
Chapter 16: Human Immunodeficiency Virus (HIV)
The human immunodeficiency retrovirus (HIV), recognized widely as the causative agent of AIDS, was discovered over 25 years ago and is one of the most highly studied viruses of all time. In the 1980s through the mid 1990s, only a few treatment options were available and contracting this virus usually resulted in death. Since the introduction in the mid-1990s of HAART (Highly Active Anti-Retroviral Therapy), the disease has become manageable for many people who can tolerate the new drugs (1, 2). This review chapter will describe the way that the virus works and will highlight the many points of intervention and the small molecules that have been developed to stop the virus from replicating.
16.1 Introduction
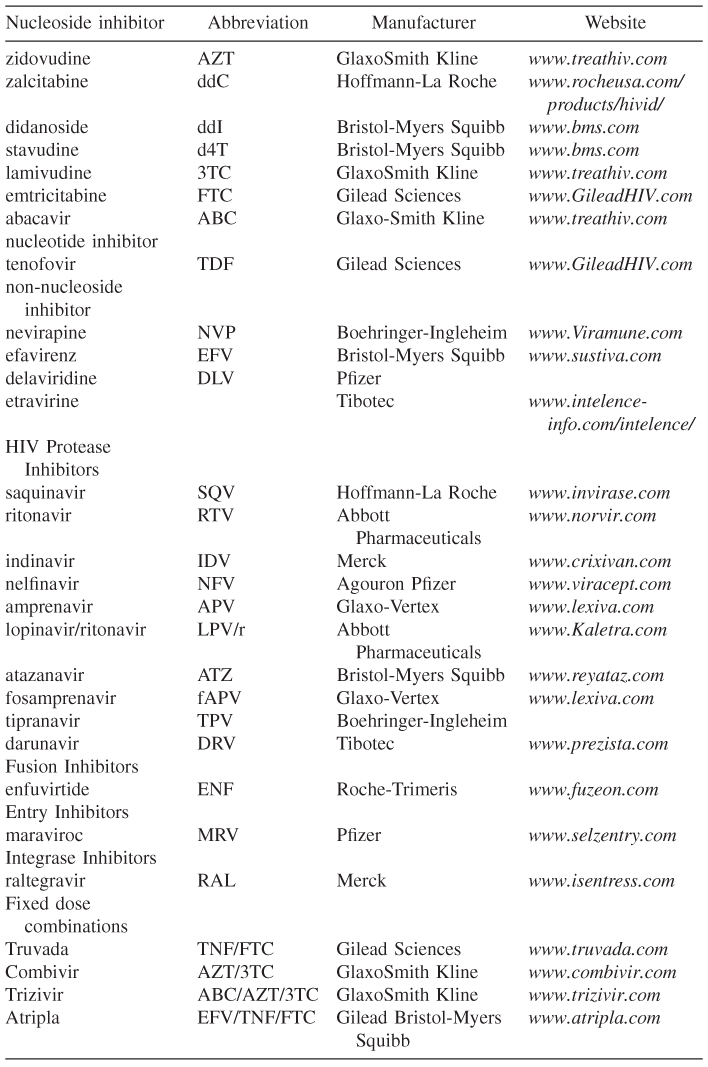
HIV is now widely recognized as the causative agent of acquired immunodeficiency syndrome (AIDS), and it is now estimated that over 30 million people are infected worldwide [3]. The discovery of the virus in 1983 and the ensuing epidemic spurred on the intense study of the mechanism of viral infectivity and how it might be incapacitated [4, 5]. This resulted in an intense effort toward the discovery and development of drugs by academic and industrial laboratories, and now over 24 approved drugs are available as either single agents or fixed-dose combination products (Table 16.1) [6, 7]. The available drugs come from four different classes: entry/fusion inhibitors, reverse transcriptase inhibitors, integrase inhibitors, and protease inhibitors. The virus has a high rate of replication and a low fidelity rate, and this leads to the development of resistance in those patients that do not adequately shut down viral replication. For this reason, all compounds must be used as part of a drug cocktail that consists of at least two different classes of drugs. Using one class of a drug (monotherapy) is never recommended because of the high risk of developing resistance against a drug class. The following chapter will describe the mechanism of viral infection, points of intervention that have been studied and the currently approved agents that have been developed against these targets.
