
4
Multivalency in Biosystems
Jens Dernedde
Institute of Laboratory Medicine, Clinical Chemistry, and Pathobiochemistry, Charité–Universitätsmedizin Berlin, 13353, Berlin, Germany
4.1 Introduction
Multivalency is the action of multiple recognition of the same type occurring simultaneously between two entities. This concept is a common principle to increase the affinity and specificity of ligand–receptor interactions in nature and results in a cooperative, over‐additive enhancement of binding affinity [1,2]. In biosystems mostly low affinity binders are clustered to achieve the required binding strength. From the evolutionary point of view, it seems to be convenient to merge multiple single low affinity interactions to generate a collectively stronger output. This strategy avoids the tedious de novo protein design and resorts to already existing interaction networks. Furthermore, a tunable multivalent receptor–ligand interaction allows for a graded response to biological signals.
Based on pioneering work from the disciplines of chemistry and biochemistry reviewed by Mammen et al. [1] and Fasting et al. [3] great progress has been achieved to describe multivalent effects in recent years, mainly from the perspective of synthetic chemistry. Nevertheless, our current understanding is still fragmentary. Novel concepts include highly interdisciplinary research to unravel the impact of multivalent molecular recognition in biological systems and to comprehend diverse complex tasks such as molecular assembly, cell–cell adhesion, and signal transduction. Although a reductionistic approach is first necessary to prove multivalent effects at the molecular level in vitro, analyses of more complex biosystems remain so far elusive. Experimental setups that meet the natural environment are therefore required.
A fascinating feature of living cells is the precise control over biological activity in space and time. The cell as a functional unit relies on such high‐fidelity spatiotemporal control of molecular interactions, wherein thousands of different components, namely lipids and proteins, self‐assemble [4]. A driving force that contributes to the recruitment of molecules and therefore to cellular organization is multivalency. As an example, the cell boundary layer, the cytoplasmic membrane, is assembled into a two‐dimensional fluid architecture and lipids and proteins seem to be heterogeneously distributed. In response to internal or external stimuli that lead to membrane protein cross‐linking, proteins can be segregated into nanometer‐sized domains referred to as lipid rafts. This clustering of biomolecules is highly dynamic and can also rapidly disassemble [5]. Inside the cell preorganization of functional units is realized by compartmentation. Membrane surrounded organelles like the nucleus, endoplasmic reticulum, and Golgi complex define functional units to perform special tasks; however, vesicles also involved in transport, storage, and degradation constitute individual subcellular reaction centers with limited diffusion and thereby foster protein–protein interaction. Recently, it was shown that also in the crowded cytoplasm areas of higher organization can develop due to phase separation, a phenomenon induced by multivalent interactions of proteins [6]. The principles that underlie control over the molecular composition of the cellular microenvironment in space and time are therefore a subject of great scientific interest as they are of essential importance for proper cell function.
4.2 Cell–Cell Adhesion
Intercellular adhesion is probably the best‐studied example of multivalency in biosystems. Cell–cell adhesion can be classified by different scenarios (Figure 4.1) to realize multivalent binding. The connection of cells from the same type in tissues is mentioned as a homotypic interaction of proteins; in contrast, a heterotypic interaction connects cells from different origin (Figure 4.1a). A homophilic interaction is caused by attraction forces between identical molecules, whereas different molecules are joined in a heterophilic manner (Figure 4.1b). Trans‐binding connects facing cell surfaces, cis‐binding connects proteins that reside on the same cell surface (Figure 4.1c), whether they cluster in a homophilic or heterophilic manner. Further, molecular interaction can be direct or bridged by additional adaptor proteins inside or outside the cell (Figure 4.1d).

Figure 4.1 Types of cell–cell adhesion and interaction of cell adhesion molecules. (a) Hetero‐ and homotypic interaction. (b–d) show the extracellular adhesion of transmembrane proteins of two adjacent cells. (b) Homophilic and heterophilic binding. (c) Trans‐ and cis‐binding. (d) Direct and bridged interaction.
4.2.1 Homotypic Interactions, Cadherins Keep Cells Together
Cell adhesion molecules that mediate lateral cell–cell contact in epithelia are organized within adhesion complexes. Individual protein clusters are segregated to defined areas and constitute tight junctions, adherens junctions, desmosomes, and gap junctions to bridge the intercellular space. This overall spatial organization leads to a polarization of epithelial cells into an apical and basal side.
Multivalent interactions at adherens junctions have been intensively studied [7–10]. The single pass transmembrane cadherin connects at adherens junctions neighboring cells by homophilic trans‐binding (Figure 4.2). Essential for adhesive function is the stabilization of the five ectodomains (ECs) of classical cadherins by Ca2+ ions. Calcium binding sites are located between the EC domains [11,12]. Calcium binding rigidifies the cadherin ectodomain overall structure and the protein retains a crescent‐shaped architecture. Homophilic binding of membrane‐distal EC1 domains from opposing cell surfaces is realized by a special mechanism. Adhesive binding arises via an intermediate X‐dimer structure through the exchange of β strands between EC1 domains of cadherins from adjacent cells [13–15]. Key to this mechanism is the vice versa docking of a hydrophobic tryptophan anchor residue into a conserved hydrophobic pocket of the partnering EC1 domain. This strand‐swap binding mode is common to classical and desmosomal cadherins and known for the oligomerization of several monomeric proteins [16] (Figure 4.2b).
