
4. How Cells Work
So far you have learned something about how cells are constructed and how enzymes function. But a cell is much more than a bag filled with lipids, amino acids, sugars, enzymes, and nucleic acids. The cell must control how these components are made and how they interact with each other. Controlling and predicting the behavior of a cell is made more challenging by the phenomenon of macromolecular crowding, a condition that occurs routinely in living cells that may be essential for the efficient operation of metabolism. For example, the cytosol of the bacterium Escherichia coli contains about 300 to 400 mg/mL of macromolecules. Crowding from these high concentrations of macromolecules reduces the volume of solvent available for other molecules in the solution, which has the result of increasing their effective concentrations. The result of this crowding effect is that molecules in cells may behave in radically different ways than in test-tube assays. Therefore, measurements of the properties of individual enzymes or metabolic processes that are made in dilute solutions in the laboratory (in vitro) may differ by many orders of magnitude from the values seen in living cells (in vivo). In this chapter, we explore some examples of metabolic regulation. It is this ability to coordinate a wide variety of chemical reactions in the face of macromolecular crowding that makes a cell a cell.
The key to metabolic regulation is the flow and control of information. The advent of computers has led many to speak of the “Information Age.” There are many analogies between human society’s need to use and exchange information and a cell’s need to use and exchange information between subcellular components. Human society depends primarily on electronic signals for information storage, processing, and transmission; cells use chemical signals for the same purposes. Molecular biology is primarily the study of information flow and control.
4.1. The Central Dogma
Fortunately, almost all living systems have the same core approach to the storage, expression, and utilization of information. Information is stored in the DNA molecule and, like information on a computer tape or disk, can be replicated. It can also be played back or transcribed to produce a message. The message must translate into some action, such as a series of calculations with the ultimate display of the results, for the message to be useful.
Cells operate with an analogous system. Figure 4.1 displays the central dogma of biology. Information is stored in the DNA molecule. That information can be replicated directly to form a second identical DNA molecule. Further segments of information on the molecule can be transcribed to yield RNAs. Using a variety of RNAs, this information is translated into proteins. The proteins then perform a structural or enzymatic role, mediating almost all the metabolic functions in the cell. The information content of the DNA molecule is static; changes occur slowly through infrequent mutations or rearrangements. Which species of RNA are present and in what amount varies with time and with changes in culture conditions. Likewise, the proteins that are present will change with time but on a different time scale than for RNA species. Some of the proteins produced in the cell bind to DNA and regulate the transcriptional process to form RNAs.
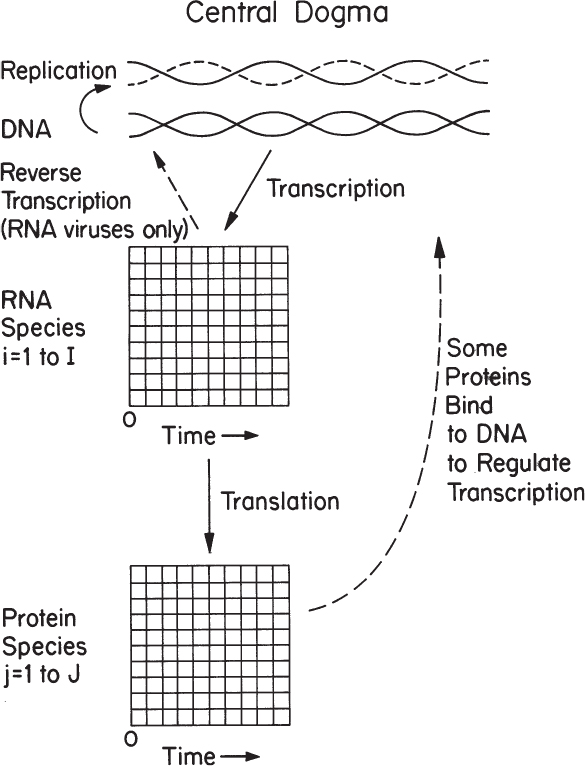
Figure 4.1. The primary tenet of molecular biology is the central dogma, which applies to all organisms.
The key feature of the central dogma is its universality from the simplest to most complex organisms. The process depicted in Figure 4.1 is essentially applicable to any cell of commercial significance. Of note is that several important but unusual transfers of biological sequential information are known to exist (i.e., occur only under specific conditions in the case of some viruses or in a laboratory). One such special transfer is known as reverse transcription (the reverse of normal transcription) and is the process by which genetic information from RNA is transcribed into new DNA. This process is known to occur in retroviruses, such as the human immunodeficiency virus (HIV), as well as in eucaryotes in the case of retrotransposons and telomere synthesis. In HIV, the virus encodes information on an RNA molecule. In the host cell, a viral enzyme called reverse transcriptase produces a viral DNA molecule using the viral RNA as a template. Such viruses are clinically important, and one approach to their treatment involves selective inhibition of reverse transcriptase. In addition to serving as an antiviral target, the reverse transcriptase enzyme is an important tool in genetic engineering. Another special transfer is RNA replication, which is the copying of one RNA to another and is used by many viruses to replicate. The enzymes that copy RNA to create new RNA are called RNA-dependent RNA polymerases. These enzymes are also found in many eucaryotes where they participate in a mechanism known as RNA silencing. Finally, another type of transfer is the direct translation from DNA to protein, a process that bypasses transcription but has only been demonstrated in a cell-free system (i.e., in a test tube using extracts from E. coli that contain ribosomes but not intact cells). At present, it is unclear whether this mechanism of translation corresponds specifically to the genetic code and whether it occurs naturally in a living cell.
Other key transfers of biological information that are not explicitly covered in the central dogma theory include posttranslational protein modification, inteins, and DNA methylation. Posttranslational modification occurs after a protein amino acid sequence has been translated from nucleic acid chains, and are catalyzed by dedicated enzymes. These modifications can occur cotranslationally in concert with the translation process (i.e., while the protein chain is still emerging from the ribosome) or posttranslationally after the protein has been fully synthesized and released from the ribosomal machinery. Important examples of such processing include the following:
• Glycosylation: The addition of a sugar group to the side chain of a specific amino acid (e.g., asparagine, serine, threonine) to form a glycoprotein; discussed in detail shortly
• Disulfide bond formation: The coupling of two thiol (—SH) groups present in cysteine residues
• Acylation: The addition of an acetyl group either at the N-terminus or at lysine residues
• Alkylation: The addition of an alkyl group (e.g., methyl, ethyl) typically at lysine or arginine residues
• Hydroxylation: The introduction of a hydroxyl group (—OH) typically to proline residues
• Lipidation: The addition of a lipid to a protein chain; specific types of lipidation include N-myristoylation, palmitoylation, and GPI-anchor addition
• Phosphorylation: The addition of a phosphate group (![]() ) usually to serine or threonine
) usually to serine or threonine
• Sulfation: The addition of a sulfate group to a tyrosine residue
• Ubiquitination: The addition of a small protein tag called ubiquitin to a lysine residue
Proteins can also change their own primary sequence from the sequence originally encoded by the DNA in a process called protein splicing. This process involves an intein, which is a “parasitic” segment of a protein that is able to excise itself from the chain of amino acids as they emerge from the ribosome and rejoin the remaining portions (exteins) with a peptide bond in such a manner that the main protein “backbone” does not fall apart. Inteins are often referred to as “protein introns” owing to their mechanistic resemblance to introns, which mediate mRNA splicing in eucaryotes (discussed shortly).
Variation in methylation states of DNA can alter gene expression levels significantly. Methylation variation of DNA is a process that usually involves the action of enzymes known as DNA methylases. Importantly, the effective information content is changed by methylation on DNA, but the primary DNA sequence is not altered. Functionally relevant changes to an organism’s genome, such as methylation or histone modification, that do not involve a change in the nucleotide sequence are known as epigenetic modifications. Unlike genetics whereby changes occur to the DNA sequence, the changes in gene expression or cellular phenotype of epigenetics have other causes, thus use of the prefix epi- from Greek meaning “over, outside of, or around.”
For information storage and exchange to take place, there must be a language. We can conceive of all life as using a four-letter alphabet made up of the nucleotides discussed in Chapter 2, “An Overview of Biological Basics” (that is, A, T, G, and C in DNA). All words are three letters long; such words are called codons. With four letters and only three-letter words, we have a maximum of 64 words. These words, when expressed, represent a particular amino acid or “stop” protein synthesis.
When these words are put into a sequence, they can make a “sentence” (i.e., a gene), which when properly transcribed and translated is a protein. Other combinations of words regulate when the gene is expressed. Carrying the analogy to the extreme, we may look at the complete set of information in an organism’s DNA (i.e., the genome) as a book. (For the human genome, it would be more than 1000 books the size of this one.)
This simple language of 64 words is all that is necessary to summarize your total physical makeup at birth and all your natural capabilities. It is essentially universal, the same for E. coli and Homo sapiens (humans). This universality has helped us to make great strides in understanding life and is a practical tool in genetic engineering and commercial biotechnology. (Recently, it has been demonstrated that this process can be manipulated to allow the incorporation of unnatural amino acids.)
Each of the steps in information storage and transfer (Figure 4.1) requires a macromolecular template. Let us examine how these templates are made and how this genetic-level language is preserved and expressed.
4.2. DNA Replication: Preserving and Propagating the Message
The double-helix structure of DNA discussed in Chapter 2 is extremely well suited to its role of preserving genetic information. Information resides simply in the linear arrangement of the four nucleotide letters (A, T, G, and C). Because G can hydrogen-bind only to C and A only to T, the strands must be complementary if an undistorted double helix is to result. Replication is semiconservative (see Figure 2.18); each daughter chromosome contains one parental strand and one newly generated strand.
To illustrate the replication process, let us briefly consider DNA replication in E. coli. The enzyme responsible for covalently linking the monomers is DNA polymerase. E. coli has three DNA polymerases (named Pol I, Pol II, and Pol III). A DNA polymerase is an enzyme that links deoxynucleotides together to form a DNA polymer. Pol III enzymatically mediates the addition of nucleotides to an RNA primer. Pol I can hydrolyze an RNA primer and duplicate single-stranded regions of DNA; it is also active in the repair of DNA molecules. The exact role of Pol II is still unclear.
