
1
Additivity of Energy Contributions in Multivalent Complexes
Hans‐Jörg Schneider
FR Organische Chemie, Universität des Saarlandes, 66123, Saarbrücken, Germany
1.1 Introduction
Additivity of individual binding contributions is the very basis of multivalency. In classical coordination chemistry such simultaneous actions are described as the chelate effect. They offer almost unlimited ways to enhance the affinity [1,2,3,4,5,6], and therefore within certain limitations also the selectivity [7] of synthetic and natural complexes. Although additivity is often implied in experimental and theoretical approaches it is subject to many limitations which will be also discussed in the present chapter.
1.2 Additivity of Single Interactions – Examples
If only one kind of interaction is present in a complex one can expect a simple linear correlation between the number n of the individual interaction free energies ΔΔGi and the total ΔGt (Equation 1.1), as illustrated in Figure 1.1 for salt bridges [8]. Even though the organic ion pair complexes are based on cations and anions of very different size and polarizability one observes essentially additive salt bridges; the slope of the correlation indicates an average of ΔΔG = (5 ± 1) kJ/mol per salt bridge. The value of (5 ± 1) kJ/mol is observed in usual buffer solution, but varies as expected from the Debye–Hückel equation with the ionic strength of the solution [9]. Scheme 1.1 shows a corresponding value of K ≈ 10 M−1 per salt bridge for typical complexes where the affinity depends as expected on the degree of protonation [7].

Figure 1.1 Additive ion pair contributions in a variety of complexes with a number nC of salt bridges. From slope: average (5 ± 1) kJ/mol per salt bridge. A,B and C,C' – complexes of a tetraphenolate cyclophane (4−) with Me4N+ and an azoniacyclophane (4+) with mono‐ and dianionic naphthalene derivatives; D – anionic (sulfonate or carboxylate) with cationic (ammonio) triphenylmethane derivatives; E – organic dianions with organic dications; F – cationic azamacrocycle (6+ charges) with aliphatic dicarboxylates; G – cationic azacrowns with adenosine mono‐, di‐ and triphosphates.
Source: Ref. [8]. Reproduced with permission of John Wiley and Sons.

Scheme 1.1 Complexation log K values of anions 1–5 with a macrocyclic amine as function of the degree of protonation of the amine; and ion pairing with some representative complexes; log K values in water; n is the estimated number of salt bridges.
The additivity depicted in Figure 1.1 and Scheme 1.1 for salt bridges is in line with the Bjerrum equation, which describes ion pair association as a function of the ion charges zA and zB; Figure 1.2 shows for over 200 ion pairs a linear dependence of log K vs. zAzB [3]. For inorganic salts one finds similar ΔΔG values of 5–6 kJ/mol per salt bridge and a similar dependence on charges [10]. At zero ionic strength the stability decreases in the order Ca2+ > Mg2+ > > Li+ > Na+ > K+ and can be described by Equation 1.2 [11]. Additivity is observed although ion pairing in water is determined entirely by entropic contributions[11], unless other contributions dominate [12].
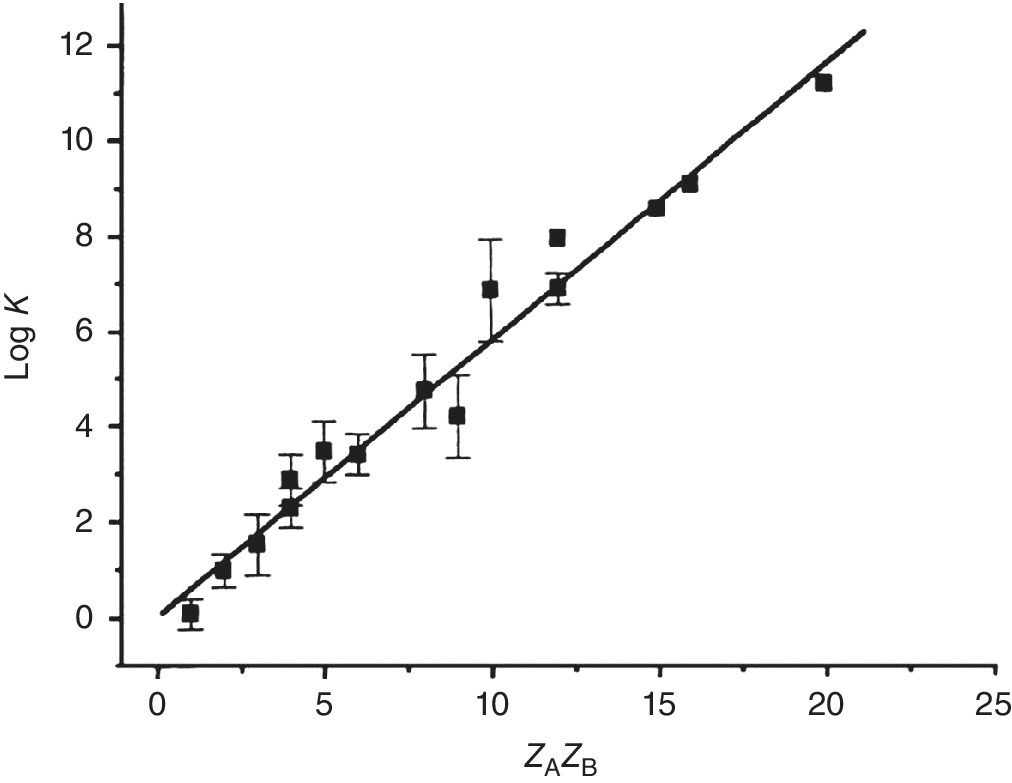
Figure 1.2 Ion pair association constants at zero ionic strength as a function of charge product, calculated for 203 ion pairs.
Source: Ref. [8]. Reproduced with permission of John Wiley and Sons.
If there is more than one kind of interaction, Equation 1.3 applies. Often however, only one of the contributions is the same, like salt bridges in complexes of nucleotides with a positively charged host (Scheme 1.2) [13]. Additivity is then observed by the constant stability difference of 2 × ΔΔG ≈ 10 kJ/mol between complexes with charged nucleotides and neutral nucleosides. The 10 kJ/mol reflects the presence of two salt bridges between the phosphate dianion and the host ammonium center, which agrees with structural analyses by NMR spectroscopy.

Scheme 1.2 Complexation free energies ΔG of nucleotides and nucleosides with the cyclophane CP66.
The complexes shown in Scheme 1.2 exhibit constant single ΔΔGA values only for the salt bridges, whereas the second contribution ΔΔGB varies as a function of the different nucleobases. Figure 1.3 illustrates a case where both ΔΔGA and ΔΔGB remain constant, the latter reflecting cation–π interactions. In principle one could use Equation 1.3 to derive both ΔΔGA and ΔΔGB, but more reliable values are obtained if for one interaction a ΔΔG value is used which is known from independent analyses, such as ΔΔGA = 5 kJ/mol for each salt bridge (see above). Then one observes a rather linear correlation with the number of phenyl units which shows a contribution of ΔΔGB ≈ 1.5 kJ/mol for the single +N–π interaction [14].