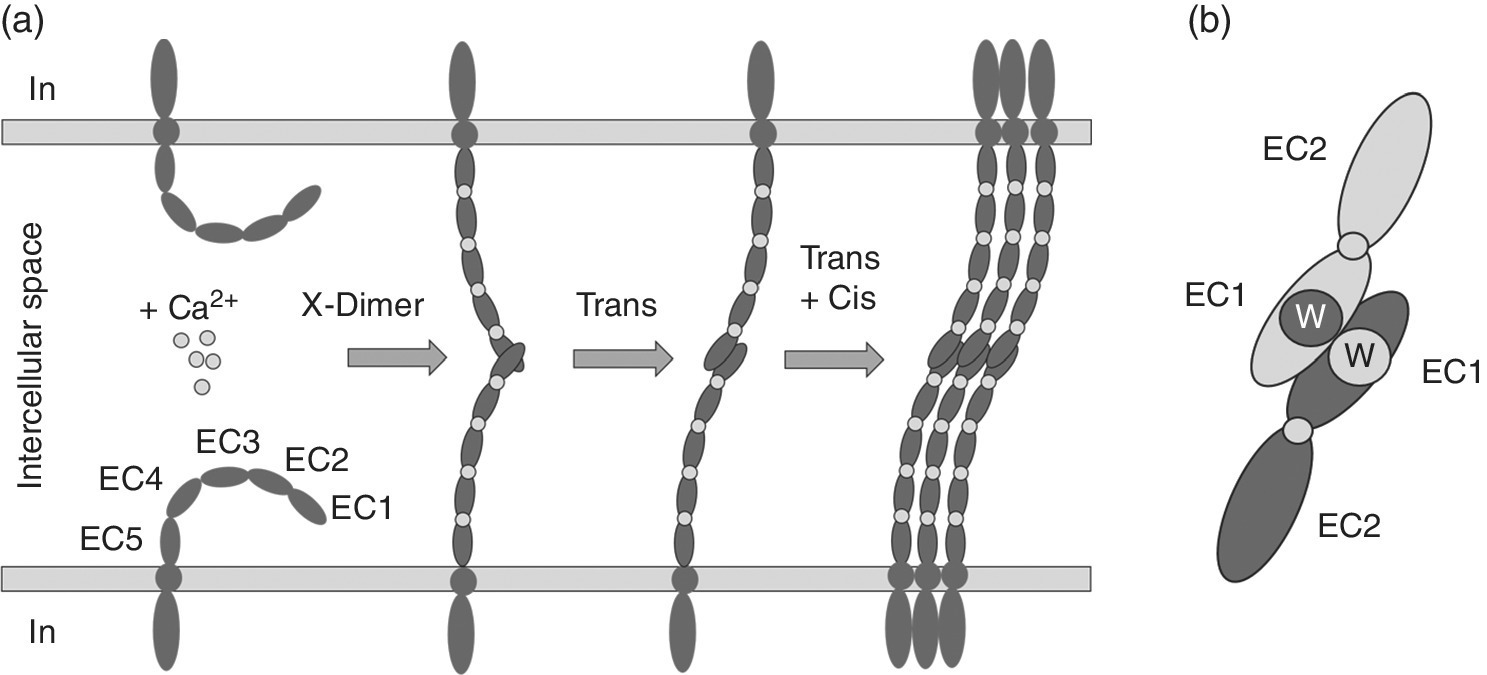
Figure 4.2 Formation of homophilic cadherin interactions at adherens junctions. (a) Calcium dependent trans‐ and cis‐binding is established via the intermediate X‐dimer structure. EC1–5 are the extracellular domains of a classical cadherin. (b) Strand swap of trans‐interacting cadherins, adjacent EC1 domains are connected via the exchange of a hydrophobic loop containing a conserved tryptophan (W).
Binding constants of homophilic cadherin interactions are of low affinity, a dissociation constant (KD) of 0.7 mM was detected for endothelial E‐cadherin [17]. For the vascular endothelial VE‐cadherin fused to the fragment crystallizable (Fc) region of an antibody, the dimeric Fc‐cadherin resulted in KD values ranging from 10−3 to 10−5 M, representing still a weak interaction [18]. Therefore, to maintain cadherin function in cell–cell adhesion, a multivalent presentation that strengthens cell‐to‐cell contact at adherens junctions is obvious. The physiological relevance of cadherin trans‐binding has been confirmed in several studies by single site mutagenesis, electron microscopy, structural and cell‐based analyses [14,19–21]. Further, cis‐interactions between neighboring cadherins seem to be important for packing the protein in adherens junctions [22–24]. Although cis‐interactions were so far not proven by determination of binding constants, they are estimated to be in the mM range. Structural data and mutational analyses point to their existence. The intracellular linkage to the actin skeleton is not essential to enable molecular interactions of EC1 domains, but solely determines density of cadherin distribution at adherens junctions. Cell culture experiments show that directed actin polymerization is the driving force for epithelial cell adhesion. A zipper‐like architecture initiated by membrane protrusions tipped with cadherin of opposing cells mediates initial attachment [25].
Cadherins constitute a diverse family of different adhesion molecules that exhibit spatial and temporal expression patterns during development and therefore contribute to tissue morphogenesis. The conclusion that cadherins are homophilic adhesion molecules came from aggregation experiments, in which cells expressing different types of cadherins were observed to sort out and form distinct aggregates [26]. Segregation of cells into specific tissue layers and the formation of tissue boundaries rely on preferred homophilic interactions of individual cadherins [27]. Furthermore, differential levels of expression of a single cadherin type also contribute to cell segregation [28–30]. In this context it is of note that a varying density of cadherin adhesive clusters in the intercellular space is a critical determinant of endothelial macromolecular sieving [31]. This task of controlling passive paracellular mass transport was earlier primarily allocated to the protein network at the apical tight junction barrier.
Given the complexity of their functions, it is not surprising that impaired cadherin expression has also been linked directly to a variety of diseases including its dominant role in metastatic cancer development. Partial or complete loss of endothelial E‐cadherin expression correlates with cancer malignancy [32]. In conclusion, a variable multivalent display of cadherins on cell surfaces determines cell function and fate.
4.2.2 Selectins, Heterotypic Cell Adhesion to Fight Infections
A classic example of glycan–receptor interaction is the leukocyte–endothelial cell interaction, which initiates leukocyte extravasation from the blood during homing to the bone marrow and in disease (Figure 4.3).

Figure 4.3 Leukocyte extravasation during inflammation. Clustered selectins and carbohydrate ligands on activated cell surfaces mediate initial contacts and slow down the velocity of leukocytes. Firm adhesion is based on integrin binding to cell adhesion molecules of the immunoglobulin family (IgCAM).
Here the family of calcium coordinating C‐type lectins, the selectins, are displayed in clusters on either specialized membrane protrusions, the microvilli of leukocytes (L‐selectin), or the flat surface of activated endothelial cells (E‐ and P‐selectin). During inflammation pro‐inflammatory stimuli prime endothelial cells and leukocytes for subsequent recognition [33,34]. The selectins interact with the common tetrasaccharide sialyl Lewis X (sLeX) binding epitope presented on proteoglycans of both cell surfaces and thereby enable leukocyte–endothelial cell interaction. Individual selectin–sLeX interactions are of low affinity in the high μM to mM range [35], but multiple interactions allow for sufficient strong binding to capture leukocytes from the blood stream to sites of inflammation and initiate the adhesion cascade. The rapid binding and release events slow down leukocyte velocity from the blood flow and mediate leukocyte rolling on the vascular endothelial cell surface [36,37]. A dominant ligand of all selectins is the proteoglycan P‐selectin glycoprotein‐ligand‐1 (PSGL‐1). The transmembrane protein is a cysteine bridged dimer and displays in addition to the carbohydrate sLeX three sulfotyrosines that contact basic residues in proximity to the glycan binding site of P‐selectin [38] and L‐selectin [39]. In the case of P‐selectin binding the bipartite protein–glycan ligand PSGL‐1 shifts the binding constant ~1000‐fold to 800 nM in comparison with sLeX and identifies PSGL‐1 as a high affinity ligand [38]. In this context it is interesting to note that during deceleration of leukocytes to a certain threshold of shear force, cell rolling appears and a primary low affinity interaction turns to high affinity binding [40]. At least P‐ and L‐selectin show this special “catch” bond behavior that relies on intramolecular structural rearrangements of the receptor. Upon ligand recognition the N‐terminal lectin domain is separated from the adjacent epidermal growth factor (EGF)‐like domain by shear force which improves ligand binding and results in longer bond lifetime [41,42]. In addition, homotypic leukocyte–leukocyte interactions, which are realized by the heterotypic interaction of P‐selectin and PSGL‐1 generate cell clusters at the site of adhesion and thereby further facilitate endothelial targeting of activated leukocytes. Furthermore, the initial carbohydrate–ligand recognition triggers intracellular signaling that activates leukocyte integrins, cell adhesion molecules essential to mediate firm adhesion [43–45] and a prerequisite for subsequent cell extravasation.