In addition to the enzyme, the enzymatic reaction requires activated monomer and the template. The activated monomers are the nucleoside triphosphates. The formation of the 5′–3′ phosphodiester bond to link a nucleotide with the growing DNA molecule results in the release of a pyrophosphate, which provides the energy for such a biosynthetic reaction. The resulting nucleoside monophosphates are the constituent monomers of the DNA molecule.
Replication of the chromosome normally begins at a predetermined site, the origin of replication, which in E. coli is attached to the plasma membrane at the start of replication. Initiator proteins bind to DNA at the origin of replication, break hydrogen bonds in the local region of the origin, and force the two DNA strands apart. When DNA replication begins, the two strands separate to form a Y-shaped structure called a replication fork. Movement of the fork must be facilitated by the energy-dependent action of DNA gyrase and unwinding enzymes. In E. coli, the chromosome is circular. In E. coli (but not all organisms), the synthesis of DNA is bidirectional. Two forks start at the origin and move in opposite directions until they meet again, approximately 180° from the origin.
To initiate DNA synthesis, an RNA primer is required; RNA polymerase requires no primer to initiate the chain-building process, whereas DNA polymerase does. (We can speculate on why this is so. In DNA replication, it is critical that no mistakes be made in the addition of each nucleotide. The DNA polymerase Pol III can proofread, in part because of the enzyme’s 3′-to-5′ exonuclease activity, which can remove mismatches by moving backward. In contrast, a mistake in RNA synthesis is not nearly so critical, so RNA polymerase lacks this proofreading capacity.) Once a short stretch of RNA complementary to one of the DNA strands is made, DNA synthesis begins with Pol III. Next, the RNA portion is degraded by Pol I, and DNA is synthesized in its place. This process is summarized in Figure 4.2.
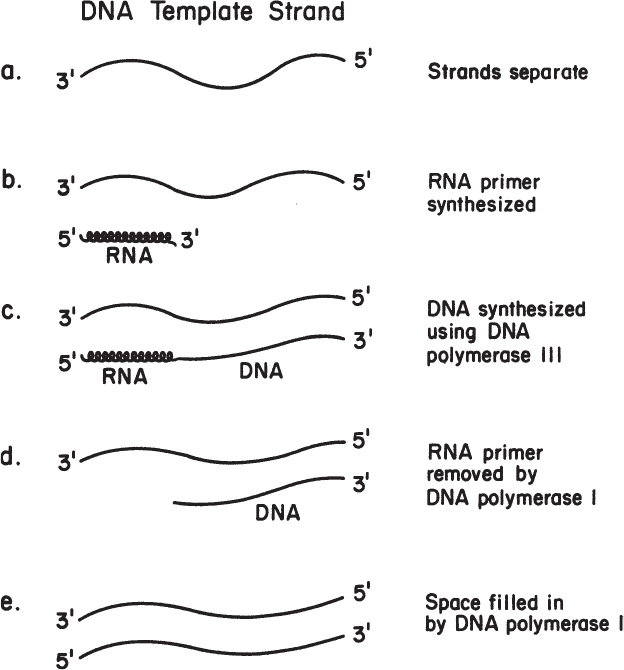
DNA polymerase works only in the 5′-to-3′ direction, which means that the next nucleotide is always added to the exposed 3′-OH group of the chain. Thus, one strand (the leading strand) can be formed continuously if it is synthesized in the same direction as the replication fork is moving. The other strand (the lagging strand) must be synthesized discontinuously. Short pieces of DNA attached to RNA are formed on the lagging strand. These fragments are called Okazaki fragments. The whole process is summarized in Figure 4.3. The enzyme DNA ligase, which joins the two short pieces of DNA on the continuous strand, is important in our discussions of genetic engineering.

Figure 4.3. Schematic representation of the steps of replication of the bacterial chromosome. The newly polymerized strands of DNA (wavy lines) are synthesized in the 5′-to-3′ direction (indicated by the arrows), using the preexisting DNA strands (solid lines) as a template. The process creates two replication forks, which travel in opposite directions until they meet on the opposite side of the circular chromosome, completing the replication process. Part (b) represents a more detailed view of one of the replicating forks and shows the process by which short lengths of DNA are synthesized and eventually joined to produce a continuous new strand of DNA. For purposes of illustration, four short segments of nucleic acid are illustrated at various stages. In (1), primer RNA (thickened area) is being synthesized by an RNA polymerase (R Pol). Then, successively, in (2) DNA is being polymerized to it by DNA polymerase III (Pol III); in (3) a preceding primer RNA is being hydrolyzed, while DNA is being polymerized in its place by the exonuclease and polymerase activities of DNA polymerase I (Pol I); finally, the completed short segment of DNA (4) is joined to the continuous strand (5) by the action of DNA ligase. (R. Y. Stainer, J. L. Ingraham, M. L. Wheelis, P. R. Painter, The Microbial World, 5th ed., © 1986. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
This brief discussion summarizes the essentials of how one DNA molecule is made from another and thus preserves and propagates the genetic information in the original molecule. Now we turn our attention to how this genetic information can be transferred.
4.3. Transcription: Sending the Message
The primary products of transcription are the three major types of RNA we introduced in Chapter 2: messenger RNA (mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA). Their rates of synthesis determine the cell’s capacity to make proteins. RNA synthesis from DNA is mediated by the enzyme RNA polymerase. To be functional, RNA polymerase must have two major subcomponents: the core enzyme and the sigma factor. The core enzyme contains the catalytic site, whereas the sigma factor is a protein essential to locating the appropriate beginning for the message. The core enzyme plus the sigma factor constitutes the holoenzyme.
One may wonder which of the two strands of DNA is actually transcribed. It turns out that either strand can be read. RNA polymerase always reads in the 3′-to-5′ direction, so the direction of reading will be opposite on each strand. On one part of the chromosome, one strand of DNA may serve as the template, or sense strand, and on another portion of the chromosome, the other strand may serve as a template.
The processes of initiation, elongation, and termination are summarized in Figure 4.4. The sigma factor is involved only in initiation. The sigma factor recognizes a specific sequence of nucleotides on a DNA strand. This sequence is the promoter region. Promoters can vary somewhat, and variations alter the affinity of the sigma factor (and consequently the holoenzyme) for a particular promoter. A strong promoter is one with a high affinity for the holoenzyme. The rate of formation of transcripts is determined primarily by the frequency of initiation of transcription, which is directly related to promoter strength. This information is important in our discussions of genetic engineering. Cells usually have one dominant sigma factor that is required to recognize the vast majority of promoters in the cell. However, other sigma factors can play important roles under different growth conditions (particularly stress) and are used to initiate transcription from promoters that encode proteins important to the cell for coping with unusual growth conditions or stress.

Figure 4.4. Transcription. Steps in messenger RNA synthesis. The start and stop sites are specific nucleotide sequences on the DNA. RNA polymerase moves down the DNA chain, causing temporary opening of the double helix and transcription of one of the DNA strands. Rho binds to the termination site and stops chain growth; termination can also occur at some sites without rho. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
After the initiation site is recognized, elongation of the transcript begins. As soon as elongation is established, the sigma factor is released so it can be reused. The synthesis of the growing RNA molecule requires energy, so activated triphosphate monomers of the ribonucleotides are required.
The transcript is made until the RNA polymerase encounters a stop signal, or transcription terminator. At this point, the RNA polymerase disassociates from the DNA template, and the RNA transcript is released. In some cases, an additional protein, the rho protein, is required for termination. Terminators can be strong or weak. If a weak terminator is coupled with a strong promoter, some of the RNA polymerase will read through the terminator, creating an artificially long transcript and possibly disrupting subsequent control regions on that DNA strand. We must consider terminator regions and their strength when constructing recombinant DNA systems.
The transcripts that are formed may be roughly lumped as either stable or unstable RNA species. The stable RNA species are rRNA and tRNA. mRNA is highly unstable (about a 1-min half-life for a typical E. coli mRNA, although mRNA may be considerably more stable in cells from higher plants and animals). Consider for a moment why mRNA is relatively unstable. The answer should become apparent as we discuss translation and regulation.
Although the general features of transcription are universal, there are some significant differences in transcription between procaryotic and eucaryotic cells. One example is that in procaryotes, related proteins are often encoded in a row without interspacing terminators. Thus, transcription from a single promoter may result in a polygenic message. Polygenic indicates many genes; each single gene encodes a separate protein. Thus, the regulation of transcription from a single promoter can provide efficient regulation of functionally related proteins; such a strategy is particularly important for relatively small and simple cells. By contrast, eucaryotic cells do not normally produce polygenic messages, although cells have been engineered to do so.
In procaryotic cells, there is no physical separation of the chromosome from the cytoplasma and ribosomes. Often, an mRNA will bind to a ribosome and begin translation immediately, even while part of it is still being transcribed! However, in eucaryotes, where the nuclear membrane separates chromosomes and ribosomes, the mRNA is often subject to processing before translation (see Figure 4.5). The DNA can encode for a transcript with an intervening sequence (called an intron) in the middle of the transcript. This intron is then cut out of the transcript at two specific sites. The ends of the remaining fragments are joined by a process called mRNA splicing. The spliced message can then be translated into an actual protein. The part of the transcript forming the intron is degraded and the monomers recycled. When mRNA is recovered from the cytosol, it will be in the mature form, while mRNA within the nucleus has introns. Many eucaryotic genes contain “nonsense DNA,” which encodes for the intronic part of the transcript. The word nonsense denotes that that particular sequence of DNA does not encode for amino acids. The role of introns is not well understood, but they likely play a role in either evolution or cellular regulation or both. The presence of introns complicates the transfer of eucaryotic genes to protein production systems in procaryotes such as E. coli.

Figure 4.5. In eucaryotes, RNA splicing is important. The presence of introns is a complication in cloning genes from eucaryotes to procaryotes.
Two other mRNA processing steps occur in eucaryotic cells that do not typically occur in procaryotes. One is RNA capping, in which the 5′ end is modified by the addition of a guanine nucleotide with a methyl group attached. The other is polyadenylation, in which a string of adenine nucleotides are added to the 3′ end. This tail of adenines is often several hundred nucleotides long. These two modifications are thought to increase mRNA stability and facilitate transport across the nuclear membrane.
