
Chapter 1: The Role of Chemical Biology in Drug Discovery
Chemical biology methods are central to the discovery of new medicines. The development of new approaches and techniques has accompanied, and been central to, major advances in understanding of disease biology and a move from pharmacology-driven to molecular-medicine-based drug discovery. This chapter will highlight some critical contributions from chemical biology methods in the context of a high-level overview of drug discovery as practiced currently.
1.1 Introduction
The discovery and development of new medicines is a highly complex, time-consuming, and expensive endeavor that has faced many changes and challenges over the last two decades [1]. On the one hand, the pace of scientific discovery within the biomedical sciences has proceeded at unprecedented levels, fueled by dramatic advances in technology (e.g., Genomics [2] and High Throughput Screening [3, 4]). On the other hand, the development of new medicines has never been more challenging in terms of cost, time, and the regulatory environment [5–8]. However, it is inevitable that societies will strive continually to improve their economic circumstances and improve human health and that discovery and access to medicines will play a critical role. The development and application of chemical biology methods are central to many aspects of the pursuit of medicines and are providing new approaches that may form the basis for a renaissance in the productivity of new medicine discovery in the future.
Drug discovery and development is a lengthy process, typically taking around 15 years from initiation of work on a target to final registration of a pharmaceutical product [9]. As a result, there is a considerable lag between the development and application of new methods and their impact on clinical practice. Consequently, most of currently prescribed drugs were discovered during a completely different era of science and technology [10]. This, coupled with the high level of attrition (i.e., failure at some point in the discovery/development process [5–9]), make it quite difficult to assess directly the impact of approaches applied in early drug discovery on the delivery of new medicines. In this chapter, the importance and potential impact of existing and emerging chemical biology methods is discussed, particularly in reference to their potential to increase the predictability and success rates of molecules in subsequent stages of the drug discovery/development process (e.g., to reduce attrition).
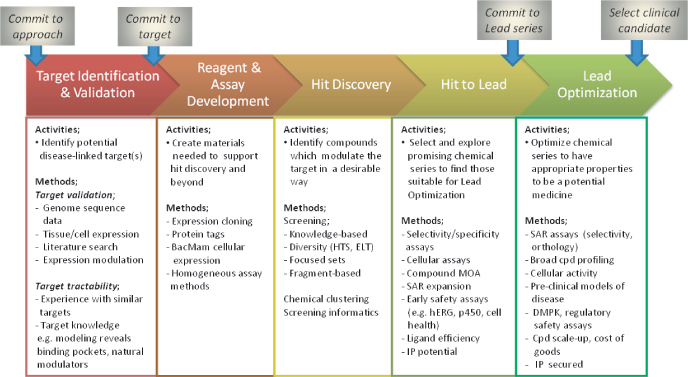
For the purposes of description, it is useful to think of drug discovery as consisting of a series of discrete steps. Figure 1.1 provides a schematic illustration that highlights some key activities underlying each phase. This chapter is organized to outline the drug discovery process from beginning to end, with an attempt to emphasize ways in which chemical biology may improve flow and success rates and, therefore, the overall delivery of new medicines. Obviously, the subject matter here is huge and this cannot provide a comprehensive overview. Many of the references provided are themselves detailed (and current) reviews of specific aspects, which should allow the reader to find more information readily. In addition, subsequent chapters cover specific aspects in much more detail.
Figure 1.1 The major stages of drug discovery. The boxes below each of the stages list some critical activities and methods used at each stage.
1.2 Target Identification and Validation
What is meant by a drug “target” and how are these identified? The generally accepted paradigm for safe and effective medicines is that they cause inhibition or activation of cellular function by binding specifically to one or more discrete protein targets and thus providing appropriate modulation of (patho)physiology. Indeed, the therapeutic targets for most currently marketed drugs, and the role of those targets in disease, is generally well established and involves specific interaction generally with a single protein target [10, 11].
Hence, most drug discovery efforts are initiated around one specific protein (or protein complex). Some exceptions to this approach are described as follows. Two major criteria need to be judged to assess the potential value of a drug discovery target:
If only the first criteria is achieved (i.e., modulation of the target by a small molecule seems unlikely to succeed), biopharmaceutical approaches may be pursued.
Target Validation. There are many ways in which evidence to link a specific gene product to disease may be obtained (for reviews, see References 12–14). Genetic studies, either from patient populations or in experimental systems such as transgenic animal models, often provide data about the pathophysiological consequences of specific proteins being modified (for example, via mutations), absent, or overexpressed [15]. These approaches are often coupled with experiments in cellular systems in which the relevant proteins are modified in similar ways [16, 17]. Often, multiple lines of evidence, each of which is relatively weak on its own, provide sufficient evidence to justify further exploration of the target. In many cases, questions of the role of the putative target can only be answered by the identification of potent and selective compounds that modulate its function (see the subsequent discussion).
Academic work reported in the scientific literature is often a source of targets for organizations that have drug discovery capabilities (i.e., pharmaceutical/biotechnology companies). Interestingly, recent studies have cast doubt on the reliability and reproducibility of this information in some cases, adding to the difficulty in selecting and validating drug targets [18].