Over recent years multiple selectin targeting compounds have been developed to counteract leukocyte adhesion [1,3]. The rationale behind an interventional strategy is that during severe injury and acute or chronic inflammatory diseases the unbalanced extravasation of leukocytes from the blood vessel contributes to further tissue destruction. Here multivalent compounds that present the pan selectin targeting natural sLeX epitope or a synthetic sLeX mimetic successfully interfere with the selectin–ligand binding. In addition, polysulfates like the dendritic polyglycerol sulfate (dPGS) that target L‐ and P‐selectin show in competitive binding assays in vitro and cell culture half‐maximal inhibitor concentration (IC50) values down to the picomolar range [46,47]. Although dPGS lacks target specificity, in vivo experiments demonstrate their overall anti‐inflammatory characteristic and indicate effective leukocyte shielding [48,49].
So far the translation of multivalent compounds targeting selectins into the clinic has not been achieved. In contrast, a successful rational design inspired by the sLeX and PSGL‐1 architectures yielded a synthetic bifunctional pan‐selectin antagonist. The molecule targets the carbohydrate binding site of all selectins and in addition basic amino acid residues present at the binding sites of L‐ and P‐selectin [50,51]. This glycomimetic GMI‐1070 (Rivipansel) is currently being tested in clinical trials.
4.2.3 Bacterial Adhesion by FimH
A second example for a multivalent lectin based cell–cell interaction is the binding of uropathogenic Escherichia coli (UPEC) to the urinary tract epithelium. Upon binding, the bacteria get internalized in an active process that is similar to phagocytosis [52] and cause tissue colonization. Urinary tract infections (UTIs) are among the most prevalent inflammatory diseases that are caused by pathogens [53,54] and are therefore of primary interest for a medical intervention.
The transmembrane protein uroplakin Ia (UPIa) is the primary cellular target of UPEC adhesion. UPIa is abundantly expressed on the superficial epithelial cells of the urinary tract, especially in the bladder. Highly mannosylated N‐glycan structures with six to nine mannose residues linked to UPIa provide an ideal surface for mannose‐specific lectin targeting [55]. Bacterial attachment relies on the highly conserved FimH lectins, which are located at the tip of the type 1 fimbriae [56]. A structure–function analysis showed that the residues of the FimH mannose binding pocket are invariant across 200 UPEC strains [57], which highlights that UPEC cause more than 80% of all infections.
A polymannose surface on one site and hundreds of FimH lectin domains on the other allow for successful adhesion. To escape clearance by urine excretion, FimH binding to mannose is a further example for adaptation to shear force. FimH binding to mannose is enhanced at higher shear stress. The molecular basis to shear adaptation relies on the interaction of the two subdomains of FimH: the N‐terminal mannose binding lectin domain FimHL and the C‐terminal pilin domain (FimHP). In the absence of shear FimHP tightly connects to FimHL and the lectin shows only low affinity to mannose [58–60]. Shear force stretches the pilus, allosterically separates the subdomains and increases thereby FimHL binding affinity to mannose. Recently, from crystallographic data, binding kinetics analyses, and molecular simulations it was found that FimHL in the separated state shows a 3300‐fold higher affinity for the model ligand heptylmannoside when compared with the associated state of full‐length FimH [61].
A second intermolecular interaction that is required for successful firm UPEC adhesion and paves the way for bacterial entry is the association with α3β1 integrins [61]. FimH is able to bind matrix proteins such as fibronectin, laminin, and collagen, which in turn recognize integrins [62,63]. Besides this mediated contact recent results also indicate a direct interaction of FimH to high mannose‐type glycans linked to α3 and β1 integrin chains [61].
In addition to UTIs, FimH‐mediated adhesion plays an important role in ileal lesions of patients suffering from Crohn´s disease (CD), a chronic and disabling inflammatory disorder of the intestine. In that case adherent‐invasive E. coli (AIEC) strains promote inflammation and lead to colitis. AIEC bacteria strongly adhere to and invade intestinal epithelial cells. As a consequence, secretion of inflammatory cytokines is induced [64]. The molecular target of FimH in the intestine are mannosylated glycans linked to the carcinoembryonic antigen‐related cell adhesion molecule 6 (CECAM6), overexpressed on intestine epithelial cells from CD patients [65].
The development of competitive FimH antagonists as anti‐adhesive compounds is a promising strategy for a potential medical application. The vast spreading of antibiotic resistances among UPEC and AIEC strains requires alternative treatment options. Different multivalent presentations of mannose derivatives have been explored and nanomolar affinities reported [66–68].
The treatment of UTIs by UPEC requires orally active FimH antagonists. Compounds should get absorbed in the intestine and renally excreted. This implies well‐defined pharmacokinetic properties to achieve an optimal balance between target affinity and retention in the bladder. Monovalent aryl mannosides seem to be well suited to target the FimH binding site by their mannose component and high affinity to a secondary binding site, the tyrosine gate, by an extended lipophilic aglycone structure that contributes to target specificity [69–73]. Oral treatment of UTIs with aryl mannosides in mouse models has already revealed promising results [74–76]. Additionally, in the context of urinary catheterization of patients, intervening concepts are proposed to reduce the bacterial load by wash out strategies with FimH antagonists [77,78].
For the treatment of Crohn´s disease an anti‐adhesive strategy that reduces the level of adherent‐invasive E. coli is also applicable [79]. A main advantage of this complementary approach to current antibiotic treatments is the maintenance of gut microbiota. In this respect it is interesting to note that a therapy with a probiotic yeast strain in a model for Crohn´s disease prevented colitis induced by AIEC [80]. Obviously the high mannose content in the yeast cell wall outcompetes FimH binding of AIEC bacteria to CEACAM6 on the epithelial cell surface.