Once we understand transcription, we can tackle translation. The translation of information on mRNA into proteins occupies a very large fraction of the cells’ resources. Like a large automobile plant, the generation of blueprints and the construction of the manufacturing machinery is worthless until product is made.
4.4. Translation: Going from Message to Product
Translation is the critical process of converting the information from RNA into peptides and proteins. After the proteins are made, they may be further modified to make the protein useful to the cell through posttranslational processing.
4.4.1. Genetic Code: Universal Message
The blueprint for any living cell is the genetic code. The code, as discussed previously, is made up of three-letter words (codons) with an alphabet of four letters. Sixty-four words are possible, but many of these words are redundant. Although such a “language” may appear to be ridiculously simple, it is sufficient to serve as a complete blueprint for the “construction” of the reader.
The dictionary for this language is given in Table 4.1, and an illustration of the relationship of nucleotides in the chromosome and mRNA to the final protein product is given in Figure 4.6. The code is degenerate in that more than one codon can specify a particular amino acid (e.g., UCU, UCC, UCA, and UCG all specify serine). Three codings, UAA, UAG, and UGA, are called nonsense codons because they do not encode normally for amino acids. These codons act as stop points in translation and are encoded at the end of each gene. It should also be pointed out that scientists have been able to artificially modify the genetic code. Such efforts have effectively expanded the genetic code by reallocating one or more specific codons, typically the nonsense codons or a four-base codon, to encode an amino acid that is not among the 20 encoded proteinogenic amino acids (e.g., O-methyltyrosine, naphthylalanine, and the photo-cross-linking benzoylphenylalanine). The ability to insert nonstandard amino acids into specific sites within proteins enriches the repertoire of useful tools available to scientists and engineers and holds great promise for a variety of biotechnological applications.
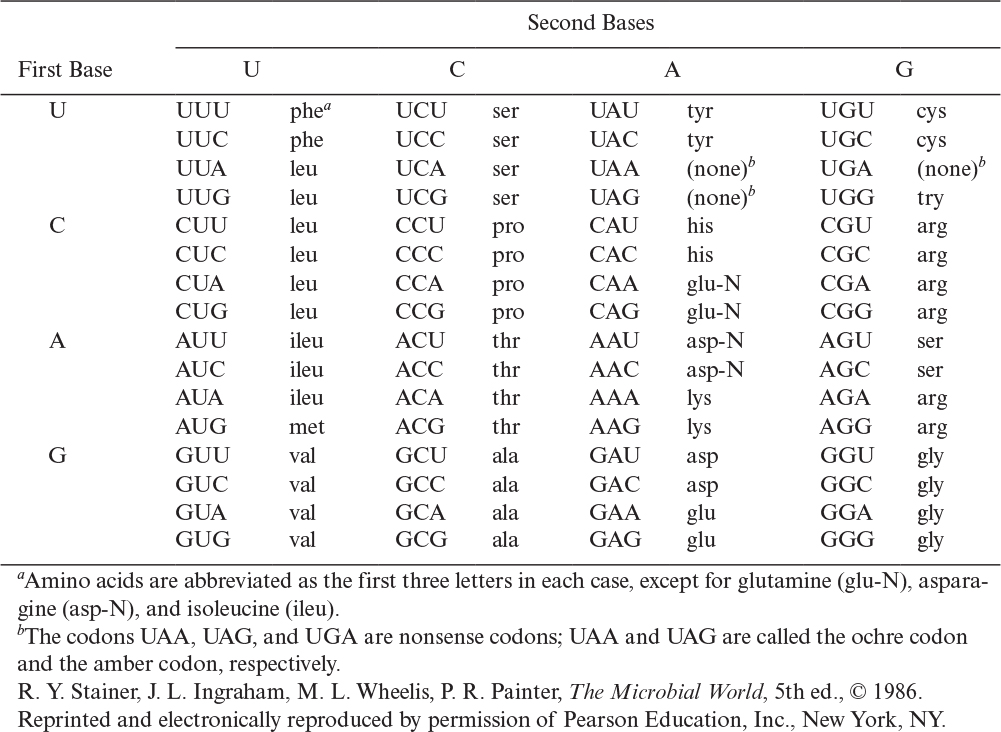
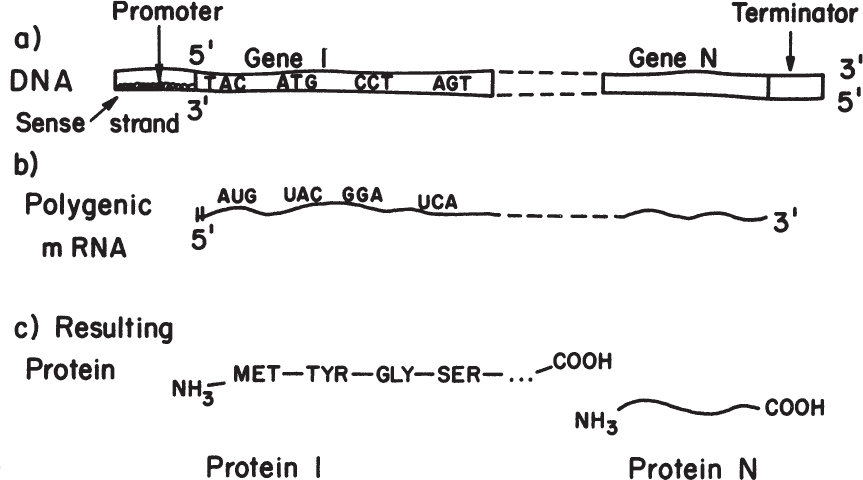
Figure 4.6. Overview of the transfer of information from codons on the DNA template to proteins. In procaryotes, messages are often polygenic, whereas in eucaryotes, polygenic messages are not made.
The genetic code is essentially universal, although some exceptions exist (particularly in the mitochondria and inclusion of rare amino acids such as selenocysteine [Sec] and pyrrolysine [Pyl], which are the 21st and 22nd amino acids in the genetic code). This essential universality greatly facilitates genetic engineering. The language used to make a human protein is understood in E. coli and yeast, and these simple cells will faithfully produce the same amino acid sequence as a human cell.
Knowing the genetic language, we may ask more about the mechanism by which proteins are actually made.
4.4.2. Translation: How the Machinery Works
The process of translation consists of three primary steps: initiation, elongation, and termination.
For initiation, mRNA must bind to the ribosome. All protein synthesis begins with an AUG codon (or GUG) on the mRNA. This AUG encodes for a modified methionine, N-formylmethionine. In the middle of a protein, AUG encodes for methionine, so the question is how the cell knows that a particular AUG is an initiation codon for N-formylmethionine. The answer lies about 10 nucleotides upstream of the AUG, where the ribosome binding site (Shine-Delgarno box) is located. Ribosome binding sites can vary in strength and are an important consideration in genetic engineering. The initiation of polymerization in procaryotes requires an initiation complex composed of a 30S ribosomal unit with an N-formylmethionine bound to its initiation region, a 50S ribosomal unit, three proteins called initiation factors (IF1, IF2, and IF3), and the phosphate bond energy from guanosine triphosphate (GTP).
The elongation of the amino acid chain uses tRNAs as decoders. One end of the tRNA contains the anticodon, which is complementary to the codon on the mRNA. The other end of the tRNA binds a specific amino acid. The tRNA is called charged when it is carrying an amino acid. The binding of an amino acid to the tRNA molecule requires the energy from two phosphate bonds and enzymes known as aminoacyl-tRNA synthetases. Figure 2.19 depicts a tRNA molecule.
The actual formation of the peptide bond between the two amino acids occurs on adjacent sites on the ribosome: the P or peptidyl site and the A or aminoacyl site (see Figure 4.7). The growing protein occupies the P site, and the next amino acid to be added occupies the A site. As the peptide bond is formed, the tRNA associated with the P site is released, and a rachet mechanism moves the mRNA down one codon so as to cause the tRNA that was in the A site to be in the P site. Then a charged tRNA with the correct anticodon can be recognized and inserted into the A site. The whole process is then repeated. The cell expends a total of four phosphate bonds to add one amino acid to each growing polypeptide (two to charge the tRNA and two in the process of elongation), and this accounts for most of the cellular energy expenditure in bacteria.