Table 16.1 List of currently approved HIV therapies
16.2 The Mechanism of Viral Infection
HIV is a retrovirus that works by weakening the immune system of its host such as depleting T-helper CD4+ cells. This virus allows opportunistic infections by other pathogens and results in death. It is an enveloped virus that contains a conical-shaped core that contains two strands of RNA and the essential proteins needed for replication. The outside of the virus contains a surface protein gp120 that is anchored to the envelope by a gp41 protein. Both of these proteins exist as a trimeric complex, and when gp120 binds to the CD4 receptor on the surface of a CD4 cell, this unmasks other coreceptors (CCR5 or CXCR4), which also bind gp120. The gp120 unfolds to reveal the gp41 helix, which then fuses with the CD4 cell surface. The fusion inhibitor enfuvirtide acts by interacting with this gp41 protein at a specific site, and this inhibits the fusion process [8–10].
The CD4 receptors CCR5 and CXCR4 also represent a drug target [11]. Cells exist with only a CCR5, or they can also have a dual CCR5 and CXCR4 receptor. A class of compounds that inhibit the binding of gp120 to the CCR5 receptor have been developed, and the first compound, maraviroc, was approved last year. Once the virus particle fuses with the T cell, then its internal core, which contains all of the essential proteins and enzymes that the virus uses for replication, is imported into the hosts' nucleus.
The core's outer proteins are dispersed, and the two strands of viral RNA are transposed into viral DNA by the essential enzyme reverse transcriptase [12]. Two classes of inhibitors work at this step, and they are known as nucleoside and nonnucleoside reverse transcriptase inhibitors (NRTIs and NNRTIs). Once the viral DNA has been made, then the enzyme integrase processes it, imports it into the host cell nucleus, and incorporates it into the host's chromosome. Until recently, no drugs were available targeted for inhibiting integrase, but one compound (raltegravir) has been approved [13]. Subsequent host cell transcription results in the formation of essential viral proteins and viral RNA. The viral proteins are processed by the enzyme HIV protease, and these proteins then aggregate at the host cell surface. Many HIV protease inhibitors have been developed and are available as effective drugs. There is a budding process where the nucleus of the newly formed viral particle is formed and a new particle is expelled from the cell.
This review will now describe in more detail the mechanism of each virus inhibition point and the drugs that have been developed as an intervention.
16.2.1 Fusion Inhibitiors
Enfuvirtide is the only available fusion inhibitor for treatment and is only approved for use in experienced patients [14, 15]. It is a 36-amino-acid peptide and works by inhibiting the fusion of the gp41 protein with the CD4 cell surface. The compound is administered via injection twice per day and is usually added to a patient's current optimized therapy.
16.2.2 CCR5 Inhibitors
As explained, cells can be described by their tropism as either having only the CCR5 or having a mixture of CCR5 and CXCR4 receptors on their surface. The CCR5 receptor has received much more attention as a target because of the discovery that deletion of the CCR5 receptor in some people has led to a natural protection from HIV infection without any deleterious effects to the host. Many compounds have been developed and studied in people as CCR5 antagonists, but the ones that have received the most attention are maraviroc and vicriviroc. Both compounds have been studied in combination cocktails and have been shown to be effective at inhibiting the virus. Maraviroc was approved for use in experienced patients in combination with nucleosides. It is an orally administered compound and is given twice per day.
16.2.3 Reverse Transcriptase Inhibitors
Reverse transcriptase (RT) is an RNA-dependent DNA polymerase and is a 66-amino-acid protein. It catalyzes the production of double-stranded proviral DNA by transcription from viral-RNA. The first inhibitors of HIV were discovered to work against RT and are listed in Table 16.1. They are nucleoside analogs of the natural substrates and work via incorporation into the growing DNA strand. These drugs are prodrugs of the active species and are activated to triphosphates via intracellular kinases. Once converted to triphosphates, they act as chain terminators of the growing DNA strand. Then once incorporated, this leads to the stoppage of viral replication.