Target Tractability. This aspect is concerned entirely with assessment of the likely success (and projected cost and time) of identifying compounds that modulate the target in the desired way and can then be further optimized, ultimately to produce a medicine. Computational methods have been successfully applied to this problem (see subsequent chapters 2 and 3). Tractability assessments are also often based on extrapolation from previous experience with related targets (e.g., from the same protein family). In the absence of any known pharmacophores against the target (or close homologs), judgments about tractability are largely subjective and based on the accrued experience and intuition of an experienced “drug hunter.”
The decoding of the human genome has allowed the potential number of tractable drug targets (independent of their linkage to disease) to be estimated. Expansion of the protein classes known to be targets for existing drugs to include all related genes was used to produce an estimate of the “druggable genome” [19, 20]. This study suggested that around 10% of all proteins encoded by the genome were potentially pharmacologically tractable, suggesting a rich potential source of novel tractable drug targets. For example, the human body contains >500 protein kinases [21], which highlights tremendous opportunities for exploitation of this class of proteins, but also caution in terms of the prospects for finding agents that only interact with a single kinase. In estimating the potential number of therapeutic targets, one must also consider the exploitation of additional new protein classes, for example, those linked to epigenetic processes [22–25]. It seems reasonable to assume that there remain large numbers of biological targets that have not been exploited pharmacologically. Therefore, the major challenges facing biomedical discovery include improving the efficiency of exploration of targets and identifying better ways to triage them in terms of potential disease linkage.
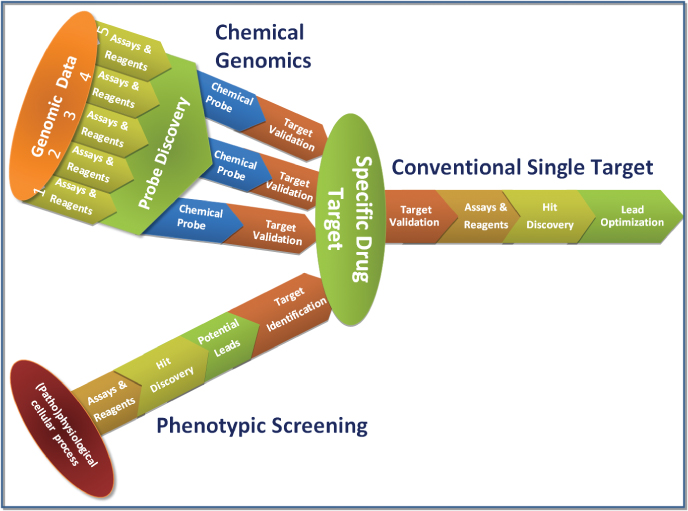
Given the fact there seems to be no shortage of potentially tractable drug targets, but that evidence from genetic/biological/translational experiments is often not as clear as desired, several alternative approaches to the problem of selection of drug targets have been developed. Two of these will be highlighted here: phenotypic screening and chemical genomics. Figure 1.2 shows a schematic comparison of these different approaches to drug discovery targets.
Phenotypic approaches to drug discovery essentially move efforts away from a reductionist focus on a specific molecular target studied in isolation (for example, at the level of a single protein domain), to a more holistic approach in which an intact biological “system” is defined as the target. In most current approaches, the “system” is defined as a population of cells [26, 27]. This strategy has some similarities to how drug discovery was performed before the development of molecular medicine approaches but uses high-tech, cell-based methods instead of primary tissue and animal pharmacology. Proponents of these methods are quick to point to studies that highlight the fact that many first-in-class medicines were discovered using approaches that may broadly be referred to a “phenotypic” [10].
Questions of chemical tractability apply as much to phenotypic approaches as to those directed toward specific protein targets, but they raise a different set of issues. As the precise mechanisms by which compounds will interact to produce the desired phenotype are unknown, at least initially, tractability can be difficult to assess [14]. First, it may be difficult to determine whether compound activities in such assays are sufficiently specific to allow further progression because there are many ways in which they can act. A second, but related issue, is how to ultimately determine the Molecular Mechanism of Action (MMOA) of the compounds. Despite these drawbacks, this approach is attractive on the basis that the pharmacology observed may be more likely to reflect compound effects in vivo than, for example, simple measurements of binding to an isolated protein domain. As most proteins exist in complexes, are expressed in very specific compartments, and are heavily post-translationally modified, there is a strong argument that the more “natural” presentation of the target can result in finding better leads. A corollary is that phenotypic approaches can be prone to nonspecific effects as a result of, for example, low-grade compound toxicity [28, 29].