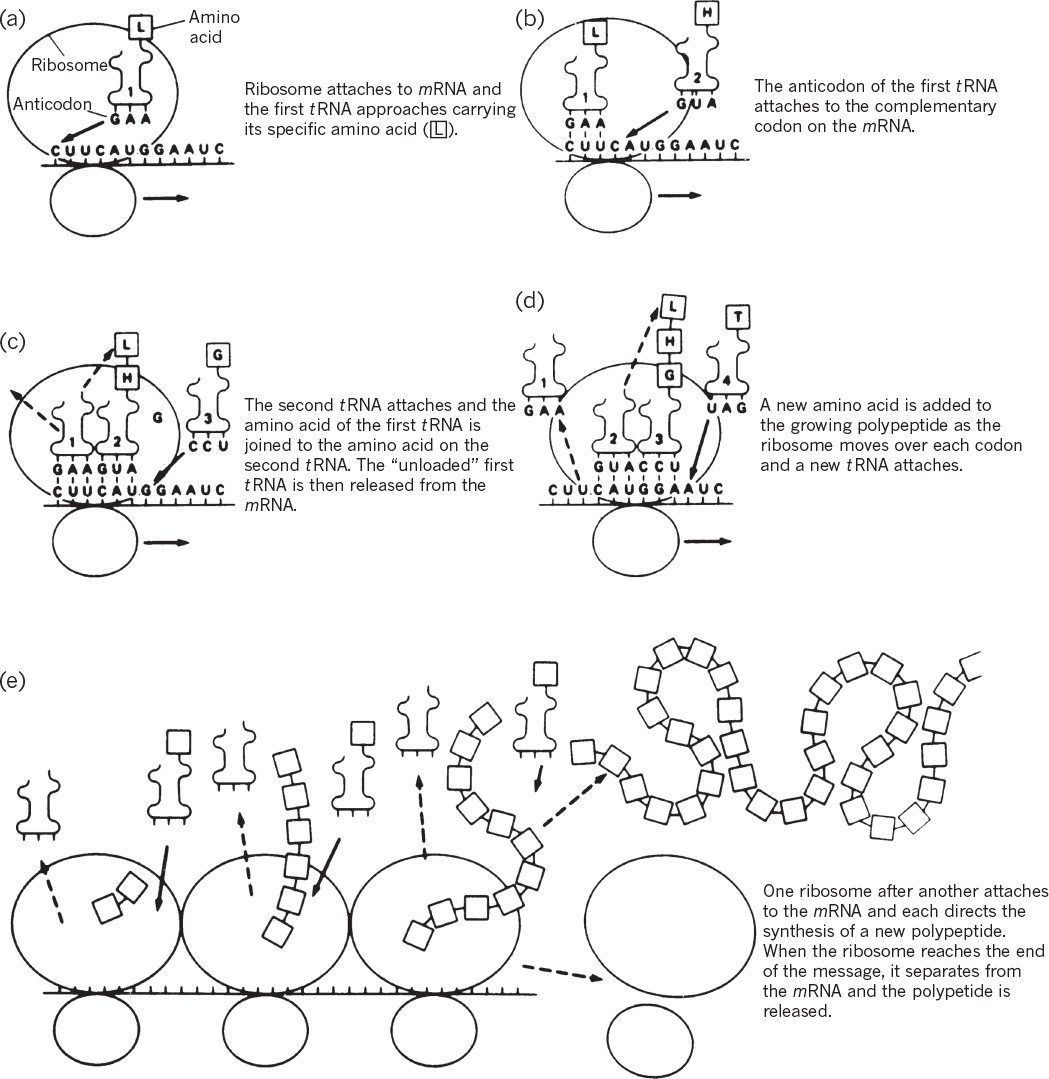
Figure 4.7. Translation of genetic information from a nucleotide sequence to an amino acid sequence. (M. W. Jensen, D. N. Wright, Introduction to Medical Microbiology, 1st ed., ©1985. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
When a nonsense or stop codon is reached, the protein is released from the ribosome with the aid of a protein release factor (RF). The 70S ribosome then dissociates into 30S and 50S subunits. An mRNA typically is being read by many (e.g., 10 to 20) ribosomes at once; as soon as one ribosome has moved sufficiently far along the message that the ribosome binding site is not physically blocked, another ribosome can bind and initiate synthesis of a new polypeptide chain.
4.4.3. Posttranslational Processing: Making the Product Useful
Often, the polypeptide formed from the ribosome must undergo further processing before it can become truly useful. First, the newly formed chain must fold into the proper structure; in some cases, several different chains must associate to form a particular enzyme or structural protein. Additionally, chaperones are an important class of proteins that assist in the proper folding of peptides. There are distinct pathways to assist in folding polypeptides. The level of chaperones in a cell increases in response to environmental stresses such as high temperature. Misfolded proteins are subject to degradation if they remain soluble. Often, misfolded proteins aggregate and form insoluble particles (i.e., inclusion bodies). High levels of expression of foreign proteins through recombinant DNA technology in E. coli often overwhelm the processing machinery, resulting in inclusion bodies. The formation of proteins in inclusion bodies greatly complicates any bioprocess, since in vitro methods to unfold and refold the protein product must be employed. Even when a cell properly folds a protein, additional cellular processing steps must occur to make a useful product.
Many proteins are secreted through a membrane. In many cases, the translocation of the protein across the membrane is done cotranslationally (during translation), whereas in some cases, posttranslation movement across the membrane occurs. When proteins move across a membrane, they have a signal peptide (about 20 to 25 amino acids). This signal peptide is clipped off during secretion. Such proteins exist in a preform and mature form. The preform is what is made from the mRNA, but the actual active form is the mature form. The preform is the signal sequence plus the mature form.
In procaryotes, secretion of proteins occurs through the cytoplasmic membrane. In E. coli and most gram-negative bacteria, the outer membrane blocks release of the secreted protein into the extracellular compartment. In gram-positive cells, secreted proteins readily pass the cell wall into the extracellular compartment. Whether a protein product is retained in a cell or released has a significant impact on bioprocess design.
In eucaryotic cells, proteins are released by two pathways. Both involve exocytosis, where transport vesicles fuse with the plasma membrane and release their contents. Transport vesicles mediate the transport of proteins and other chemicals from the endoplasmic reticulum (ER) to the Golgi apparatus and from the Golgi apparatus to other membrane-enclosed compartments. Such vesicles bud from a membrane and enclose an aqueous solution with specific proteins, lipids, or other compounds. In the secretory pathway, vesicles carrying proteins bud from the ER, enter the cis face of the Golgi apparatus, exit the Golgi trans face, and then fuse with the plasma membrane. Only proteins with a signal sequence are processed in the ER to enter the secretory pathway. Two forms of the secretary pathway exist. One is the constitutive exocytosis pathway, which operates at all times and delivers lipids and proteins to the plasma membrane. The second is the regulated exocytosis pathway, which typically is in specialized secretory cells. These cells secrete proteins or other chemicals only in response to specific chemical signals. Other modifications to proteins, known as posttranslational modifications (PTMs), can take place, as discussed earlier. These modifications involve the addition (or subtraction in the case of inteins) of components such as sugars, lipids, chemical groups, and proteinaceous tags and are typically vital to protein folding, regulation, and/or function. A bioprocess engineer must be aware that many proteins are subject to extensive processing after the initial polypeptide chain is made. These modifications can be quite complex and are important considerations in the choice of host organisms for the production of proteins.
One of the most abundant PTMs in cellular proteins is glycosylation, which is the addition of complex sugars known as glycans. The two main varieties of glycosylation are N-linked glycosylation (the glycans are attached to the nitrogen of an asparagine amino acid) and O-linked glycosylation (the glycans are attached to the hydroxyl oxygen of serine or threonine residue). The glycosylation pattern can serve to target the protein to a particular compartment or to control its degradation and removal from the organism. For therapeutic proteins injected into the human body, these issues are critical. A protein product may be ineffective if the N- or O-linked glycosylation pattern is not humanlike, as the protein may not reach the target tissue or may be cleared (i.e., removed) from the body before it exerts the desired action. Further, undesirable immunogenic responses can occur if a protein has a nonhuman glycosylation pattern. Thus, the glycan pattern (glycoform) of a protein product is a key issue in bioprocesses to make therapeutic proteins (see Chapter 14, “Utilizing Genetically Engineered Organisms”).
Because therapeutic glycoproteins must harbor human or humanlike glycans, production of therapeutic glycoproteins has traditionally been limited to mammalian cell lines that can replicate human or humanlike N-glycosylation reactions. However, not all eucaryotic cells produce proteins with humanlike N-linked glycosylation. For example, yeasts, lower fungi, and insect cells often produce partially processed products. Even mammalian cells (including human cells) will show altered patterns of glycosylation when cultured in bioreactors, and these patterns can shift upon scale-up in bioreactor size. To address the nonhuman glycan patterns produced by lower eucaryotes, scientists have engineered the N-linked glycosylation process in yeast for the production of glycoproteins bearing homogeneous, human N-glycan structures. The industrial workhorse E. coli remains the preferred host for producing therapeutic proteins such as insulin and human growth hormone that do not require glycosylation; however, the lack of glycosylation machinery in E. coli has so far restricted its use to non-glycosylated products.
The process of N-linked glycosylation in eucaryotes is depicted in Figure 4.8. The glycoform shown is “typical,” and many variants are possible. The natural proteins in the human body usually display a range of glycoforms; a single form is not observed. Eucaryotic N-linked protein glycosylation pathways span the cytosol, ER, and Golgi apparatus. Protein glycosylation can be divided into two distinct processes: the assembly of lipid-linked oligosaccharides (LLOs) at the ER membrane and the transfer of the oligosaccharide from the lipid anchor dolichyl pyrophosphate (Dol-PP) to selected asparagine residues of nascent polypeptides. The characteristics of N-linked protein glycosylation—namely, the use of Dol-PP as carrier for oligosaccharide assembly, the transfer of the completely assembled N-glycan, and the recognition of asparagine residues characterized by the sequence Asn-Xaa-Ser/Thr where X can be any amino acid except proline—are highly conserved in eucaryotes. The oligosaccharyltransferase (OST) catalyzes the transfer of a preformed branched glycan comprising 14 monosaccharides from the lipid donor Dol-PP to the acceptor protein. In yeast, eight different membrane proteins have been identified that constitute the OST complex in vivo. STT3 represents the catalytic subunit and is the most conserved subunit in the OST complex.