4.3 Phase Transition, Multivalent Intracellular Assemblies
Electron microscopy and advanced methods in light microscopy have demonstrated a subdivision and local organization of membrane‐less biomolecular organelles in the cytoplasm and nucleus at the nanometer to micrometer scale. These macromolecular dynamic aggregates include for instance P granules, Cajal bodies, promyelocytic leukemia bodies, paraspeckles, and the nucleolus [81]. Recently, it has been proposed, that these structures set up via liquid–liquid demixing phase transitions of their constituent molecules, such as proteins, DNA, and RNA [82]. Nevertheless, the molecular mechanism of how these liquid colloid particles assemble and execute specific functions is largely unknown. The interaction between multivalent molecules in a dilute solution is governed by their concentrations, and depends on the association and dissociation constants. Forces that lead to an attraction of multivalent molecules by electrostatic, hydrophobic, van der Waals interactions, or hydrogen bonding increase the local effective molarity of the resulting receptor–ligand complex and result in a higher probability of molecular recognition. Similarly, lipophilic proteins that partition into membranes, or hydrophilic proteins enclosed in an organelle or vesicle, will interact more often if not dispersed evenly throughout the system. Thus, multivalent interactions of distinct proteins might generate highly ordered assemblies in aqueous solution that phase separate at a critical concentration and lead to liquid droplet formation [83].
Characteristics of intracellular multivalent interacting proteins are their high valency and modest affinity of binding elements, which are connected via long, flexible linker elements [84]. Li et al. [83] recently followed phase separation initially in vitro on engineered proteins that contain two low affinity binding modules, the SRC homology 3 (SH3) domain and its proline‐rich motif (PRM) ligand. Both domains are widely distributed among signaling proteins and often displayed in tandem arrays [84,85]. Binding of the monomeric SH3 to PRM is of low affinity (KD = 350 μM). Titration series of oligovalent binding partners resulted in a concentration dependent sharp sol–gel transition, in which two protein solutions after reaching a critical concentration separate from the environment to produce diffractive droplets of 1 μm to >50 µm in diameter. The concentration of droplet constituent thereby increased up to ~100‐fold.
In a second in vitro experiment the authors demonstrated how phase transition can organize signaling and initiate a biochemical response. A three‐component system consisting of the proteins nephrin, NCK (non‐catalytic region of tyrosine kinase), and N‐WASP (neuronal Wiskott–Aldrich syndrome protein) is involved in actin polymerization. The proteins assemble into complexes (Figure 4.4) prone to phase transition. Nephrin is a transmembrane adhesion receptor mainly expressed in the kidney and participates in the assembly of the cortical actin skeleton necessary for proper formation of the filtration barrier in the kidney [86]. The cytoplasmic tail of nephrin contains three tyrosine residues, which can be phosphorylated upon receptor activation and bind to the SH2 domain of the adaptor protein NCK [87]. The three SH3 domains of NCK, in turn, can interact with the six PRMs of N‐WASP [88]. As a physiological consequence of protein assembly and droplet formation, actin polymerization is initiated in cooperation with the actin related protein (Arp2/3) complex [89].
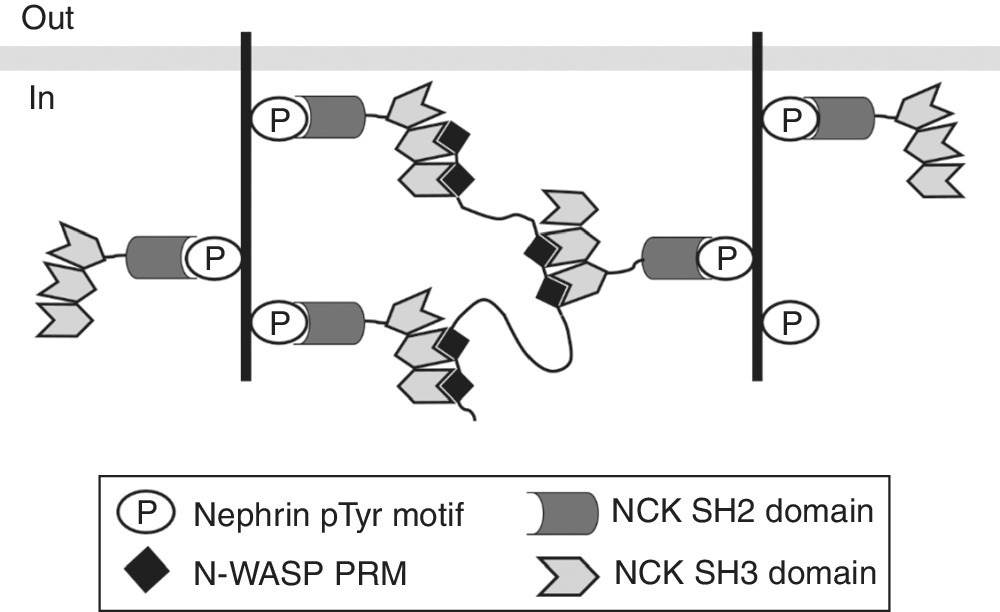
Figure 4.4 Multivalent interactions of nephrin, NCK, and N‐WASP lead to intracellular complex formation and phase separation.
Source: Adapted from Ref. [83]. Reproduced with permission of Nature Publishing Group.
Phase transition was also observed in cell culture experiments. Coexpression of pentavalent fluorescent fusion proteins [mCherry‐(SH3)5 and eGFP‐(PRM)5] in eukaryotic cells resulted in the formation of micrometer‐sized puncta in the cytoplasm where both fluorophores colocalize. High recovery rates after photobleaching indicate the dynamic behavior of these complexes. At the molecular level, this correlates to a transformation of small complexes into large polymers that were detected via dynamic light scattering, small angle X‐ray scattering, light‐ and cryo‐electron microscopy. Interestingly, further experiments indicate that not only the interacting complementary protein domains and their valency are essential, but weak interactions of disordered interdomain linkers synergistically contribute to phase separation due to homotypic self‐association. Reorganization of the linker molecules seems to condense the multivalent complexes and increase the local protein concentrations. Moreover, Pak et al. [90] have shown that non‐specific charge‐mediated interactions of the nephrin intracellular domain (NICD) and their intrinsic disordered sequences can promote phase separation in the absence of NCK and N‐WASP, when expressed in cells. The same results were obtained in vitro when they titrated a solution of the acidic NICD with highly positively charged proteins [90]. The authors explain this unexpected phenomenon with coacervation, an electrostatically driven liquid–liquid phase separation process, resulting from association of oppositely charged molecules. Thus, non‐specific charge‐mediated interactions, together with specific modular domain interactions, trigger the formation of liquid droplets by the ternary nephrin/Nck/N‐WASP complex. Since a number of transmembrane signaling proteins contain disordered cytoplasmic regions and are linked via phosphorylation to basic motifs of adaptor proteins, it is conceivable that coacervation represents a more general concept of cytoplasmic protein clustering and phase separation.
4.4 Multivalency in the Fluid Phase, Pathogen Opsonization
Invading pathogens that reach the blood have to be immediately recognized and eliminated by the immune system in order to avoid them spreading fast throughout the body. As the first line of defense, clearance of pathogen is achieved by phagocytosis from cells of the innate immune system. Mainly macrophages, dendritic cells and neutrophils are capable of recognizing conserved structures on pathogens, termed pathogen‐associated molecular patterns [91] by means of their complementary pattern recognition receptors [92]. Furthermore, the pathogen surface can be labeled by soluble plasma derived proteins and in this way prepared for uptake and degradation (opsonization). Multivalent proteins, such as antibodies and lectins accomplish this task.