Chemical genomics takes an almost exactly opposite approach to phenotypic screening. In this case, targets are selected and prosecuted on their basis of putative tractability, with the idea of discovering chemical “probes” (Fig. 1.2; [18–20]). Provided these probes have the appropriate properties (potency, selectivity, and physicochemical properties), they can then be used to understand the role of targets in disease. In some cases, screening is applied to a target or pathway that already has some basis as a drug target in an attempt to find specific probes that will allow further development of that hypothesis. This is in large part the basis for academic screening efforts [30]. Recently, attempts have been made to tackle whole classes of proteins that are currently unexploited pharmacologically, based on the ability to express and purify rapidly large numbers of proteins (structural genomics [31]). Chemical genomics is very much based around the identification of new chemical matter and therefore uses largely the same methods as those adopted for well-validated targets. These are discussed in a subsequent section (see Section 1.4).
Figure 1.2 Three approaches to the pursuit of new medicines. In conventional single target approaches, a specific protein is nominated as the target for therapeutic intervention at the outset and all activities are directed prosecution of that strategy. In chemical genomics, a range of targets is selected for discovery of chemical probes, which further enable target validation. In phenotypic screening approaches, an entire pathway of cellular process is screened, with a view to being able to tackle many potential targets simultaneously, with identification of the target mechanism of compounds occurring after they have been shown to produce a useful effect. Figure partly adapted from Reference 14.
1.3 Reagent and Assay Development
Given the starting point of a defined biological “target(s)” (which could be a specific protein, pathway, or phenotypic cellular response, or even a panel of proteins), the next objective is to identify compounds that interact with the target in a desirable way. Initial efforts to achieve this are centered around the definition, creation, and procurement of the reagents and assays required to support lead discovery efforts. This is not just concerned with primary target and hit identification methods, but also with the critical path activities required to progress such compounds successfully (e.g., understand selectivity versus related targets and orthogonal approaches to confirm initial findings). The nature of the reagents and assays deployed is obviously somewhat dependent on the target. For discrete protein targets (e.g., enzymes), an initial requirement is to clone, express, and purify the relevant protein in an appropriate form. Expression tags that aid in purification are almost universally used (for review, see Reference 32). For soluble protein targets, efforts may be focused even at an early stage on obtaining protein structural information. In some cases, virtually identical strategies may be deployed to produce protein for assays and structural work. Rapid methods for expression and purification of proteins is enabling for both single-target and multi-target “panning” approaches (Fig. 1.2 [33]).
For membrane targets, such as G-protein-coupled receptors (GPCRs) and ion channels, expression within cells is almost always used in conjunction with functional assays (for reviews, see References 34 and 35). Transient expression methods have greatly improved the speed and efficiency of generating appropriate recombinant cellular systems for assays [36]. Phenotypic approaches emphasize the presentation of the target in the most natural setting, and so the cells used are often not recombinantly engineered [27].
In addition to the appropriate biological reagents, assays are required that allow the detection of test compound effects to be determined with appropriate robustness and sensitivity. The development of a range of technologies and accompanying chemical/biochemical reagents to allow screening has been a key enabler for hit identification and profiling in the last 15 years or so (for reviews, see References 37–39). The development of assay methods, linked to specialized detection, automation, and liquid handling instrumentation is a major commercial activity and commercial systems are used by most laboratories, both in industry and academia.
1.4 Lead Discovery
Armed with suitable reagents and assays, a research team is now in a position to test compounds in earnest. However, even the most sensitive and robust screen is only as good as the compounds against which it is tested! Hence, the selection of what to screen is absolutely crucial. Table 1.1 provides a high-level summary of hit identification approaches.
Table 1.1 High-level attributes of various lead discovery methods
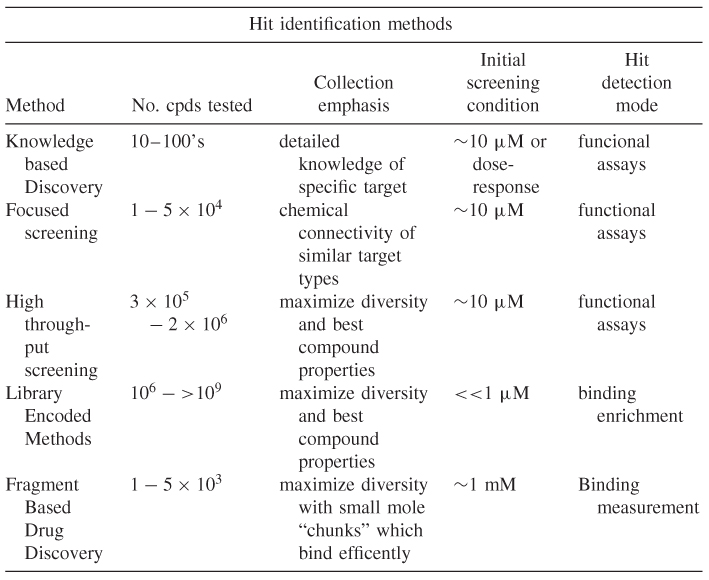
In some cases, sufficient knowledge already exists (for example from literature, knowledge of receptor ligands or substrate catalysis mechanism of an enzyme) to design compounds for testing. These types of approaches may by broadly described as knowledge-based lead discovery. It is worth noting that, prior to the advent of high-throughput methods, this (together with serendipitous observations of pharmacological effects) was the major way in which drug discovery efforts were started. Hence, together with phenotypic approaches, knowledge-based discovery is a major contributor to the origin of currently prescribed medicines [10]. In retrospect, this was greatly assisted by the types of targets pursued. For example, GPCRs, which bind small aminergic ligands such as histamine and serotonin, provided fairly straightforward approaches based on modification of cognate receptor ligands. Knowledge-based approaches still contribute significantly to current drug discovery, but they tap into different data and resources. For example, in the 1990s, virtually all compounds of interest needed to be designed and synthesized, whereas very large numbers of structures (>20M) are now available from a network of chemical suppliers [40]. Hence, electronic search of available chemical space tends to be a first line and economic way to tackle at least initial forays into knowledge-based design.
