of empirical procedures to induce competency. This procedure involves treating E. coli with high concentrations of calcium ions coupled with temperature manipulation. The competency of treated cells varies among strains of E. coli but is typically rather low (about one in a million cells becomes successfully transformed). With the use of selective markers, this frequency is still high enough to be quite useful.
Transformation is useful only when the information that enters the cell can be propagated. When performing transformation, we typically use a vector called a plasmid. This element forms the basis for most industrially important fermentations involving recombinant DNA. A plasmid is a double-stranded, circular DNA molecule that is physically separated from chromosomal DNA (i.e., extrachromosomal) and can replicate independently (i.e., self-replicating). Some plasmids are maintained as low-copy-number plasmids (only a few copies per cell), and others have a high copy number (20 to 100 copies per cell). These plasmids differ in their mechanisms for partitioning at cell division and in the control of their replication. Plasmids encode genes typically for proteins that are nonessential for growth but that can confer important advantages to their host cells under certain environmental circumstances. For example, most plasmids encode proteins that confer resistance to specific antibiotics. Such antibiotic resistance is very helpful in selecting for cells that contain a desired plasmid.
8.2.3. Transduction
Transduction is the process by which DNA is transferred from one bacterium to another by a vector, which is usually a virus, and certainly plays an important role in nature. In the most common type of transduction, generalized transduction, infection of a recipient cell results in fragmentation of the bacterial DNA into 100 or so pieces. One of these fragments can become packaged randomly into a phage particle. The altered phage particle then injects bacterial DNA into another cell, where it can recombine with that cell’s DNA. With generalized transduction, any bacterial gene may be transferred.
Another method of transfer, which is far more specific with respect to the genes that are transferred, is specialized transduction. Here the phage incorporates into specific sites in the chromosome, and the frequency of transduction of a gene is related to its distance away from the site of incorporation. This process is summarized in Figure 8.5. A lysogenic cell is one carrying a prophage or phage DNA incorporated into chromosomal DNA. Phage lambda is an example of such a temperate phage (a phage that can either lyse a cell or become incorporated into the chromosome). Such phages almost invariably insert at a specific site in the chromosome. The conversion of a prophage (the phage DNA in the chromosome) into the lytic cycle is normally a rare event (10–4 per cell division), but it can be induced in almost the whole culture upon exposure to UV light or other agents that interfere with DNA replication.

Figure 8.5. Transduction, the transfer of genetic material from donor to recipient via virus particles. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
8.2.4. Episomes and Conjugation
A third type of gene transfer involves another genetic element. This element is called an episome. It is a DNA molecule that may exist either integrated into the chromosome or separate from it. When it exists separately from the chromosome (i.e., extrachromosomally), it is essentially a plasmid. A well-known episome is the F or fertility factor. Such factors are responsible for the process known as conjugation.
Most experiments with conjugation are done with the F factor, which is present in low copy number. Direct cell-to-cell contact is required. This DNA molecule encodes at least 13 genes involved in its self-transfer from one cell to another.
A population of E. coli frequently includes some cells with the F plasmid, which are termed F+ (male). Other cells are F– (female). F+ cells encode proteins to make a sex pilus. When F+ and F– cells are mixed together, the sex pilus connects an F+ to an F– cell (see Figure 8.6). The sex pilus may act as a conduit for transferring a copy of the F plasmid to the F– cell. The actual process of transfer involves replication of the F plasmid.
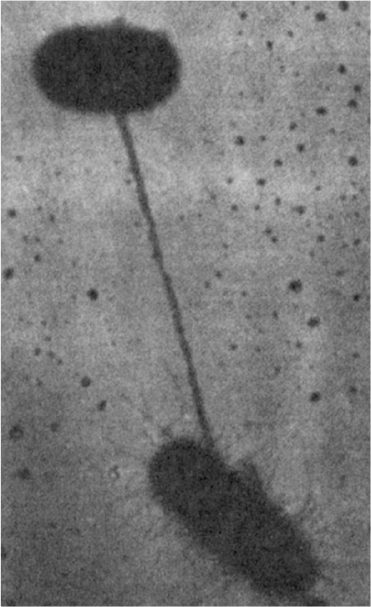
Figure 8.6. Direct contact between two conjugating bacteria is first made via a pilus. The cells are then drawn together for the actual transfer of DNA. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
This process is more typical and does not involve transfer of chromosomal genes or recombination. A less common event is when the F plasmid has been integrated into the chromosome itself to form a single, large, circular molecule. Thirteen sites for integration are known. Such cells are termed Hfr (for high-frequency recombination).
When transfer is initiated, the F plasmid moves not only itself but also the attached chromosome to the recipient cell. The time required to transfer a whole E. coli chromosome is 100 min. If contact between the two cells is broken during the transfer process, only a proportional amount of the chromosome will have been transferred (i.e., at 50 min, about 50%). Since the transfer begins at a known point, Hfr cells can be used to map the location of genes on the chromosome. This technique for gene mapping is being replaced by methods for directly sequencing nucleotide sequences in DNA. If F+ and F– cells differ in properties (e.g., the ability to make lysine), conjugation can be used to alter the properties of the F– cell.
Conjugation, transduction, and transformation all represent forms of gene transfer from one cell to another. However, gene transfer can occur within a cell.
8.2.5. Transposons: Internal Gene Transfer
Previously, we discussed the presence of IS elements on the chromosome. A closely related phenomenon is a transposon, which refers to a gene or genes that have the ability to “jump” from one piece of DNA to another or to another position on the original piece of DNA. The transposon integrates itself into the new position independently of any homology with the recipient piece of DNA. Transposons differ from IS elements in that they code for proteins. Transposons appear to arise when a gene becomes bounded on both sides by insertion sequences. Many of the transposons encode antibiotic resistance.
Transposons are important because they can induce mutations when they insert into the middle of a gene, they can bring once-separate genes together, and in combination with plasmid- or viral-mediated gene transfer, they can mediate the movement of genes between unrelated bacteria (e.g., multiple antibiotic resistance on newly formed plasmids). Transposon mutagenesis can be a very powerful tool in altering cellular properties usually by insertional inactivation of chromosomal genes.
8.3. Genetically Engineering Cells
Our description of DNA replication, mutation, and selection and the natural mechanisms for gene transfer covered all the tools necessary to genetically engineer a cell. The purposeful, predetermined manipulation of cells at the genetic level, an idea that was farfetched prior to 1970, is easily within the grasp of beginning students.
