
Chapter 15: Osteoarthritis
Osteoarthritis (OA) is considered the most common skeletal disorder and is characterized by cartilage degradation and osteophyte formation in joints. Although it is the most common joint disease, OA is often difficult to define because it is heterogeneous in its presentation style, rate of progression, and manifestations. Recently, new insights into the molecular biological basis of chondrocyte differentiation and cartilage homeostasis have been reported. The pathology of osteoarthritis is now interpreted as the disruption of balance between anabolic and catabolic signals. This chapter focuses on the risk factors, cellular and molecular mechanisms involved in OA, tools to study OA such as animal models and biomarkers, as well as treatment. Findings with regard to the physiologic and pathologic mechanisms involved in OA have made it possible to target therapeutic approaches more accurately, which allows the development of new drugs to reduce or block the progression of the disease.
15.1 Biological and Biophysical Background of Osteoarthritis
15.1.1 Diagnosis
OA is the most common chronic joint disorder. The most frequently affected sites are the hands, knees, hips, and spine [1] (Fig. 15.1). It results from degeneration of a synovial joint, which leads to a generally progressive loss of articular cartilage accompanied by attempted repair of articular cartilage, as well as remodeling and sclerosis of subchondral bone, subchondral cysts, and marginal osteophytes formation. This disease not only affects the articular cartilage but also involves the entire joint, which includes the subchondral bone, ligaments, capsule, and synovial membrane (Fig. 15.2).
Figure 15.1 Radiographs of OA in hip, knee, lumbar spine, and hand. Cartilage degradation shown as joint space narrowing and osteophyte formation at the edge of the joints, which are two major disorders of OA.
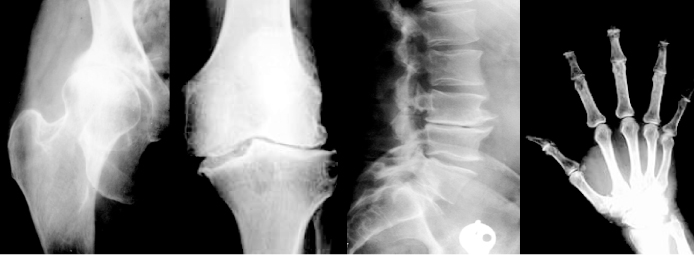
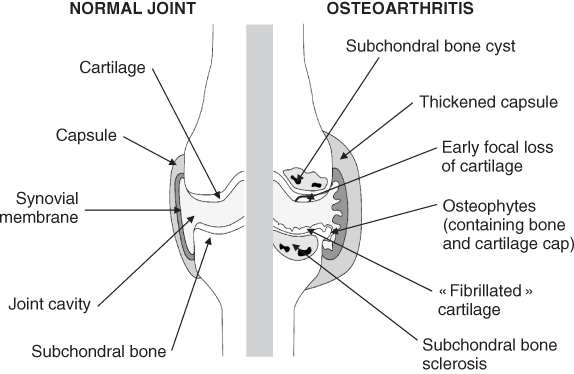
Figure 15.2 Schematic representation of the main structures of a healthy (left side) and degenerated (right side) joint in osteoarthritis. Osteoarthritic joint is characterized by the lost or severely thinned articular cartilage (“fibrillated” cartilage), the sclerosis of subchondral bone, the formation of osteophytes, the presence of subchondral bone cyst, the thickened joint capsule, and the synovial hyperplasia. From EULAR online course, J. Sellam, G. Herrero-Beaumont and F. Berenbaum.
The diagnosis of OA requires plain radiographic examination. Usually, patients present with chronic joint pain, which is the first and predominant symptom, followed by restriction of joint motion and function, crepitus with motion, and joint effusions. The most severely affected individuals develop joint deformities and subluxations [2]. The course of the disease varies but is often progressive, and the radiographic changes of OA progress inexorably (Bulletin of the World Health Organization; http://www.who.int/whr/2007/en/index.html).
15.1.2 Prevalence and Incidence
Few data are available on the incidence and prevalence of OA because of the problems of defining it and determining its onset. It is well known that OA affects people of all ethnic groups in all geographic locations in both men and women; it is the most common cause of long-term disability in most populations of elderly people (Bulletin of the World Health Organization; http://www.who.int/whr/2007/en/index.html). More than 80% of people over the age of 55 have radiological evidence of OA. Not all of these individuals are symptomatic, but 10–20% of those affected may report limitation of activity because of OA [3]. Moreover, the World Health Organization estimates that 10% of the world's people over the age of 60 suffer from OA, and that 80% of people with OA have limitation of movement and 25% cannot perform major daily activities (Bulletin of the World Health Organization; http://www.who.int/whr/2007/en/index.html). Finally, the incidence of OA increases indefinitely with age (notably after age 40) and is greater in women than in men [4, 5].
15.1.3 Risk Factors
The precise etiology of OA in most cases is unknown, but it is generally accepted that OA is a multifactorial disorder that involves both genetic and environmental components. Among factors known to affect the progression of OA include the affected joint, increasing age, obesity, excessive physical activity, joint injury, genetic predisposition, and female gender.
15.1.3.1 Age
Age is the most powerful risk factor for OA. The National Health and Nutrition Examination Survey found that the prevalence of knee OA increased from less than 0.1% in people 25–34 years old to 10–20% in people 35–74 years old [6]. A higher prevalence of 30% between 65 to 74 years old has been found in the Framingham Study [7].
Recent works suggest that the changes observed during aging in articular cartilage are caused by deterioration of chondrocyte function. With increasing age, the ability of cells to maintain and restore cartilage matrix decreases [8]. The major components of the extracellular matrix (ECM), which include type II collagen and proteoglycans, undergo changes in content, composition, and structural organization during the aging process. For example, cells synthesize smaller aggrecans and less functional link proteins, which leads to smaller more irregular proteoglycans aggregates [9]. In addition, accumulation of advanced glycation end product (AGE) in the cartilage ECM could enhance collagen cross-linking [10]. Finally, mitotic activity of chondrocytes declines with age, and cells become less responsive to anabolic cytokines and mechanical stimuli. This finding has been attributed to replicative senescence associated with reduction of telomere length [11]. Consequently, the articular surface softens, and the tensile strength and stiffness of the matrix decreases.
15.1.3.2 Obesity
Obesity increases the risk of OA, but it is unsure whether this effect is caused by excessive mechanical strains, varus malignment, or metabolic abnormalities. Increased body mass leads to abnormal strains on articular cartilage and could explain the positive association between obesity and hip and knee OA observed in many epidemiological studies [12, 13]. Interestingly, the Framingham data show that being overweight as a young adult strongly predicted the appearance of knee OA in a 36-year period of follow-up [14].
The increased fat mass in obesity may therefore alter the metabolism of articular tissues such as cartilage. Recent studies have described the association between obesity and OA in non–weight-bearing joints, such as the hand [15, 16], whereas the loss of body fat seems to be more important than the loss of body weight in improving the symptoms of OA [17]. Fat cells secrete a variety of proteins with the functional and structural properties of cytokines, which are called the adipokines. Adiponectin, leptin, resistin, and visfatin are the most abundant adipokines produced by adipose tissue, and their expression is regulated in obese individuals. They have been detected in synovial fluid from joints affected with OA. Recent evidence points to the involvement of leptin in OA and indicates that adiponectin and/or resistin mediates inflammation in arthritis. Visfatin can be produced not only by adipocytes but also by chondrocytes themselves, and it can induce catabolic activity of these cells [18]. Thus, fat tissue is an active organ whose products contribute to inflammatory and degenerative processes underlying common joint diseases [19].
15.1.3.3 Excessive Repetitive Joint Loading
Joints are highly specialized organs that allow repetitive pain-free and largely frictionless movements. This function occurs particularly because of articular cartilage and notably its ECM, which play an essential role in load transfer across the joint [20]. Strains are necessary for joint physiology, as moderate exercise stimulates cartilage homeostasis and prevents the development of OA [21]. Moreover, studies performed in space revealed the role of gravity in the chondrocyte physiology; the different steps of chondrogenesis are altered by long-term exposure to microgravity [22]. However, repetitive loading of normal joints can exceed the tolerance of a joint and cause degeneration. Occupational overuse of the knee joint in obese subjects or in jobs that require repeated kneeling, squatting, or bending, as well as lifting of heavy loads is a risk factor for OA [23]. It has been calculated that elimination of such jobs would decrease the incidence of knee OA in men by 15–30% [24]. Other studies suggested that participation in sports that repetitively expose joints to high levels of impact also increase the risk of joint degeneration [25].
Cartilage matrix submitted to mechanical stress presents an alteration in pH, osmolarity, hydrostatic pressure, and reorganization of the ECM macromolecules. Chondrocytes bear receptors that respond directly to these modifications, many of which are also receptors of ECM, such as integrins or ion channels, and are located on ciliae, which was recently identified in chondrocytes [26]. In response to strains, chondrocytes regulate the synthesis of matrix proteins, increase the production of inflammatory cytokines, or modulate their proliferation, differentiation, and apoptosis [27, 28]. Application of static compression or excessive cyclic stress could therefore alter the ECM and stimulate an inflammatory response in cartilage, which can lead to OA.
15.1.3.4 Joint Injury
Joint injuries frequently lead to progressive joint degeneration, which causes the clinical syndrome of posttraumatic OA. Studies of humans and animals models demonstrate that intra-articular fractures, dislocations, damage to the meniscus, and meniscal, ligament, and joint capsule tears lead to knee OA. Even if the cartilage is not involved at the time of injury, it will degenerate rapidly if the joint is unstable. The pathophysiology of posttraumatic OA has not been explained. Evidence indicates that first, the acute joint injury kills at least a few chondrocytes [29], and second, it increases the release of reactive oxygen species (ROS) in chondrocytes, which accelerate chondrocyte senescence and decreases the ability of the cells to maintain or restore the tissue [30]. Furthermore, the risk of posttraumatic OA varies among individuals and among joints [31].
15.1.3.5 Genetic Factors
Several lines of evidence indicate that genetic components affect the development of OA. Twin studies, segregation analyses, linkage analyses, and candidate gene-association studies have generated important information about inheritance patterns and the location in the genome of potentially causative mutations. Twin studies have shown that the influence of genetic factors vary from 39% to 74% in OA depending on the joints affected [32–34]. Genetic defects in matrix molecules, in signaling molecules, as well as in molecules that participate in the formation of cartilage matrix and patterning of skeletal elements resulting in dysplasia may determine susceptibility to OA [35–37].
15.1.3.6 Gender
Sex-specific differences are evident in OA. Before 50 years of age, the prevalence of OA in most joints is higher in men than in women. After about 50 years, women are more often affected with hand, foot, and knee OA than men. In most studies, hip OA is more frequent in men (Bulletin of the World Health Organization; http://www.who.int/whr/2007/en/index.html).
15.2 Biochemistry and Molecular Mechanism Involved in Osteoarthritis
A schematic representation of the molecular pathogenesis of osteoarthritis implicating synovial membrane, subchondral bon, and articular cartilage is shown in Figure 15.3.
15.2.1 Cartilage Damage in Osteoarthritis
OA is characterized by the slowly progressing destruction of the articular cartilage, which results from the failure of chondrocytes to maintain the balance between synthesis and degradation of the extracellular matrix [38, 39]. Moreover, phenotypic modulations as hypertrophic differentiation of chondrocytes and chondrocytes apoptosis are known to be involved in OA development [40].
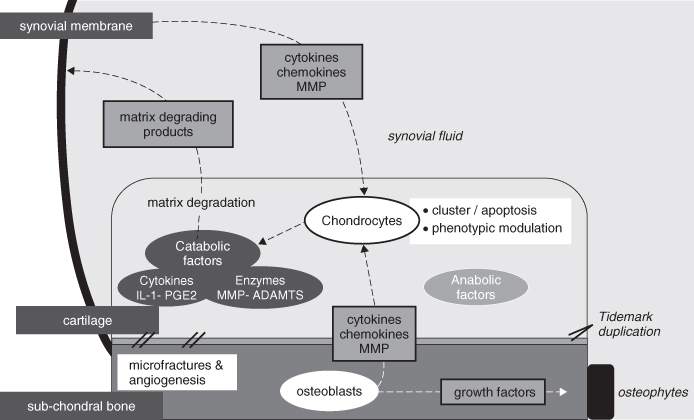
Figure 15.3 Schematic representation of the molecular pathogenesis of osteoarthritis implicating synovial membrane, subchondral bon, and articular cartilage. During OA, chondrocytes are not still quiescent cells but undergo apoptosis or proliferation and phenotypic modulation; chondrocytes synthesize inflammatory mediators and matrix-degrading enzymes. Matrix degradation products are released in synovial fluid and trigger synovial inflammation, which leads to cytokines and proteinase release by synoviocytes. Moreover, an increased cartilage calcification and microfractures with blood vessel invasions from the subchondral bone is observed. Finally, osteoblasts release growth factors, which may participate in osteophytes formation.