Lectins bind to carbohydrates present on pathogen surfaces. Abundant in human serum are the mannose binding lectin (MBL) [93,94] and members of the ficolin family [95,96] that bind to acetylated sugars (N‐acetylglucosamine). They act as pathogen sensors. Both lectins have a comparable shape, they assemble into trimers via their collagen domain and further oligomerize. Their oligomeric structure compensates for the general low binding affinity to carbohydrates that is for the trimer in the millimolar range, but can reach nanomolar affinity in the oligomeric state [97]. Pathogen binding induces intrinsic conformational changes in linked MBL‐associated serine proteases (MASPs) [98], which in turn activate the lectin pathway of the complement cascade. The final product is the membrane attack complex (MAC) [99]. A pore‐forming complex inserts into the membrane and lyses the pathogen. Comparable with MBL and ficolin in structure and function is the C1 complex that activates the classical antibody‐dependent complement pathway [100] (Figure 4.5 and see below).
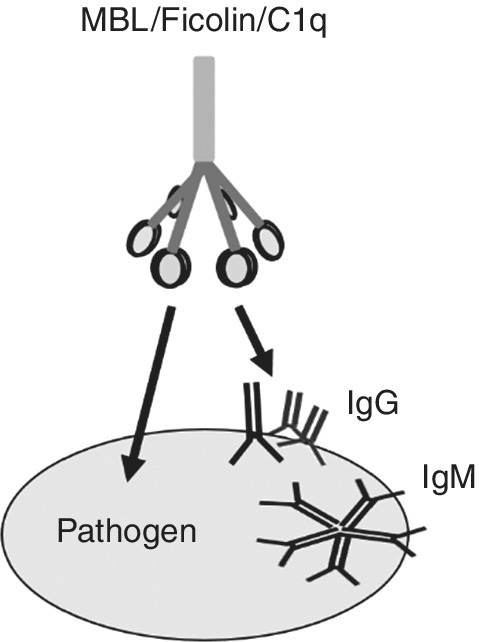
Figure 4.5 Simplified common structure of multivalent pathogen sensors. MBL and ficolin contact the pathogen directly. Targeting of C1q is mediated by IgM or IgG aggregates.
Antigen binding is at least a monovalent recognition but all immunoglobulins contain multiple receptor binding sites. The monomers IgA, IgD, IgE, and IgG have two valencies; the subclass IgA can also appear as dimer (most common), trimer, and tetramer aggregates with resulting valencies of 4, 6, and 8, respectivley. The pentamer IgM displays 10 binding sites. Multivalent epitopes will obviously strengthen binding if addressable epitopes are available. Karulin and Dzantiev [101] have shown that targeting of a bacterial surface with a bivalent antibody is more effective compared with a monovalent antibody fragment. While in this case the antibody association rates did not differ dramatically, the lower affinity of the monovalent protein originates from its enhanced dissociation rate which was about 40 times higher. At least for some immunoglobulins of the IgA subclass, multimerization even seems to be essential for affine binding [102]. The authors showed that a monomeric IgA was not able to target lipopolysaccharides (LPS) efficiently; in contrast, the dimeric IgA binds LPS with an affinity in the low nanomolar range. At least for further signaling antibody multimerization is required.
Ligand recognition by the fragment antigen binding (Fab) site of the antibody might induce conformational changes in the distant Fc part of the molecule. This can be observed by enhanced Fc reactivity, namely Fc receptor binding and complement activation. Here it is remarkable that single recognition events barely contribute to signal transduction but multivalent interactions are essential. For instance, multiple immunoglobulins attached to a pathogen surface create an immune complex that binds to several Fc receptors on a macrophage and trigger pathogen uptake. Mast cell activation only occurs when IgE cross‐linked antigens bind to the cell surface receptors and then induce degranulation of histamine and heparin containing vesicles. Moreover, classical complement activation is only mediated by binding of the C1 complex consisting of the hexavalent pattern recognition molecule C1q and a heterotetramer of proteases C1r and C1s [103]. Here the spatial arrangements of binding partners seem to play a pivotal role. C1q binds a single IgG Fc only with very low affinity (KD ~10−4 M) [104,105].
The C1 to IgG antibody stoichiometry so far remains poorly understood [106]. Potent complement activation by monoclonal antibodies is restricted to certain antigens, presumably because antigen size, epitope density and geometry affect activation [107–109]. Polyclonal antibodies in contrast appear to be less sensitive to such constraints [110,111] because binding of antibodies to a variety of epitopes facilitates clustered Fc presentation.
Recently it was shown, that some IgG species are able to self‐assemble upon ligand binding to hexamers and thereby initiate a strong complement response [112]. Certain amino acids were identified that contribute to the Fc–Fc contact formation and are obviously necessary to organize the defined well‐ordered structure. Although the hexamer arrangement was earlier detected in crystal structures [113,114] the physiological implication was not realized. The hexavalent antibody structure perfectly matches the C1q architecture and enables subsequent high affinity multivalent binding of the six C1q headpieces to the targeted antibody Fc regions. Highest binding affinities of C1q to antibody‐opsonized cells are in the low nanomolar range and binding strength positively correlates with complement‐dependent cytotoxicity (CDC) activity [112]. Due to the structural similarity, it is not surprising that the pentavalent IgM generally elicits strong complement activation and promotes equivalent signaling through conformational change.
Antibody coating of targets has been shown to mediate different potent killing mechanisms. CDC occurs via MAC formation, as described before. In the case of antibody‐dependent cellular cytotoxicity (ADCC) the antibody labeled pathogen is recognized by Natural Killer cells which consequently secrete membrane penetrating peptides and proteases to destroy the pathogen. Finally, the antibody‐dependent cellular phagocytosis (ADCP) is accomplished by macrophages and monocytes. All of these effector functions are mediated by the antibody Fc region. From the medical point of view, engineering of the Fc region to enhance the cytotoxic activity of therapeutic antibodies is currently a subject of intense investigation [115–119].
4.5 Conclusion
Multivalent interactions are widespread in biological systems. The best characterized are receptor–ligand interactions that mediate receptor clustering at cell surfaces. However, research in recent years has shown that this apparently simple study area is challenging. The dynamic character of interactions that leads to assembly and disassembly of molecular complexes in space and time is a difficult task and often remains obscure. Nevertheless, there is an increasing interest from different disciplines to unravel how multivalent interactions of biomolecules confer biological functions. Recent advances that demonstrated how membrane‐less organelles are generated are a good example to comprehend how a graded biological response relies on the multivalent intermolecular assembly of biomolecules (see Section 4.3) and there is much more to explore. Experiments aimed at interfering with biological recognition have to be validated in natural environments. Future research should address questions of how multivalency can be applied to control cell behavior or mediate specific targeting. Potential applications in the medical field are broad and include but are not limited to cell differentiation, imaging, pathogen shielding, and tumor therapy.