Genetic engineering is a set of tools and not a scientific discipline. Although difficult to define precisely, it involves the manipulation of DNA outside the cell to create artificial genes or novel combinations of genes with predesigned control elements. Because many of these manipulations can be done outside the cell, we can circumvent species limitations that constrain the age-old techniques of mutation and selection and breeding (e.g., we can express a human protein in E. coli). Learning how to use these tools is the basis of modern biotechnology.
8.3.1. Basic Elements of Genetic Engineering
An overview of the strategy typically employed in genetic engineering is given in Figure 8.7. The strategy makes use of recombinant DNA techniques, the ability to isolate genes from one organism and recombine the isolated gene with other DNA that can be propagated in a similar or unrelated host. Most of our discussion is drawn from approaches for genetically engineering bacteria.
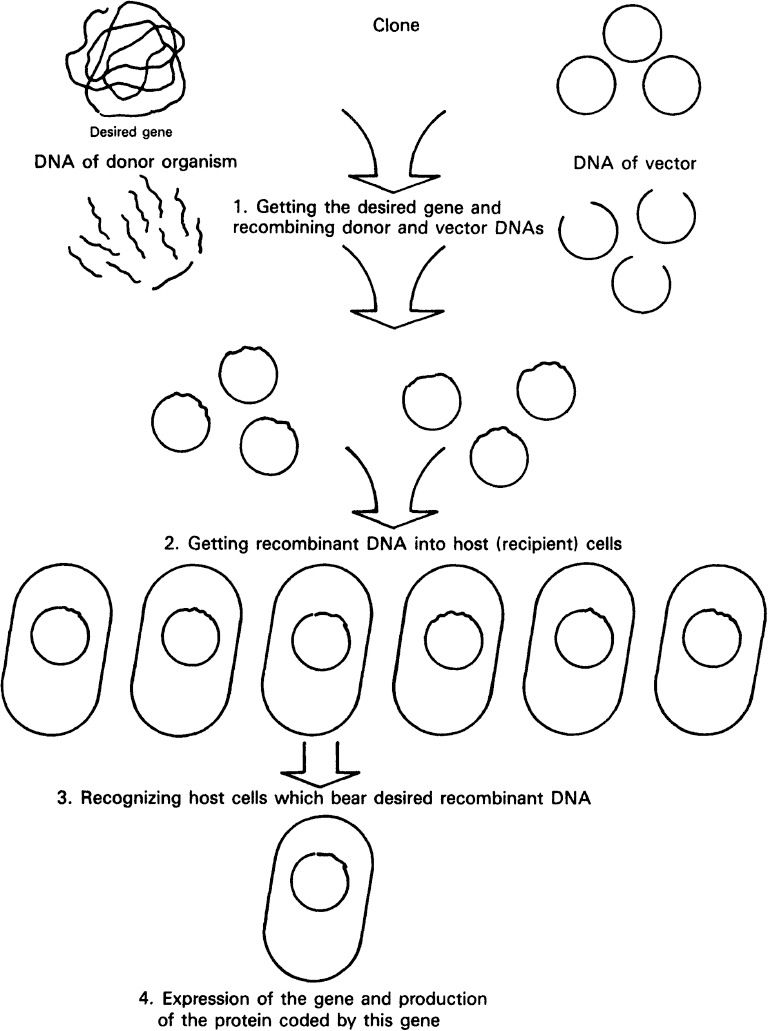
Figure 8.7. Overview of the essential steps in genetic engineering: moving a gene from one organism to another. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
8.3.1.1. Obtaining the gene of interest.
The first step is obtaining the gene of interest. A simple, brute-force approach is shotgun cloning. Here the DNA from the donor organism is cut into fragments using restriction enzymes. If an efficient screening procedure is available, large numbers of host cells with random fragments of DNA can be screened for those exhibiting a property related to the desired gene.
Most often, such a shotgun approach is very inefficient. More specific approaches use hybridization. A probe can be synthesized chemically to be complementary to a portion of the gene. The probe is usually much shorter than the gene but sufficiently long that it is unlikely for other genes to have the same complementary DNA sequence. The construction of the probe requires some knowledge of either the nucleotide sequence of the desired gene or a partial amino acid sequence for the desired gene. Since the genetic code is degenerate, the deduction of the actual nucleotide sequence is ambiguous. This ambiguity requires that a variety of probes be generated. Hybridization reactions require the donor DNA to be both fragmented and converted into single strands that can react with the single-stranded probes.
An alternative to hybridization is total chemical synthesis of a gene that corresponds to the desired protein. This approach requires knowledge of the amino acid sequence of the desired protein and commonly involves column-based, solid-phase phosphoramidite chemistry to synthesize DNA. While gene-length DNA molecules of up to ~200 nucleotide (nt) residues can be routinely synthesized by this method, the cost and error rate can be prohibitive for sequences >200 nt. Thus, a number of methods have emerged to address the cost of making longer DNA molecules.
One approach uses chemical synthesis to make small sets of shorter oligos (typically 5–50 oligos) that are subsequently assembled into larger synthetic fragments (typically 200–3,000 nt) via a variety of methods that leverage biological mechanisms such as DNA ligase-based annealing, DNA polymerase-based assembly, or homologous recombination. More recent methods leverage DNA microarray technology (see Section 8.4.1), whereby standard mask-based photolithographic techniques are used to spatially localize DNA oligo synthesis on surfaces using light-activated chemistries. Microarray-derived oligos are subsequently cleaved and harvested as one “oligo pool,” and the pool of oligos are assembled by a variety of techniques to yield full-length genes. Together, these methods allow us to produce specifically modified natural proteins or potentially totally human-designed proteins. Moreover, an artificial gene may be created that codes for exactly the same protein as in nature but with a sequence of nucleotides on the artificial gene that is not identical to the natural gene. For example, strategic use of synonymous mutations in the coding gene can optimize the expression of a recombinant gene in the target organism, a process known as codon optimization or codon harmonization. This approach takes advantage of natural codon biases that exist between organisms. Because the genetic code is degenerate (i.e., many amino acids are encoded by more than one codon), the genes of different organisms are often biased toward using one of the several codons that encode the same amino acid over the others.
