
9
Blocking Pathogens by Multivalent Inhibitors
Sumati Bhatia, Benjamin Ziem, and Rainer Haag
Institute of Chemistry and Biochemistry, Freie Universität Berlin, Takustraβe 3, 14195, Berlin, Germany
9.1 Introduction
Multivalency is a unique concept in nature to achieve strong interfacial reversible interactions between m‐valent ligands and n‐valent receptors of the participating binding partners (with m, n > 1) to increase the binding strength and the kinetic stability. Furthermore, multivalency plays an essential role in biological systems for recognition, adhesion, and signaling processes involving antibodies, membranes, molecules, cells, and pathogens such as viruses and bacteria [1–4]. Understanding the mode of action at the molecular level is the first priority for designing multivalent scaffolds, which can play a huge role in the fields of medicine, bio‐ and supramolecular chemistry, or materials science. The velcro fastener represents one example of these decoding and developing processes, which are necessary to achieve a marketable system. Inspired by the natural burr (Figure 9.1a), the velcro fastener (Figure 9.1b) also uses hooks and loops for reversible multivalent ligand–receptor interactions.
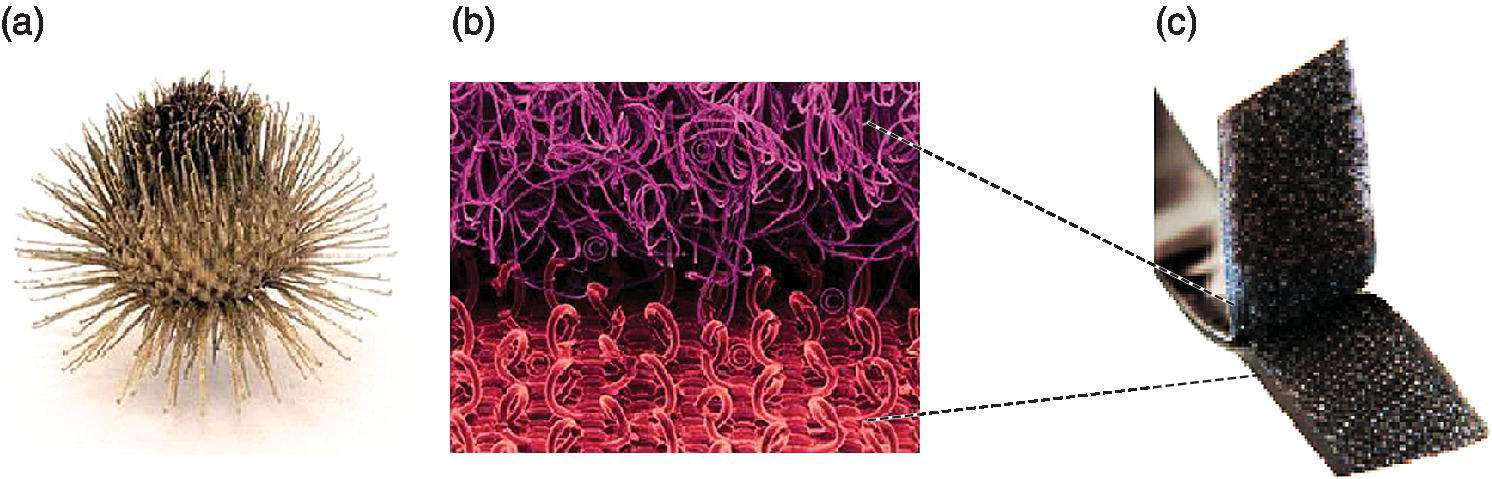
Figure 9.1 (a) A natural burr. (b) The binding mode of velcro on a molecular level. (c) A velcro fastener.
Source: Adapted from Ref. [2]. Reproduced with permission of John Wiley and Sons.
Another example for natural multivalency is the pathogen adhesion, for example, of bacteria, or virus particles to the host cells. This adsorption process is based on non‐covalent bindings towards the favored glycan or protein structure on the extra cellular matrix or directly on the cell surface (Figure 9.2a). Influenza virus, for example, initially binds to sialic acid in the mucous while herpesviridae‐beta prefers heparansulfate as an interaction partner in the extracellular matrix [1]. After cellular uptake, the genetic material is released to initiate pathogen reproduction followed by the spreading of new virus particles. So far, only small monovalent drugs (Figure 9.2b), for example, Tamiflu as neuraminidase inhibitor against influenza infections and other antibiotics for different bacterial and fungal infections, have been applied in high doses for medical treatment, which can lead to resistance of the pathogens [5,6]. A new approach is, therefore, required, which not only binds with several receptors on the target but also prevents cell adhesion through steric shielding (Figure 9.2c–f). This strategy can lower the dose of multivalent drugs to achieve an improved efficacy in therapies as compared with monovalent analogs.

Figure 9.2 (a) Multivalent interaction of a pathogen particle to the cell surface. (b) A standard, highly dosed monovalent drug cannot efficiently prevent pathogen adhesion by blocking single pathogen receptors. (c) A globular multivalent inhibitor decorated with multiple ligands is able to bind several receptors simultaneously and additionally influence the operability of further receptors due to its shape and size. (d) In contrast to the more rigid globular architectures, dendritic or starlike polymer‐based structures can be disposed as highly adaptive inhibitors to enhance the binding and shielding efficiency. (e) Linear polymeric inhibitors offer a more flexible and higher surface area compared with their globular equivalents. Due to this fact, they are capable of stretching and coiling to access more receptor sites simultaneously, and additionally can sterically shield further receptors. (f) Since a flexible two‐dimensional architecture functionalized by numerous multivalent ligands is not only able to strongly interact with the pathogen and shield several binding sites, it should ideally be able to wrap the whole infectious particle.