Given sufficient knowledge of the target (e.g., a high-resolution crystal structure), it is possible to approach the question of which compounds should be selected, designed, and screened entirely using computational methods. These approaches are the subject of subsequent chapters and so not covered in any detail here (see chapters 2 and 3). Such methods often play a crucial role once compounds have been identified from other methods (see the subsequent discussion). In most cases, chemical structures suggested by computational methods require confirmation of their effects in a biological assay. Hence, computational methods to understand and refine compound binding to a drug target tend to be used in conjunction with other approaches.
Another way in which knowledge is applied to lead discovery is what may be broadly referred to as focused screening. In this case, knowledge of the drug target is applied to select which compounds to test. Perhaps the most successful example of this approach is for protein kinases. This large and important class of proteins has a canonical role in cell signaling and thus provides potential targets for intervention in several diseases [41]. Kinases share common structural/functional features (e.g., use of adenosine triphosphate (ATP) to transfer a phosphate group to a protein amino acid residue), which results in considerable “chemical connectivity” between kinase inhibitors and their targets. Therefore, a much higher level of hit enrichment is obtained when testing compounds designed against kinases than, say, a random set of compounds with similar chemical properties. For this reason, a set of ∼10,000 carefully selected kinase compounds can provide a relatively good chance of obtaining potent hits (perhaps equivalent to ∼ 106 randomly selected compounds; a 100-fold enrichment). This approach yields a rich source of starting points for this class of target, but it does have important limitations. As only a relatively narrow set of chemical space is interrogated, novelty (e.g., in compound structure, mechanism of binding, and scope for intellectual property creation) is more difficult to achieve than for other methods. Given that there are several hundred protein kinases in the human body, inhibitor selectivity may be a critical factor to achieve safe and efficacious medicines. It seems reasonable that the same chemical connectivity factors that make focused screening against kinases successful may make it more difficult to achieve high levels of target selectivity.
At least in principle, focused screening approaches can be applied to any target or attribute (e.g., predicted blood–brain penetration for compounds targeted toward neuroscience indications). However, even the best design principles do not guarantee successful hit enrichment, so focused sets should be continually refined based on their actual performance. Personal experience at GlaxoSmithKline suggests that many focused sets do not provide very large hit enrichment over “diverse” (see the subsequent discussion) compounds sets with similar properties (<10-fold).
The most widely adopted method for lead discovery during the last 10 years or so is high throughput screening (HTS). This approach grew out of the development of methods to test large numbers of natural product samples (the first form of “random” screening). Once capabilities were developed to test large numbers of natural product extracts, groups turned their attention to curating and testing internal collection of compounds synthesized in house. The development of this capability was contemporary with an explosion in combinatorial chemistry methods, which in turn drove the building and acquisition of large HTS libraries. Natural-product–based drug discovery is discussed in detail in chapter 4.
At the time of writing, HTS is currently the commonly practiced form of what can be broadly categorized as diversity screening methods. All of these types of methods operate on the same basic principle—which is to test a target against the broadest possible range of appropriate chemical space with the intent of finding novel pharmacophores. In some ways, this approach represents the antithesis of focused screening, in that selection of the set of targets does not take into account the nature of the target (i.e., the same set of compounds is applied to each target, irrespective of other information). However, it is important to emphasize that the diversity screening methods do not ever use “random” selection of compounds. HTS compound sets are often highly curated to maximize the quality of the hits discovered.
This needs to take several factors into account [42], including:
- Chemical properties (e.g., compounds must meet Lipinski, or more stringent, criteria [43])
- Chemical diversity coverage (e.g., compounds are spread in many diversity clusters [44])
- Compound quality (e.g., purity, stability, reactivity, and solubility [42])
- Known hit properties (e.g., promiscuity)
- Cost of acquisition, maintenance, and dispensing compounds (which, along with the cost of screening, places an upper limit on library size)
These factors mean that diversity screening strategies are often driven largely by pragmatic and economic factors and therefore vary considerably in scale. In large pharmaceutical companies, HTS collections typically currently contain 1–2M compounds [3]. Smaller biotech and contract research organizations (CRO) typically use collections in the range of 100–500 K compounds. In most cases, all compounds in the library are tested initially at a single concentration (typically 10 μM), although some groups have proposed that there is additional value in testing the entire library at multiple doses [45]. All other things being equal (although they never are!), the success of diversity screening should increase in direct proportion to the library size, but given the economic limits on the scale of HTS, the design of diversity collections and careful tracking of their performance are critical [44].