Figure 1.3 Ion pairs exhibiting both salt bridges and cation–π interactions; if ΔΔGA = 5 kJ/mol for each salt bridge are subtracted from ΔGt of each complex. Outliers (open circles) are due to conformational mismatch.
Source: Ref. [14]. Reproduced with permission of American Chemical Society.
The effect of nitro substituents on dispersive interactions is another example of additive energy contributions (Figure 1.4) [15,16]. Additivity with respect to substituent effects is observed in Hammett‐type linear free energy relationship correlations; Figure 1.5 shows an example for hydrogen bonds with C─H bonds as donor and with hexamethylphosphoramide as acceptor [17].

Figure 1.4 Additive ΔΔGX increments in complexes of porphyrins bearing cationic or anionic substituents R in meso position (TPyP or TPS) in water, after deduction of 5 kJ/mol for ion pair contribution where applicable. ΔΔGX increments in TPyP complexes for nitro substituents as an example (deviation for ortho‐dinitro due to steric hindrance); correlation between measured complexation energies ΔGexp and ΔGcalc calculated on the basis of experimentally determined averaged single contributions ΔGS. Filled circles, complexes with TPyP; open circles, complexes with TPS.
Source: Ref. [15]. Reproduced with permission of John Wiley and Sons.

Figure 1.5 Hammett‐type correlation of equilibria of hydrogen bonds with hexamethylphosphoramide as acceptor and para‐substituted tetrafluorobenzenes or phenylacetonitriles as donor; log K versus Hammett substituent constants.
Source: Ref. [17]. Reproduced with permission of John Wiley and Sons.
1.3 Limitations of Additivity
1.3.1 Free Energy Values ΔG Instead of Enthalpic and Entropic Values ΔH, TΔS
The examples shown above as well as most others in the literature rely on free energy values ΔG, although consideration of the corresponding ΔH and TΔS parameters could shed more light on the underlying binding mechanisms. As pointed out earlier by Jencks, the empirical use of ΔG “avoids the difficult or insoluble problem of interpreting observed ΔH and TΔS values for aqueous solution” [18]. Furthermore, according to Jencks, there is often an additional “connection Gibbs energy, ΔGS” (Equation 1.4) which he ascribed largely to changes in translational and rotational entropy. These connection ΔGS can be either negative or positive and will be discussed as major liming factors for additivity below in the context of cooperativity and allostery.
The success of using free energy values instead of enthalpic and entropic values is in an essential part due to entropy–enthalpy compensation which has empirically been found to hold with many complexations, although it is theoretically not well‐founded [19,20,21]. Another factor is that in typical supramolecular complexes the loss of translatory freedom is already paid by a single association step. The loss of rotational freedom upon complex formation has been experimentally [9] found to be smaller than theoretically expected (see below).
Entropy contributions pose particular problems, not only for the precise experimental determination, which in the past often relied on the temperature dependence of equilibrium constants (the Van ‘tHoff method) instead of on more reliable calorimetry techniques. Also their theoretical interpretation is hampered by several factors, for instance because ΔS values depend on the choice of the standard concentration, in contrast to ΔH [8]. Configurational entropy, which refers also to solute motions has been addressed in several papers [22,23,24]. Data for the loss of translatory degrees of freedom in complex formation range from TΔS = 3 to 9 kJ/mol, and depend also on the reaction medium [25]. In multivalent associations this TΔS penalty plays, as mentioned above, a minor role as it is paid already by a single interaction. For the loss of rotatory degrees of freedom in complex formation values from TΔS = 1.5 to 6 kJ/mol were proposed [26], which also should depend on the nature of the bond involved in the rotation [27]. Measurements of complexes involving an increasing number n of single bonds between two binding units furnished values of only ΔΔG = 0.5 to 1.3 kJ/mol per single bond (e.g. from the slope in Figure 1.6) [9,28]. Similar small numbers have been found in complexes involving peptide‐ ß‐sheets [29], with calcium‐EDTA complexes [30], and for example in the coordination of nickel or copper with either trans‐1,2‐diaminocyclohexane or the more flexible ethylene diamine [31]. In line with these rather small numbers it has been found that preorganization of a linker in host molecules has no or a small effect on supramolecular effective molarities [32,33].
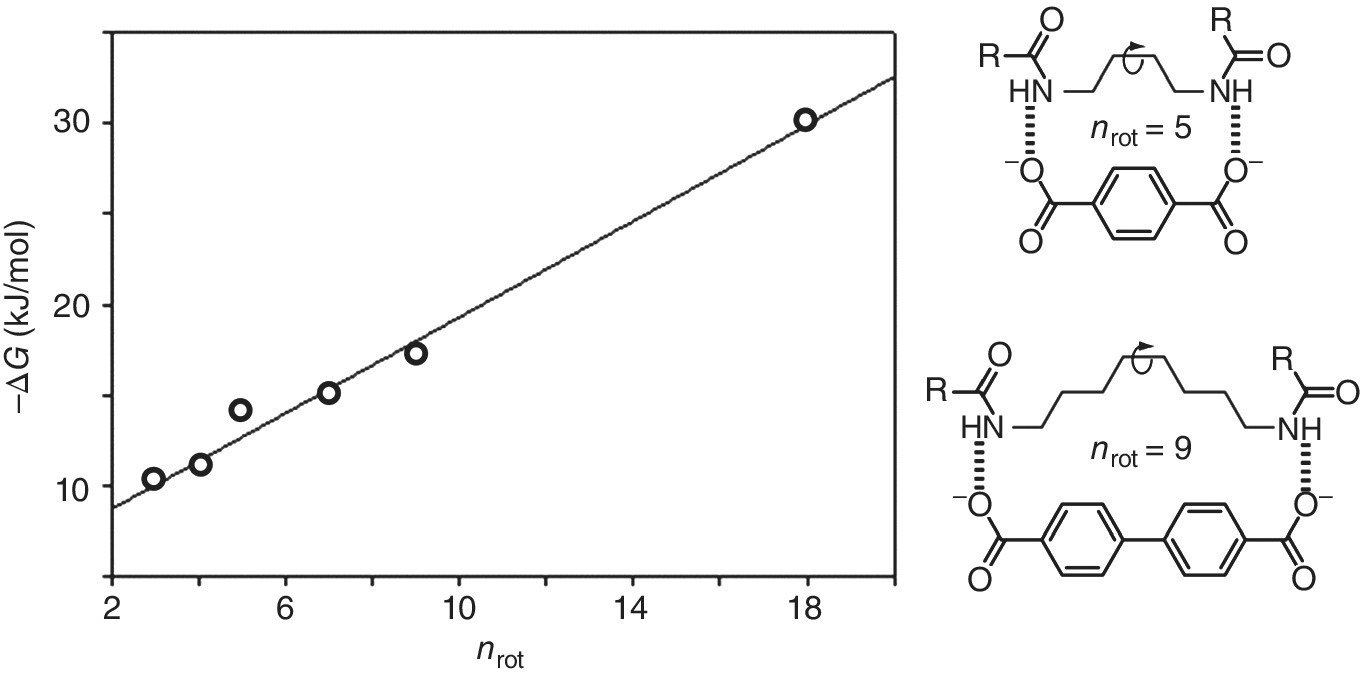
Figure 1.6 Free energies of complex formation between α,ω‐diamides and α,ω‐dicarboxylates in CHCl3 as a function of the number of rotatable single bonds (nrot) between the terminal amide and carboxylate functions.