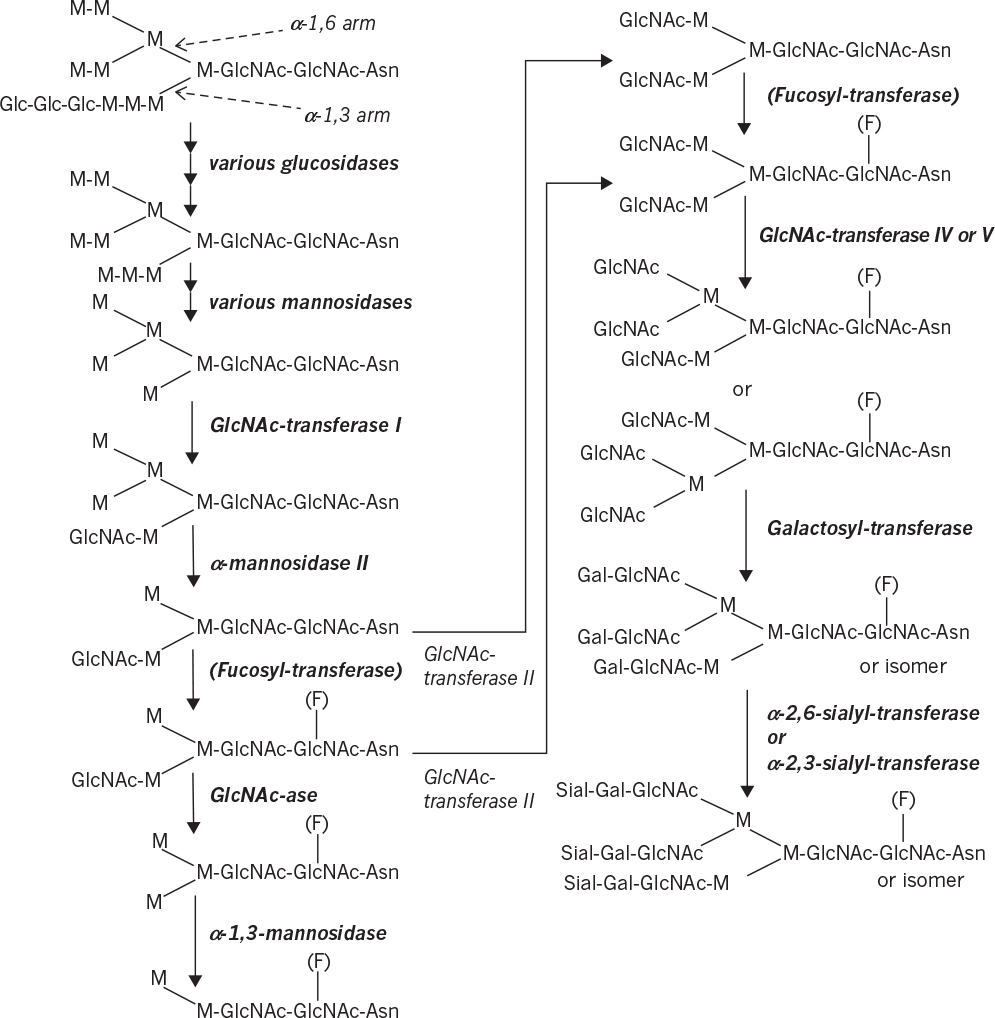
Figure 4.8. Example of an N-linked glycosylation pathway (Glc = glucose, M = mannose, GlcNAc = N-acetylglucosamine, F = fucose, Gal = galactose, Sial = sialic acid). The oligosaccharide side chain is bound to an asparagine (Asn) of the protein. The upper arm represents the α-1,6 arm and the lower one the α-1,3 arm. The parentheses refer to an optional fucosylation. The GlcNAc-ase step is important in insect cells, but not mammalian cells. (Courtesy of C. Joosten.)
Following transfer to the protein, the 14-residue glycan is first “trimmed” by a set of specific glycosidases. In yeast, oligosaccharide processing often stops in the ER, leading to simple glycoforms (or high mannose or oligomannose forms). The initial trimming takes place in the ER, followed by transfer to the Golgi apparatus where final trimming occurs, followed by addition of various sugars or aminosugars. These units are added through the action of various glycosyltransferases using nucleotide-activated sugar cosubstrates as sugar donors. In insect cells, high levels of N-acetylglucosaminidase activity results typically in dead-end structures with a mannose cap. Complex glycoforms have sugar residues (N-acetylglucosamine, galactose, and/or sialic acid) added to all branches of the oligosaccharide structure. Hybrid glycoforms have at least one branch modified with one of these sugar residues and one or more with mannose as the terminal residue.
Until recently, N-linked protein glycosylation was thought to be the exclusive domain of eucaryotes. However, bona fide N-linked glycosylation pathways are now known to exist in all domains of life. In the case of procaryotes, N-linked glycosylation was first discovered in the pathogenic bacterium Campylobacter jejuni and subsequently transferred to laboratory strains of E. coli. This procaryotic glycosylation system shares many functional similarities with the eucaryotic process, such as assembly of glycans on a lipid carrier in the bacterial cytoplasmic membrane (the equivalent of the eucaryotic ER membrane) and transfer of the glycans to asparagines in secretory or membrane proteins by an integral membrane OST. There are also several major differences between the systems. Most notably, the structures of bacterial N-linked glycans are significantly divergent from those produced in eucaryotes. Hence, glycoproteins derived from natural procaryotic glycosylation systems are likely to be immunogenic in humans and thus of limited therapeutic value. However, bottom-up engineering of a synthetic pathway for performing eucaryotic N-linked glycosylation reactions in E. coli has been described recently. This development paves the way for using laboratory strains of E. coli for production of therapeutically relevant glycoproteins.
4.5. Metabolic Regulation
Metabolic regulation is the heart of any living cell. Regulation takes place principally at the genetic level and at the cellular level (principally, control of enzyme activity and through cell surface receptors). Let us first consider genetic-level changes, as these fit most closely with our discussion of transcription and translation.
4.5.1. Genetic-Level Control: Which Proteins Are Synthesized?
Recall that the formation of a protein requires transcription of a gene. Transcriptional control of protein synthesis is the most common control strategy used in bacteria. Control of protein synthesis in eucaryotes can be more complex, but the same basic concepts hold. In the simplest terms, the cell senses that it has too much or too little of a particular protein and responds by increasing or decreasing the rate of transcription of that gene. One form of regulation is feedback repression; in this case, the end product of enzymatic activity accumulates and blocks transcription. Another form of regulation is induction; a metabolite (often a substrate for a pathway) accumulates and acts as an inducer of transcription. These concepts are summarized in Figures 4.9 and 4.10. In both cases, a repressor protein is required. The repressor can bind to the operator region and hinder RNA polymerase binding. For repression, a corepressor (typically the end product of the pathway) is required, and the repressor can block transcription only when bound to the corepressor. For induction, the inducer (typically a substrate for a reaction) will combine with the repressor, and the complex is inactive as a repressor.
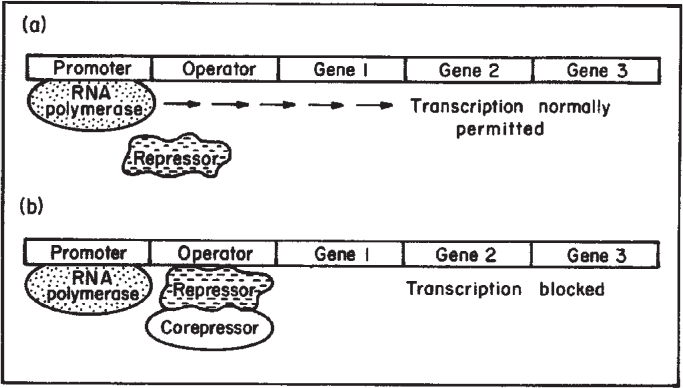
Figure 4.9. Process of enzyme repression. (a) Transcription of the operon occurs because the repressor is unable to bind to the operator. (b) After a corepressor (small molecule) binds to the repressor, the repressor now binds to the operator and blocks transcription. mRNA and the proteins it codes for are not made. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
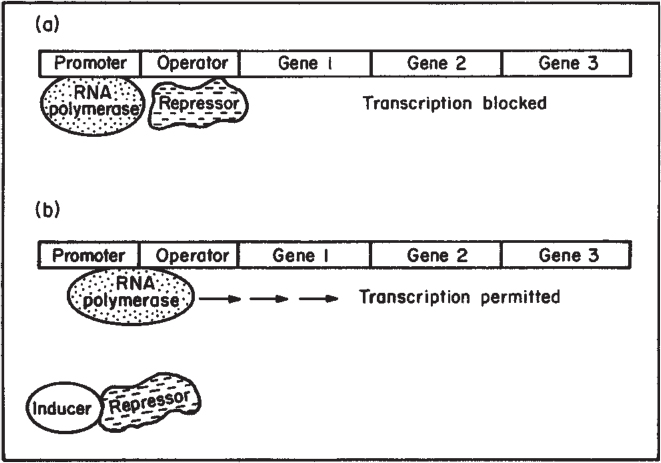
Figure 4.10. Process of enzyme induction. (a) A repressor protein binds to the operator region and blocks the action of RNA polymerase. (b) Inducer molecule binds to the repressor and inactivates it. Transcription by RNA polymerase occurs, and an mRNA for that operon is formed. In the lac operon, gene 1 is lacZ, gene 2 is lacY, and gene 3 is lacA. The repressor is made on a separate gene called lac i. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
Note in Figures 4.9 and 4.10 that several genes are under the control of a single promoter. A set of contiguous genes, encoding proteins with related functions, under the control of a single promoter-operator, is called an operon. The operon concept is central to understanding microbial regulation.
Control can be even more complex than indicated in Figures 4.9 and 4.10. The lactose (or lac) operon controls the synthesis of three proteins involved in lactose utilization as a carbon and energy source in E. coli. These genes are lacZ, lacY, and lacA. The lacZ gene encodes the LacZ protein, known as β-galactosidase (or lactase), which is an enzyme that cleaves lactose to glucose and galactose. The lacY gene encodes the LacY enzyme, a permease that acts to increase the rate of uptake of lactose into the cell. Lactose is modified in the cell to allolactose, which acts as the inducer. The conversion of lactose to allolactose is through a secondary activity of the enzyme β-galactosidase. Repression of transcription in uninduced cells is incomplete, and a low level (basal level) of proteins from the operon is made. Allolactose acts as indicated in Figure 4.10, but induction by allolactose is necessary but not sufficient for maximum transcription. Further regulation is exerted through catabolite repression (also called the glucose effect).
When E. coli senses the presence of a carbon–energy source preferred to lactose, it will not use the lactose until the preferred substrate (e.g., glucose) is fully consumed. This control mechanism is exercised through a protein called CAP (cyclic-AMP-activating protein). Cyclic AMP (cAMP) levels increase as the amount of energy available to the cell decreases. Thus, if glucose or a preferred substrate is depleted, the level of cAMP will increase. Under these conditions, cAMP will readily bind to CAP to form a complex that binds near the lac promoter. This complex greatly enhances RNA polymerase binding to the lac promoter. Enhancer regions exist in both procaryotes and eucaryotes.
The reader should now see how the cellular control strategy emerges. If the cell has an energetically favorable carbon–energy source available, it will not expend significant energy to create a pathway for utilization of a less favorable carbon–energy source. If, however, energy levels are low, then it seeks an alternative carbon–energy source. If and only if lactose is present will it activate the pathway necessary to utilize it.