15.2.1.1 Anabolic and Catabolic Signals of Cartilage in Osteoarthritis
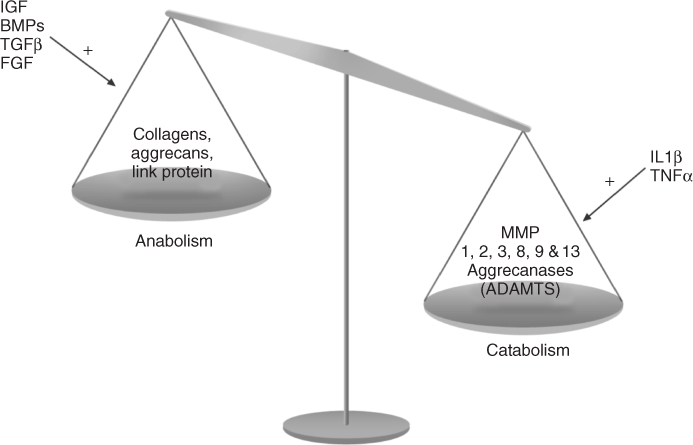
A balance between anabolic and catabolic signals maintains the homeostasis of cartilage. In normal cartilage, the balance between anabolism and catabolism is equivalent. In OA cartilage, catabolism becomes more dominant than anabolism, which leads to the degradation of cartilage [39] (Fig. 15.4).
Figure 15.4 Osteoarthritis exhibits an imbalance in cartilage matrix turnover. Anabolic and catabolic stimulatory factors are synthesized by the chondrocytes themselves in an automatic and paracrine manner. In OA cartilage, catabolism (characterized by MMPs and ADAMTS production) becomes more dominant than anabolism (characterized by collagens, aggrecans and link protein expressions), which leads to the degradation of cartilage. (FGF, fibroblast growth factor)
Anabolic Signals in Osteoarthritis
Insulin Growth Factors (IGFs)
In articular cartilage, IGF-1 plays a crucial role in the maintenance of homeostasis by stimulating the production of matrix proteins by chondrocytes, which counteracts their degradation [41–43]. Interestingly, despite increased cartilage degradation in OA, the expression of IGF-1 messenger RNA (mRNA) is upregulated in OA cartilage, and IGF-1 protein increases in synovial fluid [44–46]. The increased production of IGF-1 in OA reflects an attempt to restore cartilage homeostasis, but it is inefficacious because of the lack of bioavailability. The accessibility of IGF-1 to its receptor is regulated by extracellular IGF-binding proteins (IGFBPs), which are also key players in the failure of cartilage homeostasis during OA. IGFBP-3 is secreted by articular cartilage and chondrocytes [47], and its expression has been found to be correlated with the severity of OA [48, 49]. Anabolic effects on chondrocytes by IGF-1 depend on the level of secreted IGFBPs.
Transforming Growth Factor (TGF)-β
TGF-β is an important anabolic factor in OA because it promotes type II collagen and proteoglycan synthesis and downregulates cartilage-degrading enzymes [50–54]. Moreover, TGF-β can counteract the suppression of proteoglycan synthesis by interleukin 1 (IL-1) [53]. In aging and OA, the expression of TGF-β receptors and Smad2 phosphorylation are reduced in chondrocytes [55]. The reduced response of chondrocytes to TGF-β may play a pivotal role in OA.
Bone Morphogenetic Proteins (BMPs)
BMPs belong to the TGF-β superfamily and are considered to be one of the most important anabolic factors for articular cartilage. Recent studies showed that BMPs play a crucial role in adult articular cartilage homeostasis [56, 57]. BMPs enhance proteoglycan and type II collagen synthesis in vitro and in vivo [58, 59]. BMPs also counteract catabolic cytokines such as IL-1. BMP7 has been shown to counteract matrix metalloproteinases-13 (MMP-13) induction by IL-1β. BMPs signal through Smad pathways, whereas IL-1 signals through extracellular signal regulated kinase (ERK)-p38- Jun terminal kinase(JNK)/NF-kB pathways [60]. These pathways may seem to be separated but are often cross-linked in several ways. Therefore, intracellular crosstalk between the BMPs and IL-1 signaling pathways plays a part in balancing anabolism–catabolism in articular cartilage.
Fibroblast Growth Factors (FGFs)
Several studies have shown that bFGF is the most potent mitogen for chondrocytes and that it stimulates or stabilizes the synthesis of the cartilage matrix [61–63]. Intra-articular administration of FGF2 enhances cartilage extracellular matrix formation in vivo [64, 65]. However, others report that it was inhibitory or had no effect [66]. Recent studies have shown that bFGF stored in the articular cartilage matrix is released with mechanical injury or with loading [67, 68]. Moreover, bFGF has stimulated production of MMP-1, -3, and -13 instead of proteoglycan synthesis. The excessive release of bFGF from cartilage matrix may be partly responsible for cartilage matrix degradation during OA.
Catabolic Signals in Osteoarthritis
MMPs
The MMP family members are the major enzymes that degrade the extracellular matrix of cartilage. Twenty-three members of this family of neutral Zn2+ metalloproteinases have been identified [69, 70]. The MMP family is broadly classified into secreted-type MMPs and membrane-type MMPs (MT-MMPs). Because they are active at neutral pH, the MMPs can act on the cartilaginous matrix at some distances of the chondrocytes. They can be synthesized by chondrocytes, synoviocytes and osteoblasts after stimulation by cytokines or mechanical stress. Among MMPs, at least five are elevated in OA: collagenase-1 (MMP-1), stromelysin (MMP-3), gelatinase-92kd (MMP-9), matrisylin (MMP-7) and collagenase-3 (MMP-13). In OA, MMP-1 (also known as collagenase-1) and MMP-13 (also known as collagenase-3) have a major role in cartilage degradation. MMP-13 is usually produced by chondrocytes in OA [71, 72]. The expressions of these MMPs are low in normal cells, which contributes to normal connective tissue re-modeling. However, excessive MMP expression results in aberrant connective tissue destruction such as observed in arthritic diseases, periodontitis, and tumor invasion [73]. MMPs are induced in response to cytokines and growth factors. In OA, the mechanical stress causes cytokine expression such as IL-1beta by articular chondrocytes, which subsequently express MMPs [74]. MMP-1, MMP-8 (also known as neutrophil collagenase), and MMP-13 have the ability to cleave the triple helix of collagen and unwind the chains. After that, the chains become susceptible to even more degradation by other MMPs, such as MMP-9. MMP-13 plays a crucial role in collagen degradation because it is expressed by chondrocytes and it hydrolyzes type II collagen more efficiently than other collagenases [75]. In an animal model, the postnatal expression of constitutively active human MMP-13 in the articular cartilage of transgenic mice leads to the degeneration of articular cartilage and joint pathology of the kind observed in OA [76].
Aggrecanases
The ADAMTS (a disintegrin and metalloproteinase domain with thrombospondin motifs) family contains 19 members [77, 78]. Family members ADAMTS-2, ADAMTS-4, ADAMTS-5, ADAMTS-9, and ADAMTS-15 are called aggrecanases. They degrade the interglobular domain separating G1 and G2 of aggrecan at a specific Glu373-Ala374 bond [78–81]. Aggrecanases are active in the early phase of OA, with a later increase in MMPs activity. In surgically induced OA model mice, however, the degeneration of articular cartilage and joint pathology of OA were significantly reduced in ADAMTS-5 knockout mice with no difference in severity in ADAMTS-4 knockout mice [82, 83]. In an inflammatory arthritis model, the deletion of ADAMTS-5 also prevented the progression of aggrecan degradation, whereas severe cartilage degradation was induced in ADAMTS-4 knockout mice [84]. These findings suggest that ADAMTS-5 might be the key player for aggrecan degradation in arthritic disease.
Proinflammatory Factors
In many joint diseases, proinflammatory factors such as cytokines and prostaglandins are released at sites of inflammation, together with ROS and nitric oxide (NO) [85, 86].
Cytokines
The cytokines such as IL-1β and tumor necrosis factor-α (TNF-α) also play a key role in cartilage degeneration in OA. IL-1beta is one of the most prominent catabolic cytokines that plays a pivotal role in OA [87], and its level is increased in the OA cartilage [88]. IL-1 not only promotes the release of degenerative enzymes including MMP-1, MMP-3, MMP-13, or ADAMTS-4 and ADAMTS-5, but also it inhibits the synthesis of extracellular matrix proteins by chondrocytes. IL-1 also induces other catabolic cytokines, such as IL-6, with synergistic effects [89]. IL-6 family members have been shown to induce the JAK/STAT signaling pathways [90]. The effects of IL-6 are similar to IL-1; however, its effects are weaker than that of IL-1 in inhibiting proteoglycan synthesis. IL-6 is required for prolonged proteoglycan suppression by IL-1, and it enhances the expression and activity of MMPs and aggrecanase in articular cartilage [91]. IL-1 acts through nuclear factor-kappa beta (NF-kB) and the following three classic MAPK-signaling pathways: ERK, p38, and JNK. Although IL-1beta can activate all four pathways, the ERK pathway is especially important for the induction of other cytokines such as IL-6 [89].
Prostaglandin E2 (PGE2)
PGE2 is one of the major catabolic mediators involved in cartilage degradation and the progression of OA [92–94]. The synthesis of PGE2 is the endpoint of a sequence of enzymatic reactions, which includes the release of arachidonic acid from membrane phospholipids by soluble phospholipase A2 and conversion of this substrate to prostaglandin H2 (PGH2) by cyclooxygenase (COX)-1 and COX-2, which is also known as prostaglandin H synthase 1 and 2. Then, the PGE synthase catalyzes the conversion of PGH2 to PGE2. PGE2 is a mediator of inflammation, which has numerous actions during OA. PGE2 modulates the activity of synovial cells, macrophages, and chondrocytes, and it induces bone resorption [95]. The role of prostaglandins in the metabolism of articular cartilage is still a matter of debate. Some reports indicate that prostaglandins participate in the destruction of articular cartilage by degrading cartilage ECM [96, 97], whereas others show that they promote chondrogenesis and terminal differentiation [98, 99]. The opposing biologic roles attributed to these compounds are a direct reflection of the molecular complexity of prostaglandins (PGs) and their unique cognate receptors [100]. PGE2 is known to bind to four distinct cell surface receptors (EP1, EP2, EP3, and EP4), each of which is a “serpentine seven” G-protein–coupled receptor; the ligation of individual receptors mediates distinct intracellular signaling pathways that may contribute to the pleiotropic effects of PGs in different tissues [101, 102]. Recently, two different studies report that the predominant effects of PGE2 in OA chondrocytes are catabolic and that these effects are mediated via EP4 receptor signaling [103, 104].
Oxydative Stress
ROS are normal by-products of cellular metabolism. In some physiological and pathological circumstances, O2 may be transformed into ROS and then may be involved in the control of various aspects of biological processes, which include cell activation, proliferation, and death. Especially low levels of ROS have been reported to act as one of the different intracellular second messenger molecules involved in the regulation of the expression of a wide variety of gene products, which include cytokines, MMPs, adhesion molecules, and matrix components [105, 106]. The elevated production of ROS and/or depletion of antioxidants has been observed in a variety of pathological conditions, including inflammatory joint diseases. The intracellular and extracellular reduction-oxidation state may be causally implicated in the progression of these diseases (see review 107, 108). When the imbalance between oxidants and antioxidants is large enough to induce cellular and/or tissue structural and/or functional changes, the situation is called “oxidative stress” and is considered as an abnormal catabolic event. The overproduction of ROS in pathological cartilage has been evidenced indirectly by the accumulation of lipid peroxidation products and nitrotyrosine in situ [109, 110]. Recently, Henrotin et al. [111] have found elevated nitrated type II collagen peptides in sera of patients with OA, which suggests the formation of ONOO- in OA cartilage. Chondrocytes produce basically cNO and O2c− that generate derivative radicals including ONOO- and H2O2. ROS may directly oxidize nucleic acids, transcriptional factors, and membrane phospholipids, as well as intracellular and extracellular components, which leads to impaired biological activity, cell death, and breakdown of matrix components [106, 107, 112–115]. ROS are highly reactive compounds with a short half-life. However, nitrotyrosine, which is formed when tyrosine is oxidized in the presence of nitrous oxide (NOO), can serve as a measure of oxidative damage in vivo. The presence of nitrotyrosine in cartilage was associated with older age and with osteoarthritis, which suggests a role of oxidative stress in cartilage aging and degeneration [116]. Thus, oxidative stress affects chondrocyte function in joint cartilage [117, 118].