Source: Ref. [28]. Reproduced with permission of VCH/Wiley.
1.3.2 Mismatch as Limitation of Additivity
The most obvious limitation for additivity of non‐covalent interactions and therefore also for the lock‐and‐key principle is the necessary geometric fit between host and guest [34]. Insufficient fit between receptor and ligand is a major factor, in particular for a conformationally more rigid polyvalent entity [1]. The steric requirements for an optimal binding between host and guest depend on the nature of the non‐covalent bonds. In particular, electrostatic interactions fall off with only with r−1 between binding sites whereas dispersive interactions fall off with r−6. In addition, the latter interactions have no or only a small directional dependence, whereas for example the strength of hydrogen or halogen bonds depends on the orientation of donor and acceptor. Exceptions are molecular containers [35] in which the binding of substrates is in most cases controlled by the size of the portals. However, here as in other supramolecular complexes another important restriction is the presence of solvent molecules in a ligand‐containing cavity, so that the guest molecule can only use a limited number of interactions which are possible, again depending on the binding mechanism. Thermal motions as well as vibrational and translatory freedom of movement of host and guest are also responsible for the limited fitting; moreover, the surfaces of interacting molecules are characterized by corners and dimples. Recent studies with cryptophanes composed of two bowl‐shaped cyclotriveratrylene units showed large solvent molecules such as tetrachloroethane inside the cavity [36]. It has been found earlier [37] that for example some cryptophanes bind, say, chloroform better than methane, although methane fits geometrically as well in the cavity. An occupancy factor or packing coefficient (PC) of 0.886 was calculated for the chloroform complex, similar to that in a closely packed crystal. For methane the occupancy factor amounts to a PC of only 0.35. These values are in the range with later systematic evaluations with many container‐ and capsule‐type hosts [38], which were leading to generally observed 55 ± 9% occupancy of the space available.
Even small geometric changes can have a dramatic impact on the stability of supramolecular complexes, such as in recently described associations with crown‐ammonium pseudorotaxanes [39] (Scheme 1.3). Here insertion of just one methylene group in the spacer leads to a drop from K = 25 000 M−1 for the optimal spacer (n = 0) to K = 1100 M−1 with the longer spacer (n = 1), due to differences in both ΔH (−4.8 kJ/mol) and TΔS (2.9 kJ/mol).

Scheme 1.3 Complex with crown‐ammonium pseudorotaxanes [39], with a very large affinity difference between spacer length of either n = 0 or n = 1.
Frequently one interaction in a supramolecular complex is significantly larger than another one, which then can lead to an induced misfit. Figure 1.7 illustrates schematically the consequences for cyclodextrin complexes as an example [40]. Only in ideal situations like in Case I (Figure 1.7a) one can expect additivity (as for example with the nucleotide complexes in Scheme 1.2). In Case II (Figure 1.7b) the force between D and A is so strong that the second interaction is severely diminished, with an ensuing loss of additivity. Such situations have been seen for example with complexes of nucleotides and cyclodextrins, which bear a different number n of aminoalkyl substituents at the rim [41,42]. With the monosubstituted cyclodextrin CDI (n = 1) the affinity increases from AMP to ATP by only ΔΔG = 4.7 kJ/mol (Scheme 1.4), much less than expected by the possible increase of salt bridges between the phosphate residue and the CDI cation, and in contrast to observations with cyclophane complexes (Scheme 1.2). This indicates that the nucleoside residue seeks a sufficient contact with the CDI moiety, resulting in diminished ion pair contacts. Furthermore, there is a moderate selectivity with respect to the nucleobase, but the differences between AMP, GMP, CMP and UMP become smaller with the stronger binder CDII (n = 7), for example the ΔΔG between AMP and CMP diminishes from 7 to 4 kJ/mol (Scheme 1.4). This is the result of the then much stronger D…A salt bridge, which allows less contact between the cyclodextrin moiety and the nucleoside residue.
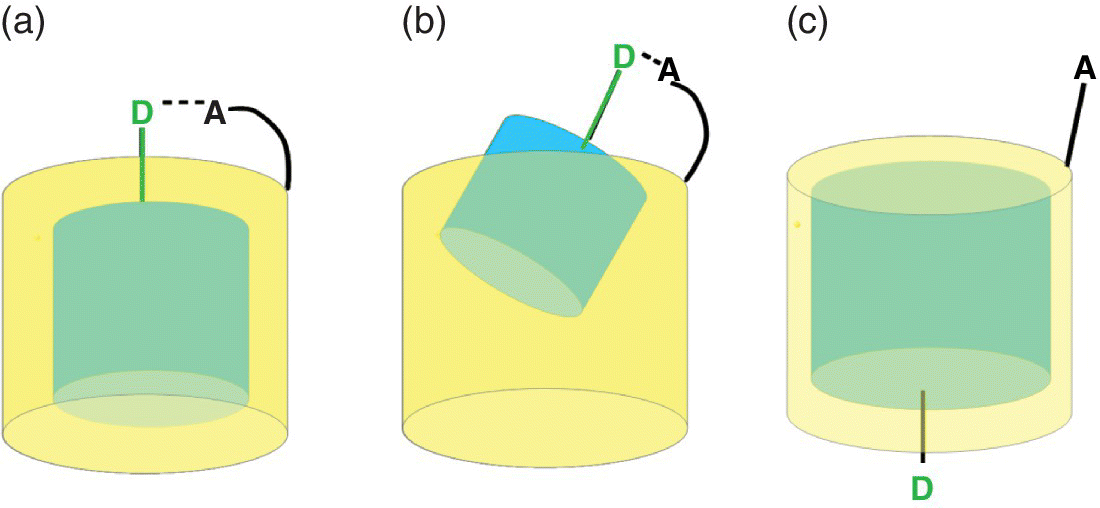
Figure 1.7 Schematic consequences of mismatch: (a) similar interaction in‐ and outside and sufficient matching (e.g. Case I); (b) stronger interaction outside (Case II); (c) stronger interaction inside cavity (e.g. Case III).