Acknowledgment
I gratefully acknowledge the German Research Foundation (DFG) for financial support within the Collaborative Research Centre 765.
References
- 1 Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998; 37(20):2755–94.
- 2 Hunter CA, Anderson HL. What is cooperativity? Angew. Chem. Int. Ed. 2009; 48(41):7488–99.
- 3 Fasting C, Schalley CA, Weber M, et al. Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl. 2012; 51(42):10472–98.
- 4 Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science 1972; 175(4023):720–31.
- 5 Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011; 3(10):a004697.
- 6 Banjade S, Wu Q, Mittal A, et al. Conserved interdomain linker promotes phase separation of the multivalent adaptor protein Nck. Proc. Natl Acad. Sci. USA 2015; 112(47):E6426–35.
- 7 Tomschy A, Fauser C, Landwehr R, Engel J. Homophilic adhesion of E‐cadherin occurs by a co‐operative two‐step interaction of N‐terminal domains. EMBO J. 1996; 15(14):3507–14.
- 8 Brieher WM, Yap AS, Gumbiner BM. Lateral dimerization is required for the homophilic binding activity of C‐cadherin. J. Cell Biol. 1996; 135(2):487–96.
- 9 Ahrens T, Lambert M, Pertz O, et al. Homoassociation of VE‐cadherin follows a mechanism common to “classical” cadherins. J. Mol. Biol. 2003; 325(4):733–42.
- 10 Leckband D, Sivasankar S. Mechanism of homophilic cadherin adhesion. Curr. Opin. Cell Biol. 2000; 12(5):587–92.
- 11 Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium‐induced E‐cadherin rigidification and dimerization. Nature 1996; 380(6572):360–4.
- 12 Harrison OJ, Jin X, Hong S, et al. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 2011; 19(2):244–56.
- 13 Shapiro L, Fannon AM, Kwong PD, et al. Structural basis of cell–cell adhesion by cadherins. Nature 1995; 374(6520):327–37.
- 14 Harrison OJ, Corps EM, Berge T, Kilshaw PJ. The mechanism of cell adhesion by classical cadherins: the role of domain 1. J. Cell Sci. 2005; 118(Pt 4):711–21.
- 15 Posy S, Shapiro L, Honig B. Sequence and structural determinants of strand swapping in cadherin domains: do all cadherins bind through the same adhesive interface? J. Mol. Biol. 2008; 378(4):954–68.
- 16 Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995; 4(12):2455–68.
- 17 Haussinger D, Ahrens T, Aberle T, et al. Proteolytic E‐cadherin activation followed by solution NMR and X‐ray crystallography. EMBO J. 2004; 23(8):1699–708.
- 18 Baumgartner W, Hinterdorfer P, Ness W, et al. Cadherin interaction probed by atomic force microscopy. Proc. Natl Acad. Sci. USA 2000; 97(8):4005–10.
- 19 Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 2009; 1(3):a003053.
- 20 Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009; 1(6):a002899.
- 21 Patel SD, Chen CP, Bahna F, et al. Cadherin‐mediated cell‐cell adhesion: sticking together as a family. Curr. Opin. Struct. Biol. 2003; 13(6):690–8.
- 22 Troyanovsky RB, Sokolov E, Troyanovsky SM. Adhesive and lateral E‐cadherin dimers are mediated by the same interface. Mol. Cell Biol. 2003; 23(22):7965–72.
- 23 Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010; 11(7):502–14.
- 24 Hong S, Troyanovsky RB, Troyanovsky SM. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc. Natl Acad. Sci. USA 2010; 107(8):3528–33.
- 25 Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell. 2000; 100(2):209–19.
- 26 Nose A, Nagafuchi A, Takeichi M. Expressed recombinant cadherins mediate cell sorting in model systems. Cell 1988; 54(7):993–1001.
- 27 Takeichi M. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 1995; 7(5):619–27.
- 28 Steinberg MS. Does differential adhesion govern self‐assembly processes in histogenesis? Equilibrium configurations and the emergence of a hierarchy among populations of embryonic cells. J. Exp. Zool. 1970; 173(4):395–433.
- 29 Friedlander DR, Mege RM, Cunningham BA, Edelman GM. Cell sorting‐out is modulated by both the specificity and amount of different cell adhesion molecules (CAMs) expressed on cell surfaces. Proc. Natl Acad. Sci. USA 1989; 86(18):7043–7.
- 30 Steinberg MS, Takeichi M. Experimental specification of cell sorting, tissue spreading, and specific spatial patterning by quantitative differences in cadherin expression. Proc. Natl Acad. Sci. USA 1994; 91(1):206–9.
- 31 Quadri SK, Sun L, Islam MN, et al. Cadherin selectivity filter regulates endothelial sieving properties. Nat. Commun. 2012; 3:1099.
- 32 Berx G, Cleton‐Jansen AM, Nollet F, et al. E‐cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995; 14(24):6107–15.
- 33 Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007; 7(9):678–89.
- 34 Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013; 13(3):159–75.
- 35 Poppe L, Brown GS, Philo JS, et al. Conformation of sLe(x) tetrasaccharide, free in solution and bound to E‐, P‐, and L‐selectin. J. Am. Chem. Soc. 1997; 119(7):1727–36.
- 36 Varki A. Selectin ligands. Proc. Natl Acad. Sci. USA 1994; 91(16):7390–7.
- 37 Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999; 79(1):181–213.
- 38 Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P‐ and E‐selectin bound to SLe(X) and PSGL‐1. Cell 2000; 103(3):467–79.
- 39 Woelke AL, Kuehne C, Meyer T, et al. Understanding selectin counter‐receptor binding from electrostatic energy computations and experimental binding studies. J. Phys. Chem. B 2013; 117(51):16443–54.
- 40 McEver RP. Selectins: lectins that initiate cell adhesion under flow. Curr. Opin. Cell Biol. 2002; 14(5):581–6.
- 41 Marshall BT, Long M, Piper JW, et al. Direct observation of catch bonds involving cell‐adhesion molecules. Nature 2003; 423(6936):190–3.
- 42 Sarangapani KK, Yago T, Klopocki AG, et al. Low force decelerates L‐selectin dissociation from P‐selectin glycoprotein ligand‐1 and endoglycan. J. Biol. Chem. 2004; 279(3):2291–8.
- 43 Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E‐selectin‐induced alpha(L)beta(2) integrin‐mediated rolling on intercellular adhesion molecule‐1. Immunity 2007; 26(6):773–83.
- 44 Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011;118(26):6743–51.