Another method to obtain the desired gene is particularly useful for genes with introns. Since bacteria lack the cellular machinery to cut out introns and perform mRNA splicing, eucaryotic genes with introns cannot be directly placed in bacteria to make a desired protein. Often, we wish to make these proteins in bacteria, since the bacteria grow much more rapidly and are much easier and cheaper to culture. Often the processed mRNA for the desired gene can be isolated directly from the donor organism’s cytoplasm (using hybridization probes that are complementary to the poly[A] tails found on eucaryotic mRNA for total mRNA isolation). Once the mRNA is isolated, the enzyme reverse transcriptase (see Chapter 4) can be used to synthesize a DNA molecule with the corresponding nucleotide sequence; this molecule is called complementary DNA (cDNA).
Large amounts of a target gene can be generated by a technique called the polymerase chain reaction (PCR). PCR is the preferred method to amplify DNA. This technique uses a target sequence of interest on double-stranded DNA (dsDNA) such as cDNA or genomic DNA (gDNA).† In addition, the technique requires that two short primer sequences (<20 nt) on either side of the target be known and chemically synthesized. If heat is applied, the complementary strands of dsDNA separate. While the strands are separated, two pieces of chemically synthesized DNA (the primers) are added. Each primer binds to complementary sequences. A heat-stable DNA polymerase from a bacterium that grows in hot springs (the Taq polymerase) is added and quickly synthesizes from the primers the complementary DNA strands using the four nucleotides (A, G, T, and C) added to the reaction mixture. At the end of the cycle, there are two copies of the original gene. If the cycle is repeated, those two copies become four. Thirty cycles can be done over the course of a few hours. Thus, from a single gene copy, 230 copies of the gene can be generated, representing more than a billion-fold increase. Thermal cyclers and PCR kits are commercially available.
† For forensic studies or phylogenic studies on relationships of organisms, a nonprotein coding sequence may be the target.
It should be noted that although our focus is on amplifying the number of genes as a basis for producing target proteins or altering pathways, gene cloning is often done to obtain many copies of a particular gene. The amplification of the gene number facilitates gene sequencing and analysis. This amplification is particularly important for mapping genomes (e.g., the human genome project), for diagnosis of disease-causing organisms (both microbial and viral), for biologists studying evolution, and for forensic scientists.
8.3.1.2. Inserting the gene into DNA.
Once the desired gene is isolated or made, it can be inserted into a small piece of carrier DNA called a vector. Typically, vectors are plasmids, although temperate viruses can be used. The process for preparing the donor DNA and vector for recombination and the actual joining of the DNA segments usually requires special enzymes (see Figure 8.8). We discussed these enzymes in our previous consideration of DNA replication and genetic recombination. A wide variety of restriction enzymes exist that will cut DNA at a different prespecified site. Most vectors have maps showing the various restriction sites; important examples are EcoR1 from E. coli and Bam H1 from Bacillus amyloliquefaciens. Many restriction enzymes leave “sticky ends,” a few nucleotides of single-stranded DNA projecting from the cut site. Pieces of DNA with complementary sticky ends naturally associate, and in a mixture of cut vector and donor DNA, some pieces of donor DNA will associate with vector DNA. DNA ligase can permanently join these ends.
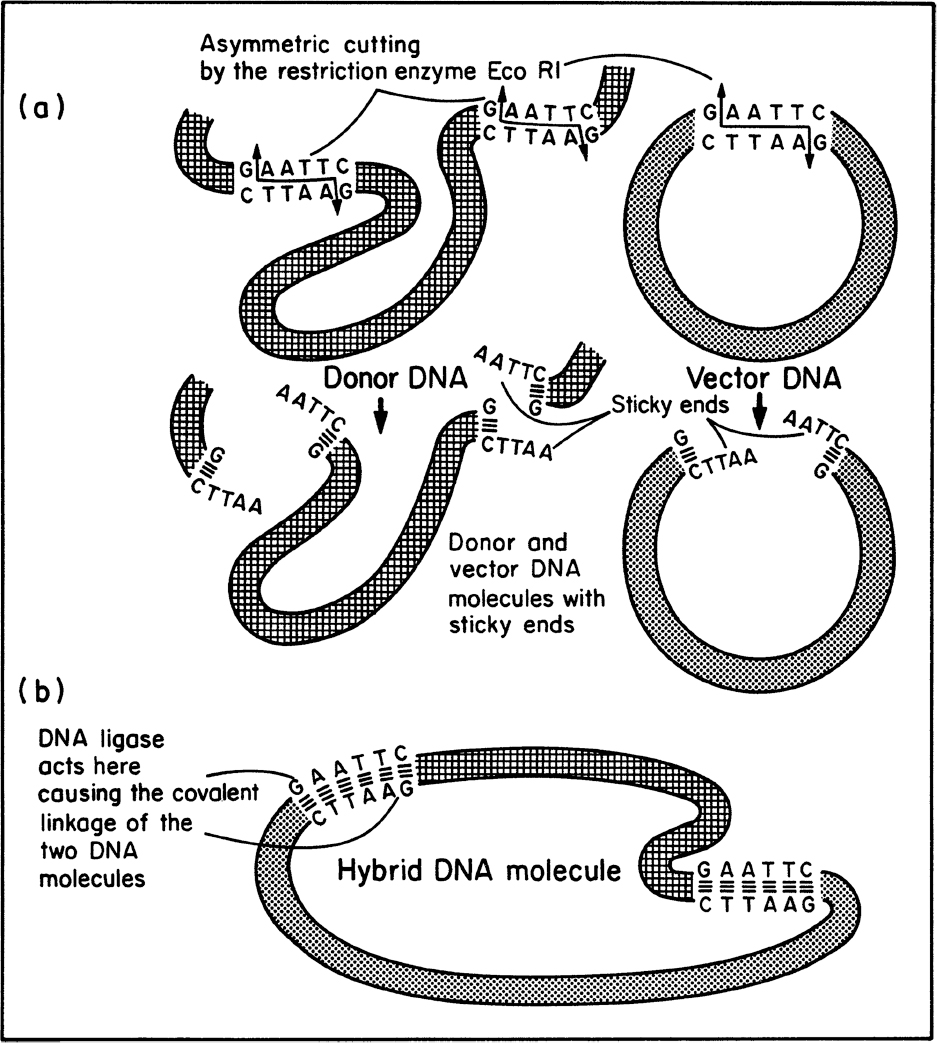
Figure 8.8. Use of special enzymes in genetic engineering. (a) The specific cutting of DNA by a restriction enzyme results in the formation of ends that contain small, single-stranded complementary sequences (“sticky ends”). (b) The enzyme DNA ligase links pieces of DNA that have become associated by their sticky ends. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 3rd ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
The mixture bearing the desired vector–donor combinations is then moved into the recipient or host cell. In most cases, this is done by transformation, although other techniques can be used if transformation of the host is difficult. Note that the construction of the desired vector–donor DNA usually results in a mixture (e.g., some vector molecules may be opened and rejoined without donor DNA being inserted, or multiple copies of donor DNA may be inserted, or DNA contaminants of the donor DNA mixture may become inserted into the vector). Consequently, an efficient method to screen transformants for those with the desired vector–donor DNA combination is important.