Nitric Oxide
NO is synthesized by way of the oxidation of L-arginine by the NO synthase; one result is constitutive NOS and the other is inducible NOS. Some studies have reported a local production of NO in the OA joint by chondrocytes and their presence in synovial fluid [119]. NO synthesis is implicated as a catabolic factor in OA. The various roles of NO as a mediator of other IL-1–induced responses, which include the inhibition of aggrecan and collagen synthesis [120, 121], enhancement of MMP activity and chondrocyte apoptosis [122–124], and reduction of the production of IL-1 receptor antagonist (IL-1RA) [125], have also been suggested [126]. NO may also increase chondrocyte susceptibility to injury by other oxidants such as H2O2 and contribute to resistance against the anabolic effects of IGF-I [127]. Nitric oxide also has been implicated as an important mediator in chondrocyte apoptosis, and an association between NO production and apoptosis in OA cartilage has been proposed [122].
Transcriptional Factors in Osteoarthritis
Sox9 is a transcriptional factor with a high-mobility group DNA-binding domain and is required for cartilage formation and for expression of chondrocyte-specific genes, such as COL2A1 [128]. In OA cartilage, several studies have shown that Sox9 is downregulated and that IL-1β and TNF-α inhibit the expression of cartilage-specific genes, such as COL2A1, COLL11A2, COL9A2, and aggrecan [129–131]. The major mechanism by which IL-1 and TNF-α inhibit the chondrocyte phenotype is by downregulating the expression of Sox9 [132]. IL-1 and TNF-α inhibit Sox9 binding to the COL2A1 enhancer, and these effects are mediated by NF-kB [133]. Another study showed that Sox9 is downregulated by IL-6/sIL-6R associations at both mRNA and protein levels through the JAK/STAT signaling pathway [134]. However, other studies reported that in the early phase of OA, the expressions of type II collagen and type I collagen are upregulated, which suggested that the Sox9 type II collagen master regulation gene might be also upregulated [135]. The evidence for upregulation or downregulation is mixed regarding the alteration of Sox9 gene expression during the progression of OA. It is possible that anabolic and catabolic signals might be abnormally intermingled in OA cartilage. IL-1 induction of MMP-13, MMP-1, and MMP-9 requires NF-kB [136, 137]. Because inhibition of NF-kB results in a beneficial effect on OA synovial fibroblasts, such as the decreased expression of MMPs and ADAMTS-4 aggrecanase [138], the inhibition of NF-kB is anticipated as an alternative therapeutic target. Moreover, the key role of NF-kB in regulating mechanical stress in chondrocytes has been described recently in different studies [139, 140].
15.2.1.2 Chondrocyte Proliferation and Apoptosis in Osteoarthritis
The chondrocyte, which is the only cell type that resides in the adult cartilage matrix, has a low metabolic activity, and it survives under relatively hypoxic conditions and in the absence of a vascular supply. This cell, which is ultimately responsible for remodeling and maintaining the structural and functional integrity of the cartilage matrix, possesses poor regenerative capacity. The adult chondrocyte plays a critical role in the pathogenesis of OA in responding to adverse environmental stimuli by promoting matrix degradation and downregulating processes essential for cartilage repair. Physiologically, the chondrocytes maintain a low turnover rate of replacement of cartilage matrix proteins with a half-life for collagen of greater than 100 years [141]. In contrast, the glycosaminoglycan constituents on the aggrecan core protein are more readily replaced, and the half-life of aggrecan subfractions has been estimated in the range of 3–24 years [142]. The half-life of proteoglycan has been estimated in the range of 7–200 days [143]. In early OA, evidence suggests the presence increased synthetic activity, which is viewed as an attempt to regenerate the matrix with cartilage-specific components, including types II, IX, and XI collagens; aggrecan; and pericellular type IV collagen [144]. The aberrant behavior of OA chondrocytes is reflected in the appearance of fibrillations, matrix depletion, and cell clusters, as well as in changes in quantity, distribution, or composition of matrix proteins [145]. Evidence of phenotypic modulation is reflected in the presence of collagens not normally found in adult articular cartilage, which includes the hypertrophic chondrocyte marker, type X collagen, as well as other chondrocyte differentiation genes; this finding suggests a recapitulation of a developmental program [146, 147]. Several studies [148, 149] have clearly shown that (low) proliferative activity occurs in osteoarthritic chondrocytes in contrast to normal articular chondrocytes, which do not show any proliferative activity. A proliferative activity is also observed in the early stage, at a low level, which leads to the chondrocyte clustering [150–152]. This increased proliferative activity, especially in the upper cartilage zone, could be explained by the exposition of chondrocytes to growth factors released by the cartilage itself, which are present in the synovial fluid. This proliferative activity is visualized as a cellular reaction to cartilage destruction to repair cartilage.
In addition to hypertrophic differentiation of chondrocytes, chondrocyte apoptosis is known to be involved in OA development [153]. The intra-articular injection of a pan-caspase inhibitor has been reported to suppress cartilage degradation under OA induction in a rabbit ACLT model [154]. Many studies have considered whether cell death plays a role in the pathology of OA [for review, see Aigner et al. [155]], because articular chondrocytes cannot self-renew, and therefore cell loss would be permanent. However, opinions on the prevalence the importance and the relevance of chondrocyte death for OA pathology differ widely. Early studies [156, 157] estimated the incidence of cell death as 25–50%, but the data were obtained with isolated chondrocytes and therefore may have measured a predisposition to death rather than actual incidence. Estimates of cell death based on in vivo staining also vary widely, from 21% [158] to less than 1% [159]. Opinions also vary as to the type of cell death. Most authors classify chondrocyte death as caused by apoptosis, even though injury or mechanical damage by themselves would lead to a high level of necrosis.
15.2.2 Synovium Involvement in Osteoarthritis
The clinical presentation in OA joints (such as swelling, effusions, and stiffness) clearly reflects synovial inflammation lower than in rheumatoid arthritis, as a low-grade contribution to disease pathogenesis. This synovitis occurs even in early OA and can be subclinical, as arthroscopic studies suggest that localized proliferative and inflammatory changes of the synovium occur in up to 50% of OA patients (many of whom do not seem to have active inflammation) [160]. Synovial histological changes include synovial hypertrophy and hyperplasia, with an increased number of lining cells, which are often accompanied by infiltration of the sublining tissue, with scattered foci of lymphocytes. In contrast to rheumatoid arthritis, synovial inflammation in OA is mostly confined to areas adjacent to pathologically damaged cartilage and bone. This activated synovium can release proteinases and cytokines that may accelerate the destruction of nearby cartilage. The synovium produces some of the chemokines and metalloproteinases that degrade cartilage, even though the cartilage itself produces most of these destructive molecules in a vicious autocrine and paracrine fashion. In turn, cartilage breakdown products, which result from mechanical or enzymatic destruction, can provoke the release of collagenase and other hydrolytic enzymes from synovial cells and lead to vascular hyperplasia in OA synovial membranes. This cascade sequentially results in the induction of synovial IL-1β and TNF-α, which extend the inflammatory outcome [161]. This cytokine storm may be more likely to occur in earlier stages of the disease before end-stage damage. As shown by a recent study of 10 patients with early OA (arthroscopic specimens) and 15 patients undergoing total knee arthroplasty; synovial tissues from early OA had higher levels of IL-1β and TNF-α and increased mononuclear cell infiltration compared with late OA [162]. Erosive OA likely represents a more inflammatory process, as evidenced by higher proteinase and cytokine levels One study of rapidly destructive hip OA demonstrated MMP-3 and -9 levels that were especially elevated, not only in patients' synovial cells but also in their synovial fluid, plasma, and sera [163, 164]. In vitro studies from diseased human OA tissue have implicated MMP-10 expression in synovial fibroblasts, as well as in OA synovial fluid and chondrocytes stimulated with catabolic IL-1 and oncostatin M [165].
15.2.3 Role of Subchondral Bone in Osteoarthritis
In addition to the progressive loss of articular cartilage, OA is characterized by increased subchondral plate thickness, formation of new bone at the joint margins (osteophytes), and the development of subchondral bone cysts [166, 167]. Interestingly, these abnormalities take place not only during the final stage of OA but also earlier at the onset of the disease, maybe before the cartilage degradation [168].
Subchondral bone remodeling is responsible for an alteration in mechanical load absorbance. This tissue is characterized by the accumulation of osteoid substance, which is decreased mineralization related to the production of an abnormal trimeric type I collagen that has a low affinity for calcium [169]. Several studies have shown that osteoarthritic subchondral osteoblasts exhibit specific pathologic characteristics, such as overproduction of alkaline phosphate and osteocalcin, as well as transforming growth factor-beta 1 (TGF-β1), IGF-1, and urokinase [170].
Moreover, it has been shown recently that mature sclerotic osteoblasts exhibit an increased expression of osteocalcin, IL-6, IL-8, C-terminal type I procollagen propeptide, TGF-β1, osteopontin, and accumulation of osteoid substance. The expression of parathyroid hormone receptor is decreased, which explains the resistance of subchondral bone to resist to parathyroid hormone stimulation [171].
The subchondral bone is involved in the OA process [172] at the following two different levels:
Of note, the subchondral bone in OA is in a state of hypercoagulation, hypofibrinolysis, and thrombosis, which is related to reduced blood flow and increased production of procoagulant factors. These factors lead to venous stasis and hypertension, which are associated with thrombosis and focal ischemic bone necrosis [176].
All these modifications of structure and phenotype of osteoblasts in the subchondral bone can modify strength load on the cartilage and enhance the alteration of chondrocytes and ECM as well as the cartilage breakdown.
Angiogenesis phenomenon is observed at the junction of the articular hyaline cartilage and adjacent subchondral bone. Evidence exists to suggest vascular invasion and advancement in the zone of calcified cartilage in the region of the so-called tidemark that contributes to a decrease in articular cartilage thickness [177]. A recent study showed that angiogenesis in the osteochondral junction is independent of synovial angiogenesis and synovitis but is associated with cartilage changes and clinical disease activity [178]. These structural alterations in the articular cartilage and periarticular bone may lead to modification of the contours of the adjacent articulating surfaces [179, 180]. The accompanying alterations in subchondral bone remodeling may contribute to the development of an adverse biomechanical environment and enhance the progression of the articular cartilage deterioration. Changes in the mineral content and thickness of the calcified cartilage and the associated tidemark advancement may be related to the localization of COL10A1, MMP-13, and Runx2 in the deep zone of OA cartilage, where the chondrocytes may attempt a defective repair response by recapitulation of the hypertrophic phenotype [181].
The link between cartilage and subchondral bone is still debated. Mechanical stress on weight-bearing joints is responsible for local lesions of cartilage, which increase stress to the local area of bone underneath this area of cartilage and enhance microfractures and sclerosis of the subchondral bone. These modifications of the bone are responsible for mechanical disturbance, which increases the lesions of the cartilage and the subchondral bone itself. Another possibility is that because of repeated microfractures, subchondral bone becomes stiffer. This structural alteration occurs before cartilage changes and can modify joint conformation and thus induce secondary cartilage alteration under load [182]. Indeed, studies in animal models suggest that subchondral bone changes may precede cartilage degradation and loss. This finding has been confirmed in magnetic resonance imaging (MRI) studies in humans. Likewise, these bone changes are not a secondary manifestation in OA but may be an active part in the initiation of the OA process. Indeed, subchondral bone tissue in OA could provide growth factors, cytokines, and eicosanoids to the overlying cartilage and could promote its abnormal remodeling and metabolism, which leads to ECM degradation [183].