Catabolite repression is a global response that affects more than lactose utilization. Furthermore, even for lactose, the glucose effect can work at levels other than genetic. The presence of glucose inhibits the uptake of lactose, even when an active uptake system exists. This is called inducer exclusion.
The role of global regulatory systems is still emerging. One concept is that of a regulon. Many noncontiguous gene products under the control of separate promoters can be coordinately expressed in a regulon. The best studied regulon is the heat shock regulon. The cell has a specific response to a sudden increase in temperature (or other stresses that result in abnormal protein formation or membrane disruption), which results in the elevated synthesis of specific proteins. Evidence now exists that this regulon works by employing the induction of an alternative sigma factor, which leads to high levels of transcription from promoters that do not readily recognize the normal E. coli sigma factor. Examples of other regulons involve nitrogen and phosphate starvation, as well as a switch from aerobic to anaerobic conditions.
Although many genes are regulated, others are not. Unregulated genes are termed constitutive, which means that their gene products are made at a relatively constant rate irrespective of changes in growth conditions. Constitutive gene products are those that a cell expects to utilize under almost any condition; the enzymes involved in glycolysis are an example.

Irrespective of whether an enzyme is made from a regulated or constitutive gene, its activity in the cell is regulated. Let us now consider control at the enzymatic level.
4.5.2. Metabolic Pathway Control
In Chapter 3, “Enzymes,” we learned how enzyme activity could be modulated by inhibitors or activators. Here we discuss how the activities of a group of enzymes (a pathway) can be controlled. The cell will attempt to make the most efficient use of its resources; the fermentation specialist tries to disrupt the cell’s control strategy so as to cause the cell to overproduce the product of commercial interest. An understanding of how cells control their pathways is vital to the development of many bioprocesses.
First, consider the very simple case of a linear pathway making a product, P1. Most often the first reaction in the pathway is inhibited by accumulation of the product (feedback inhibition or end-product inhibition). The enzyme for the entry of substrate into the pathway would be an allosteric enzyme (as described in Chapter 3), where the binding of the end product in a secondary site distorts the enzyme so as to render the primary active site ineffective. Thus, if the cell has a sufficient supply of P1 (perhaps through an addition to the growth medium), it will deactivate the pathway so that the substrates normally used to make P1 can be utilized elsewhere.
This simple concept can be extended to more complicated pathways with many branch points (see Figure 4.12). Assuming that P1 and P2 are both essential metabolites, the cell may use one of several strategies to ensure adequate levels of P1 and P2 with efficient utilization of substrates.
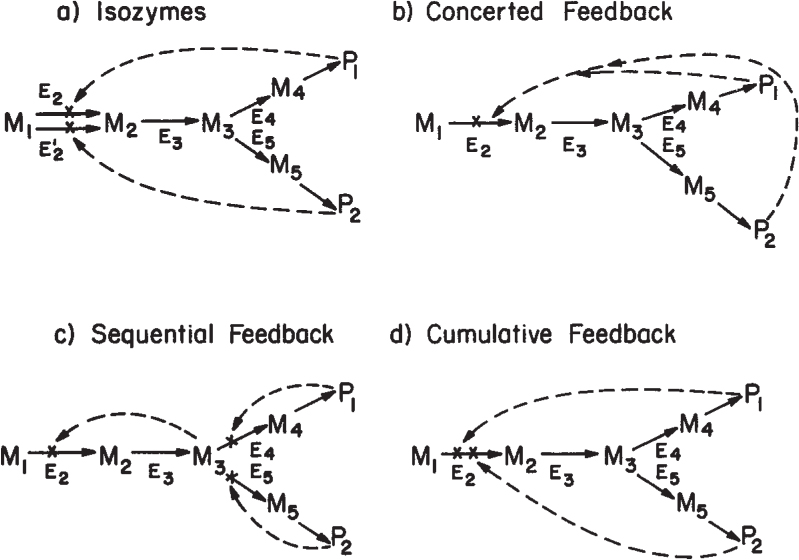
Figure 4.12. Examples of feedback control of branched pathways. P1 and P2 are the desired end products. M1, M2,.. . , Mj are intermediates, and Ej is the enzyme involved in converting metabolite Mj-1 to Mj. Possible paths of inhibition are shown by dashed lines.
One strategy is the use of isofunctional enzymes (isozymes). Two separate enzymes are made to carry out the same conversion, while each is sensitive to inhibition by a different end product. Thus, if P1 is added in excess in the growth medium, it inhibits one of the isozymes (E2), while the other enzyme (E′2) is fully active. Sufficient activity remains through isozyme E′2 to ensure adequate synthesis of P2.
An alternative approach is concerted feedback inhibition. Here a single enzyme with two allosteric binding sites (for P1 and P2) controls entry into the pathway. A high level of either P1 or P2 is not sufficient by itself to inhibit enzyme E2, while a high level of both P1 and P2 will result in full inhibition.
A third possibility is sequential feedback inhibition, by which an intermediate at the branch point can accumulate and act as the inhibitor of metabolic flux into the pathway. High levels of P1 and P2 inhibit enzymes E4 and E5, respectively. If either E4 or E5 is blocked, M3 will accumulate, but not as rapidly as when both E4 or E5 are blocked. Thus, intermediate flux levels are allowed if either P1 or P2 is high, but the pathway is inactivated if both P1 and P2 are high.
Other effects are possible in more complex pathways. A single allosteric enzyme may have effector sites for several end products of a pathway; each effector causes only partial inhibition. Full inhibition is a cumulative effect, and such control is called cumulative feedback inhibition or cooperative feedback inhibition. In other cases, effectors from related pathways may also act as activators. Typically, this situation occurs when the product of one pathway was the substrate for another pathway. An example of control of a complex pathway (for aspartate) is described in Section 4.8.
Let us pause to consider the differences between feedback inhibition and repression. Inhibition occurs at the enzyme level and is rapid; repression occurs at the genetic level and is slower and more difficult to reverse. In bacteria where growth rates are high, unwanted enzymes are diluted out by growth. Would such a strategy work for higher cells in differentiated structures? Clearly not, since growth rates would be nearly zero. In higher cells (animals and plants), the control of enzyme levels is done primarily through the control of protein degradation rather than at the level of synthesis. Most of our discussion has centered on procaryotes; the extension of these concepts to higher organisms must be done carefully.
Another caution is that the control strategy that one organism adopts for a particular pathway may differ greatly from that adopted by even a closely related organism with an identical pathway. Even if an industrial organism is closely related to a well-studied organism, it is prudent to check whether the same regulatory strategy has been adopted by both organisms. Knowing the cellular regulatory strategy facilitates choosing optimal fermenter operating strategy, as well as guiding strain improvement programs.
4.6. How the Cell Senses its Extracellular Environment
We have touched on some aspects of cellular metabolic regulation. A related form of regulation that we are just now beginning to appreciate has to do with the cell-surface receptors and how they sense the extracellular environment and cause the cell to respond to extracellular changes.
4.6.1. Transporting Small Molecules across Cellular Membranes
A cell must take nutrients from its extracellular environment if it is to grow or retain metabolic activity. As we discussed in Example 4.1, which nutrients enter the cell and at what rate can be important in regulating metabolic activity.
Molecules enter the cell through either energy-independent or energy-dependent mechanisms. The two primary examples of energy-independent uptake are passive diffusion and facilitated diffusion. Energy-dependent uptake mechanisms include active transport and group translocation.
In passive diffusion, molecules move down a concentration gradient (from high to low concentration) that is thermodynamically favorable, as follows:
where JA is the flux of species A across the membrane (mol/cm2-s), Kp is the permeability (cm/s), CAE is the extracellular concentration of species A (mol/cm3), and CAI is the intracellular concentration. The cytoplasmic membrane consists of a lipid core with perhaps very small pores. For charged or large molecules, the value Kp is very low and the flow of material across the membrane is negligible. The cellular uptake of water and oxygen appears to be due to passive diffusion. Furthermore, lipids or other highly hydrophobic compounds have relatively high diffusivities (10–8 cm2/s) in cellular membranes, and passive diffusion can be a mechanism of quantitative importance in their transport.
With facilitated transport, a carrier molecule (protein) can combine specifically and reversibly with the molecule of interest. The carrier protein is considered embedded in the membrane. By mechanisms that are not yet understood, the carrier protein, after binding the target molecule, undergoes conformational changes, which result in release of the molecule on the intracellular side of the membrane. The carrier can bind to the target molecule on the intracellular side of the membrane, resulting in the efflux or exit of the molecule from the cell. Thus, the net flux of a molecule depends on its concentration gradient.
The carrier protein effectively increases the solubility of the target molecule in the membrane. Because the binding of the molecule to the carrier protein is saturable (just as the active sites in the enzyme solution can be saturated), the flux rate of the target molecule into the cell depends on concentration differently than indicated in equation 4.1. Following is a simple equation to represent uptake by facilitated transport:

where KMT is related to the binding affinity of the substrate (mol/cm3) and JA MAX is the maximum flux rate of A (mol/cm2 – s). When CAE > CAI, the net flux will be into the cell. If CAI > CAE, there will be a net efflux of A from the cell. The transport is down a concentration gradient and is thermodynamically favorable. Facilitated transport of sugars and other low-molecular-weight organic compounds is common in eucaryotic cells but infrequent in procaryotes. However, the uptake of glycerol in enteric bacteria (such as E. coli) is a good example of facilitated transport.
Active transport is similar to facilitated transport in that proteins embedded in the cellular membrane are necessary components. The primary difference is that active transport occurs against a concentration gradient. The intracellular concentration of a molecule may be a hundred-fold (or more) greater than the extracellular concentration. The movement of a molecule up a concentration gradient is thermodynamically unfavorable and will not occur spontaneously; energy must be supplied. In active transport, several energy sources are possible: the electrostatic or pH gradients of the proton-motive force, and secondary gradients (e.g., of Na+ or other ions) derived from the proton-motive force by other active transport systems and by the hydrolysis of ATP.