Most vectors contain selectable markers such as antibiotic resistance or the genes to make essential growth factors that have been removed by mutation from the host cell. In the latter case, growth in minimal medium is possible only in the presence of the plasmid. These selectable markers allow the isolation of genetic clones that have been successfully transformed. A further screening step is then necessary to ensure that the donor DNA is present and being expressed (i.e., a functional protein is being made from the donor DNA).
8.3.1.3. Alternative strategies for gene cloning.
Over the years, a number of molecular cloning strategies have emerged that provide alternatives to the “classic” restriction enzyme-cloning method previously described. Usually, several different approaches can be used for a specific cloning project; however, for any given project, there may be an ideal approach. The choice of approach may be based on speed, cost, availability of starting materials, or just personal preference.
One of these approaches, TOPO cloning (or TA cloning) takes advantage of the fact that the Taq polymerase enzyme (the same one used in PCR reactions, as discussed earlier) leaves a single adenosine (A) overhang on the 3′ end of PCR products. Using commercially available TOPO vectors, the PCR products are efficiently hybridized between the 3′ A overhang of the PCR product and the 5′ T overhang of the TOPO backbone vector. The major disadvantage of this approach is that very few plasmid backbones are available TOPO ready, and it is difficult to create a TOPO vector yourself.
Another strategy, Gateway cloning, is a bacterial recombination–based method whereby a fragment of DNA is first cloned into a “donor” plasmid, a process that is still most often achieved by traditional restriction enzyme cloning. The DNA fragment in the donor plasmid can then be rapidly shuttled into any compatible Gateway “destination” vector. The advantage of this method is that you can clone your gene of interest one time by restriction enzyme cloning into a donor plasmid, and then leverage bacterial recombination to easily move it into a series of plasmids that allow you to do many different molecular biology techniques (such as fusing it with different tags, putting it under a variety of promoters and into backbones with different selection cassettes).
Isothermal cloning, more commonly known as Gibson assembly, is a method that uses three common molecular biology enzymes—5′ exonuclease, polymerase, and ligase—to reliably assemble multiple fragments of DNA in the chosen orientation. Consequently, there is no need for any unwanted sequence at the junctions (such as restriction enzyme or Gateway recombination sites). The method works with any dsDNA fragments, PCR products, or synthesized oligos with appropriately designed overhangs (i.e., DNA extensions that will hybridize with a target sequence) that can be efficiently ligated into any plasmid backbone.
A particular type of restriction endonuclease, known as a type IIS enzyme, has unique properties that have been harnessed to create molecular cloning methods such as Golden Gate, GreenGate, and modular cloning (MoClo). In these methods, the type IIS enzyme cuts dsDNA at a specified distance from the recognition sequence, which creates custom overhangs, something that is not possible using traditional restriction enzymes. One advantage of this system is that the entire cloning step (digestion and ligation) is carried out in a single pot containing a single restriction enzyme, which is possible because the resulting overhangs are distinct and maintain the directionality of the cloning reaction. Another advantage is that all recognition sequences are removed from the final product (i.e., no undesired sequence or scar remain).
Ligation independent cloning (LIC) employs the 3′ → 5′ exonuclease activity of T4 DNA polymerase to create overhangs with complementarity between the vector and insert. Because of the relatively long stretches of base pairing in the hybridized product, ligation is rendered unnecessary. The product may be transformed directly into E. coli, where the nicks (discontinuity in the dsDNA molecule) are repaired by the normal replication process.
A final method is yeast-mediated cloning, which is very similar in principle to Gibson cloning, but rather than an in vitro assembly reaction using purified enzymes, it takes advantage of the native recombination abilities of yeast. Like Gibson cloning, this method efficiently assembles two (or more) fragments of dsDNA that have overlapping homology. The advantage of this method over others is that much larger final products can be generated (up to 100 kb), which is significant because cloning in bacterial hosts can become progressively more difficult for plasmids larger than 10 kb.
8.3.1.4. Expression of cloned genes.
Obtaining good expression from the donor DNA is often a difficult challenge. Careful selection of stably propagating vectors and of promoters, checks to ensure that the correct reading frame is being used, and the selection of host cell backgrounds that do not interact unfavorably with the “foreign” protein are all important considerations. Discussions of how to obtain and maintain high levels of expression occupy much of Chapter 14, “Utilizing Genetically Engineered Organisms.” We can screen for the expression of donor DNA, for example, if the product confers a selectable marker itself (e.g., the ability to grow on a substrate not normally utilized by the host). Also, antibodies to the target protein when tagged with a radiolabel or fluorescent label can be used to identify colonies expressing the target protein.
An important tool in working with both proteins and DNA fragments or plasmids is electrophoresis. In protein electrophoresis, an electric field is applied to a solution containing proteins placed at the top of a gel (typically made of polyacrylamide). The proteins migrate through the porous structure of the gel in a direction and at a speed that reflect both the size and net charge of the molecule. The gel reduces the effects of convection, although thermally induced convection can be problematic in large gels. For proteins, SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) is commonly used. In this technique, the proteins are denatured by heating with SDS and mercaptoethanol. Mercaptoethanol is a reducing agent used to break disulfide bonds. Individual polypeptide chains form a complex with SDS, which is negatively charged. The negatively charged complex then migrates through the gel at a rate that reflects the molecular weight of the polypeptide. In a typical SDS-PAGE gel, one lane is loaded with a standard that contains polymers of known molecular weight. Other lanes will have samples with unknown proteins. After a defined period of time (typically a few hours), the process is stopped and the gel is examined. Each protein forms a band. With some types of electrophoresis, the band is highlighted by use of stains such as Coomassie blue or silver stain. Smaller molecules travel a greater distance. The molecular weight of a protein band in the unknown sample can be estimated by comparing to the lane with the molecular weight standard. The relative amount of a protein can be determined by the intensity of the band.