15.3 Tools and Methods for Studying Osteoarthritis
15.3.1 Animal Models
In an effort to clarify the pathophysiological mechanisms that lead to OA, experimental animal models have been developed. The classic models of OA have involved the induction of OA-like changes in many animal species, which include dogs, rabbits, guinea pigs, sheep, and rats using surgical intervention. Transection of ligaments combined with resection of the meniscus is used to produce instability in the knee joint. Interestingly, spontaneous forms of OA exist in various species, such as mice. But even if spontaneous models of OA are more relevant, they are more difficult and less practical [184].
Recently, transgenic and knockout mouse models, which are the most ideal animal for molecular study, have provided new tools to elucidate the mechanisms implicated in OA pathophysiology [185]. The application of surgical models on these mice has been developed to characterize the in vivo significance of a particular gene [186–188].
Although these models are excellent for studying the time course of OA and the implication of different molecules in this event, no experimental model is totally equivalent to OA in humans.
15.3.2 Biomarkers
15.3.2.1 Imaging as a “Biomarker”
Standards radiographs (X rays) are used to establish diagnosis. The radiographic hallmarks of the most common form of OA include localized joint space narrowing, subchondral bone sclerosis, formation of osteophytes, and bone cysts. However, clinical symptoms and radiographic findings are poorly correlated; many joints with radiographic evidence of OA remain asymptomatic and, conversely, the joints of many patients with severe symptoms can seem only marginally affected on X rays [189]. Moreover, X rays have shortcomings with respect to the assessment of progressive disease. For example, they are insensitive to early changes within cartilage and bone and do not report synovial pathology [190].
MRI is a powerful technique that provides precocious alterations of cartilage, synovium, and bone. It represents a sensitive and specific technique for measuring disease progression. Traditional MRI is now being used for the quantitative assessment of hyaline cartilage thickness and volume, synovial hypertrophy, and bone marrow edema. Most recently, functional MRI studies (dGEMRIC, NaMRI, or T1ro), which detect biochemical changes of ECM proteins in cartilage, could be used to assess the improvement from the treatment. This study is of particular interest because cartilage thinning in OA is preceded by modification of the composition and structural organization of the collagen–proteoglycan matrix [191–193].
15.3.2.2 Biochemical Markers
Because of the increasing knowledge of the cartilage matrix composition, molecular markers have been identified for monitoring changes in cartilage metabolism and for assessing joint damage in OA. These biochemical markers could be useful to establish state of disease, to understand the pathophysiology of OA, to predict progression, and to assess the treatment efficacy. Candidate biochemical markers of OA joint tissues present in synovial fluids, blood, or urine can reflect different actors of OA (bone, cartilage, synovitis, and systemic inflammation) as well as the different states of these tissues (i.e., degradation or synthesis). These biomarkers involve components of matrix proteins, which include several collagens as well as cross-linked derivative peptides and matrix metalloproteinases. For example, serum cartilage oligomatrix protein, urinary C-terminal cross-linking telopeptide of type II collagen, and serum hyaluronan denote the severity and extent of disease in joints [190, 194, 195]. Moreover, various type II collagen epitopes have been described, including Coll 2-1, which was characterized as a disease-specific marker that is sensitive to the structural changes occurring in OA [196]. Interestingly, the urinary levels of Coll 2-1 or Coll 2-1NO2 over 1 year was found to be predictive of the progression joint space narrowing in OA patients [197, 198]. Finally, myeloperoxidase is an innovative marker that allows oxidative stress assessment in OA: Its decrease after joint replacement indicates that neutrophil activation occurs during this disease [196]. Moreover, plasma high-sensitivity C reactive protein levels reflect synovial inflammation in OA patients [199], and its levels probably reflect the disease activity of OA [200]. But actually, no consensus exists to what is the optimal biomarker, and at the time of writing in 2008, there is not any indication to apply them in daily practice. Although a single marker may not be sufficient, it may be possible to use a combination of biomarkers that may discriminate between different stages of OA in different populations [201].
15.3.2.2 Clinical Markers
Nonmodifiable risk factors, such as age, gender, body mass index, injury, or genetic background could be considered to be predictors of disease progression. The first-degree relatives of patients with generalized OA have a twofold risk of developing the disease compared with the general population [202]. Moreover, monozygotic twins are significantly more concordant for hand and knee osteoarthritis than dizygotic twins [33].
Clinical endpoints can also serve as markers of disease progression. Factors that have consistently been reported as associated with radiographic OA progression are obesity, generalized OA, alignment, and synovitis [203]. For example, in a 1-year follow-up study, people who suffered from OA with synovitis were found to have more rapid progression of cartilage damage [204]. But, even if clinical signs can potentially be used to predict progression in OA, they are only observed in a subset of patients.
15.3.2.3 Use of Biomarkers in Drug Development
Because the knowledge of the pathophysiological mechanisms of OA has advanced, efforts were made to identify new drugs to treat OA. Validation of clinical, chemical and imaging biomarkers should aid in this field of research. They could be used in clinical assay to track improvement, stabilization, or even progression of OA with the use of new therapeutic agents; to guide patient selection; and to reduce the number of participants needed to treat and strengthen the power of these studies [190].
15.4 Therapeutic Management of Osteoarthritis
The aims of the treatment of OA are to reduce joint pain and stiffness, to maintain and improve joint mobility, to reduce physical disability and handicap, to improve health-related quality of life, to limit the progression of joint damage, and to educate patients about the nature of the disorder and its management [205].
Experts in the field of OA have developed concise, patient-focused, up to date, evidence-based recommendations for the management of hip and knee OA. These recommendations are classified in three fields: nonpharmacological therapies, surgical therapy, and drug therapy. Experts agree that optimal management of OA requires a combination of nonpharmacological and pharmacological modalities (Fig. 15.5) [205].
Figure 15.5 Medical treatment modalities of osteoarthritis. Current treatments of OA include a wide range of nonpharmacological and pharmacological treatment to decrease pain, improve function, and prevent the progression of damage to joint tissues. In each category, the major therapies are listed.

15.4.1 Nonpharmacological Therapy
15.4.1.1 Nonchirurgical Therapy
Regular physical activities are important for patients with knee and hip OA to reduce joint pain and improve functional articular ability. Patients should be encouraged to undertake regular aerobic, muscle strengthening, and range of motion exercises, such as walking and swimming [206]. Moreover, exercises in water can be effective [207]. However, the effect sizes are small to moderate, with no long term benefit [208].
It is well known that obesity is a risk factor for OA (see Section 15.1). Data also indicate that weight reduction and stabilization at a lower level in patients who are overweight may result in reduction in pain and improvement in function of weight-bearing joints [14]. Even a small amount of weight loss may be beneficial in patients with knee OA [209].
Some thermal modalities may be effective for relieving symptoms. Application of superficial heat, cold, or both has been widely employed for pain relief. Less-conventional treatments such as acupuncture and massage are emerging as promising options for OA treatment, but the magnitude of benefit has varied between studies [207].
Finally, patients should be given information access and education about the objectives of treatment and the importance of changes in lifestyle, exercise, pacing of activities, weight reduction, and other measures to unload the damaged joint(s).
15.4.1.2 Surgical Therapy
Surgical interventions in OA can be categorized in the following three groups based on the rationale and/or the main objective of the treatment [210]:
15.4.2 Drug Therapy
15.4.2.1 Analgesic Treatment Modalities
Acetaminophen is the treatment recommended as first-line by experts (recommendation of the European League Against Rheumatism, the American College of Rheumatology, and the Osteoarthritis Research Society International) [206, 215]. Studies demonstrated the efficacy of acetaminophen (3–4 g/day) on pain relief. It could be combined with narcotic analgesics such as codeine. However, hepatic toxicity was reported in patients using acetaminophen greater than 4 g per day or with liver disease or concomitant medications. Acetaminophen may be the most appropriate initial therapy, but if pain persists, then prescription-strength nonsteroidal anti-inflammatory drugs (NSAIDs) or a more specific cyclooxygenase type 2 (COX-2) inhibitor may prove useful [216].
NSAIDs that block prostaglandin synthesis are widely used for treating OA. It includes aspirin, nonselective NSAIDS, and cyclooxygenase-2 inhibitors (coxibs), which selectively block the synthesis of the prostaglandin E2. NSAIDs provide short-term pain relief in patients with OA [217]. However, adverse effects such as gastrointestinal toxicity, congestive heart failure, and renal failure were observed, and these risks increased in frequency in elderly patients. Moreover, two coxibs, rofecoxib and valdecoxib, were withdrawn from the market in 2004 because they were associated with increased incidence of cardiovascular adverse events [218]. Therefore, NSAIDs, which include both nonselective and COX-2 selective agents, should be used with caution in patients in accord with concomitant medications and comorbidities, especially in case of long-term prescription.
Compounds called symptomatic slow-acting disease osteoarthritis (SYSADOA) have been developed for the management of OA. Compared with NSAIDs and acetaminophen, they exhibit lower, delayed, and carryover effects on symptoms. Interestingly, the use of these drugs may associate with less NSAIDs requirement and could have a weak beneficial structural effect. The most frequently used SYSADOA is glucosamine sulfate, either alone or in combination with chondroïtine sulfate. Glucosamine is obtained from shrimp exoskeletons and is a component of many macromolecules, such as hyaluronic acid (an important substance in collagen formation). Chondroïtine sulfate is a macromolecule that contributes to, like hyaluronic acid, a framework for collagen formation. Solid evidence of their effectiveness and safety is available for OA treatment [219, 220]. Although many studies have examined potential mechanisms of action for glucosamine and chondroïtine in OA, the exact nature of these mechanisms remains unclear. They are available commercially as nutritional supplements [221]. Other examples of SYSADOA are diacerein, which may inhibit IL-1β production and reduce cartilage breakdown [222], and avocado/soybean unsaponifiables, which may counteract deleterious processes triggered by IL-1β and mechanical stress [140] and reduce pain and Lesquene index in OA patients [223]. These treatments are purchased as a prescribed drug. The choice of the OA treatment depends on the benefit/risk ratio for the patient in addition to its availability, cost, and patient acceptance. Even if the effect size of acetaminophen is modest, this drug is considered as a safe treatment of OA and remains the first-line oral analgesic in the management of OA [206, 215]. NSAIDs, which are more efficient to improve pain relief, are prescribed in a second time because it is not as good concerning the safety profile. Moreover, classic NSAID and coxib are similar for the symptomatic relief of OA [224]. They should be used at the lowest effective dose for the shortest possible duration of treatment. However, it is not rare that the highest dose is necessary to achieve the clinical goal and sometimes on a chronic use design. The choice between these two treatments depends on comorbidities and patient history [225]. Finally, SYDASOA's global effect size reveals lower efficacy, as glucosamine has shown to provide a low to medium response rate [226] and chondrotine sulfate provides a small response rate.
Topical treatments are of interest especially in patients intolerant or unwilling to take oral analgesics and especially for superficial joints. The most widely used types of topical treatment include capsaicin, topical lidocaine, and topical NSAIDs [227].
Finally, intra-articular injections of corticosteroids and hyaluronans may be useful in patients with OA. Intra-articular corticosteroids have been used for decades as adjunctive therapy, especially when local inflammation is present as indicated by the presence of a synovitis [228]. Intra-articular viscosupplementations of hyaluronic acid, which is a high-molecular-weight polysaccharide that is a major component of synovial fluid and cartilage, assures relief of OA-related pain and improves joint function. Moreover, intra-articular hyaluronan formulations are well tolerated and are associated with a low incidence of adverse effects usually localized to the injected joint [229].
15.4.2.2 Disease-modifying Osteoarthritis Drugs (DMOADs)
DMOADs are a group of therapeutic agents that would, ideally, both control symptoms and provide structure modification. They inhibit one or more OA pathophysiological processes, such as synovial inflammation, cartilage destruction, or subchondral bone remodeling. It is a large family of molecules, the most documented of which are listed in this chapter. The following agents are under clinical investigations for the treatment of OA and are not currently used for treating OA (for review see 230).
IL-1β Blockade
Of the cytokines thought to be involved in the pathogenesis of OA, IL-1β has attracted the most interest as a target for disease modification. Because the IL-1 receptor antagonist (IL-1Ra) anakinra has been used successfully in the treatment of the inflammation and bone destruction of rheumatoid arthritis, some have suggested that it can possibly retard the disease progression of OA. A randomized placebo-controlled study has revealed improvement in knee pain and function with intra-articular injection of anakinra treatment compared with placebo [231]. The clinical benefit of a local injection of IL-1Ra in knee osteoarthritis may be limited by the antagonist's short half-life (for review, see Reference 232).