The proton-motive force results from the extrusion of hydrogen as protons. The respiratory system of cells (see Chapter 5, “Major Metabolic Pathways,” section 5.3) is configured to ensure the formation of such gradients. Hydrogen atoms, removed from hydrogen carriers (most commonly nicotinamide adenine dinucleotide plus hydrogen [NADH]) on the inside of the membrane, are carried to the outside of the membrane, while the electrons removed from these hydrogen atoms return to the cytoplasmic side of the membrane. These electrons are passed to a final electron acceptor, such as O2. When O2 is reduced, it combines with H+ from the cytoplasm, causing the net formation of OH– on the inside. Because the flow of H+ and OH– across the cellular membrane by passive diffusion is negligible, the concentration of chemical species cannot equilibrate. This process generates a pH gradient and an electrical potential across the cell. The inside of the cell is alkaline compared to the extracellular compartment. The cytoplasmic side of the membrane is electrically negative, and the outside is electrically positive. The proton-motive force is essential to the transport of many species across the membrane, and any defect in the cellular membrane that allows free movement of H+ and OH– across the cell boundary can collapse the proton-motive force and lead to cell death.
Some molecules are actively transported into the cell without coupling to the ion gradients generated by the proton-motive force. By a mechanism that is not fully understood, the hydrolysis of ATP to release phosphate bond energy is utilized directly in transport (e.g., the transport of maltose in E. coli).
For these mechanisms of active transport, irrespective of energy source, we can write an equation analogous to Michaelis–Menten kinetics to describe uptake:

The use of equation 4.3 is meaningful only when the cell is in an energy-sufficient state.
Another energy-dependent approach to the uptake of nutrients is group translocation. The key factor here is the chemical modification of the substrate during the process of transport. The best-studied system of this type is the phosphotransferase system. This system is important in the uptake of many sugars in bacteria. The biological system itself is complex, consisting of four separate phosphate-carrying proteins. The source of energy is phosphoenolpyruvate (PEP).
Effectively, the process can be represented as follows:
By converting the sugar to the phosphorylated form, the sugar is trapped inside the cell. The asymmetric nature of the cellular membrane and this process make the process essentially irreversible. Because the phosphorylation of sugars is a key step in their metabolism, nutrient uptake of these compounds by group translocation is energetically preferable to active transport. In active transport, energy would be expended to move the unmodified substrate into the cell, and then further energy would be expended to phosphorylate it.
Certainly, the control of nutrient uptake is a critical cellular interface with its extracellular environment. In some cases, however, cells can sense their external environment without the direct uptake of nutrients.
4.6.2. Role of Cell Receptors in Metabolism and Cellular Differentiation
Almost all cells have receptors on their surfaces. These receptors can bind a chemical in the extracellular space. Such receptors are important in providing a cell with information about its environment. Receptors are particularly important in animals in facilitating cell-to-cell communication. Animal cell-surface receptors are important in transducing signals for growth or cellular differentiation. These receptors are also prime targets for the development of therapeutic drugs. Many viruses mimic certain chemicals (e.g., a growth factor) and use cell-surface receptors as a means to entering a cell.
Simpler examples exist with bacteria. Some motile bacteria have been observed to move up concentration gradients for nutrients or down gradients of toxic compounds. This response is called chemotaxis. Some microbes also respond to gradients in oxygen (aerotaxis) or light (phototaxis). Such tactic phenomena are only partially understood. However, the mechanism involves receptors binding to specific compounds, and this binding reaction results in changes in the direction of movement of the flagella. Motile cells move in a random-walk fashion; the binding of an attractant extends the length of time the cell moves on a “run” toward the attractant. Similarly, repellents decrease the length of runs up the concentration gradient. Chemotaxis is described in Figure 4.13. These drawings are two-dimensional projections of the three-dimensional movement.
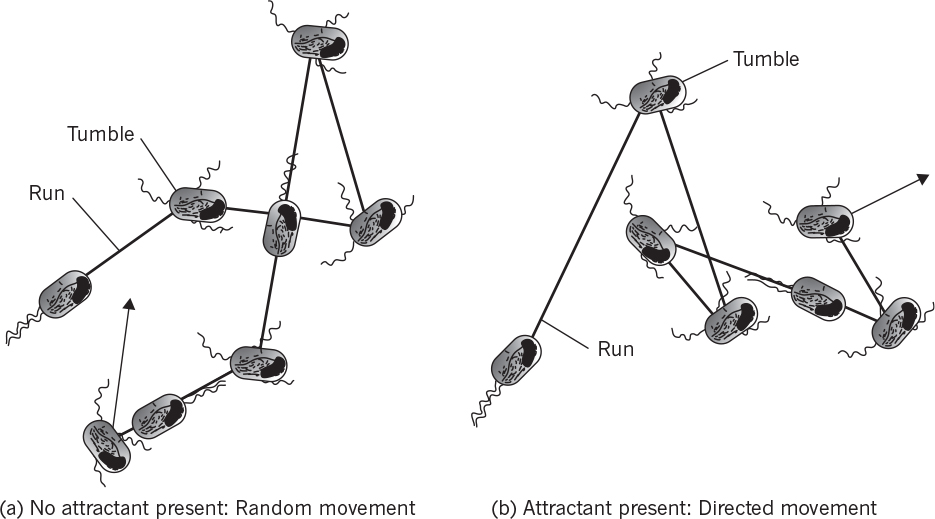
Figure 4.13. Chemotaxis in a peritrichously flagellated bacterium such as Escherichia coli. (Brock, Thomas D., Brock, Katherine M., Ward, David M., Basic Microbiology with Applications, 14th ed., © 2015. Reprinted and electronically reproduced by permission of Pearson Education, Inc., New York, NY.)
Microbial communities can be highly structured (e.g., biofilms), and cell-to-cell communication is important in the physical structure of the biofilm. Cell-to-cell communication is also important in microbial phenomena such as bioluminescence, exoenzyme synthesis, and virulence factor production. Basically, these phenomena depend on local cell concentration. How do bacteria count? They produce a chemical known as quorum sensing molecule, whose accumulation is related to cell concentration. When the quorum sensing molecule reaches a critical concentration, it activates a response in all of the cells present. A typical quorum sensing molecule is an acylated homoserine lactone. The mechanism of quorum sensing depends on an intracellular receptor protein, while chemotaxis depends on surface receptor proteins.
With higher cells, the timing of events in cellular differentiation and development is associated with surface receptors. With higher organisms, these receptors are highly evolved. Some receptors respond to steroids (steroid hormone receptors). Steroids do not act by themselves in cells, but rather the hormone-receptor complex interacts with specific gene loci to activate the transcription of a target gene.
A host of other animal receptors respond to a variety of small proteins that act as hormones or growth factors. These growth factors are normally required for the cell to initiate DNA synthesis and replication. Such factors are a critical component in the large-scale use of animal tissue cultures. Other cell-surface receptors are important in the attachment of cells to surfaces. Cell adhesion can lead to changes in cell morphology, which are often critical to animal cell growth and normal physiological function. The exact mechanism by which receptors work is only now starting to emerge. One possibility for growth factors that stimulate cell division is that binding of the growth factor to the receptor causes an alteration in the structure of the receptor. This altered structure possesses catalytic activity (e.g., tyrosine kinase activity), which begins a cascade of reactions leading to cellular division. Surface receptors are continuously internalized, complexes degraded, and receptors recycled to supplement newly formed receptors. Thus, the ability of cells to respond to changes in environmental signals is continuously renewed. Such receptors will be important in our later discussions on animal cell culture in Chapter 12, “Bioprocess Considerations in Using Animal Cell Cultures.”
4.7. Summary
In this chapter, you learned some of the elementary concepts of how cells control their composition in response to an ever-changing environment. The essence of an organism resides in the chromosome as a linear sequence of nucleotides that form a language (genetic code) to describe the production of cellular components. The cell controls the storage and transmission of such information, using macromolecular templates. DNA is responsible for its own replication and is also a template for transcription of information into RNA species that serve both as machinery and template to translate genetic information into proteins. Proteins often must undergo posttranslational processing to perform their intended functions.
The cell controls both the amount and activity of proteins it produces. Many proteins are made on regulated genes (e.g., repressible or inducible), although other genes are constitutive. With regulated genes, small effector molecules alter the binding of regulatory proteins to specific sequences of nucleotides in the operator or promoter regions. Such regulatory proteins can block transcription or in other cases enhance it. A group of contiguous genes under the control of a single promoter-operator is called an operon. More global control through regulons is also evident. Some gene products are not regulated, and their synthesis is constitutive.
Once a protein is formed, its activity may be continuously modulated through feedback inhibition. A number of alternative strategies are employed by the cell to control the flux of material through a pathway. Another form of regulation occurs through the interaction of extracellular compounds with cell surface protein receptors.
4.8. Appendix: Example Regulation of Complex Pathways†
† With permission, from R. Y. Stanier and others, The Microbial World, 5th ed., Prentice Hall, Englewood Cliffs, NJ, 1986.
In E. coli, the conversion of aspartic acid to aspartyl phosphate is mediated by three isofunctional enzymes, of which two (designated as a and c in Figure 4A.1) also mediate the conversion of aspartic acid semialdehyde to homoserine. Enzyme a, possessing both these functions, is feedback inhibited, and its synthesis is repressed by threonine. Enzyme c, which similarly possesses both functions, is inhibited and is repressed by lysine. The third, aspartokinase (enzyme b), is not subject to end-product inhibition, but its synthesis is repressed by methionine (Table 4A.1).