Various forms of gel electrophoresis can be used to determine if a protein from a clone is being produced and at what relative level. A particularly useful procedure is an immunoblot or Western blot. Here, a nondenaturing gel is run to separate proteins, they are blotted onto nitrocellulose paper, and the protein identified by binding to a specific antibody (primary antibody) followed by a secondary antibody that binds specifically to the first antibody. This secondary antibody is usually conjugated with a radioactive label or enzyme (e.g., horseradish peroxidase, HRP) that permits detection of the target protein band. A similarly conjugated primary antibody can be used instead, which eliminates the need for a secondary antibody but at the expense of the signal amplification that comes from the second antibody labeling step. The band is visualized by exposure of the nitrocellulose paper to x-ray film when a radioactive label is used or by reaction with a substrate that, when oxidized by HRP using hydrogen peroxide as the oxidizing agent, yields a characteristic change that is detectable by spectrophotometric methods. HRP catalyzes the conversion of chromogenic substrates into colored products and produces light when acting on chemiluminescent substrates (The use of phosphoimaging is replacing the use of x-ray film). Because of the high degree of specificity of antibodies, a positive band is good confirmation that the protein band represents a target product rather than another protein of similar molecular weight.
Similarly, DNA molecules can be separated by gel electrophoresis using agarose or polyacrylamide. DNA is negatively charged. DNA fragments (e.g., from digestion with restriction enzymes) can be separated by molecular weight as well as plasmids from larger DNA elements. The DNA separates on the basis of molecular weight. The DNA can be recovered by simply cutting out the part of the gel corresponding to the band for the desired DNA and eluting. The DNA is invisible unless stained. One approach is to expose DNA to an intercalating dye that fits itself in between base pairs of DNA and fluoresces when under ultraviolet light (e.g., ethidium bromide, propidium iodide).
With shotgun cloning (which produces a large gene library or gene bank), a desired colony can be isolated using radiolabeled RNA or DNA probes complementary to the cloned gene. Such a procedure involves transferring colonies to nitrocellulose filter paper, where they are lysed. After lysis, the DNA is released and binds to the filter paper. The paper is flooded with the probe; the probe only binds to the DNA with a complementary sequence (hybridization), and excess probe is washed away. If the filter is covered with x-ray film, radioactivity will expose the film (visible as a black spot), identifying which colonies contained the donor DNA. This procedure identifies colonies that have been transformed with the desired DNA; expression has to be established via separate experiments.
Another approach to using a gene library to find genes that express proteins with certain functions is the use of display technologies. Most common among these are phage display and cell-surface display (e.g., bacterial display, yeast display). For example, a bacteriophage or bacterium may display on its exterior surface proteins encoded from genes derived from the library. Each cell may display at most only a few of these gene products. An example of the use of such a system is the isolation of a cell producing a gene product that will bind to a particular molecule (ligand). The ligand can be bound to a surface, and only those cells expressing a protein on their surface that binds to that ligand can “stick” to the surface. These cells that stick can be recovered and propagated to make more copies of the gene and protein.
This simple summary does not cover all the intricacies investigators often face in obtaining industrially useful clones. However, the procedures discussed here are applicable in most cases where plasmids are used as vectors to transform bacteria such as E. coli.
8.3.2. Genetic Engineering of Higher Organisms
The direct genetic engineering of higher organisms can be a great deal more difficult than for bacteria because of a lack of good effective techniques to introduce foreign DNA and lack of an understanding of host cell genetics. Be aware of the techniques being developed to work with some of these host systems. The introduction of foreign DNA into higher organisms is usually termed transfection.
Some plants are subject to infection with the bacterium Agrobacterium tumefaciens. A. tumefaciens contains a plasmid that contains a section known as T-DNA. This T-DNA can integrate into the plant chromosome. If genes are inserted into the T-DNA region, they can be incorporated ultimately in the plant chromosome. Unfortunately, most cereal plants are not readily susceptible to Agrobacterium infection.
The biolistic process for obtaining plant transformation is to coat small (1-μm diameter) projectiles (e.g., of tungsten) and shoot them into cells at high velocity. Results with this approach have been remarkably successful, and this technique is fairly general.
Another fairly general approach is electroporation, which involves a brief highvoltage electric discharge that renders cells permeable to DNA. Electroporation can be used with animal, plant, fungal, and bacterial systems. The formation of protoplasts can enhance transfection but is not essential. A protoplast is a cell in which the outer cell envelope has been removed so that only the cytoplasmic membrane remains.
Chemically or electrically mediated protoplast fusion is another technique for transferring genetic information from one cell to another. Such approaches have been particularly useful with some fungi for which few or no plasmids have been identified. Protoplast fusion can be interspecies and can result in stable hybrids with desired properties due to recombination events between the two genomes or extrachromosomal pieces of DNA.
For most animal cells, genetic manipulation can be accomplished by modifying viruses to become vectors. For example, in the insect cell system, a baculovirus can be modified so as to place a desired gene under the control of a very strong promoter at the expense of a gene product that is unessential for viral replication in cell culture.
Although the basic conceptual approach to genetic engineering is rather straightforward, its implementation can vary widely in difficulty. The level of difficulty depends on the nature of the gene product and its corresponding gene, as well as the character of the desired host cell. Techniques to improve the ease of obtaining desirable genetic modification will undoubtedly continue to be developed. The ultimate limitation will be human imagination and wisdom.
8.3.3. Genome Engineering
Genome engineering, or genome editing, refers to the collection of strategies and techniques developed in recent years for the targeted, specific modification of the genetic information—or genome—of living organisms. Genome engineering has the potential to impact a wide range of applications, particularly in the areas of human health, agricultural and industrial biotechnology, and research tool development. Examples include the inactivation or modification of a specific gene for understanding its function, the correction of a gene carrying a harmful mutation, the production of therapeutic proteins, the elimination of persistent viral sequences, and the development of new generations of genetically modified crops. A number of techniques have been developed for these purposes, several of which are discussed in this section.
Multiplex automated genome engineering (MAGE) is a method for introducing targeted modifications directly into the chromosome of E. coli using synthetic single-stranded DNA (ssDNA). The process is cyclical and begins with transformation of ssDNA by electroporation followed by outgrowth, during which bacteriophage homologous recombination enzymes mediate annealing of ssDNAs to their genomic targets. With MAGE, many independent or combinatorial mutations can be made in genes, gene networks, or full genomes. MAGE can generate combinatorial genetic diversity in a cell population either by targeting one ssDNA per site (locus) in the genome (i.e., many target sites, single genetic mutations) or by targeting more than one ssDNA per locus (i.e., single target site, many genetic mutations). Using MAGE, researchers have engineered E. coli strains that produce five times the normal amount of lycopene, an antioxidant normally found in tomato seeds and linked to anticancer properties. The iterative approach took only 3 days and about $1000 in materials.