MMP Inhibitor
Developing effective and specific inhibitors of the catabolic enzymes that suspend cartilage degradation in OA, which include MMP and ADAMTS, has been the focus of several efforts. Specific MMP inhibitors were tested in clinical trials that studied joint destruction occurring in RA, but some trials were terminated because of lack of efficacy or safety concerns. Increased understanding of the structure, regulation, and function of individual MMPs may lead to more effective strategies. Actually, research focused on the development of a novel selective MMP-13 inhibitor [233, 234].
Biphosphonates and Strontium Ranelate
Biphosphonates (risedronate, alendronate, and zodrenate) can suppress bone turnover because of their antiosteoclastic activity. The rationale to evaluate this compound in OA is based on the potential role of subchondral bone in both the occurrence and the progression of OA. Recently, it has been shown that risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or retard radiographic progression in patients with knee OA [235]. The elucidation of risedronate's effects will require additional studies. A recent study has shown in secondary analysis that alendronate could have a structural effect on spinal OA [236]. Strontium ranelate, which is another antiosteoporotic drug that specifically targets the osteoblast, could have also a beneficial effect on spinal OA with a structural and analgesic beneficial effect [237]. However, all these results need additional studies to establish a potent effect in OA.
Treatment that targets NO synthesis by specific inhibitor of the inducible NO synthase and acts on bone remodeling as calcitonin represents other potential treatments in the future. For example, it has been shown that calcitonin reduces Lequesne's algofunctional index scores and decreases urinary and serum levels of biomarkers of joint metabolism, such as type II collagen neoepitopes, MMP-3, and MMP-13, in knee osteoarthritis [238].
15.4.3 Future Directions
Articular cartilage has limited repair and regeneration potential. The scarcity of treatment modalities for large chondral defects has motivated attempts to engineer cartilage tissue constructs with chondrocytes distributed in a three-dimensional structure, which could integrated the resident tissue. Transplantation of autologous chondrocytes has been used to repair focal cartilage defects, but this procedure could lead to variable results with the frequently formation of fibrous cartilage. In experimental studies, several differentiation factors, such as TGF-β, BMPs, or IGF-1, have been shown to stimulate chondrogenesis, which promotes the formation of functionally cartilage-like tissue repair. Therefore, the gene transfer of anabolic factors to promote chondrocytes differentiation as the production of such factors directly at the site of the lesions has been developed. However, the clinical application of such strategies is limited because of the short half-lives of the proteins in vivo [239, 240]. Actually, various approaches are tested in vitro for the use of mesenchymal stem cells (MSCs) to generate tissue-engineered cartilage. MSCs are considered the cell type of choice for tissue engineering because of their multipotence and ability to differentiate into mesenchyme-derived cells, such as osteoblasts and chondrocytes. Moreover, they can be easily isolated from various adult tissues, such as bone marrow, synovium, or adipose tissue, and can be expanded. However, differentiation of MSCs into articular chondrocytes and reparation of injured cartilage are still poorly understood on the molecular level. Therefore, successful regeneration or replacement of damaged or diseased cartilage will depend on future advances in our understanding of the biology of cartilage and stem cells and technological development in engineering [241–243].
References
1. Brandt KD. Osteoarthritis. In: Internal Medicine 4th Edition. Stein J, ed. 1994. St. Louis, MO: Mosby Year Book, Inc.
2. Buckwalter JA, Martin JA. Osteoarthritis. Adv. Drug Deliv. Rev. 2006;58:150–167.
3. Loeser RF. Aging and the etiopathogenesis and treatment of osteoarthritis. Rheum. Dis. Clin. North Am. 2000;26:547–567.
4. Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman BN, Aliabadi P, Levy D. The incidence and natural history of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum. 1995;38:1500–1505.
5. Buckwalter JA, Heckman JD, Petrie DP. AOA. An AOA critical issue: aging of the North American population: new challenges for orthopaedics. J Bone Joint Surg. Am. 2003;85:748–758.
6. Davis MA, Ettinger WH, Neuhaus JM, Mallon KP. Knee osteoarthritis and physical functioning: evidence from the NHANES I Epidemiologic Followup Study. J. Rheumatol. 1991;18:591–598.
7. Felson DT, Naimark A, Anderson J, Kazis L, Castelli W, Meenan RF. The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum. 1987;30:914–918.
8. Aigner T, Haag J, Martin J, Buckwalter J. Osteoarthritis: aging of matrix and cells—going for a remedy. Curr. Drug Targets 2007;8:325–331.
9. Dudhia J. Aggrecan, aging and assembly in articular cartilage. Cell Mol. Life Sci. 2005;62:2241–2256.
10. Verzijl N, Bank RA, TeKoppele JM, DeGroot J. AGEing and osteoarthritis: a different perspective. Curr. Opin. Rheumatol. 2003;15:616–22.
11. Martin JA, Brown TD, Heiner AD, Buckwalter JA. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Relat. Res. 2004; S96–103.
12. Lievense AM, Bierma-Zeinstra SM, Verhagen AP, van Baar ME, Verhaar JA, Koes BW. Influence of obesity on the development of osteoarthritis of the hip: a systematic review. Rheumatology 2002;41:1155–1162.
13. Manek NJ, Hart D, Spector TD, MacGregor AJ. The association of body mass index and osteoarthritis of the knee joint: an examination of genetic and environmental influences. Arthritis Rheum. 2003;48:1024–9.
14. Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF. Obesity and knee osteoarthritis. The Framingham Study. Ann. Intern. Med. 1988;109:18–24.
15. Cicuttini FM, Baker JR, Spector TD. The association of obesity with osteoarthritis of the hand and knee in women: a twin study. J. Rheumatol. 1996;23:1221–6.
16. Haara MM, Manninen P, Kröger H, Arokoski JP, Kärkkäinen A, Knekt P, Aromaa A, Heliövaara M. Osteoarthritis of finger joints in Finns aged 30 or over: prevalence, determinants, and association with mortality. Ann. Rheum. Dis. 2003;62:151–158.
17. Toda Y, Toda T, Takemura S, Wada T, Morimoto T, Ogawa R. Change in body fat, but not body weight or metabolic correlates of obesity, is related to symptomatic relief of obese patients with knee osteoarthritis after a weight control program. J. Rheumatol. 1998;25:2181–2186.
18. Gosset M, Berenbaum F, Salvat C, Sautet A, Pigenet A, Jacques C. Crucial role of Visfatin in matrix degradation and PGE2 synthesis in chondrocytes: possible role in osteoarthritis process. Arthritis Rheum. 2008;58:1399–1409.
19. Toussirot E, Streit G, Wendling D. The contribution of adipose tissue and adipokines to inflammation in joint diseases. Curr. Med. Chem. 2007;14:1095–1100.
20. Aigner T, Sachse A, Gebhard PM, Roach HI. Osteoarthritis: pathobiology-targets and ways for therapeutic intervention. Adv. Drug Deliv. Rev. 2006;58:128–149.
21. Roos EM, Dahlberg L. Positive effects of moderate exercise on glycosaminoglycan content in knee cartilage: a four-month, randomized, controlled trial in patients at risk of osteoarthritis. Arthritis Rheum. 2005;52:3507–3514.
22. Duke PJ, Montufar-Solis D. Exposure to altered gravity affects all stages of endochondral cartilage differentiation. Adv. Space Res. 1999;24:821–827.
23. Maetzel A, Mäkelä M, Hawker G, Bombardier C. Osteoarthritis of the hip and knee and mechanical occupational exposure--a systematic overview of the evidence. J. Rheumatol. 1997;24:1599–1607.
24. Felson DT. Preventing knee and hip osteoarthritis. Bull. Rheum. Dis. 1998;47:1–4.
25. Buckwalter JA, Martin JA. Sports and osteoarthritis. Curr. Opin. Rheumatol. 2004;16:634–639.
26. McGlashan SR, Jensen CG, Poole CA. Localization of extracellular matrix receptors on the chondrocyte primary cilium. J. Histochem. Cytochem. 2006;54:1005–1014.
27. Adams MA. The mechanical environment of chondrocytes in articular cartilage. Biorheology 2006;43:537–545.
28. Smith RL, Carter DR, Schurman DJ. Pressure and shear differentially alter human articular chondrocyte metabolism: a review. Clin. Orthop. Relat. Res. 2004; S89–95.
29. Kim HT, Lo MY, Pillarisetty R. Chondrocyte apoptosis following intraarticular fracture in humans. Osteoarth. Cartil. 2002;10:747–749.
30. Martin JA, Brown T, Heiner A, Buckwalter JA. Post-traumatic osteoarthritis: the role of accelerated chondrocyte senescence. Biorheology 2004;41:479–491.
31. Marsh JL, Buckwalter J, Gelberman R, Dirschl D, Olson S, Brown T, Llinias A. Articular fractures: does an anatomic reduction really change the result? J. Bone Joint Surg. Am. 2002;84:1259–1271.
32. MacGregor AJ, Antoniades L, Matson M, Andrew T, Spector TD. The genetic contribution to radiographic hip osteoarthritis in women: results of a classic twin study. Arthritis Rheum. 2000;43:2410–2416.
33. Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D. Genetic influences on osteoarthritis in women: a twin study. BMJ 1996;312:940–943.
34. Sambrook PN, MacGregor AJ, Spector TD. Genetic influences on cervical and lumbar disc degeneration: a magnetic resonance imaging study in twins. Arthritis Rheum. 1999;42:366–372.
35. Loughlin J. Polymorphism in signal transduction is a major route through which osteoarthritis susceptibility is acting. Curr. Opin. Rheumatol. 2005;17:629–633.
36. Valdes AM, Loughlin J, Oene MV, Chapman K, Surdulescu GL, Doherty M, Spector TD. Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007;56:137–146.
37. Li Y, Xu L, Olsen BR. Lessons from genetic forms of osteoarthritis for the pathogenesis of the disease. Osteoarth. Cartil. 2007;15:1101–1105.
38. Poole AR. Cartilage in health and disease. In Arthritis and Allied Conditions: A Textbook of Rheumatology. 14th Edition. Koopman W, ed. 2001. Philadelphia, PA: Lippincott Williams & Wilkins.
39. Aigner T, Soeder S, Haag J. IL-1beta and BMPs—Interactive players of cartilage matrix degradation and regeneration. Eur. Cell. Mater. 2006;12:49–56.
40. Kühn K, D'Lima DD, Hashimoto S, Lotz M. Cell death in cartilage. Osteoarth. Cartil. 2004;12:1–16.
41. McQuillan DJ, Handley CJ, Campbell MA, Bolis S, Milway VE, Herington AC. Stimulation of proteoglycan biosynthesis by serum and insulin-like growth factor-I in cultured bovine articular cartilage. Biochem. J. 1986;240:423–430.
42. Trippel SB, Corvol MT, Dumontier MF, Rappaport R, Hung HH, Mankin HJ. Effect of somatomedin-C/ insulin-like growth factor I and growth hormone on cultured growth plate and articular chondrocytes. Pediatr. Res. 1989;25:76–82.
43. Luyten FP, Hascall VC, Nissley SP, Morales TI, Reddi AH. Insulin-like growth factors maintain steady state metabolism of proteoglycans in bovine articular cartilage explants. Arch. Biochem. Biophys. 1988;267:416–425.
44. Matsumoto T, Gargosky SE, Iwasaki K, Rosenfeld RG. Identification and characterization of insulin-like growth factors (IGFs), IGF-binding proteins (IGFBPs), and IGFBP proteases in human synovial fluid. J. Clin. Endocrinol. Metab. 1996;81:150–155.
45. Fernihough JK, Billingham ME, Cwyfan-Hughes S, Holly JM. Local disruption of the insulin-like growth factor system in the arthritic joint. Arthritis Rheum. 1996;39:1556–1565.
46. Tavera C, Abribat T, Reboul P, Dore S, Brazeau P, Pelletier JP, Martel-Pelletier J. IGF and IGF-binding protein system in the synovial fluid of osteoarthritic and rheumatoid arthritic patients. Osteoarth. Cartil. 1996;4:263–274.
47. Chevalier X, Tyler JA. Production of binding proteins and role of the insulin-like growth factor I binding protein 3 in human articular cartilage explants. Br. J. Rheumatol. 1996;35:515–522.
48. Morales TI. The insulin-like growth factor binding proteins in uncultured human cartilage: increases in insulin-like growth factor binding protein 3 during osteoarthritis. Arthritis Rheum. 2002;46:2358–2367.