In recent years, the value of diversity screening to identify chemical probes has been recognized by academic institutions, with the development of an infrastructure similar to that of the pharmaceutical industry [46, 47]. The most prominent example of this is the NIH-funded Molecular Libraries Screening Center (MLSCN) and Molecular Libraries Production Centers Network (MLPCN) [30]. These centers were established with a mission of providing access to chemical probes to academics investigating putative disease mechanisms and putative intervention approaches, in some ways directly analogous to the early stage activities pursued by pharmaceutical companies. The MLPCN library consists, at the time of writing, of around 350,000 discrete compounds. All data produced from these screens are made publically available [48].
Given the massive numbers of possible chemical structures within “lead-like” chemical space that can be envisioned [49], it seems sensible to assume the success of HTS may be limited by its economic limits of scale. For this reason, methods allowing larger number of compounds to be tested provide the potential to complement, or even replace, HTS. Clearly, a different approach to the problem is required. This has been successfully achieved in some cases where large numbers of compounds are synthesized by using combinatorial chemistry methods employing compound tagging strategies that allow identification of which compounds bind to a target when many are tested simultaneously [50, 51]. Recently, a method that uses DNA-barcode sequences to code for chemistry and binding affinity selection using DNA sequencing methodology was described [52]. This allows the generation and testing of massive libraries (>109 compounds) and is now being exploited as a mainstream hit identification method at GlaxoSmithKline.
Another approach to provide coverage of chemical space is applied in fragment-based drug discovery (FBDD [53–55]. In this method, small “chunks” of molecules (e.g., MW <200) are screened at a relatively high concentration (e.g., 1 mM) to identify compounds that bind weakly but with high ligand efficiency [52]. By using this approach, quite a large coverage of chemical space can be achieved with relatively small numbers of compounds (<10,000). The number of possible drug “fragments” (i.e., <200 Da) is around 10 orders of magnitude smaller than the equivalent value for drug-sized molecules (i.e., <450 Da) [55].
Because fragments bind weakly, screening is usually used in conjunction with biophysical and protein structure methods to provide detailed characterization of binding to confirm that fragments are binding productively and to guide subsequent optimization [54]. The ability to determine bound ligand structures is normally thought to be a prerequisite for effective use of these methods, so they are mainly applied to soluble targets such as enzymes. However, recent reports suggest that, given appropriate engineering of the target proteins, this technique has recently also been applied to GPCRs [56].
Hit Confirmation. Whatever hit identification method is used, a critical next step is to confirm that compounds identified as apparently active in screens are not a result of artifacts [57, 58]. Often a first step is to eliminate false-positive signals by confirming that the initial result can be reproduced under the same conditions. This is then normally followed by dose response testing, to produce a candidate list of preliminary hits. At this point, the structures of hits are typically examined to determine whether preliminary patterns of structure activity relationships (SAR) are present (e.g., multiple hits occur within related chemical families).
Given a list of preliminary hits, a critical next step is to identify those that have promise for further exploration. Several factors are typically taken into account in this phase. First, it is important to determine which classes of compound bind to the target in a productive manner (e.g., with defined stoichiometry and mechanism). Several “nuisance” mechanisms exist that can result in apparent activity in assays but where the compounds are not progressable (e.g. 66, 67). For biochemical screening approaches, biophysical methods such as surface plasmon resonance and isothermal calorimetry of nuclear magnetic resonance may be useful in determining binding specificity, kinetics, and stoichiometry [59].
For cellular approaches, demonstration that positive compounds interact with the target is more difficult because direct methods to measure binding cannot generally be used. Hit confirmation for these targets is usually concerned with identifying those compounds that act in a specific manner without causing cellular toxicity. Specificity is usually tested by counter screening hits against a closely related system in which the signal is not dependent on the target. In the case of phenotypic approaches where, by definition, useful hits are not restricted to activity at a single specific target, the definition of specific hit activity may represent the starting point for experiments to elucidate compound MMOA. Proteomic methods, such as stable-isotope labeling by amino acids in cell culture (SILAC) have proved extremely powerful as a means to profile the interaction targets for compounds [60, 61].
Another important step in hit confirmation is to check that the observed activity is actually caused by the specific chemical structure of interest because activity in assays can occur as a result of compound breakdown products or minor contaminants. For this reason, putative hits are often confirmed by using a fresh sample of compound or, in some cases, via resynthesis of compounds of interest.