Another drug that is now used widely (tenofovir) is known as a nucleotide RT inhibitor (nRTI). This drug works via a similar mechanism as the NRTIs but exists as a mono-phosphonate prodrug. It readily undergoes uptake into the cell and is converted into a triphosphate via cellular kinases. This drug then works via a similar chain terminator mechanism and leads to inhibition of replication [16].
Another class of RT inhibitors are known as nonnucleoside inhibitors (NNRTIs) [17–19]. These inhibitors are not chain terminators but work via binding to an allosteric site on RT. This binding then leads to inhibition of the enzyme that then leads to the stoppage of virus replication. For many years there were three approved NNRTIs, and two of them (efavirenz and nevirapine) were widely used in combination with nucleosides. These resulted in very potent regimens, but the NNRTis had a low barrier to resistance because one amino acid change within the allosteric site could lead to loss of activity. The most prevalent mutations are found with the residues K103N and Y181C. Several new compounds that work against the most prevalent mutations have been discovered and studied in patients whose virus contains the NNRTI resistant single and double mutations. One of these compounds (etravirine) has been approved for use in NNRTI treatment-experienced patients who fail their current regimens. This compound has a good antiviral effect in patients who harbor one or two documented mutations. Many other compounds are currently being studied in patients and should offer an alternative regimen for those who have exhausted other options.
16.2.4 HIV Integrase
HIV integrase is a 288-amino-acid enzyme that consists of three main complexes: a zinc finger region, the catalytic region, and a DNA binding domain [20, 21]. The enzyme works via a metal catalyzed active site mechanism, and the site has three essential amino acids (D64;D121;E154) that are involved in metal coordination. Integrase processes the 5′′ and 3′′ ends of the proviral DNA to form a preintegration complex [22–24]. This complex is then imported into the cell's nucleus and then splices the pro-viral DNA into the host cell's chromosome. Cellular enzymes then repair the gaps that were formed, and this results in a copy of the viral DNA incorporated into the host cell. Transcriptional replication of the cell then leads to production of the newly formed virus particles. Integrase was recognized for many years as a key target for inhibiting replication; however, the search for new drugs was not successful until recently. In early 2000, novel inhibitors of integrase were reported by several laboratories, but the early inhibitors were weak and were not effective in cell culture because of poor passage into the nucleus [25]. Subsequent compounds were developed, and the potency was greatly improved by changing the chemical structure [26]. Two compounds, raltegravir and elvitegravir, have advanced far into human clinical trials and showed good antiviral activity in patients as part of a cocktail regimen. Raltegravir was approved for use in treatment-experienced patients in combination with nucleosides and is given orally twice per day. Several other compounds are also being studied, and this class of inhibitors represents a new treatment option for many experienced patients.
16.2.5 HIV Protease
HIV protease is a 99-amino-acid protein that exists as a homodimer and belongs to the aspartic acid family of enzymes. The protein works by cleaving viral proteins into smaller proteins that then form the new virus particle. Inhibition of the protease leads to the production of noninfectious virus particles [27–29]. The mechanism of inhibition was based on the discovery of inhibitors of other aspartic acid proteases such as pepsin and renin. During the hydrolysis process, a water molecule is added to a peptide bond and this results in a tetrahedral intermediate that then breaks down. Inhibitors have been designed that incorporate nonhydrolyzable versions of these tetrahedral intermediates, and this leads to inhibition of the enzyme. Several compounds were first licensed in 1995 and 1996 (saquinavir, ritonavir, and indinavir), and these were combined with two nucleosides (usually AZT and 3TC or d4T) to produce very potent regimens. The use of these drug cocktails revolutionized therapy for patients, and for the first time, the death rate for AIDS patients was reduced. Although the cocktails were very potent, they also had many side effects related to them, and this limited their use. They also had complicated dosing regimens and were very expensive. This led to nonadherence by many patients, and the prevalence of resistance to the nucleosides and protease inhibitors began to emerge. This led to the discovery and development of subsequent regimens that were easier to take and had fewer side effects.