49. Eviatar T, Kauffman H, Maroudas A. Synthesis of insulin-like growth factor binding protein 3 in vitro in human articular cartilage cultures. Arthritis Rheum. 2003;48:410–417.
50. Ballock RT, Heydemann A, Izumi T, Reddi AH. Regulation of the expression of the type-II collagen gene in periosteum-derived cells by three members of the transforming growth factor-beta superfamily. J. Orthop. Res. 1997;15:463–467.
51. Izumi T, Scully SP, Heydemann A, Bolander ME. Transforming growth factor beta 1 stimulates type II collagen expression in cultured periosteum-derived cells. J. Bone Miner. Res. 1992;7:115–121.
52. van Beuningen HM, van der Kraan PM, Arntz OJ, van den Berg WB. In vivo protection against interleukin- 1-induced articular cartilage damage by transforming growth factor-beta 1: Age-related differences. Ann. Rheum. Dis. 1994;53:593–600.
53. van Beuningen HM, van der Kraan PM, Arntz OJ, van den Berg WB. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Invest. 1994;71:279–290.
54. Edwards DR, Murphy G, Reynolds JJ, Whitham SE, Docherty AJ, Angel P, Heath JK. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J. 1987;6:1899–1904.
55. Blaney Davidson EN, Scharstuhl A, Vitters EL, van der Kraan PM, van den Berg WB. Reduced transforming growth factor-beta signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther. 2005;7:R1338–1347.
56. Rountree RB, Schoor M, Chen H, Marks ME, Harley V, Mishina Y, Kingsley DM. BMPreceptor signaling is required for postnatal maintenance of articular cartilage. PloS. Biol. 2004;2:e355.
57. Soder S, Hakimiyan A, Rueger DC, Kuettner KE, Aigner T, Chubinskaya S. Antisense inhibition of osteogenic protein 1 disturbs human articular cartilage integrity. Arthritis Rheum. 2005;52:468–478.
58. Goldring MB. Osteoarthritis and cartilage: the role of cytokines. Curr. Rheumatol. Rep. 2000;2:459–465.
59. Glansbeek HL, van Beuningen HM, Vitters EL, Morris EA, van der Kraan PM, van den Berg WB. Bone morphogenetic protein 2 stimulates articular cartilage proteoglycan synthesis in vivo but does not counteract interleukin-1alpha effects on proteoglycan synthesis and content. Arthritis Rheum 1997;40:1020–1028.
60. Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev 2005;19:2783–2810.
61. Kato Y, Gospodarowicz D. Sulfated proteoglycan synthesis by confluent cultures of rabbit costal chondrocytes grown in the presence of fibroblast growth factor. J. Cell. Biol. 1985;100:477–485.
62. Kato Y, Nomura Y, Daikuhara Y, Nasu N, Tsuji M, Asada A, Suzuki F. Cartilage-derived factor (CDF) I. Stimulation of proteoglycan synthesis in rat and rabbit costal chondrocytes in culture. Exp. Cell Res. 1980;130:73–81.
63. Kato Y, Hiraki Y, Inoue H, Kinoshita M, Yutani Y, Suzuki F. Differential and synergistic actions of somatomedin-like growth factors, fibroblast growth factor and epidermal growth factor in rabbit costal chondrocytes. Eur. J. Biochem. 1983;129:685–690.
64. Jentzsch KD, Wellmitz G, Heder G, Petzold E, Buntrock P, Oehme P. A bovine brain fraction with fibroblast growth factor activity inducing articular cartilage regeneration in vivo. Acta Biol. Med. Ger. 1980;39:967–971.
65. Cuevas P, Burgos J, Baird A. Basic fibroblast growth factor (FGF) promotes cartilage repair in vivo. Biochem. Biophys. Res. Commun. 1988;156:611–618.
66. Jones KL, Addison J. Pituitary fibroblast growth factor as a stimulator of growth in cultured rabbit articular chondrocytes. Endocrinology 1975;97:359–365.
67. Vincent T, Hermansson M, Bolton M, Wait R, Saklatvala J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc. Natl. Acad. Sci. U. S. A. 2002;99:8259–8264.
68. Vincent TL, Hermansson MA, Hansen UN, Amis AA, Saklatvala J. Basic fibroblast growth factor mediates transduction of mechanical signals when articular cartilage is loaded. Arthritis Rheum 2004;50:526–533.
69. Stetler-Stevenson WG, Yu AE. Proteases in invasion: matrix metalloproteinases. Semin Cancer Biol 2001;11:143–152.
70. Murphy G, Knauper V, Atkinson S, Butler G, English W, Hutton M, Stracke J, Clark I. Matrix metalloproteinases in arthritic disease. Arthritis Res 2002;4:S39–S49.
71. Borden P, Solymar D, Sucharczuk A, Lindman B, Cannon P, Heller RA. Cytokine control of interstitial collagenase and collagenase-3 gene expression in human chondrocytes. J. Biol. Chem. 1996;271:23577–23581.
72. Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun Nterminal kinase, and nuclear factor kappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–811.
73. Brinckerhoff CE, Rutter JL, Benbow U. Interstitial collagenases as markers of tumor progression. Clin. Cancer Res. 2000;6:4823–4830.
74. Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997;40:2065–2074.
75. Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF, Hambor JE. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Invest. 1996;97:761–768.
76. Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, DeGennaro LJ. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001;107:35–44.
77. Cal S, Obaya AJ, Llamazares M, Garabaya C, Quesada V, Lopez-Otin C. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene 2002;283:49–62.
78. Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, Wight TN, Leduc R, Apte SS. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem. 2003;278:9503–9513.
79. Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, Liu R, Rosenfeld SA, Copeland RA, Decicco CP, Wynn R, et al. Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science 1999;284:1664–1666.
80. Abbaszade I, Liu RQ, Yang F, Rosenfeld SA, Ross OH, Link JR, Ellis DM, Tortorella MD, Pratta MA, Hollis JM, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J. Biol. Chem. 1999;274:23443–23450.
81. Kuno K, Okada Y, Kawashima H, Nakamura H, Miyasaka M, Ohno H, Matsushima K. ADAMTS-1 cleaves a cartilage proteoglycan, aggrecan. FEBS Lett. 2000;478:241–245.
82. Glasson SS, Askew R, Sheppard B, Carito BA, Blanchet T, Ma HL, Flannery CR, Kanki K, Wang E, Peluso D, et al. Characterization of and osteoarthritis susceptibility in ADAMTS- 4-knockout mice. Arthritis Rheum. 2004;50:2547–2558.
83. Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005;434:644–648.
84. Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005;434:648–652.
85. Henrotin Y, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarth. Cartil. 2003;11:747–755.
86. Sakurai H, Kohsaka H, Liu MF, Higashiyama H, Hirata T, Kanno K Saito I, Miyasaka N. Nitric oxide production and inducible nitric oxide synthase expression in inflammatory arthritides. J. Clin. Invest. 1995;96:2357-2363.
87. Goldring MB. The role of cytokines as inflammatory mediators in osteoarthritis: lessons from animal models. Connect Tissue Res. 1999;40:1–11.
88. Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–594.
89. Fan Z, Bau B, Yang H, Aigner T. IL-1beta induction of IL-6 and LIF in normal articular human chondrocytes involves the ERK, p38 and NFkappaB signaling pathways. Cytokine 2004;28:17–24.
90. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334:297–314.
91. Flannery CR, Little CB, Hughes CE, Curtis CL, Caterson B, Jones SA. IL-6 and its soluble receptor augment aggrecanase-mediated proteoglycan catabolism in articular cartilage. Matrix Biol. 2000;19:549–553.
92. Hardy MM, Seibert K, Manning PT, Currie MG, Woerner BM, Edwards D, Koki A, Tripp CS. Cyclooxygenase 2–dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheum. 2002;46:1789–803.
93. Martel-Pelletier J, Pelletier JP, Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin Arthritis Rheum. 2003;33:155–167.
94. Mathy-Hartert M, Burton S, Deby-Dupont G, Devel P, Reginster JY, Henrotin Y. Influence of oxygen tension on nitric oxide and prostaglandin E2 synthesis by bovine chondrocytes. Osteoarth Cartil 2005;13:74–9.
95. Goldring MB, Suen LF, Yamin R, Lai WF. Regulation of collagen gene expression by prostaglandins and interleukin-1beta in cultured chondrocytes and fibroblasts. Am. J. Ther. 1996;3:9–16.
96. Fulkerson JP, Damiano P. Effect of prostaglandin E2 on adult pig articular cartilage slices in culture. Clin. Orthop. Relat. Res. 1983: 266–269.
97. Lippiello L, Yamamoto K, Robinson D, Mankin HJ. Involvement of prostaglandins from rheumatoid synovium in inhibition of articular cartilage metabolism. Arthritis Rheum. 1978;21:909–917.
98. Clark CA, Schwarz EM, Zhang X, Ziran NM, Drissi H, O'Keefe RJ, Zuscik MJ. Differential regulation of EP receptor isoforms during chondrogenesis and chondrocyte maturation. Biochem. Biophys. Res. Commun. 2005;328:764–776.
99. Miyamoto M, Ito H, Mukai S, Kobayashi T, Yamamoto H, Kobayashi M, Maruyama T, Akiyama H, Nakamura T. Simultaneous stimulation of EP2 and EP4 is essential to the effect of prostaglandin E2 in chondrocyte differentiation. Osteoarth. Cartil. 2003;11:644–652.
100. Aoyama T, Liang B, Okamoto T, Matsusaki T, Nishijo K, Ishibe T, Yasura K, Nagayama S, Nakayama T, Nakamura T, et al. PGE2 signal through EP2 promotes the growth of articular chondrocytes. J. Bone Miner. Res. 2005;20:377–389.
101. Sugimoto Y, Narumiya S. 2007. Prostaglandin E receptors. J. Biol. Chem. 282:11613–11617.
102. Hata, A. N., and R. M. Breyer. 2004. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 103:147–166.
103. Attur M, Al-Mussawir HE, Patel J, Kitay A, Dave M, Palmer G, Pillinger MH, Abramson SB. Prostaglandin E2 exerts catabolic effects in osteoarthritis cartilage: evidence for signaling via the EP4 receptor. J. Immunol. 2008;181:5082–5088.
104. Li X, Ellman M, Muddasani P, Wang JH, Cs-Szabo G, van Wijnen AJ, Im HJ. Prostaglandin E[2] and its cognate EP receptors control human adult articular cartilage homeostasis and are linked to the pathophysiology of osteoarthritis. Arthritis Rheum. 2009; 60:513–523.
105. Clancy RM, Gomez PF, Abramson SB. Nitric oxide sustains nuclear factor kappaB activation in cytokine stimulated chondrocytes. Osteoarth. Cartil. 2004;12:552–558.
106. Lo YY, Conquer JA, Grinstein S, Cruz TF. Interleukin-1 beta induction of c-fos and collagenase expression in articular chondrocytes: involvement of reactive oxygen species. J. Cell Biochem. 1998;69:19–29.
107. Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarth. Cartil. 2003;11:747–755.
108. Mates JM, Perez-Gomez C, Nunez de Castro I. Antioxidant enzymes and human diseases. Clin. Biochem. 1999;32:595–603.
109. Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and OA cartilage: correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–57.
110. Bae SC, Kim SJ, Sung MK. Inadequate antioxidant nutrient intake and altered plasma antioxidant status of rheumatoid arthritis patients. J. Am. Coll. Nutr. 2003;22:311–315.
111. Henrotin Y, Deberg M, Dubuc JE, Quettier E, Christgau S, Reginster JY. Type II collagen peptides for measuring cartilage degradation. Biorheology 2004;41:543–547.
112. Tiku ML, Gupta S, Deshmukh D. Aggrecan degradation in chondrocytes is mediated by reactive oxygen species and protected by antioxidants. Free Radic. Res. 1999;30:395–405.
113. Tiku ML, Shah R, Allison GT. Evidence linking chondrocyte lipid peroxidation to cartilage matrix protein degradation. Possible role in cartilage aging and the pathogenesis of osteoarthritis. J. Biol. Chem. 2000;275:20069–20076.
114. Mendes AF, Caramona MM, Carvalho AP, Lopes MC. Hydrogen peroxide mediates interleukin-1beta-induced AP-1 activation in articular chondrocytes: implications for the regulation of iNOS expression. Cell Biol. Toxicol. 2003;19:203–214.