Source: Ref. [40]. Reproduced with permission of Royal Society of Chemistry.
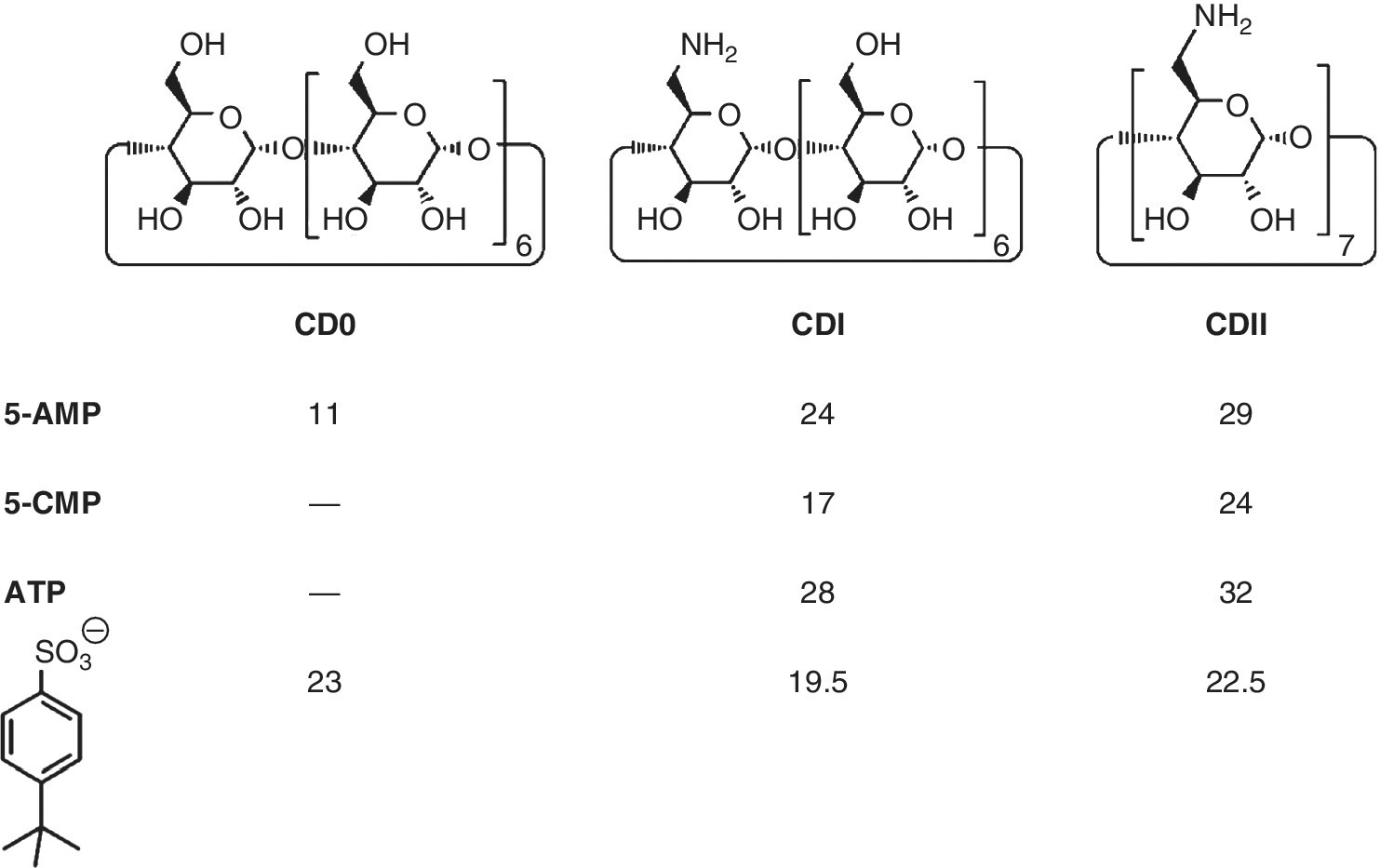
Scheme 1.4 Complexation free energies ΔG (kJ/mol) of ß‐cyclodextrin derivatives bearing zero, one or seven charges at the rim (CD0, CDI, CDII) with AMP, ATP and p‐tert‐butylphenyl compounds.
Data from Ref. [42].
In Case III (Figure 1.7c) one interaction is so strong that the second one can barely materialize. The strong interaction of the butylphenyl residue in the cyclodextrins dominates the binding mode, and prohibits a contact between the anion and cation. This is obvious from the affinity with the positively charged host CDI which strikingly is even smaller in comparison with the neutral CD0, and from the negligible difference between CD0 and CDII complexes [42].
Stereoelectronic effects are also difficult to count as additive contribution, since they strongly depend on orientation, as shown for example for complexes between 1.10‐diaza‐crown and potassium ions [43]. Here, only after introduction of methyl groups at the nitrogen atoms are the lone pairs enforced towards a diequatorial orientation, and the binding energy increases to much larger affinity (Figure 1.8). A similar situation holds for other directional enforcers, in particular for hydrogen bonds, and makes it difficult to simply summarize the number of interactions.

Figure 1.8 Stereoelectronics: the 1.10‐diaza‐crown with R = H (diaxial lone pair orientation, (a) binds K+ ions with only ΔG = 10 kJ/mol, with R = Me (diequatorial lone pair orientation, (b) ΔG increases to 26 kJ/mol (in methanol)
Source: Ref. [43]. Reproduced with permission of John Wiley and Sons.
1.3.3 Medium Effects as Limiting Factor
Solvent effects can also significantly limit the possible additivity in multivalent complexes. First, they can decisively change the binding mechanism. Thus, dispersive interactions can be large in water, but are negligible in most organic solvents [16]. The energy for desolvation of host and guest prior to complex formation depends critically on the nature of binding elements, and thus can obscure additivity. In addition, solvophobic contributions can lead to a complete independence of specific non‐covalent forces. In particular, water as medium, but also other solvents of low polarizability [44] can lead to dominating solvophobic forces. Especially cucurbituril hosts, which lack binding sites inside their cavity, complex with unsurpassed affinity with many ligands [45,46,47]. It has been shown that these cucurbiturils contain a sizeable number of water molecules which usually can exert only a few inter‐water hydrogen bonds. If these are replaced by a suitable guest and freed to the bulk, they enjoy close to four hydrogen bonds. High energy water inside cavities is also present in for example cyclodextrins, cyclophanes, some tweezer or cleft hosts, and so on, and contributes to binding which is difficult to separate from direct non‐covalent interactions [48] (Figure 1.9). Crystal structures of cyclodextrin hydrates have indicated the presence of such less coordinated water inside the cavity [49].