The main problem with protease inhibitors were that the compounds were complicated to synthesize and were metabolized extensively by the patients' oxidative mechanisms. This led to variable pharmacokinetics and the need to administer the compounds two or three times per day. It was subsequently found that one compound, ritonavir, was a very potent P450 inhibitor and could be administered in a low dose with other protease inhibitors to produce a regimen with high protease compound exposure. Researchers at Abbott Pharmaceuticals were the first to coadminister this agent with a protease inhibitor (lopinavir) to produce a potent regimen [30]. Subsequent clinicians studied the use of ritonavir (RTV) to “boost” other protease inhibitors. This revolutionized the use of PIs and led to the use of lower doses and less frequent administration. The result was that patients experienced lower side effects and were more adherent to their regimens.
One compound approved for exclusive use in patients harboring resistant mutations is darunavir [31]. This compound is potent against wild-type (WT) and resistant forms of the protease and is effective in experienced patients when coadministered with RTV. At the time of writing, this compound is currently being studied in combination with many of the new classes of drugs such as integrase and second-generation NNRTIs.
16.2.6 Resistance
All of the clinically effective drug combinations lose their effectiveness if the patient develops resistance against one or more of the components. HIV replicates quickly and makes many mistakes as it does so. This replication results in the probability of very low levels of resistance virus being harbored in a patient. If the drug regimen is not used properly, then this can lead to insufficient suppression of virus replication. The result is the virus replicates in the presence of suboptimal levels of a drug, and this can lead to the emergence of resistant strains. The common mutations found against the nucleoside inhibitor classes of compounds are found in the reverse transcriptase active site and are indicated with residues such as K55R and L74V. Several mutations have been identified with the use of nonnucleoside RT inhibitors and reside in the allosteric binding site where the inhibitor sits. The most common ones found are K103N, Y181C, V106A, and other various double and triple mutants. Resistance toward HIV protease inhibitors is usually a little hard to achieve, and it is thought that this class has a higher genetic barrier. Nonetheless, many mutations have been identified, usually in combination with each other, and are associated with residues found in the active site and outside of it. It is thought that the nonactive site mutations are developed to increase the enzyme's fitness. Finally, the development of HIV integrase inhibitors is just starting to show patterns of resistance toward the agents, both in clinical use and in laboratory experiments.
16.2.7 Fixed-Dose Combinations
One limitation to taking a three-drug combination is the number of pills that a person has to take. As the drug regimens improved, the frequency of dosing has been reduced that has led to better adherence and a lower incidence of resistance developing. To simplify the regimens even more, many drug manufacturers have started to combine two and even three drugs into one pill. Table 16.1 lists the fixed-dose combination products available at the time of writing with AtriplaTM being the only regimen that contains three drugs in one pill. Several other combinations have been proposed and should be on the market within the next ten years.
16.3 Conclusion
Since the discovery of HIV over 25 years ago, many ways to attack the virus have been studied and the result has been the introduction of five classes of drugs. These compounds can be used in many different combinations and can offer the patient several different options before the same class has to be reused. This offers the patient and their families great hope that this infection can be a manageable disease. This is evident from the lowering of the death rate of patients, and now many are living full and productive lives.
References
1. Vermund SH. Millions of life-years saved with potent antiretroviral drugs in the United States: a celebration, with challenges. J. Infect. Dis. 2006;194:1–5.
2. Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackan BR, Sax PE, Weinstein MC, Freedburg KA. The survival benefits of AIDS treatment in the United States. J. Infect. Dis. 2006;194:11–19.
3. Uniting the World Against AIDS. UNAIDS Epidemic report. Available: www.unaids.org.
4. Gallo RC, Montagnier L. The discovery of HIV as the cause of AIDS. N. Engl. J. Med. 2003;349:2283–2285.
5. Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature, 1985;313:277–284.