115. Tiku ML, Allison GT, Naik K, Karry SK. Malondialdehyde oxidation of cartilage collagen by chondrocytes. Osteoarth. Cartil. 2003;11:159–166.
116. Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–2357.
117. Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarth. Cartil. 2005;13:643–654.
118. Afonso V, Champy R, Mitrovic D, Collin P, Lomri A. Reactive oxygen species and superoxide dismutases: role in joint diseases. Joint Bone Spine 2007;74:324–329.
119. Farrell AJ, Blake DR. Nitric oxide. Ann. Rheum. Dis. 1996;55:7–20.
120. Cao M, Westerhausen-Larson A, Niyibizi C, Hebda PA, Stefanovic-Racic M, Evans CH. Nitric oxide inhibits the synthesis of type-II collagen without altering Col2A1mRNA abundance: prolyl hydroxylase as a possible target. Biochem. J. 1997;324:305–310.
121. Taskiran D, Stefanovic-Racic M, Georgescu H, Evans C. Nitric oxide mediates suppression of cartilage proteoglycan synthesis by interleukin-1. Biochem. Biophys. Res. Commun. 1994;200:142–148.
122. Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995;146:75–85.
123. Clancy RM, Amin AR, Abramson SB. The role of nitric oxide in inflammation and immunity. Arthritis Rheum. 1998;41:1141–1151.
124. Sasaki K, Hattori T, Fujisawa T, Takahashi K, Inoue H, Takigawa M. Nitric oxide mediates interleukin-1-induced gene expression of matrix metalloproteinases and basic fibroblast growth factor in cultured rabbit articular chondrocytes. J. Biochem. 1998;123:431–439.
125. Pelletier JP, Mineau F, Ranger P, Tardif G, Martel-Pelletier J. The increased synthesis of inducible nitric oxide inhibits IL-1Ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarth. Cartil. 1996;4:77–84.
126. Abramson SB, Attur M, Amin AR, Clancy R. Nitric oxide and inflammatory mediators in the perpetuation of osteoarthritis. Curr. Rheumatol. Rep. 2001;3:535–541.
127. Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1− and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–2357.
128. Zhao Q, Eberspaecher H, Lefebvre V, De Crombrugghe B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 1997;209:377–386.
129. Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM. Interleukin 1 suppresses expression of cartilage-specific types II and IX collagens and increases types I and III collagens in human chondrocytes. J. Clin. Invest. 1988;82:2026–2037.
130. Lefebvre V, Peeters-Joris C, Vaes G. Modulation by interleukin 1 and tumor necrosis factor alpha of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim. Biophys. Acta 1990;1052:366–378.
131. Lum ZP, Hakala BE, Mort JS, Recklies AD. Modulation of the catabolic effects of interleukin-1 beta on human articular chondrocytes by transforming growth factor-beta. J. Cell Physiol. 1996;166:351–359.
132. Murakami S, Lefebvre V, de Crombrugghe B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha. J. Biol. Chem. 2000;275:3687–3692.
133. Sitcheran R, Cogswell PC, Baldwin AS, Jr. NF-kappaB mediates inhibition of mesenchymal cell differentiation through a posttranscriptional gene silencing mechanism. Genes Dev. 2003;17:2368–2373.
134. Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J. Biol. Chem. 2003;278:2903–2912.
135. Lorenz H, Wenz W, Ivancic M, Steck E, Richter W. Early and stable upregulation of collagen type II, collagen type I and YKL40 expression levels in cartilage during early experimental osteoarthritis occurs independent of joint location and histological grading. Arthritis Res. Ther. 2005;7:R156–R165.
136. Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4:157–164.
137. Lianxu C, Hongti J, Changlong Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarth. Cartil. 2006;14:367–376.
138. Bondeson J, Lauder S, Wainwright S, Amos N, Evans A, Hughes CE, Feldmann M, Caterson B. Adenoviral gene transfer of the endogenous inhibitor IkappaBalpha into human osteoarthritis synovial fibroblasts demonstrates that several matrix metalloproteinases and aggrecanases are nuclear factor-kappaB-dependent. J. Rheumatol. 2007;34:523–533.
139. Chowdhury TT, Salter DM, Bader DL, Lee DA. Signal transduction pathways involving p38 MAPK, JNK, NFkappaB and AP-1 influences the response of chondrocytes cultured in agarose constructs to IL-1beta and dynamic compression. Inflamm. Res. 2008;57:306–313.
140. Gabay O, Gosset M, Levy A, Salvat C, Sanchez C, Pigenet A, Sautet A, Jacques C, Berenbaum F. Stress-induced signaling pathways in hyalin chondrocytes: inhibition by avocado-soybean unsaponifiables (ASU). Osteoarth. Cartil. 2008;16:373–384.
141. Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. Effect of collagen turnover on the accumulation of advanced glycation end products. J. Biol. Chem. 2000;275:39027–39031.
142. Maroudas A, Bayliss MT, Uchitel-Kaushansky N, Schneiderman R, Gilav E. Aggrecan turnover in human articular cartilage: use of aspartic acid racemization as a marker of molecular age. Arch. Biochem. Biophys. 1998;350:61–71.
143. Corvol MT. the chondrocyte: from cell aging to osteoarthritis. Joint Bone Spine 2000;67:557–560.
144. Poole AR, Guilak F, Abramson SB. Etiopathogenesis of osteoarthritis. In: Osteoarthritis: Diagnosis and Medical/Surgical Management. 4th edition. Moskowitz RW, Altman RW, Hochberg MC, Buckwalter JA, Goldberg VM, eds. 2007. Philadelphia, PA: Lippincott, Williams, and Wilkins.
145. Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB. Osteoarthritis cartilage histopathology: grading and staging. Osteoarth. Cartil. 2006;14:13–29.
146. Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–113.
147. Tchetina EV, Squires G, Poole AR. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol. 2005;32:876–886.
148. A.G. Rothwell, G. Bentley, Chondrocyte multiplication in osteoarthritic articular cartilage. J. Bone Jt. Surg. 1973;588–594.
149. A. Hulth, L. Lindberg, H. Telhag, Mitosis in human articular osteoarthritic cartilage, Clin. Orthop. 84; 1972;197–199.
150. Aigner T, Hemmel M, Neureiter D, Gebhard PM, Zeiler G, Kirchner T, McKenna L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001;44:1304–1312.
151. Meachim G, Collins DH. Cell counts of normal and osteoarthritic articular cartilage in relation to the uptake of sulphate (35SO4) in vitro. Ann. Rheum. Dis. 1962;21:45–50.
152. Rothwell AG, Bentley G. Chondrocyte multiplication in osteoarthritic articular cartilage. J. Bone Joint Surg. Br. 1973;55:588–894.
153. Kühn K, D'Lima DD, Hashimoto S, Lotz M. Cell death in cartilage. Osteoarth. Cartil. 2004;12:1–16.
154. D'Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54, 1814–1821.
155. Aigner T, Kim HA, Roach H. Apoptosis in osteoarthritis, Rheum. Dis. Clin. North Am. 2004;30:639–653.
156. Blanco FJ, Guttian R, Va′zquez-Martul E, de Toro FJ, Galdo F. Osteoarthritic chondrocytes die by apoptosis. Arthritis Rheum. 1998;41:284–289.
157. Hashimoto S, Ochs RL, Komiya S, Lotz M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 1998;41:1632–1638.
158. Heraud F, Heraud A, Harmand MF. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000;59:959–965.
159. Aigner T, Hemmel M, Neureiter D, Gebhard PM, Zeiler G, Kirchner T, McKenna, LA. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritic human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage, Arthritis Rheum. 2001;44:1304–1312.
160. Ayral X, Dougados M, Listrat V, Bonvarlet JP, Simonnet J, Amor B. Arthroscopic evaluation of chondropathy in osteoarthritis of the knee. J. Rheumatol. 1996;3:698–706.
161. Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001;44:1237–1247.
162. Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis. 2005;64:1263–1267.
163. Masuhara K, Nakai T, Yamaguchi K, Yamasaki S, Sasaguri Y. Significant increases in serum and plasma concentrations of matrix metalloproteinases 3 and 9 in patients with rapidly destructive osteoarthritis of the hip. Arthritis Rheum. 2002;46:2625–2631.
164. Tchetverikov I, Lohmander LS, Verzijl N, Huizinga TW, TeKoppele JM, Hanemaaijer R, DeGroot J. MMP protein and activity levels in synovial fluid from patients with joint injury, inflammatory arthritis, and osteoarthritis. Ann. Rheum. Dis. 2005;64:694–698.
165. Barksby HE, Milner JM, Patterson AM, Peake NJ, Hui W, Robson T, Lakey R, Middleton J, Cawston TE, Richards CD, et al. Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: implications for cartilage degradation in arthritis. Arthritis Rheum. 2006;54:3244–3253.
166. Hill CL, Gale DG, Chaisson CE, Skinner K, Kazis L, Gale ME, Felson DT. Knee effusions, popliteal cysts, and synovial thickening: association with knee pain in osteoarthritis. J. Rheumatol. 2001;28:1330–1337.
167. Burr DB. Anatomy and physiology of the mineralized tissues: role in the pathogenesis of osteoarthrosis. Osteoarth. Cartil. 2004;12:S20–S30.
168. Lajeunesse D, Hilal G, Pelletier JP, Martel-Pelletier J. Subchondral bone morphological and biochemical alterations in osteoarthritis. Osteoarth. Cartil. 1999;7:321–322.
169. Dequeker J, Mohan S, Finkelman RD, Aerssens J, Baylink DJ. Generalized osteoarthritis associated with increased insulin-like growth factor types I and II and transforming growth factor beta in cortical bone from the iliac crest. Possible mechanism of increased bone density and protection against osteoporosis. Arthritis Rheum. 1993;36:1702–1708.
170. Mansell JP, Bailey AJ. Abnormal cancellous bone collagen metabolism in osteoarthritis. J Clin. Invest. 1998;101:1596–1603.
171. Sanchez C, Deberg MA, Bellahcene A, Castronovo V, Msika P, Delcour JP, Crielaard JM, Henrotin YE. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum. 2008;58:442–455.
172. Sanchez C, Deberg MA, Piccardi N, Msika P, Reginster JY, Henrotin YE. Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarth. Cartil. 2005;13:988–997.
173. Sanchez C, Gabay O, Salvat C, Henrotin Y, Berenbaum F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarth. Cartil. 2009;17:473–481.
174. Westacott CI, Webb GR, Warnock MG, Sims JV, Elson CJ. Alteration of cartilage metabolism by cells from osteoarthritic bone. Arthritis Rheum. 1997;40:1282–1291.
175. van Beuningen HM, van der Kraan PM, Arntz OJ, van den Berg WB. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. /??/Laboratory investigation; a journal of technical methods and pathology. 1994;71:279–290.
176. Ghosh P, Cheras PA. Vascular mechanisms in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2001;15:693–709.
177. Burr DB, Schaffler MB. The involvement of subchondral mineralized tissues in osteoarthrosis: quantitative microscopic evidence. Microsc. Res. Tech. 1997;37:343–357.
178. Walsh DA, Bonnet CS, Turner EL, Wilson D, Situ M, McWilliams DF. Angiogenesis in the synovium and at the osteochondral junction in osteoarthritis. Osteoarth. Cartil. 2007;15:743–751.
179. Bullough PG. The role of joint architecture in the etiology of arthritis. Osteoarth. Cartil. 2004;12:S2–S9.
180. Messent EA, Ward RJ, Tonkin CJ, Buckland-Wright C. Differences in trabecular structure between knees with and without osteoarthritis quantified by macro and standard radiography, respectively. Osteoarth. Cartil. 2006;14:1302–1305.
181. Aigner T, Bartnik E, Sohler F, Zimmer R. Functional genomics of osteoarthritis: on the way to evaluate disease hypotheses. Clin. Orthop. Relat. Res. 2004;427:S138–143.
182. Brown AN, McKinley TO, Bay BK. Trabecular bone strain changes associated with subchondral bone defects of the tibial plateau. J. Orth. Traum. 2002;16:638–643.
183. Carlson CS, Loeser RF, Purser CB, Gardin JF, Jerome CP. Osteoarthritis in cynomolgus macaques. III: effects of age, gender, and subchondral bone thickness on the severity of disease. J. Bone Miner. Res. 1996;11:1209–1217.
184. Wollheim FA, Lohmander LS. Pathology and animal models of osteoarthritis. In: Osteoarthritis. A Companion to Rheumatology. 1st edition. Sharma L, Berenbaum F, eds. 2007. New York: Elsevier.