- 45 Pruenster M, Kurz AR, Chung KJ, et al. Extracellular MRP8/14 is a regulator of beta2 integrin‐dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015; 6:6915.
- 46 Weinhart M, Groger D, Enders S, et al. The role of dimension in multivalent binding events: structure–activity relationship of dendritic polyglycerol sulfate binding to L‐selectin in correlation with size and surface charge density. Macromol. Biosci. 2011; 11(8):1088–98.
- 47 Weinhart M, Groger D, Enders S, et al. Synthesis of dendritic polyglycerol anions and their efficiency toward L‐selectin inhibition. Biomacromolecules 2011; 12(7):2502–11.
- 48 Dernedde J, Rausch A, Weinhart M, et al. Dendritic polyglycerol sulfates as multivalent inhibitors of inflammation. Proc. Natl Acad. Sci. USA 2010; 107(46):19679–84.
- 49 Oishi K, Hamaguchi Y, Matsushita T, et al. A crucial role of L‐selectin in C protein‐induced experimental polymyositis in mice. Arthritis Rheumatol. 2014; 66(7):1864–71.
- 50 Magnani JL, Ernst B. Glycomimetic drugs – a new source of therapeutic opportunities. Discov. Med. 2009; 8(43):247–52.
- 51 Chang J, Patton JT, Sarkar A, et al. GMI‐1070, a novel pan‐selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010; 116(10):1779–86.
- 52 Palmer LM, Reilly TJ, Utsalo SJ, Donnenberg MS. Internalization of Escherichia coli by human renal epithelial cells is associated with tyrosine phosphorylation of specific host cell proteins. Infect. Immun. 1997; 65(7):2570–5.
- 53 Fihn SD. Clinical practice. Acute uncomplicated urinary tract infection in women. N. Engl. J. Med. 2003; 349(3):259–66.
- 54 Mak RH, Kuo HJ. Pathogenesis of urinary tract infection: an update. Curr. Opin. Pediatr. 2006; 18(2):148–52.
- 55 Zhou G, Mo WJ, Sebbel P, et al. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J. Cell Sci. 2001; 114(Pt 22):4095–103.
- 56 Krogfelt KA, Bergmans H, Klemm P. Direct evidence that the FimH protein is the mannose‐specific adhesin of Escherichia coli type 1 fimbriae. Infect. Immun. 1990; 58(6):1995–8.
- 57 Hung CS, Bouckaert J, Hung D, et al. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 2002; 44(4):903–15.
- 58 Thomas WE, Trintchina E, Forero M, et al. Bacterial adhesion to target cells enhanced by shear force. Cell 2002; 109(7):913–23.
- 59 Sokurenko EV, Vogel V, Thomas WE. Catch‐bond mechanism of force‐enhanced adhesion: counterintuitive, elusive, but … widespread? Cell Host Microbe 2008; 4(4):314–23.
- 60 Le Trong I, Aprikian P, Kidd BA, et al. Structural basis for mechanical force regulation of the adhesin FimH via finger trap‐like beta sheet twisting. Cell 2010; 141(4):645–55.
- 61 Eto DS, Jones TA, Sundsbak JL, Mulvey MA. Integrin‐mediated host cell invasion by type 1‐piliated uropathogenic Escherichia coli. PLoS Pathog. 2007; 3(7):e100.
- 62 Pouttu R, Puustinen T, Virkola R, et al. Amino acid residue Ala‐62 in the FimH fimbrial adhesin is critical for the adhesiveness of meningitis‐associated Escherichia coli to collagens. Mol. Microbiol. 1999; 31(6):1747–57.
- 63 Kukkonen M, Raunio T, Virkola R, et al. Basement membrane carbohydrate as a target for bacterial adhesion: binding of type I fimbriae of Salmonella enterica and Escherichia coli to laminin. Mol. Microbiol. 1993; 7(2):229–37.
- 64 Eaves‐Pyles T, Allen CA, Taormina J, et al. Escherichia coli isolated from a Crohn's disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int. J. Med. Microbiol. 2008; 298(5–6):397–409.
- 65 Barnich N, Carvalho FA, Glasser AL, et al. CEACAM6 acts as a receptor for adherent‐invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest. 2007; 117(6):1566–74.
- 66 Touaibia M, Wellens A, Shiao TC, et al. Mannosylated G(0) dendrimers with nanomolar affinities to Escherichia coli FimH. ChemMedChem 2007; 2(8):1190–201.
- 67 Almant M, Moreau V, Kovensky J, et al. Clustering of Escherichia coli type‐1 fimbrial adhesins by using multimeric heptyl alpha‐D‐mannoside probes with a carbohydrate core. Chemistry 2011; 17(36):10029–38.
- 68 Gouin SG, Wellens A, Bouckaert J, Kovensky J. Synthetic multimeric heptyl mannosides as potent antiadhesives of uropathogenic Escherichia coli. ChemMedChem 2009; 4(5):749–55.
- 69 Bouckaert J, Berglund J, Schembri M, et al. Receptor binding studies disclose a novel class of high‐affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005; 55(2):441–55.
- 70 Sperling O, Fuchs A, Lindhorst TK. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: Design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006; 4(21):3913–22.
- 71 Han Z, Pinkner JS, Ford B, et al. Structure‐based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010; 53(12):4779–92.
- 72 Jiang X, Abgottspon D, Kleeb S, et al. Antiadhesion therapy for urinary tract infections – a balanced PK/PD profile proved to be key for success. J. Med. Chem. 2012; 55(10):4700–13.
- 73 Scharenberg M, Schwardt O, Rabbani S, Ernst B. Target selectivity of FimH antagonists. J. Med. Chem. 2012; 55(22):9810–6.
- 74 Klein T, Abgottspon D, Wittwer M, et al. FimH antagonists for the oral treatment of urinary tract infections: from design and synthesis to in vitro and in vivo evaluation. J. Med. Chem. 2010; 53(24):8627–41.
- 75 Cusumano CK, Pinkner JS, Han Z, et al. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci. Transl. Med. 2011; 3(109):109ra15.
- 76 Kleeb S, Pang L, Mayer K, et al. FimH antagonists: bioisosteres to improve the in vitro and in vivo PK/PD profile. J. Med. Chem. 2015; 58(5):2221–39.
- 77 Wellens A, Garofalo C, Nguyen H, et al. Intervening with urinary tract infections using anti‐adhesives based on the crystal structure of the FimH‐oligomannose‐3 complex. PLoS One 2008; 3(4):e2040.
- 78 Guiton PS, Cusumano CK, Kline KA, et al. Combinatorial small‐molecule therapy prevents uropathogenic Escherichia coli catheter‐associated urinary tract infections in mice. Antimicrob. Agents Chemother. 2012; 56(9):4738–45.
- 79 Alvarez Dorta D, Sivignon A, Chalopin T, et al. The antiadhesive strategy in Crohn's disease: Orally active mannosides to decolonize pathogenic Escherichia coli from the gut. ChemBioChem 2016; 17(10):936–52.