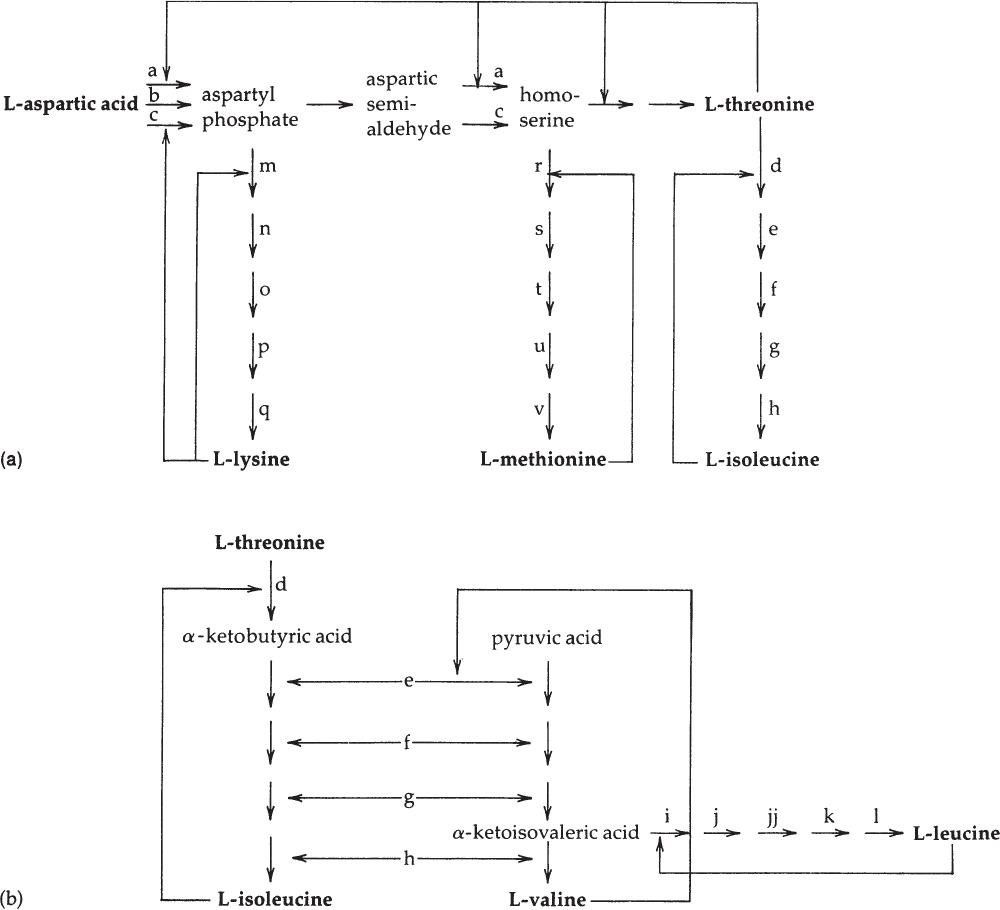
Figure 4A.1. A simplified diagram of the aspartate pathway in E. coli. Each solid arrow designates a reaction catalyzed by one enzyme. The biosynthetic products of the pathway (in boldface) are all allosteric inhibitors of one or more reactions. Careful study of this diagram reveals that with a single exception (the inhibition exerted by valine) the inhibition imposed by one amino acid does not cause starvation for a different amino acid. (a) The regulatory interrelationships of the L-lysine, L-methionine, and L-isoleucine branches of the pathway. (b) The regulatory interrelationships of the L-isoleucine, L-valine, and L-leucine branches.

The enzymes of the L-lysine branch (m–q) and the L-methionine branch (r–v) catalyze reactions leading in each case to a single end product and are subject to specific repression by that end product (L-lysine and L-methionine, respectively).
The third branch of the aspartate pathway is subject to much more complex regulation, for two reasons. First, L-threonine, formed through this branch, is both a component of proteins and an intermediate in the synthesis of another amino acid, L-isoleucine. Second, four of the five enzymes (e–h) that catalyze L-isoleucine synthesis from L-threonine also catalyze analogous steps in the completely separate biosynthetic pathway by which L-valine is synthesized from pyruvic acid. The intermediate of this latter pathway, α-ketoisovaleric acid, is also a precursor of the amino acid L-leucine. These interrelationships are shown in Figure 4A.1(b).
L-isoleucine is an end-product inhibitor of the enzyme d, catalyzing the first step in its synthesis from L-threonine; this enzyme has no other biosynthetic role. L-valine is an end-product inhibitor of an enzyme (e) that has a dual metabolic role, since it catalyzes steps in both isoleucine and valine biosynthesis. In certain strains of E. coli, this enzyme is extremely sensitive to valine inhibition, with the result that exogenous valine prevents growth, an effect that can be reversed by the simultaneous provision of exogenous isoleucine. The L-leucine branch of the valine pathway is regulated by L-leucine, which is an end-product inhibitor of the first enzyme, i, specific to this branch. These interrelationships are shown in Figure 4A.1(b).
As shown in Table 4A.2, many of the enzymes that catalyze steps in the synthesis of L-isoleucine, L-valine, and L-leucine are subject to repression only by a mixture of the three end products, a phenomenon known as multivalent repression. However, the five enzymes specific to L-leucine synthesis are specifically repressed by this amino acid alone.

TABLE 4A.2. Repressive Control of the Enzymes (See Figure 4A.1)
Suggestions for Further Reading
ALBERTS, B., D. BRAY, K. HOPKIN, A. JOHNSON, J. LEWIS, M. RAFF, K. ROBERTS, AND P. WALTER, Essential Cell Biology, 4th ed., Garland Science, New York, 2013.
BLACK, J. G., Microbiology: Principles and Applications, 3d ed., Prentice Hall, Upper Saddle River, NJ, 1996.
CAIN, J. A., N. SOLIS, AND S. J. CORDWELL, “Beyond Gene Expression: The Impact of Protein Post-translational Modifications in Bacteria,” J. Proteomics 97: 268–286, 2014.
CHUBUKOV, V., L. GEROSA, K. KOCHANOWSKI, AND U. SAUER, “Coordination of Microbial Metabolism,” Nature Rev. Microbiol. 12(5): 327–340, 2014.
COOK, P. R., “The Organization of Replication and Transcription,” Science 284(5421): 1790–1795, 1999.
ELGARD, L., M. MOLINARI, AND A. HELENIUS, “Setting the Standards: Quality Control in the Secretory Pathway,” Science 286(5446): 1882–1888, 1999.
HARDIN, J., G. BERTONI, AND L. J. KLEINSMITH, Becker’s World of the Cell, 9th ed., Pearson, New York, 2015.
KELLY, M. T., AND T. R. HOOVER, “Bacterial Enhancers Function at a Distance,” ASM News 65: 484–489, 1999.
KOLTER, R., AND R. LOSICK, “One for All and All for One,” Science 280(5361): 226–227, 1998. (This article is an overview related to a more detailed article on quorum sensing in biofilms; see DAVIES, D. G., AND OTHERS, “The Involvement of Cell-to-Cell Signals in the Development of a Bacterial Film,” Science 280: 295–298, 1998).
MADIGAN, M. T., J. M. MARTINKO, K. S. BENDER, D. H. BUCKLEY, AND D. A. STAHL, Brock Biology of Microorganisms, 14th ed., Pearson, Boston, 2015.
MORAN, L. A., K. G. SCRIMGEOUR, H. R. HORTON, R. S. OCHS, AND J. D. ROWN, Biochemistry, 2d ed., Prentice Hall, Upper Saddle River, NJ, 1994.
NOTHAFT, H., AND C. M. SZYMANSKI, “Protein Glycosylation in Bacteria: Sweeter Than Ever,” Nature Rev. Microbiol. 8(11): 765–778, 2010.
STANIER, R. Y., AND OTHERS, The Microbial World, 5th ed., Prentice Hall, Englewood Cliffs, NJ, 1986.
VON HIPPEL, P. H., “An Integrated Model of the Transcription Complex in Elongation, Termination, and Editing,” Science 281(5377): 660–665, 1998.
WADHAMS, G. H., AND J. P. ARMITAGE, “Making Sense of It All: Bacterial Chemotaxis,” Nature Rev. Mol. Cell Biol. 5(12): 1024–1037, 2004.
WEERAPANA, E., AND B. IMPERIALI, “Asparagine-Linked Protein Glycosylation: From Eukaryotic to Prokaryotic Systems,” Glycobiology 16(6): 91R–101R, 2006.
Problems
4.1. Consider the aspartic acid pathway shown in Figure 4A.1. Assume you have been asked to develop a high-lysine-producing mutant. What strategy would you pursue? (That is, which steps would you modify by removing feedback inhibition, and what changes in medium composition would you make over a simple mineral salts–glucose base medium?)
4.2. Why is mRNA so unstable in most bacteria (half-life of about 1 min)? In many higher organisms, mRNA half-lives are much longer (>1 h). Why?
4.3. What would be the consequence of one base deletion at the beginning of the message for a protein?
4.4. How many ribosomes are actively synthesizing proteins at any instant in an E. coli cell growing with a 45-min doubling time? The birth size of E. coli is 1-μm diameter and 2-μm length. The water content is 75%. About 60% of the dry material is protein, and the rate of amino acid addition per ribosome is 20 amino acids per second. The average molecular weight of free amino acids in E. coli is 126.
4.5. Describe simple experiments to determine if the uptake of a nutrient is by passive diffusion, facilitated diffusion, active transport, or group translocation.
4.6. Consider the following mRNA nucleotide code:
CCG UAU CGA CUU GUA ACA ACG CGC
a. Deduce the corresponding sequence of amino acids.
b. What is the corresponding nucleotide sequence on the chromosome? This sequence codes for a part of insulin.
4.7. Consider the pathway in Figure 4A.1 for production of lysine, methionine, isoleucine, and threonine. You need to produce lysine. Describe a strategy for making large amounts of lysine. Your strategy can consist of adding various amino acids to the medium and choosing the mutant cells altered in regulation. Say you can identify up to two points of mutation (e.g., removal of feedback inhibition).
4.8. Suggest an experiment to determine if the uptake of a compound is by either facilitated or active transport.
4.9. What is catabolite repression, and how does it affect the level of protein expression from the lac operon?
4.10. Explain the difference between feedback inhibition and feedback repression.
4.11. You are asked to produce a human protein in E. coli. Because you have learned some of the differences in the way that procaryotes and eucaryotes make proteins, you worry about at least two factors that could complicate production of an authentic protein for human use.
a. What complication might you worry about if the human DNA encoding the protein were placed directly in E. coli?
b. Assume that the correct primary sequence of amino acids has been produced. What posttranslational steps do you worry about, and why?
4.12. Consider the process of N-linked glycosylation.
a. What organelles are required?
b. What is the residual sugar on a glycoprotein that has simple glycosylation?
c. If glycosylation is complete, what will be the final sugar on the glycoform?