185. Goldring MB, Goldring SR. Osteoarthritis. J. Cell Physiol. 2007;213:626–634.
186. Glasson SS. In vivo osteoarthritis target validation utilizing genetically-modified mice. Curr. Drug Targets 2007;8:367–376.
187. Clements KM, Price JS, Chambers MG, Visco DM, Poole AR, Mason RM. Gene deletion of either interleukin-1beta, interleukin-1beta-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum. 2003;48:3452–3463.
188. Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, Uchida M, Ogata N, Seichi A, Nakamura K, et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarth. Cartil. 2005;13:632–641.
189. Peterfy CG. Scratching the surface: articular cartilage disorders in the knee. Magn. Reson. Imaging Clin. North Am. 2000;8:409–430.
190. Abramson S, Krasnokutsky S. Biomarkers in osteoarthritis. Bull. NYU Hosp. Jt. Dis. 2006;64:77–81.
191. Blumenkrantz G, Majumdar S. Quantitative magnetic resonance imaging of articular cartilage in osteoarthritis. Eur. Cell Mater. 2007;13:76–86.
192. Burstein D, Gray ML. Is MRI fulfilling its promise for molecular imaging of cartilage in arthritis? Osteoarth. Cartil. 2006;14:1087–1090.
193. Krasnokutsky S, Samuels J, Abramson SB. Osteoarthritis in 2007. Bull. NYU Hosp. Jt. Dis. 2007;65:222–228.
194. Charni-Ben Tabassi N, Garnero P. Monitoring cartilage turnover. Curr. Rheumatol. Rep. 2007;9:16–24.
195. Rousseau JC, Delmas PD. Biological markers in osteoarthritis. Nat. Clin. Pract. Rheumatol. 2007;3:346–356.
196. Deberg M, Dubuc JE, Labasse A, Sanchez C, Quettier E, Bosseloir A, Crielaard JM, Henrotin Y. One-year follow-up of Coll2-1, Coll2-1NO2 and myeloperoxydase serum levels in osteoarthritis patients after hip or knee replacement. Ann. Rheum. Dis. 2008;67:168–174.
197. Henrotin Y, Deberg M, Dubuc JE, Quettier E, Christgau S, Reginster JY. Type II collagen peptides for measuring cartilage degradation. Biorheology 2004;41:543–547.
198. Henrotin Y, Addison S, Kraus V, Deberg M. Type II collagen markers in osteoarthritis: what do they indicate? Curr. Opin. Rheumatol. 2007;19:444–450.
199. Pearle AD, Scanzello CR, George S, Mandl LA, DiCarlo EF, Peterson M, Sculco TP, Crow MK. Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarth. Cartil. 2007;15:516–523.
200. Punzi L, Ramonda R, Oliviero F, Sfriso P, Mussap M, Plebani M, Podswiadek M, Todesco S. Value of C reactive protein in the assessment of erosive osteoarthritis of the hand. Ann. Rheum. Dis. 2005;64:955–957.
201. Davis CR, Karl J, Granell R, Kirwan JR, Fasham J, Johansen J, Garnero P, Sharif M. Can biochemical markers serve as surrogates for imaging in knee osteoarthritis? Arthritis Rheum. 2007;56:4038–4047.
202. Kellgren JH, Lawrence JS, Bier F. Genetic factors in generalized osteo-arthrosis. Ann. Rheum. Dis. 1963;22:237–255.
203. Lohmander LS, Felson D. Can we identify a ‘high risk’ patient profile to determine who will experience rapid progression of osteoarthritis? Osteoarth. Cartil. 2004;12: S49–52.
204. Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: a potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis—results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarth. Cartil. 2005;13:361–367.
205. Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, Bierma-Zeinstra S, Brandt KD, Croft P, Doherty M, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treatment guidelines and systematic review of current research evidence. Osteoarth. Cartil. 2007;15:981–1000.
206. Lee YC, Shmerling RH. The benefit of nonpharmacologic therapy to treat symptomatic osteoarthritis. Curr. Rheumatol. Rep. 2008;10:5–10.
207. Bartels EM, Lund H, Hagen KB, Dagfinrud H, Christensen R, Danneskiold-Samsøe B. Aquatic exercise for the treatment of knee and hip osteoarthritis. Cochrane Database Syst Rev. 2007: CD005523.
208. Pisters MF, Veenhof C, van Meeteren NL, Ostelo RW, de Bakker DH, Schellevis FG, Dekker J. Long-term effectiveness of exercise therapy in patients with osteoarthritis of the hip or knee: a systematic review. Arthritis Rheum. 2007;57:1245–1253.
209. Felson DT, Chaisson CE. Understanding the relationship between body weight and osteoarthritis. Baillieres Clin. Rheumatol. 1997;11:671–681.
210. Segal NA, Buckwalter JA, Amendola A. Other surgical techniques for osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2006;20:155–176.
211. Ayral X. Arthroscopy and joint lavage. Best Pract. Res. Clin. Rheumatol. 2005;19:401–415.
212. Laupattarakasem W, Laopaiboon M, Laupattarakasem P, Sumananont C. Arthroscopic debridement for knee osteoarthritis. Cochrane Database Syst. Rev. 2008: CD005118.
213. Kirkley A, Birmingham TB, Litchfield RB, Giffin JR, Willits KR, Wong CJ, Feagan BG, Donner A, Griffin SH, D'Ascanio LM, et al. A randomized trial of arthroscopic surgery for osteoarthritis of the knee. N. Engl. J. Med. 2008;359:1097–1107.
214. Brouwer RW, Raaij van TM, Bierma-Zeinstra SM, Verhagen AP, Jakma TS, Verhaar JA. Osteotomy for treating knee osteoarthritis. Cochrane Database Syst. Rev. 2007;18: CD004019.
215. Zhang W, Doherty M, Arden N, Bannwarth B, Bijlsma J, Gunther KP, Hauselmann HJ, Herrero-Beaumont G, Jordan K, Kaklamanis P, et al. EULAR evidence based recommendations for the management of hip osteoarthritis: report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann. Rheum. Dis. 2005;64:669–681.
216. Bannwarth B. Acetaminophen or NSAIDs for the treatment of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2006;20:117–129.
217. Bjordal JM, Ljunggren AE, Klovning A, Slørdal L. Non-steroidal anti-inflammatory drugs, including cyclo-oxygenase-2 inhibitors, in osteoarthritic knee pain: meta-analysis of randomised placebo controlled trials. BMJ 2004;329:1317.
218. Scanzello CR, Moskowitz NK, Gibofsky A. The post-NSAID era: what to use now for the pharmacologic treatment of pain and inflammation in osteoarthritis. Curr. Rheumatol. Rep. 2008;10:49–56.
219. Towheed TE, Anastassiades TP, Shea B, Houpt J, Welch V, Hochberg MC. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst. Rev. 2001: CD002946.
220. Monfort J, Martel-Pelletier J, Pelletier JP. Chondroitin sulphate for symptomatic osteoarthritis: critical appraisal of meta-analyses. Curr. Med. Res. Opin. 2008;24:1303–1308.
221. Dougados M. Management of limb osteoarthritis. In: Osteoarthritis. A Companion to Rheumatology. 1st edition. Sharma L, Berenbaum F, eds. 2007. New York: Elsevier.
222. Moldovan F, Pelletier JP, Jolicoeur FC, Cloutier JM, Martel-Pelletier J. Diacerhein and rhein reduce the ICE-induced IL-1beta and IL-18 activation in human osteoarthritic cartilage. Osteoarth. Cartil. 2000;8:186–196.
223. Christensen R, Bartels EM, Altman RD, Astrup A, Bliddal H. Does the hip powder of Rosa canina (rosehip) reduce pain in osteoarthritis patients?—a meta-analysis of randomized controlled trials. Osteoarth. Cartil. 2008;16:965–972.
224. Chen YF, Jobanputra P, Barton P, Bryan S, Fry-Smith A, Harris G, Taylor RS. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib and lumiracoxib) for osteoarthritis and rheumatoid arthritis: a systematic review and economic evaluation. Health Technol. Assess. 2008;12:1–278.
225. Jones R, Rubin G, Berenbaum F, Scheiman J. Gastrointestinal and cardiovascular risks of nonsteroidal anti-inflammatory drugs. Am. J. Med. 2008;121:464–474.
226. Towheed TE, Maxwell L, Anastassiades TP, Shea B, Houpt J, Robinson V, Hochberg MC, Wells G. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst. Rev. 2005:CD002946.
227. Barron MC, Rubin BR. Managing osteoarthritic knee pain. J. Am. Osteopath. Assoc. 2007;107: ES21–7.
228. Palacios LC, Jones WY, Mayo HG, Malaty W. Clinical inquiries. Do steroid injections help with osteoarthritis of the knee? J. Fam. Pract. 2004;53:921–922.
229. Waddell DD. Viscosupplementation with hyaluronans for osteoarthritis of the knee: clinical efficacy and economic implications. Drugs Aging 2007;24:629–642.
230. Pelletier JP, Martel-Pelletier J. DMOAD developments: present and future. Bull NYU Hosp. Jt. Dis. 2007;65:242–248.
231. Chevalier X, Giraudeau B, Conrozier T, Marliere J, Kiefer P, Goupille P. Safety study of intraarticular injection of interleukin 1 receptor antagonist in patients with painful knee osteoarthritis: a multicenter study. J. Rheumatol. 2005;32:1317–1323.
232. Iqbal I, Fleischmann R. Treatment of osteoarthritis with anakinra. Curr. Rheumatol. Rep. 2007;9:31–5.
233. Milner JM, Cawston TE. Matrix metalloproteinase knockout studies and the potential use of matrix metalloproteinase inhibitors in the rheumatic diseases. Curr. Drug Targets Inflamm. Allergy 2005;4:363–375.
234. Takaishi H, Kimura T, Dalal S, Okada Y, D'Armiento J. Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr. Pharm. Biotechnol. 2008;9:47–54.
235. Bingham CO 3rd, Buckland-Wright JC, Garnero P, Cohen SB, Dougados M, Adami S, Clauw DJ, Spector TD, Pelletier JP, Raynauld JP, et al. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: results of the two-year multinational knee osteoarthritis structural arthritis study. Arthritis Rheum. 2006;54:3494–3507.
236. Neogi T, Nevitt MC, Ensrud KE, Bauer D, Felson DT. The effect of alendronate on progression of spinal osteophytes and disc-space narrowing. Ann. Rheum. Dis. 2008;67:1427–1430.
237. Bruyere O, Delferriere D, Roux C, Wark JD, Spector T, Devogelaer JP, Brixen K, Adami S, Fechtenbaum J, Kolta S, Reginster JY. Effects of strontium ranelate on spinal osteoarthritis progression. Ann. Rheum. Dis. 2008;67:335–339.
238. Manicourt DH, Azria M, Mindeholm L, Thonar EJ, Devogelaer JP. Oral salmon calcitonin reduces Lequesne's algofunctional index scores and decreases urinary and serum levels of biomarkers of joint metabolism in knee osteoarthritis. Arthritis Rheum. 2006;54:3205–3211.
239. Evans CH, Gouze E, Gouze JN, Robbins PD, Ghivizzani SC. Gene therapeutic approaches-transfer in vivo. Adv. Drug Deliv. Rev. 2006;58:243–258.
240. Kuo CK, Li WJ, Mauck RL, Tuan RS. Cartilage tissue engineering: its potential and uses. Curr. Opin. Rheumatol. 2006;18:64–73.
241. Raghunath J, Salacinski HJ, Sales KM, Butler PE, Seifalian AM. Advancing cartilage tissue engineering: the application of stem cell technology. Curr. Opin. Biotechnol. 2005;16:503–509.
242. Jorgensen C, Djouad F, Bouffi C, Mrugala D, Noël D. Multipotent mesenchymal stromal cells in articular diseases. Best Pract. Res. Clin. Rheumatol. 2008;22:269–284.
243. Chen FH, Rousche KT, Tuan RS. Technology Insight: adult stem cells in cartilage regeneration and tissue engineering. Nat. Clin. Pract. Rheumatol. 2006;2:373–382.
Further Reading
Bulletin of the world Health organization. http://www.who.int/whr/2007/en/index.html.
Sharma L, Berenbaum F. Osteoarthritis. A Companion to Rheumatology. 1st Edition. 2007. New York: Elsevier.