1.5 Hit to Lead/Probe
Once activity in a screening assay has been confirmed and shown to be appropriately specific, potential compound series are selected for further characterization and expansion. The following types of characteristics may be used:
- The number of chemically related compounds identified, which can give initial evidence for the emergence of SAR
- Good affinity and/or ligand efficiency for the target [62, 63]
- Good selectivity (i.e., relative potency for the target versus related proteins)
- Productive binding mechanism (e.g., reversible, substrate competitive binders)
- Good chemical properties (e.g., MW <400, cLogP <4) and solubility
- Good chemical tractability (i.e., the ability to procure and/or synthesize analogs)
- Intellectual property (i.e., scope to generate intellectual property from subsequent optimization)
- Identification of liabilities that may affect subsequent progression (e.g., inhibiting P450 enzymes, drug transporters, and cardiac ion channels)
As a result of this triage, a small number (<10) of potential compound series is typically selected for further characterization and expansion. This normally starts with an exhaustive search of available analogs [40] coupled to some limited exploratory synthesis. The expansion of potential chemical lead series is also accompanied by further characterization of the mechanism and specificity of interaction with the target. For phenotypic approaches, this is the point at which experiments to determine the binding target often need to have produced clear results for the effort to be progressed further [14]. In the case of chemical genomic approaches, compounds will have now met the criteria for use in further biological experiments to investigate the role of the target(s)—i.e., they are completed “chemical probes” (e.g., see References 64–67). For mainstream drug discovery programs, this phase of activity culminates in lead series, which meet all of the desired criteria for further optimization and thus allow the initiation of lead optimization and the investment of chemistry resources.
1.6 Lead Optimization
According to a recent SBS/IUPAC glossary of terms [68], a lead is defined as “compound (or compound series) that satisfies predefined minimum criteria for further structure and activity optimization.” In the general case, this refers to a series of compounds for which there is already demonstrated SAR against the target of interest, scope for further modification, and at least the potential to create new intellectual property. These attributes must be combined with appropriate biological (i.e., potency, specificity, and selectivity) and physicochemical properties to provide a useful prospect for further optimization to a clinical candidate.
Lead optimization processes are discussed in detail in chapter 4. Fundamentally, this stage consists of iterative cycles of optimization of chemistry, each of which is associated with the measurement of particular aspects of the biological properties of compounds that aim to move continually closer to the selection of a single compound as a clinical candidate. Lead optimization is characterized by a complex balancing of a number of critical factors, some of which are listed in Table 1.2. The precise challenges presented in any particular lead optimization campaign are to some extent unique to that target; therefore, a highly flexible approach is required. Given the high levels of attrition that occur during drug clinical development [5–7], the identification of issues that may cause problems in development as early as possible is a key area of focus.
Table 1.2 Some key properties of lead that are optimized on the pathway from during lead optimization to produce clinical candidates
| Optimization property | Rationale | Measured how? |
| Potency and ligand efficiency against primary target(s) | Ensure optimal engagement of therapeutic target and balance of physical properties | Dose response profiling, biophysical assays, calculated properties |
| Selectivity for primary target(s) verus related biological systems | Minimize off-target effects, which could result in toxicity | As above |
| Activity in predictive models of disease | Increase confidence in therapeutic target approach and predict potential clinical dose | cellular and animal models |
| Potency in animal orthologues | Asses predictvity of pre-clinical studies for efficacy and safety | As above |
| Specificity relative to broad panels of pharmacologically relevant targets | Minimize off-target effects, which could result in toxicity | Broad profiling of compounds against panels of targets |
| Compound physico-chemical properties (solubility, lipophilicity etc.) | Good properties required for ADME | Some simple properties can be calculated, other are physically measured |
| ADME/Pharmacokinetic properties | Optimize bioavailability and subsequent pharmacokinetics | In vitro models, animal DMPK |
| Early safety assessment | Identify potential drug safety issues as early as possible | In vitro models (e.g. hERG channel assays, cell health measurements, AMES/genotox testing) |
| Intellectual Property generation/protection | Protect value of discovery | patent literature and authorship |
Optimization of a lead series to the point of nomination of a clinical candidate typically takes 1–3 years of work and involves around 1000 compounds made over perhaps 30–100 iterations of chemistry optimization. One critical factor is therefore supplying data to support the next round of chemical synthesis as rapidly as possible for the characterization of previous SAR to be built into subsequent chemistry plans (chapter 4). Subsequent chapters discuss other critical factors that need to be taken into account during lead optimization, such as ADME and pharmacokinetic properties (chapters 5 and 6), drug transport processes (chapter 7), and blood–brain barrier penetration (chapter 8). Several recent studies have pointed out the importance of focusing effort during this phase not only on biological data but also on compound physicochemical properties (for reviews, see References 69 and 70).
Naturally, the major focus in lead optimization is to find a molecule that has good potential to be useful therapeutically in terms of safety and efficacy. Loss of a drug candidate during clinical development can be highly costly and disruptive commercially [8], so an absolutely key activity during lead optimization is to attempt to minimize the risk of subsequent termination. Excluding commercial and strategic considerations, there are only two ways in which a drug candidate can fail: either inadequate effectiveness or safety concerns. Therefore, as compounds are optimized to improve their overall properties, more predictive information is sought around how target modulation by those molecules could translate into useful disease modification in the clinic [71]. Inevitably, this is often linked to consideration of the potential commercial environment for such a medicine (e.g., what is the size of the unmet medical need? how does this medicine compare with others currently on the market or in development, and what end-points will the medicine need to achieve to add value and therefore maximize likelihood of reimbursement?).