Figure 1.9 Examples of high energy water. (a) Cucurbit[8]uril (CB8) and 14 water molecules. (b) CB8 with viologen as guest and 6 water molecules in the cavity. (c) ß‐Cyclodextrin with 5 water molecules, all from molecular dynamics simulations.
Source: Ref. [48]. Reproduced with permission of John Wiley and Sons.
(d) ß‐Cyclodextrin dodecahydrate structure derived from neutron diffraction.
Source: Ref. [49]. Reproduced with permission of American Chemical Society.
1.3.4 Strain and Induced Fit
Many, if not most complex formations occur with some conformational changes for maximizing the pertinent non‐covalent interactions. Such an induced fit necessarily costs some strain energy, leading to weaker affinities than they would be if all possible interactions would be simply additive. This poses limits to the evaluation of additive single free energies from the observed total complexation free energies. Such strain effects play a particular role in cooperativity and allostery in multivalent complexes, which are dealt with in the following sections.
1.4 Cooperativity
Positive cooperativity implies that the binding of one ligand to one of several binding sites in a receptor enhances the affinity at other sites, while negative cooperativity diminishes the affinity [1,4,5,50,51,52]. In classical allosteric systems this is due to conformational coupling between binding sites, as will be discussed in Section 1.5. Cooperativity also occurs if there are direct interactions between the complexed guest molecules. This is typical for ion pair complexation [53,54,55] where the electrostatic forces between anion and cation can lead to significantly enhanced binding constants K (Scheme 1.5). In Case A [56] the presence of Na+ increases the value of K from 20 to 620 M−1, in the crown ether host (Case B) the K increases from 50 to 470 M−1 in presence of Na+ [57].

Scheme 1.5 Positive cooperativity: enhanced anion binding constants in heterotopic complexes.
The cyclopeptide A shown in Scheme 1.6 binds BuNMe3X salts in chloroform for X = I with K = 300 M−1, while for the tosylate (X = OTs) a K increase by 104 was observed compared with the iodide, explained also by a tosylate‐induced conformational change of the host [58]. A related host B [59] binds very efficiently N‐methyl‐quinuclidinium iodide as ion pair in chloroform with K = 8.3 × 104 M−1. With the host C a 260‐fold affinity increase to K = 1.8 × 104 M−1 was observed, with +H3NCH(Bn)CO2Me as the cation and nitrate as anion, while tetraalkylammonium salts bind weakly due the steric hindrance of the tetraalkyl residue, with for example K = 70 M−1 with nitrate as the anion [60].

Scheme 1.6 Cyclopeptide hosts receptors with cooperativity between binding of anions and cations.
1.5 Allostery
Typical allosteric systems exhibit cooperativity due to conformational coupling between binding sites [61,62]. Case A shown in Scheme 1.6 [59] exemplifies that changes in flexible host structures may often play a large role in limitation of additivity rules. Extreme limitations of observable additivity occur in allosteric systems, which form a binding cavity only in the presence of strongly bound effector, such as metal ions in complexes A and B in Scheme 1.7. In complexes A and B the affinity of the fluorescent dye DNSA (dansylamide) in the absence of the zinc ion is so weak that it cannot be measured, so that the cooperativity ratio amounts to Krel = KZn/0 > 100 [63,64].
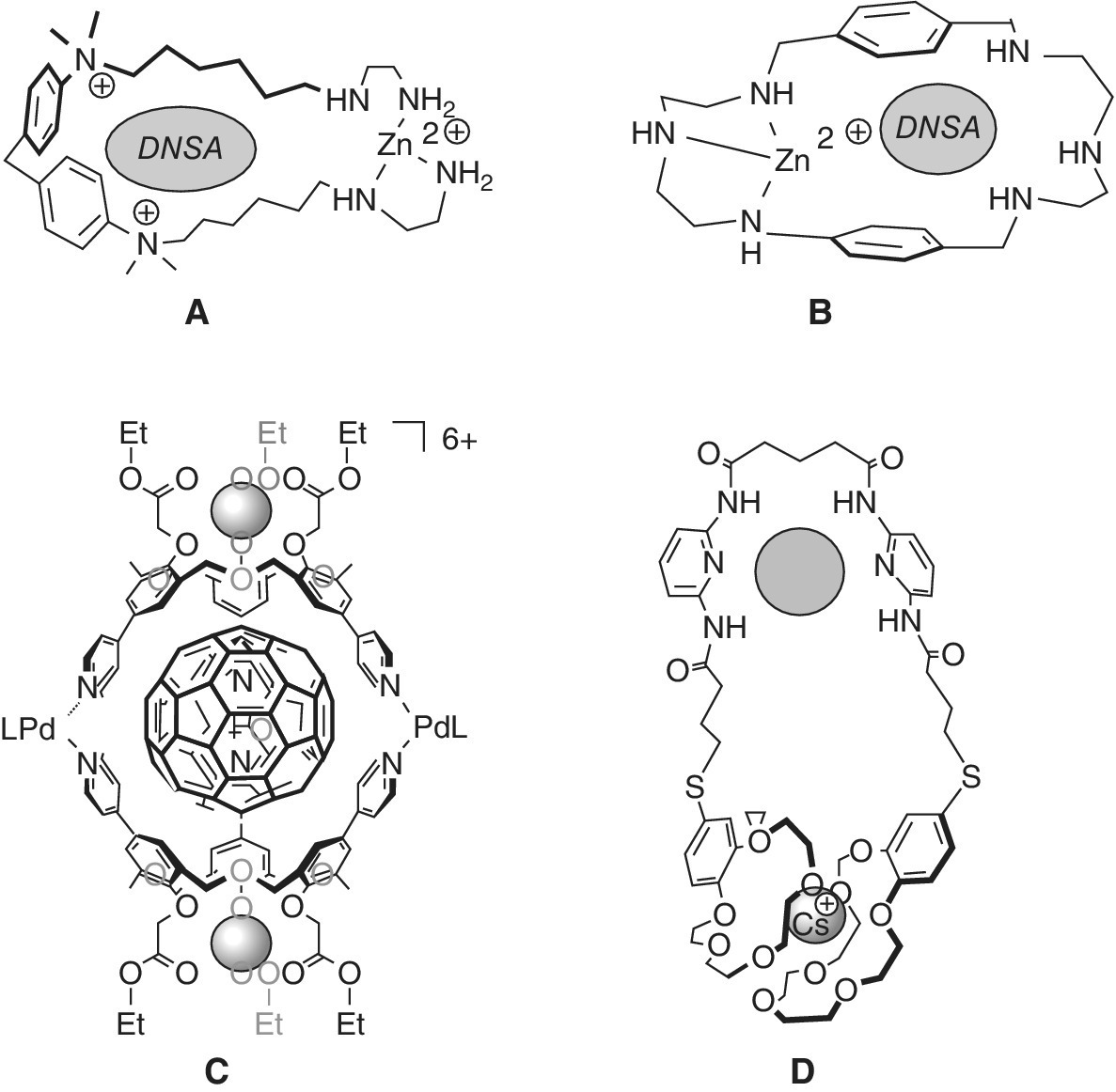
Scheme 1.7 Cooperativity with allosteric ditopic receptor complexes, see text.