- 80 Sivignon A, de Vallee A, Barnich N, et al. Saccharomyces cerevisiae CNCM I‐3856 prevents colitis induced by AIEC bacteria in the transgenic mouse model mimicking Crohn's disease. Inflamm. Bowel Dis. 2015; 21(2):276–86.
- 81 Spector DL. SnapShot: Cellular bodies. Cell 2006;127(5):1071.
- 82 Hyman AA, Simons K. Cell biology. Beyond oil and water – phase transitions in cells. Science 2012; 337(6098):1047–9.
- 83 Li P, Banjade S, Cheng HC, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012; 483(7389):336–40.
- 84 Jin J, Xie X, Chen C, Park JG, et al. Eukaryotic protein domains as functional units of cellular evolution. Sci. Signal 2009; 2(98):ra76.
- 85 Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science 2003; 300(5618):445–52.
- 86 Jones N, Blasutig IM, Eremina V, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006; 440(7085):818–23.
- 87 Blasutig IM, New LA, Thanabalasuriar A, et al. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol. Cell Biol. 2008; 28(6):2035–46.
- 88 Rohatgi R, Nollau P, Ho HY, et al. Nck and phosphatidylinositol 4,5‐bisphosphate synergistically activate actin polymerization through the N‐WASP‐Arp2/3 pathway. J. Biol. Chem. 2001; 276(28):26448–52.
- 89 Banjade S, Wu Q, Mittal A, et al. Conserved interdomain linker promotes phase separation of the multivalent adaptor protein Nck. Proc. Natl Acad. Sci. USA 2015; 112(47):E6426–35.
- 90 Pak CW, Kosno M, Holehouse AS, et al. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell 2016; 63(1):72–85.
- 91 Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009; 22(2):240–73.
- 92 Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem. J. 2009; 420(1):1–16.
- 93 Kawasaki T, Etoh R, Yamashina I. Isolation and characterization of a mannan‐binding protein from rabbit liver. Biochem. Biophys. Res. Commun. 1978; 81(3):1018–24.
- 94 Drickamer K, Dordal MS, Reynolds L. Mannose‐binding proteins isolated from rat liver contain carbohydrate‐recognition domains linked to collagenous tails. Complete primary structures and homology with pulmonary surfactant apoprotein. J. Biol. Chem. 1986; 261(15):6878–87.
- 95 Ichijo H, Hellman U, Wernstedt C, et al. Molecular cloning and characterization of ficolin, a multimeric protein with fibrinogen‐ and collagen‐like domains. J. Biol. Chem. 1993; 268(19):14505–13.
- 96 Lu J, Teh C, Kishore U, Reid KB. Collectins and ficolins: sugar pattern recognition molecules of the mammalian innate immune system. Biochim. Biophys. Acta 2002; 1572(2–3):387–400.
- 97 Kawasaki N, Kawasaki T, Yamashina I. Isolation and characterization of a mannan‐binding protein from human serum. J. Biochem. 1983; 94(3):937–47.
- 98 Takahashi M, Mori S, Shigeta S, Fujita T. Role of MBL‐associated serine protease (MASP) on activation of the lectin complement pathway. Adv. Exp. Med. Biol. 2007; 598:93–104.
- 99 Kolb WP, Haxby JA, Arroyave CM, Muller‐Eberhard HJ. The membrane attack mechanism of complement. Reversible interactions among the five native components in free solution. J. Exp. Med. 1973; 138(2):428–37.
- 100 Gaboriaud C, Teillet F, Gregory LA, et al. Assembly of C1 and the MBL‐ and ficolin‐MASP complexes: structural insights. Immunobiology 2007; 212(4–5):279–88.
- 101 Karulin A, Dzantiev BB. Polyvalent interaction of antibodies with bacterial cells. Mol. Immunol. 1990; 27(10):965–71.
- 102 Lullau E, Heyse S, Vogel H, et al. Antigen binding properties of purified immunoglobulin A and reconstituted secretory immunoglobulin A antibodies. J. Biol. Chem. 1996; 271(27):16300–9.
- 103 Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology 2000; 49(1‐2):159–70.
- 104 Hughes‐Jones NC, Gardner B. Reaction between the isolated globular sub‐units of the complement component C1q and IgG‐complexes. Mol. Immunol. 1979; 16(9):697–701.
- 105 Feinstein A, Richardson N, Taussig MI. Immunoglobulin flexibility in complement activation. Immunol. Today 1986; 7(6):169–74.
- 106 Gaboriaud C, Thielens NM, Gregory LA, et al. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 2004; 25(7):368–73.
- 107 Bindon CI, Hale G, Waldmann H. Importance of antigen specificity for complement‐mediated lysis by monoclonal antibodies. Eur. J. Immunol. 1988; 18(10):1507–14.
- 108 Cragg MS, Morgan SM, Chan HT, et al. Complement‐mediated lysis by anti‐CD20 mAb correlates with segregation into lipid rafts. Blood 2003; 101(3):1045–52.
- 109 de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011; 186(3):1840–8.
- 110 Hughes‐Jones NC, Gorick BD, Howard JC, Feinstein A. Antibody density on rat red cells determines the rate of activation of the complement component C1. Eur. J. Immunol. 1985; 15(10):976–80.
- 111 Dechant M, Weisner W, Berger S, et al. Complement‐dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008; 68(13):4998–5003.
- 112 Diebolder CA, Beurskens FJ, de Jong RN, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014; 343(6176):1260–3.
- 113 Saphire EO, Parren PW, Pantophlet R, et al. Crystal structure of a neutralizing human IGG against HIV‐1: a template for vaccine design. Science 2001; 293(5532):1155–9.
- 114 Wu Y, West AP, Jr, Kim HJ, Thornton ME, Ward AB, Bjorkman PJ. Structural basis for enhanced HIV‐1 neutralization by a dimeric immunoglobulin G form of the glycan‐recognizing antibody 2G12. Cell Rep. 2013; 5(5):1443–55.
- 115 Ying T, Gong R, Ju TW, Prabakaran P, Dimitrov DS. Engineered Fc based antibody domains and fragments as novel scaffolds. Biochim. Biophys. Acta 2014; 1844(11):1977–82.
- 116 Caaveiro JM, Kiyoshi M, Tsumoto K. Structural analysis of Fc/FcgammaR complexes: a blueprint for antibody design. Immunol. Rev. 2015; 268(1):201–21.
- 117 Park HI, Yoon HW, Jung ST. The highly evolvable antibody Fc domain. Trends Biotechnol. 2016; 34(11):895–908.
- 118 Lobner E, Traxlmayr MW, Obinger C, Hasenhindl C. Engineered IgG1‐Fc – one fragment to bind them all. Immunol. Rev. 2016; 270(1):113–31.
- 119 Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr. Opin. Immunol. 2016; 40:62–9.