Several genome engineering techniques seek to efficiently eliminate a gene using a “molecular scissors” approach, with the goal of generating breaks in dsDNA along the sequence to be modified. Most standard restriction enzymes recognize very short (1 to 10 base pairs) and generally palindromic sequences, which often occur at several sites in the genome of an organism. As a result, restriction enzymes are likely to cut the DNA molecule multiple times. To gain more precise targeting for genome engineering/editing applications requires enzymes that recognize and interact with DNA sequences that are sufficiently long so as to occur only once, with high probability, in any given genome. In this way, the DNA modification (e.g., cutting) is narrowly restricted to the site of the target sequence. Because they recognize sites that are greater than 12 base pairs, meganucleases, zinc finger nucleases (ZFN), and transcription activator-like effector nuclease (TALEN) fusions offer a level of precision that is suitable for genome engineering/editing.
A drawback of these methods is the significant effort required for each new genome engineering target. Typically, a technique known as protein engineering (see Section 14.9) is used to produce a custom meganuclease, ZFN, or TALEN that recognizes the sequence of interest. The need for engineering customized proteins can be circumvented using CRISPR (clustered regularly interspaced short palindromic repeat) technology. CRISPR is a naturally occurring procaryotic immune system that functions to identify and destroy foreign DNA such as bacteriophage viruses and plasmids. However, the CRISPR mechanism can be leveraged for genome engineering/editing by delivering the CRISPR-associated endonuclease (Cas9) protein and appropriate guide RNAs (gRNAs) into a target cell. Once internalized, the gRNAs direct Cas9 to the target sequence where it unwinds and cleaves the dsDNA, enabling an organism’s genome to be specifically cut at any desired location. An advantage of the technique is that users need to design only gRNAs that target their DNA sequence of interest; no laborious protein engineering is required. Another advantage is that CRISPR technology has been adapted such that genome engineering/editing can be performed in cell types throughout the three domains of life. CRISPR technology has enormous potential in a number of application areas, most notably (and perhaps controversially) in altering the germline of humans, animals, and other organisms and modifying the genes of food crops.
8.4. Genomics
Genomics is the set of experimental and computational tools that allow the genetic blueprints of life to be read. A genome is an organism’s total inheritable DNA. Most important, we have the genomic sequence for humans. This sequence information is simply a string of nucleotide letters. Functional genomics is the process of relating genetic blueprints to the structure and behavior of an organism. To completely relate physiological behavior to this sequence of nucleotides is an extremely challenging problem—one to which bioengineers can and have made significant contributions.
Much of the progress in molecular biology has been due to a reductionist approach in which a subcomponent has been isolated and studied in detail. This approach has been fruitful in learning about the detailed mechanisms at the heart of living systems. The detailed sequence information now available is the ultimate limit in reduction in biology. There is increasing interest in asking how the individual subcellular components work together. Function arises from the complex interactions of the components. A systems engineering approach allows one to integrate component parts into a functional whole. This integrative approach is often known as systems biology.
Whereas genetic information is linear and static, cellular systems are highly nonlinear, dynamic systems that respond to their environment and regulate gene expression. Over the next decades, a focus of biochemical engineering, in conjunction with other disciplines, will be relating this linear sequence information to those nonlinear dynamical systems.
The role of the bioengineer is twofold. One role is as an enabler, by making better tools for rapid analysis of DNA sequences, of expression of mRNAs, and of a cell’s total proteins (proteomics). A second role is as an interpreter and organizer of genetic information; this role usually involves mathematical modeling.
8.4.1. Experimental Techniques
The primary tools of genomics are used for DNA sequencing, detecting which mRNAs are expressed, and determining which proteins are present in a cell or tissue. Historically, the nucleotide sequence of DNA fragments was determined on a sequencing gel by a process called Sanger sequencing, named after its inventor Frederick Sanger. The key to this method is the use of dideoxyribonucleoside triphosphates, which are derivatives of the natural deoxyribonucleoside triphosphates and lack the hydroxyl (OH) group at the 3′ position. For example, 2′,3′-dideoxyadenosine-5′-triphosphate (ddATP) is the derivative deoxyadenosine triphosphate (dATP). If a strand of DNA is being replicated and if ddATP is inserted into the position normally occupied by dATP, replication is stopped. The OH group at the 3′ position is essential for continued replication. The basic process for this type of sequencing is shown in Figure 8.9.

Figure 8.9. Example of a sequencing gel to obtain the nucleotide sequence of a DNA fragment. See text for details.
It is easiest to imagine reactions in four separate tubes labeled A, T, C, and G. To each tube is added the DNA fragment to be sequenced; DNA polymerase; a stoichiometric excess of dATP, deoxythymidine triphosphate (dTTP), deoxycytidine triphosphate (dCTP), and deoxyguanosine triphosphate (dGTP); and an oligonu-cleotide primer for the DNA polymerase to use. The reaction is analogous to the first step in the PCR reaction. Only one strand of the DNA fragment to be sequenced is read. In addition to these reactants, in each tube a small amount of ddNTP is added where N is any of the four nucleotides. For example, in tube A, ddATP is added; in tube T, ddTTP is added; and so on. Since the amount of ddNTP added is small in tube N, several reaction products are formed. In tube N, the first time dNTP or ddNTP must be added to the growing copy of the original DNA fragment, there is a high probability that dNTP will be added and the chain can be extended. However, there is also a finite probability (determined by the ratio of ddNTP to dNTP) that ddNTP will be added and the chain extension will be terminated. At the second position where N is required, either ddNTP or dNTP will be added. Again, some chains will be terminated, and others will continue to extend. This reaction continues, and tube N will generate fragments of different sizes, all of which end in the letter N. These fragments are separated by gel electrophoresis, and the sequence of the DNA fragment can be read directly from the gel (as shown in Figure 8.9). This technique is limited to relatively short DNA fragments (a few hundred nucleotides).
For sequencing genomes, the Sanger technique is too slow and must be modified and automated. The basic approach to sequence a large genome is to cut it into millions of overlapping fragments of 2000 to 10,000 base pairs in length. Each fragment is ligated into a plasmid, which is transformed into E. coli. The E. coli form a living genomic library. These colonies are robotically picked, identified with a barcode, and placed in a 384-well plate. The amount of the cloned DNA fragment is amplified using PCR. From each end of the fragment, 500 letters are replicated using the ddNTP approach previously described. For the purposes of automation, a fluorescent dye is used for each ddNTP. Four different dyes are used to distinguish A, T, C, and G. These labeled fragments are then read in automated sequencing machines. These machines rely on a large number (>100) of glass capillaries; capillary electrophoresis is used to separate the fragments by size. As the fragments exit the capillary, a laser beam detects the color of the dye at the end of the fragment. The information is fed to a computer, and the 500-letter sequence is determined. Reading 100 million letters of DNA sequence is possible. These sequences are stored in a computer; computer algorithms are then used to align the overlapping sequences.