In complex C (Scheme 1.7) a conformational change induced by Li+ ions leads to strong binding of [60]fullerene with K = 2.1 × 103 M−1, in comparison with K = 39 M−1 without the metal [65]. A negative cooperativity is seen with Na+, with Krel < 10. The association of anions such as chloride with amide functions in complex D (Scheme 1.7) is significantly enhanced by complexation with Cs+ ions, due to interaction with the crown ether units by a conformational rearrangement [66]. In s‐hydrindacenes conformational changes of binding group orientation and polarity is observed upon association with substituted resorcinols, with a cooperativity ratio K2/K1 of up to 30 (Scheme 1.8) [67].

Scheme 1.8 Positive cooperativity in s‐hydrindacene complexes: conformational changes lead to association with substituted resorcinols with a cooperativity ratio K2/K1 of up to 30 [68].
Conformational changes within a receptor, induced by an effector molecule, can lead to reinforced binding at different receptor locations [68]. In flexible proteins correlated rearrangement allows allosteric communication between different locations [69,70,71]; the ensuing entropic factors will limit the additivity of single binding contributions [72]. The host CER (Figure 1.10) exhibits a related allosteric complexation; it bears anionic binding groups attached at the end of cholic acid arms which by hydrophobic interactions between them fold back and complex by ion pairing 1,3,5‐tris(amino methyl) benzene as guest G with K = 138 M−1, in comparison with only 24 M−1 with a parent receptor R lacking the steroidal arms. Both enthalpic and entropic contributions are responsible for the different complexations [73].

Figure 1.10 (a) Receptor CER with and R without steroidal arms, tricationic guest G3+. (b) Hydrophobic interactions between the steroidal arms of CER preorganizes the CER for binding of guest G3+.
Source: Ref. [73]. Reproduced with permission of American Chemical Society.
Finally, we note that the decisive factor for the efficiency of allosteric systems with positive cooperativity is the conformational energy ΔGC required for the formation of a suitable cavity for ligand binding in the absence of an effector. ΔGC is usually dominated by an increase of strain in a folded conformer and/or by high energy solvents within a cavity. A recent analysis of the thermodynamics in synthetic allosteric systems exemplifies how the binding strength of an effector molecule at a second binding site must pay for the energy ΔGC needed for the binding of the first substrate [74]. Additivity of the binding contributions can only be expected if ΔGC could be determined independently. Negative cooperativity depends only on the difference ΔΔGA,B between the binding energies at the two sites, which are enhanced or lowered by concomitant changes in ΔGC. The often small efficiency of synthetic allosteric receptors [61,62], measured by the binding constant ratio KA/KA(B), in which KA refers to association of ligand A in the absence of the effector ligand B, and KA(B), is due to small ΔGC values. Larger efficiency can be expected with increased ΔGC values, for example by introduction of alkyl substituents in the ortho‐position of pyridine in the often used [61,62] bipyridyl‐based allosteric systems.
1.6 Conclusions
For efficient multivalent complexes it is desirable to preserve as much as possible additivity of all possible binding contributions. Ideally not only the affinity and therefore sensitivity but also the selectivity of such complexes is optimal if additivity of the single binding free energies is materialized. To what degree a geometric fit in the sense of the lock‐and‐key principle or preorganization is required depends first on the binding mechanism. The distance dependence of the interaction increases distinctly from electrostatic effects or ion pairing to dispersive forces. Mismatch between binding partners leads to a strong decrease of both affinity and selectivity particularly if the binding mechanism is characterized by a steeper distance dependence, and if the components are less flexible. Solvents can strongly influence the binding mechanisms; hydrophobic effects of high energy water inside cavities or clefts can make intermolecular binding contributions unimportant.
If one binding contribution is much stronger than others, the second interaction is often severely weakened due to mismatch; even a complete change of binding modes can occur. High selectivity combined with high affinity, which both require optimal fit, is difficult to attain if binding sites in a receptor are rigidly connected. In principle one can overcome this problem by flexible connections between a primary binding site securing high affinity with another site securing selectivity, provided such sites are available. If a multivalent complex should operate in for example a nanomolar solution the primary interactions should be worth about 50 kJ/mol, while at the secondary site values of, say, ΔGX = 15 and ΔGY = 5 kJ/mol are enough to achieve a sizeable selectivity for distinction of two compounds X and Y (ΔΔG ≈ 10 kJ/mol or KX/KY ≈ 100).
In systems with positive cooperativity larger affinity than that predicted by additive single interactions is possible either by attractive forces between nearby bound substrates, or in the classical case by conformational change at one site A induced by occupation at another site B. The efficiency of related allosteric systems can be defined as the ligand A concentration needed for complexation in the absence of the effector binding at B; it depends on the strain energy which would be needed to form an optimal conformation for binding A in the absence of occupation at B. Instead of conformational strain unfavorable solvents in cavities or clefts, such as high energy water, can enhance the efficiency of allosteric systems. It is hoped that the design of synthetic systems for, say, highly sensitive and selective new sensors as well as for drug design can be facilitated by taking into account some of the limitations and possibilities outlined in this chapter.
References
- 1 Mammen, M.; Choi, S.‐K.; Whitesides, G. M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794.
- 2 Persch, E.; Dumele, O.; Diederich, F. Molecular recognition in chemical and biological systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327.
- 3 Schneider, H.‐J. Binding mechanisms in supramolecular complexes Angew. Chem. Int. Ed. 2009, 48, 3924–3977.
- 4 Hunter, C. A. Quantifying intermolecular interactions: Guidelines for the molecular recognition toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324.
- 5 Biedermann, F.; Schneider, H.‐J. Experimental binding energies in supramolecular complexes, Chem. Rev. 2016, 116, 5216–5300.
- 6 Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E. W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. 2012, 51, 10472–10498.
- 7 Schneider, H.‐J.; Yatsimirsky, A. Selectivity in supramolecular host–guest complexes Chem. Soc. Rev. 2008, 263–277.
- 8 Schneider, H.‐J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, 2000.
- 9 Hossain, M. A.; Schneider, H.‐J. Flexibility, association constants, and salt effects in organic ion pairs: How single bonds affect molecular recognition. Chemistry Eur. J. 1999, 5, 1284–1290.