6. De Clercq E. The design of drugs for HIV and HCV. Nature Rev. 2007;6:1001–1018.
7. De Clercq E. Antiviral drugs in current clinical use. J. Clin. Vir. 2004;30:115–133.
8. Meanwell NA, Kadow JF. Inhibitors of the entry of HIV into host cells. Curr. Opin. Drug Disc. Dev. 2003;6:451–461.
9. Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 Lymphocytes. Nat. Rev. Drug Disc. 2004;3:215–25.
10. Debnath AK. Novel uses for current and future direct thrombin inhibitors: focus on ximelagatran and bivalirudin. Expert Opin. Invest. Drugs 2006;15:465–478.
11. Biswas P, Nozza S, Scartlatti G, Lazzarin A, Tambussi G. Oral CCR5 inhibitors: will they make it through? Expert Opin. Invest. Drugs 2006;15:451–464.
12. Demeter LM, Doamaoal RA. Structural and biochemical effects of human immunodeficiency virus mutants resistant to non-nucleoside reverse transcriptase inhibitors. Int. J. Biochem. Cell Biol. 2004;36:1735–1751.
13. Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, Isaacs RD. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 2007;369:1261–1269.
14. Buti M, Esteban R. Adefovir Dipivoxil. Drugs of Today 2003;39:127–35.
15. LaBonte J, Lebbos J, Kirkpatrick P. Enfuvirtide. Nature Rev. Drug Disc. 2003; 345–346.
16. Srinivas RV, Robbins BL, Connelly MC, Gong Y-F, Bischofberger N, Fridland A. Metabolism and in vitro antiretroviral activities of bis(pivaloyloxymethyl) prodrugs of acyclic nucleoside phosphonates. Antimicrob. Agents Chemother. 1993;37:2247–2250.
17. Fortin C, Joly V, Yeni P. Emerging reverse transcriptase inhibitors for the treatment of HIV infection in adults. Expert Opin. Emerging Drugs 2006;11:217–230.
18. Tarby CM. Recent advances in the development of next generation non-nucleoside reverse transcriptase inhibitors. Curr. Top. Med. Chem. 2004;4:1045–1057.
19. Zhao Z, et al. Novel indole-3-sulfonamides as potent HIV nonnulceoside reverse transcriptase inhibitors (NNRT's). Bioorg. Med. Chem. Lett. 2008;18:554–559.
20. Anthony NJ. HIV-1 integrase: a target for new AIDS chemotherapeutics. Curr Top. Med Chem. 2004;4:979–990.
21. Ellison V, Brown PO. A stable complex between integrase and viral DNA ends mediates human immunodeficiency virus integration in vitro. Proc. Natl. Acad. Sci. USA. 1994;91:7316–7320.
22. Parrill AL. HIV-1 Integrase inhibition: binding sites, structure activity relationships and future perspectives. Curr. Med. Chem. 2003;10:1811–1824.
23. Pommier Y, Neamati N. Inhibitors of human immunodeficiency virus integrase. Advances in Virus Res. 1999;52:427–458.
24. Hazuda DJ, Felock PJ, Hastings JC, Pramanik B, Wolfe AL. Differential divalent cation requirements uncouple the assembly and catalytic reactions of human immunodeficiency virus type 1 integrase. J. Virol. 1997;71:7005–7011.
25. Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000;287:646–650.
26. Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Paz OG, Hazuda DJ, et al. Discovery of raltegravir a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV AIDS infection. J. Med. Chem. 2008. In-press.
27. Toh H, Ono M, Saigo K, Miyata T. Retroviral protease-like sequence in the yeast transposon ty 1. Nature 1985;315:691.
28. Seelmeier S, Schmidt H, Turk V, von der Helm K. Human immunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proc. Natl. Acad. Sci. USA. 1988;85:6612–6616.
29. Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RAF, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA. 1988;85:4686–4690.
30. Sham HL et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998;42:3218–3223.
31. Ghosh AK, Chapsal BD, Weber IT, Mitsuya. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. H. Acc. Chem. Res. 2008;41:78–86.