The drug discovery process essentially ends at the point of identification of a clinical candidate. This is the nomination of a specific compound that will be the subject of drug development processes, which is outside of the scope of this chapter.
1.7 Conclusions
Chemical biology methods play a central role in drug discovery, particularly as it is practiced currently. In addition, new and established methods are allowing further exploration of interactions between compounds and biological processes at an unprecedented scale and quality. It is too soon to tell in most cases how current methods will contribute to the rate of successful development of new medicines. However, the combination of current and future (e.g., nano-engineering) technologies together with careful analysis of the lessons from the past seems poised to drive many new advances in the most noble of causes, that of improving our understanding, treatment, and prevention of disease.
References
1. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004;3:711–715.
2. Lander ES. Initial impact of the sequencing of the human genome. Nature 2011;470:187–197.
3. Macarron R, Banks NM, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DVS, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam SG. Impact of high throughput screening on biomedical research. Nat. Rev. Drug Discov. 2009;10:188–195.
4. Mayr LM, Fuerst P (2008) The future of high-throughput screening. J. Biomol. Scr. 2008;13:443.
5. DiMasi JA, Feldman L, Seckler A, Wilson A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Therapeut. 2010;87(3):272–277.
6. Arrowsmith J. Phase III and submission failures: 2007–2010. Nat. Rev. Drug Discov. 2011;10:1.
7. Paul SM, Mytelka, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: The pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 2010;9:203–214.
8. Morgan S, Grootendorst P, Lexchin J, Cunningham C, Greyson D. The cost of drug development: A systematic review. Health Policy 2011;100:4–17.
9. Dickson M, Gagnon JP. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004;3:417–429.
10. Swinney DC, Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011;10(7):507–512.
11. Inning P, Sining C, Meyer A. Drugs, their targets and the nature and number of drugs. Nat. Rev. Drug Discov. 2006;5:821–834.
12. Chan JNY, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol. Sci. 2010;31(2):82–88.
13. Rask-Anderson M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011;10:579–590.
14. Tertappen GC, Schluipen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 2007;6:891–903.
15. Hardy J, Singleton A. Genomewide association studies and human disease. N. Engl. J Med. 2009;360:1759–1768.
16. Prokop A, Michelson S. Springer Briefs in Pharmaceutical Science & Drug Development. 2m 69–76. 2012. Springer, New York.
17. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67.
18. Prinz F, Schlange T, Asadullah. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov 2011;10:712.
19. Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug Discov 2002;1:727–730.
20. Russ AP, Lampel S. The druggable genome: An update. Drug Discov. Today 2005;10(23–24):1607–1610.
21. Manning G, Whyte DB, Martinez R, Hunter T, Sudersanam S. The protein kinase complement of the human genome. Science 2002;298(5600):1912–1934.
22. Heightman T, Pope AJ. Epigenetic drug discovery. J. Biomol. Scr. 2011;16:1135–1136.
23. Lu Q, Quinn AM, Patel MP, Semus SF, Graves AP, Bandyopadhyay D, Pope AJ, Thrall SH. Perspectives on the discovery of small-molecule modulators for epigenetic processes. J. Biomol. Scr. 2012;17(5):1–17.
24. Carey N. Epigenetics—an emerging and highly promising source of new drug targets. Med. Chem. Commun. 2012;3:162–166.
25. Sippl W, Jung Manfred M, Eds. Epigenetic Targets in Drug Discovery. Methods and Principles in Medicinal Chemistry. Vol. 42. Wiley-VCH, New York, 2009.
26. Frank A, Tolliday N. Cell-based assays for high-throughput screening. Mol. Biotechnol. 2010;45(2):180–186.
27. Lee JA, et al. Open innovation for phenotypic drug discovery. J. Biomol. Scr. 2011;16(6):588–602.
28. Shukla SJ, Huang R, Austin CP, Xia M. The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today 2010;15(23–24):997–1007.
29. Thorne N, Auld DS, Inglese J. Apparent activity in high-throughput screening: Origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 2010;14(3):315–324.
31. Weigelt J. Structural genomics—impact on biomedicine and drug discovery. Exp. Cell Res. 2010;316(8):1332–1338.
32. Koehn J, Hunt I. High throughput protein production (HTTP): A review of enabling technologies to expedite protein production. Methods Mol. Biol. 2009;498:1–18.
33. Hunt I. From gene to protein: A review of new an enabling technologies for multi-parallel protein expression. Protein Expr. Purif. 2005;40(1):1–22.
34. Wang D, Li Y, Zhang Y, Liu Y, Shi G. High throughput screening (HTS) in identification new ligands and drugable targets of G protein-coupled receptors (GPCRs) combinatorial chemistry & high throughput screening. Comb. Chem. High T. Scr. 2012;15(3):232–242.
35. Terstappen GC, Roncarati R, Dunlop, Peri R. Screening technologies for ion channel drug discovery. Future Med. Chem. 2010;2(5):715–730.