- 10 De Robertis, A.; De Stefano, C.; Foti, C.; Giuffrè, O.; Sammartano, S. Thermodynamic parameters for the binding of inorganic and organic anions by biogenic polyammonium cations. Talanta 2001, 54, 1135–1152, and references cited therein.
- 11 Daniele, P. G.; Foti, C.; Gianguzza, A.; Prenesti, E.; Sammartano, S. Weak alkali and alkaline earth metal complexes of low molecular weight ligands in aqueous solution. Coord. Chem. Rev. 2008, 252, 1093–1107.
- 12 Guo, D. S.; Wang, L. H.; Liu, Y. Highly effective binding of methyl viologen dication and its radical cation by p‐sulfonatocalix[4,5]arenes. J. Org. Chem. 2007, 72, 7775–7778.
- 13 Schneider, H.‐J.; Blatter, T.; Palm, B.; Pfingstag, U.; Rüdiger, V.; Theis, I. Complexation of nucleosides, nucleotides, and analogs in an azoniacyclophane J. Am. Chem. Soc. 1992, 114, 7704–7708.
- 14 Schneider, H.‐J.; Schiestel, T.; Zimmermann, P. The incremental approach to noncovalent interactions: Coulomb and van der Waals effects in organic ion pairs. J. Am. Chem. Soc. 1992, 114, 7698–7703.
- 15 Liu, T.J.; Schneider H.‐J. Additivity and quantification of dispersive interactions – from cyclopropyl‐ to nitrogroups. Angew. Chem. Int. Ed. 2002, 41, 1368–1370.
- 16 Schneider, H.‐J. Dispersive interactions in solution complexes Acc. Chem. Res. 2015, 48, 1815–1822.
- 17 Lorand, J. P. Hammett correlations for phenylacetonitriles and tetrafluorobenzenes with HMPA. J. Phys. Org. Chem. 2011, 24, 267–273.
- 18 Jencks, W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 1981, 78, 4046–4050.
- 19 Cornish‐Bowden, A. Enthalpy–entropy compensation: A phantom phenomenon. J. Biosci. 2002, 27, 121–126.
- 20 Ford, D. M. Enthalpy − entropy compensation is not a general feature of weak association. J. Am. Chem. Soc. 2005, 127, 16167–16170.
- 21 Zhou, H.‐X.; Gilson, M. K. Theory of free energy and entropy in noncovalent binding. Chem. Rev. 2009, 109, 4092–4107.
- 22 Dunitz, J. D. Win some, lose some: Enthalpy–entropy compensation in weak intermolecular interactions. Chem. Biol. 1995, 2, 709–712.
- 23 Chodera, J. D.; Mobley, D. L. Entropy–‐enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142.
- 24 Sharp, K. Entropy—enthalpy compensation: Fact or artifact? Protein Sci. 2001, 10, 661–667.
- 25 Camara‐Campos, A.; Musumeci, D.; Hunter, C. A.; Turega, S. Chemical double mutant cycles for the quantification of cooperativity in H‐bonded complexes. J. Am. Chem. Soc. 2009, 131, 18518–18524.
- 26 Searle, M. S.; Williams, D. H. The cost of conformational order: entropy changes in molecular associations. J. Am. Chem. Soc. 1992, 114, 10690–10697; Groves, P.; Searle, M. S.; Westwell, M. S.; Williams, D. H. Expression of electrostatic binding cooperativity in the recognition of cell‐wall peptide analogues by vancomycin group antibiotics. J. Chem. Soc., Chem. Commun. 1994, 1519–1520.
- 27 Mammen, M.; Shakhnovich, E. I.; Whitesides, G. M. Using a convenient, quantitative model for torsional entropy to establish qualitative trends for molecular processes that restrict conformational freedom. J. Org. Chem. 1998, 63, 3168–3175.
- 28 Eblinger, F.; Schneider, H.‐J. Stabilities of hydrogen‐bonded supramolecular complexes with various numbers of single bonds. Angew. Chem. Int. Ed. 1998, 37, 826–829.
- 29 Nowick, J. S.; Cary, J. M.; Tsai, J. H. A Triply Templated artificial β‐sheet. J. Am. Chem. Soc. 2001, 123, 5176–5180.
- 30 Christensen, T.; Gooden, D. M.; Kung, J. E.; Toone, E. J. Additivity and the physical basis of multivalency effects: A thermodynamic investigation of the calcium EDTA interaction. J. Am. Chem. Soc. 2003, 125, 7357–7366.
- 31 Smith, R.; Martell, A. Critical Stability Constants, Vol. 2, Plenum Press, New York, 1975.
- 32 Sun, H.; Navarro, C.; Hunter, C. A. Influence of non‐covalent preorganization on supramolecular effective molarities. Org. Biomol. Chem. 2015, 13, 4981–4992.
- 33 Sun, H.; Hunter, C. A.; Llamas, E. M. The flexibility‐complementarity dichotomy in receptor‐ligand interactions. Chem. Sci. 2015, 6, 1444–1453, and references cited therein.
- 34 Schneider, H.‐J. Limitations and extensions of the lock‐and‐key principle: differences between gas state, solution and solid state structures. Int. J. Mol. Sci. 2015, 16, 6694.
- 35 Cram, D. J.; Cram, J. M. Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, 1997.
- 36 Haberhauer G.; Woitschetzki, S.; Bandmann, H. Strongly underestimated dispersion energy in cryptophanes and their complexes Nature Commun. 2014, 5, Article 3542, 1–7.
- 37 Garel, L.; Dutasta, J.; Collet, A. Complexation of methane and chlorofluorocarbons by cryptophane‐A in organic solution Angew. Chem. Int. Ed. 1993, 32, 1169–1171; Collet, A. in Comprehensive Supramolecular Chemistry, Vol. 2 (Eds J. L. Atwood, J. E. Davies, D. D. MacNicol, and F. Vögtle), Elsevier, Dordrecht, 1996, 325–365.
- 38 Mecozzi, S.; Rebek Jr, J. The 55% solution: A formula for molecular recognition in the liquid state. Chem. Eur. J., 1998, 4, 1016–1022, and references cited therein.
- 39 Schalley, C. A.; Beizai, K.; Vögtle, F. On the way to rotaxane‐based molecular motors: Studies in molecular mobility and topological chirality. Acc. Chem. Res. 2001, 34, 465–476.
- 40 Schneider, H.‐J. Spatial mismatch, non‐additive binding energies and selectivity in supramolecular complexes. Org. Biomol. Chem. 2017, 15, 2146–2151.
- 41 Eliseev, A. V.; Schneider, H.‐J. Molecular recognition of nucleotides, nucleosides, and sugars by aminocyclodextrins. J. Am. Chem. Soc. 1994, 116, 6081–6088.