Using the capillary-electrophoresis-based Sanger sequencing method, the Human Genome Project took more than 10 years to complete and cost nearly $3 billion. In contrast, next-generation sequencing (NGS), also known as high-throughput sequencing, is a catch-all term that describes a number of recently developed sequencing methods, such as Illumina sequencing and Roche 454 sequencing that both leverage the sequencing by synthesis (SBS) principle. These and other related technologies make large-scale whole-genome sequencing accessible and practical for the average researcher and as such have revolutionized the study of genomics and molecular biology.
Another approach, which increases throughput, is the use of nanopores. A strand of DNA is forced to pass through a nanopore as a long string using, for example, electrophoresis. As each nucleotide passes through the pore, it obstructs the passage of electrical current. Each base (A, T, G, C) blocks current in its own characteristic manner, enabling a direct reading of the DNA sequences.
With recent advances in several technologies, the cost of sequencing a complete human genome decreased dramatically, and the cost to sequence the genome of a microbe and the time necessary to do so are low enough that such an effort is now routine.
Another technology that is having a large impact on genomics is microarrays for measuring which genes are being expressed by measuring the corresponding mRNA levels. These microarrays are high-density oligonucleotide arrays. These oligonucleotides hybridize with the corresponding mRNA. For a known gene or protein, an oligonucleotide can be synthesized that binds the corresponding mRNA. Using photolithography, the manufacturing of arrays containing 280,000 individual oligonucleotides on glass substrates of 1.64 cm2 is now routine. Transcriptomics describes the measurement of all the transcripts in a cell. Such arrays can simultaneously analyze expression from 6000 to 10,000 genes. For bioprocesses, the array can be used to determine which genes are upregulated or downregulated in response to a process change (e.g., temperature).
An increase in mRNA levels does not immediately correspond to a change in protein level. Because different mRNAs have different rates of degradation, efficiency of translation, and so on, changes in mRNA expression may not translate directly into changes in the protein content of a cell or tissue. Because the proteins are the primary components responsible for biological activity, a knowledge of the protein content of a cell is highly valuable. Because proteins cannot be easily amplified, as can nucleic acids, measurements of all of the proteins in a cell (i.e., proteomics) is very difficult. Also, proteins vary greatly in properties such as hydrophobicity. Additionally, changes in splicing and posttranslational processing alter protein properties significantly.
The traditional technique for identifying proteins in a complex mixture has been two-dimensional gel electrophoresis. This technique is derived from the SDS-PAGE technique described earlier in this chapter. For two-dimensional gel electrophoresis, two methods are combined. The first is to separate proteins in one direction (say, the x-axis) according to their isoelectric point—the pH at which the protein has no net charge. Using a special set of buffers, a pH gradient can be established in a polyacrylamide gel. When the proteins are subjected to an electric field, they will move to the pH equal to their isoelectric point and remain there. After equilibration with an anionic surfactant, the proteins are subjected to an electric field in the other direction (say, the y-axis). The proteins will remain at the same pH but move down the y-axis according to their molecular weight. By separating protein mixtures using these two parameters (isoelectric point and molecular weight), a complex protein mixture can be resolved into separate spots. Often, those “spots” can be removed and analyzed by tandem mass spectrometry to identify the protein’s sequence.
The technique of two-dimensional gel electrophoresis is time consuming and expensive. Improvements in techniques in mass spectrometry have had a large impact, although it still depends on off-line two-dimensional gel electrophoresis to prepare samples for analysis. Significant improvements will be necessary for routine analysis of cell and tissue protein content. Promising approaches combine microtechnology, liquid chromatographies, and mass spectrometry techniques. The engineering challenges in making a practical device are significant.
8.4.2. Computational Techniques
The wealth of data emerging from these new experimental techniques is overwhelming. Generally, DNA sequence information could be used to identify which sequences are genes that encode for proteins, what these proteins are, what their biological function is, and ultimately how these genes are regulated.
Deciding what nucleotide sequence corresponds to a gene is not always a clear-cut process. Investigators look for open reading frames, or ORFs. An ORF is a nucleotide sequence without a “stop” signal that encodes some minimal number of amino acids (about 100). In procaryotes, identifying ORFs is fairly straightforward. In eucaryotes, because of introns and exons, assignment of ORFs is complicated. Some computer programs can recognize probable (consensus) intron–exon boundaries.
When a prospective gene is identified, we need to know the function of the corresponding protein. In some cases, the function can be deduced by comparison to databases with amino acid sequence information on known proteins. Occasionally, the amino acid sequence and function of a protein is conserved across species.
With a highly conserved protein, we might find similar amino acid sequences—for example, in the fruit fly and humans. In this case, we would determine how homologous the two amino acid sequences were. We then might infer that the human gene encodes a protein with similar function. Conservation of amino acids near a catalytic or binding site is particularly critical. Efficient computer algorithms to do such searches are under development. BLAST, or basic local alignment search tool, is an example of an algorithm for the comparison of amino acid sequence information.
Ideally, one would like to predict protein structure and function solely from the amino acid sequence. Heroic efforts have been made to accomplish this goal, but there is no good general solution. Understanding the folding of proteins into their three-dimensional configuration is a computationally difficult problem.
Even with well-studied organisms, such as E. coli, we generally can suggest the function of about 70% of the genes. Clearly, knowing the full genome sequence has told us how little we really understand. The process of identifying single genes and the function of the corresponding genes has been the focus of bioinformatics.
Even if we knew the identity of every gene in a cell and the function of each corresponding protein, we would have an incomplete understanding of function and cellular physiology. A list of the proteins and their functions needs to be supplemented by an understanding of cellular structure and regulation. A combination of proteins can form metabolic pathways, and there will be a corresponding genetic circuit. Relating the cell’s “parts list” to its dynamic, physiological state is an unmet challenge that provides exciting, long-term opportunities for bioengineers.