36. Kost A, Condrea P, Ames R. Baculovirus gene delivery: A flexible assay development tool. Curr. Gene Ther. 2010;10(3):168–173.
37. Inglese J. A Practical Guide to Assay Development and High Throughput Screening in Drug Discovery. 2010. CRC Press, Boca Raton, FL.
38. An WE, Tolliday N. Cell-based assays for high throughput screening. Mol. Biotechnol. 2010;45(2):180–186.
39. Pope AJ, Haupts UM, Moore KJ. Homogeneous fluorescence readouts for miniaturized high-throughput screening: Theory and practice. Drug Discov. Today. 1999;4(8):350–362.
40. http://www.emolecules.com/.
41. Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009;21(2):317–324.
42. Wigglesworth M, Wood T, eds. Management of Chemical and Biological Samples for Screening Applications. 2012. Wiley-VCH, New York.
43. Lipinski C. Lead and drug-like compounds: The rule of five revolution. Drug Discov. Today: Technologies 2004;1(4):337–341.
44. Harper G, Pickett SD, Green DVS. (2004) Design of a compound screening collection for use in high throughput screening. Comb. Chem. High T. Scr. 2004;7(1):63–70.
45. Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11473–11478.
46. Dove A. High throughput screening goes to school. Nat. Methods 2007;4(6):523–532.
47. Frearson JA, Collie IT. HTS and hit finding in academia—from chemical genomics to drug discovery. Drug Discov. Today 2009;14:1150–1158.
48. http://www.ncbi.nlm.nih.gov/pcassay.
49. Medina-Franco JL, et al. Visualization of chemical space in drug discovery. Curr. Comput. Aided Drug Design 2008;4:322–333.
50. Baldwin, JJ, Horlbeck, EG. U.S. Patent 5 663 046, 1997.
51. Rokosz LL, Huang CY, Reader JC, Stauffer TM, Southwick EC, Li G, Chelsky D, Baldwin JJ. Exploring structure-activity relationships of tricyclic farnesyltransferase inhibitors using ECLiPS libraries. Comb. Chem. High T. Scr. 2006;9:545–558.
52. Clark MA, Acharya RA, Arico-Muendel CC, Belyanskaya SL, Benjamin DR, Carlson NR, Centrella PA, Chiu CH, Creaser SP, Cuozzo JW, Davie CP, Ding Y, Franklin GJ, Franzen KD, Gefter ML, Hale SP, Hansen NJV, Israel DI, Jiang J, Kavarana MJ, Kelley MS, Kollmann CS, Li F, Lind K, Mataruse S, Medeiros PF, Messer JA, Myers P, O'Keefe H, Oliff MC, Rise CE, Satz AL, Skinner SR, Svendsen JL, Tang L, Vloten KV, Wagner RW, Yao G, Zhao B, Morgan BA. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 2009;5(9):647–654.
53. Bembenek SD, Tounge BA, Reynolds CH. Ligand efficiency and fragment-based drug discovery. Drug Discov. Today 2009;14(5/6):278–283.
54. Edfeldt FNB, Folmer RHA, Breeze AL. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov. Today 2011;16(7/8):284–287.
55. Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat. Chem. 2009;1:187–192.
56. Congreve M, Rich R, Myszka DG, Figaroa, Siegal G, Marshall F. Fragment screening of stabilized g-protein-coupled receptors using biophysical methods. Methods Enzymol. 2011;495:115–136.
57. Johnson P. Redox cycling compounds produce H2O2 in HTS buffers containing strong reducing agents—real hits of promiscuous artefacts. Curr. Opin. Chem. Biol. 2011;15(1):174–182.
58. Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005;1(2):146–148.
59. Lundquvist T. The devil is still in the details—driving early drug discovery forward with biophysical experimental methods. Curr. Opin Drug Discov. Develop. 2005;8(4):513–519.
60. Drewes G. Chemical proteomics in drug discovery. Chemical proteomics: Methods and protocols. Methods Mol Biol 2012;803:15–21.
61. Ong SE, Mann M. Stable isotope labelling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 2007;359:37–52.
62. Perola E. 2010. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J. Med. Chem. 2010;53:2986–2997.
63. Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005;10(7):464–469.
64. Cong F, Cheung AK, Huang SM. Chemical genetics-based target identification in drug discovery. Annu. Rev. Pharmcol. Toxicol. 2012;52:57–78.
65. Rix U, Superti-Furga. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009;5:616–624.
66. Edwards AE, Bountra C, Kerr DJ, Wilson TM. Open access chemical and clinical probes to support drug discovery. Nat. Chem. Biol. 2009;5:436–440.
67. Hauser AT, Jung M. Chemical probes: Sharpen your epigenetic tools. Nat. Chem. Biol. 2011;7:499–500.
68. http://www.slas.org/education/glossary.cfm.
69. Meanwell NA. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011;24:1420–1456.
70. Waring MJ. Lipophilicity in drug discovery. Expert. Opin. Drug Discov. 2010;5(3):235–248.
71. Littman BH, Krishma R. Translational Medicine and Drug Discovery. 2011. Cambridge University Press, New York.