- 42 Wenz, G.; Strassnig, C.; Thiele, C.; Engelke, A.; Morgenstern, B.; Hegetschweiler, K. Recognition of ionic guests by ionic ß‐cyclodextrin derivatives. Chem. Eur. J. 2008, 14, 7202–7211.
- 43 Solovev, V. P.; Strakhova, N. N.; Kazachenko, V. P.; Solotnov, A. F.; Baulin, V. E.; Raevsky, O. A.; Rüdiger, V.; Eblinger, F.; Schneider, H.J. Steric and stereoelectronic effects in aza crown ether complexes. Eur. J. Org. Chem. 1998, 1379–1389.
- 44 Yang, L.; Adam, C.; Cockroft, S. L. Quantifying solvophobic effects in nonpolar cohesive interactions. J. Am. Chem. Soc. 2015, 137, 10084–10087.
- 45 Barrow, S. J.; Kasera, S.; Rowland, M. J.; Del Barrio, J.; Scherman, O. A. Cucurbituril‐based molecular recognition. Chem. Rev. 2015, 115, 12320–12406.
- 46 Shetty, D.; Khedkar, J. K.; Park, K. M; Kim, K. Can we beat the biotin‐avidin pair?: Cucurbit[7]uril‐based ultrahigh affinity host–guest complexes and their applications Chem. Soc. Rev. 2015, 44, 8747–8761.
- 47 Isaacs, L. Stimuli responsive systems constructed using cucurbit[n]uril‐type molecular containers. Acc. Chem Res. 2014, 47, 2052–2062.
- 48 Biedermann, F.; Nau, W. M.; Schneider, H.‐J. The hydrophobic effect revisited Angew. Chem. Int. Ed. 2014, 53, 11158–11171.
- 49 Betzel, C.; Saenger, W.; Hingerty, B. E.; Brown, G. M. Circular and flip‐flop hydrogen bonding in ß‐cyclodextrin undecahydrate: A neutron diffraction study. J. Am. Chem. Soc. 1984, 106, 7545–7557.
- 50 Di Cera, E. Site‐specific thermodynamics: understanding cooperativity in molecular recognition. Chem. Rev. 1998, 98, 1563–1592.
- 51 Hunter, C. A.; Anderson, H. L. What is cooperativity? Angew. Chem., Int. Ed. 2009, 48, 7488–7499.
- 52 Mahadevi, A.S.; Sastry, G.N. Cooperativity in noncovalent interactions. Chem. Rev. 2016, 116, 2775–2825.
- 53 McConnell, A. J.; Beer, P. D. Heteroditopic receptors for ion‐pair recognition. Angew. Chem. Int. Ed. 2012, 51, 5052–5061
- 54 Chrisstoffels, L. A. J.; de Jong, F.; Reinhoudt, D. N.; Sivelli, S.; Gazzola, L.; Casnati, A.; Ungaro, R. Facilitated transport of hydrophilic salts by mixtures of anion and cation carriers and by ditopic carriers. J. Am. Chem. Soc. 1999, 121, 10142–10151.
- 55 Kim, S. K.; Sessler, J. L. Ion pair receptors. Chem. Soc. Rev. 2010, 39, 3784–3809.
- 56 Webber, P. R.; Beer, P. D. Ion‐pair recognition by a ditopic calix [4] semitube receptor. Dalton Trans. 2003, 2249–2252.
- 57 Deetz, M. J.; Shang, M.; Smith, B. D. A macrobicyclic receptor with versatile recognition properties: simultaneous binding of an ion pair and selective complexation of dimethylsulfoxide. J. Am. Chem. Soc. 2000, 122, 6201–6207.
- 58 Kubik, S. Large increase in cation binding affinity of artificial cyclopeptide receptors by an allosteric effect. J. Am. Chem. Soc. 1999, 121, 5846–5855.
- 59 Kubik, S.; Goddard, R. A new cyclic pseudopeptide composed of (L)‐proline and 3‐aminobenzoic acid subunits as a ditopic receptor for the simultaneous complexation of cations and anions. J. Org. Chem. 1999, 64, 9475–9486.
- 60 Gong, J.; Gibb, B. C. A new macrocycle demonstrates ditopic recognition properties. Chem. Commun. 2005, 1393–1395.
- 61 Kremer, C.; Lützen, A. Artificial allosteric receptors. Chem. Eur. J. 2013, 19, 6162–6196.
- 62 Kovbasyuk, L.; Krämer, R. Allosteric supramolecular receptors and catalysts. Chem. Rev. 2004, 104, 3161–3188.
- 63 Schneider, H. J.; Ruf, D. A synthetic allosteric system with high cooperativity between polar and hydrophobic binding sites. Angew. Chem. Int. Ed. 1990, 29, 1159–1160.
- 64 Baldes, R.; Schneider, H. J. Complexes from polyazacyclophanes, fluorescence indicators, and metal cations Angew. Chem. Int. Ed. 1995, 34, 321–323.
- 65 Ikeda, A.; Udzu, H.; Yoshimura, M.; Shinkai, S. Inclusion of [60]fullerene in a self‐assembled homooxacalix[3]arene‐based dimeric capsule constructed by a PdII–pyridine interaction. Tetrahedron 2000, 56, 1825–1832.
- 66 Nabeshima, T.; Hanami, T.; Akine, S.; Saiki, T. Control of ion binding by cooperative ion‐pair recognition using a flexible heterotopic receptor. Chem. Lett. 2001, 30, 560–561.
- 67 Kawai, H. Hydrindacenes as versatile supramolecular scaffolds. Bull. Chem. Soc. Jpn. 2015, 88, 399–409.
- 68 Otto, S. Reinforced molecular recognition as an alternative to rigid receptors. Dalton Trans. 2006, 2861–2864.
- 69 DuBay, K. H.; Bowman, G. R.; Geissler, P. L. Fluctuations within folded proteins: Implications for thermodynamic and allosteric regulation. Acc. Chem. Res. 2015, 48, 1098–1105.
- 70 Frederick, K. K.; Marlow, M. S.; Valentine, K. G.; Wand, A. J. Conformational entropy in molecular recognition by proteins. Nature 2007, 448, 325–329.
- 71 Motlagh, H. N.; Wrabl, J. O.; Li, J.; Hilkser, V. J. The ensemble nature of allostery. Nature 2014, 508, 331–339.
- 72 Williams, D. H.; Stephens, E.; O'Brien, D. P.; Zhou, M. Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand‐induced reductions in motion within receptors and enzymes. Angew. Chem. Int. Ed. 2004, 43, 6596–6616.
- 73 Gunasekara, R. W.; Zhao, Y. Rationally designed cooperatively enhanced receptors to magnify host–guest binding in water. J. Am. Chem. Soc. 2015, 137, 843–849.
- 74 Schneider, H.‐J. Efficiency parameters in artificial allosteric systems. Org. Biomol. Chem., 2016, 14, 7994–8001.