In particular, models of cells and metabolic circuits, as discussed in Chapter 6, provide tools with which to organize data and to understand biological function. Simple stoichiometric models of central metabolism have been used with good success to identify which genes are essential to a cell in a particular environment. Models that also incorporate kinetics and regulatory structure are in development. Such models will be key to relating cellular physiology to the genome. Such an understanding will provide important guidance to development of future bioprocesses.
8.5. Summary
A cell’s genotype represents the cell’s genetic potential, whereas its phenotype represents the expression of a culture’s potential. The genotype of a cell can be altered by mutations. Examples of mutations are point mutations, deletions, and additions. Additions are usually the result of insertion sequences that “jump” from one position to another.
Mutations may be selectable or unselectable. The rate of mutation can be enhanced by the addition of chemicals called mutagens or by radiation. Auxotrophs are of particular use in genetic analysis and as a basis for some bioprocesses. Another useful class of mutants is conditional mutants.
Gene transfer from one cell to another augments genetic information in ways that are not possible through mutation only. Genetic recombination of different DNA molecules occurs within most cells. Thus, genetic information transferred from another organism may become a permanent part of the recipient cell. The three primary modes of gene transfer in bacteria are transformation, transduction, and conjugation. Self-replicating, autonomous, extrachromosomal pieces of DNA called plasmids play important roles in transformation. Episomes, which are closely related to plasmids, are the key elements in conjugation. Bacteriophages are critical to generalized transduction, and temperate phages are key to specialized transduction. Internal gene transfer can occur due to the presence of transposons, which probably also play a role in the assembly of new plasmids.
We can use gene transfer in conjunction with restriction enzymes and ligases to genetically engineer cells. In vitro procedures to recombine isolated donor DNA genes with vector DNA (e.g., plasmids, temperate phages, or modified viruses) are called recombinant DNA techniques. Once the vector with the DNA donor insert has been constructed, it can be moved to a recipient cell through any natural or artificial method of gene transfer. Although transformation is the most common technique in bacteria, a large variety of artificial methods have been developed (e.g., biolistic process, electroporation, modification of infective agents, and protoplast fusion) to insert foreign DNA into a host cell.
Genomics is the set of experimental and computational tools that allows the genetic blueprints of a whole organism to be read. Functional genomics is the process of relating genetic blueprints to the behavior and structure of an organism.
Suggestions For Further Reading
Many of the references cited at the end of Chapter 4 have selections dealing with mutation and selection, gene transfer, and genetic engineering.
General Textbooks
ALBERTS, B., D. BRAY, K. HOPKIN, A. JOHNSON, J. LEWIS, M. RAFF, K. ROBERTS, AND P. WALTER, Essential Cell Biology, 4th ed., Garland Science, New York, 2014.
HARDIN, J., G. BERTONI, AND L. J. KLEINSMITH, Beckers World of the Cell, 9th ed., Pearson Education, New York, 2015.
MADIGAN, M. T., J. M. MARTINKO, K. S. BENDER, D. H. BUCKLEY, AND D. A. STAHL, Brock Biology of Microorganisms, 14th ed., Pearson Education, New York, 2014.
Basic Techniques
GLICK, B. R., J. J. PASTERNAK, AND C. L. PATTEN, Molecular Biotechnology: Principles and Applications of Recombinant DNA, 4th ed., ASM Press, Washington, D.C., 2010.
GREEN, M. R., AND J. SAMBROOK, Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2012.
Articles
BURNS, M. A., AND OTHERS, “An Integrated Nanoliter DNA Analysis Device,” Science 282: 484–487, 1998.
CIPRIANY, B. R., AND OTHERS, “Real Time Analysis and Selection of Methylated DNA by Fluorescence-Activated Single Molecule Sorting in a Nanofluidic Channel,” PNAS 109: 8477–8482, 2012.
DUTT, M. J., AND K. H. LEE, “Proteomic Analysis,” Curr. Opin. Biotechnol. 11: 176–179, 2000.
Problems
8.1. Would a cell with a point mutation or a deletion be more likely to revert back to the original phenotype? Why?
8.2. Consider the metabolic pathway based on aspartic acid shown in Chapter 4, Section 4.8, Figure 4A.1. Describe the procedure you would use to obtain a methionine overproducer. Use mutation-selection procedures, detailing the experiments to be done and their sequence.
8.3. You wish to isolate temperature-sensitive mutants (e.g., those able to grow at 30°C but not at 37°C). Describe experiments to isolate such a cell.
8.4. An important method for screening for carcinogens is called the Ames test. The test is based on the potential for mutant cells of a microorganism to revert to a phenotype similar to the nonmutant. The rate of reversion increases in the presence of a mutagen. Many compounds that are mutagens are also carcinogens, and vice versa. Describe how you would set up an experiment and analyze the data to determine if nicotine is mutagenic.
8.5. How many different hybridization probes must you make to ensure that at least one corresponds to a set of four codons encoding the amino acid sequence val-leu-trp-lys?
8.6. You wish to develop a genetically engineered E. coli producing a peptide hormone. You know the amino acid sequence of the peptide. Describe the sequence of steps you would use to obtain a culture expressing the gene as a peptide hormone.
8.7. You wish to produce a small protein using E. coli. You know the amino acid sequence of the protein. The protein converts a colorless substrate into a blue product. You have access to a high-copy-number plasmid with a penicillin-resistant gene and normal reagents for genetic engineering. Describe how you would engineer E. coli to produce this protein. Consider the source of donor DNA, regulatory elements that need to be included, how the donor and vector DNA are combined, how the vector DNA is inserted, and how you would select for a genetically engineered cell to use in production.
8.8. You wish to express a particular peptide in E. coli using a high-copy-number plasmid. You have the amino acid sequence for the peptide.
a. Explain the experimental process for generating and selecting the genetically engineered E. coli using restriction enzymes, ligase, E. coli, plasmid with neomycin resistance, and the known amino acid sequence.
b. What control elements would you place on the plasmid to regulate expression and to prevent read-through?
8.9. There are three primary methods for obtaining donor DNA when doing genetic engineering.
a. What are those methods (two- to six-word descriptions of each are acceptable)?
b. You need to produce a protein from humans in E. coli. You do not know the primary amino acid sequence. You suspect that introns are present. Which method will you use to obtain the donor DNA?
8.10. What is the difference between transduction and transformation when discussing genes transfer to bacteria?
8.11. You wish to isolate a thymidine auxotrophic mutant of E. coli. Describe briefly what experiments you would do to accomplish this.
8.12. For the DNA sequence TAGGATCATAAGCCA and using a primer ATCC, sketch what the corresponding sequencing gel should look like.