Source: (a and b) Adapted from Ref. [2]. Reproduced with permission of John Wiley and Sons. (c–f) Adapted from Ref. [9]. Reproduced with permission of American Chemical Society.
Developing an efficient multivalent pathogen inhibitor is a complex process. First of all, the binding mode of the target pathogen has to be decrypted using thermodynamic and kinetic simulations. An accurate theoretical modeling [7,8] is, however, very difficult, due to the complexity of biological systems, which often prevents good theoretical predictions. Furthermore, toxicity as well as biocompatibility also play a major role in the development process, since the intended inhibitors should not be harmful to the treated organisms. Although constant efforts have brought up a number of multivalent inhibitors with great potential, even against drug‐resistant pathogens, the pharmaceutical industry has not yet translated this concept. The reason might be that the production of polymeric multivalent ligand architectures is more challenging in terms of polydispersity and reproducible therapeutic efficacy than that of monovalent drug molecules [9]. In spite of these challenges a few systems [10,11] have been successfully tested in vivo and further studies are required to explore the full potential of multivalency for pathogen blocking.
9.2 Design of Multivalent Ligand Architectures
The monovalent ligand–receptor interaction is mostly an enthalpy driven process where a ligand diffuses in solution to the target and binds a receptor with a free energy of interaction ∆G = ∆H − T∆S, where ∆G is the free energy of binding and is the sum of an enthalpic (∆H) and an entropic (−T∆S) contributions. Only two states (bound and unbound) exist in a monovalent system from which the corresponding free enthalpy difference can be calculated. In the multivalent system, significant entropic penalties are incorporated by the conformational flexibility of the scaffold itself and the spacer linking the multiple ligands to the backbone. An additional entropic contribution is imposed by the release of water molecules from the binding site during hydrophobic interactions.
To assess the multivalent binding effect, Whitesides and coworkers [1] proposed an enhancement factor β, which is the ratio of the binding constant for the multivalent binding (Kmulti) of a multivalent ligand to a multivalent receptor and the binding constant for the monovalent binding (Kmono) of a monovalent ligand to a multivalent receptor. Multivalent binding interactions are complex and dynamic in nature, which makes the kinetic evaluation of the binding process inevitable. The first binding event between a multivalent ligand and a multivalent receptor produces the spatial proximity at the interface that leads to high local concentration. The preorganization of ligands makes the following binding events faster. The overall association rate (kon) of several binding events during multivalent interactions is expressed kinetically. It is a diffusion limited parameter and a weighted average quantity for many elementary binding process. The kon is not always significantly affected in multivalent interactions. In contrast, the lower dissociation rates koff in multivalent systems usually determine the large differences in dissociation constant (Kd) values of the multivalent and the corresponding monovalent systems. Kim et al. [12] have recently reported a binding kinetic study for monovalent and bivalent 15‐base aptamers (15‐Apt) for thrombin binding. The relative koff value for bivalent 15‐Apt was 50 times lower than that for monovalent 15‐Apt. The relative kon of bivalent 15‐Apt was found to be similar to the free 15‐Apt. The bivalent 15‐Apt exhibited ~60 times higher association constant (Ka) value than the single 15‐Apt for thrombin binding [12]. An efficient design of a multivalent inhibitor requires the optimization of the scaffold size and flexibility, spacer length, and ligand density to reduce the thermodynamic penalties and to match the receptor’s topological distribution. An optimized multivalent inhibitor should ideally increase its on‐rate due to a faster binding to the multivalent receptor surface and decrease its off‐rate through continuous rebinding of the ligands in close proximity. Consequently, the multivalent ligand construct will possess a longer life‐time during the binding with the receptor surface than the monovalent ligand.
The first step in desiging a multivalent ligand inhibitor is knowledge about the receptor’s surface. This includes an intrinsic affinity of the monovalent ligand–receptor pair, size, charge, and mechanical property of the pathogen and inhibitor architecture, and topological distribution profile of receptors on the target/pathogen surface. Multivalent ligand architecture needs to be optimized in terms of ligand density. This can be achieved by varying the ratio of the active functional groups to the non‐active functional groups on the carrier backbone. Too much deviation from the optimum ligand density can significantly affect the inhibitory potential of the scaffold. For example, the most abundant trimeric hemagglutinin (HA) glycoprotein (400–500 copies, 10–12 nm apart from each other per virus) each having three binding sites for sialic acid (SA), on the influenza virus surface mediates virus attachment with the cell surface [13–16] (Figure 9.3). X‐ray crystallographic studies [17] showed that SA binding sites are located in shallow pockets on the globular head domain of HA trimers. An extensive study was carried out by Roy and coworkers and Whitesides and coworkers [18–20] to inhibit the erythrocyte agglutination by the influenza A virus by different multivalent polyacrylamide sialosides (PAA‐SA). The multivalent PAA‐o‐SA ligand architectures bearing intermediate mole fractions of SA residues (χSA = 0.2–0.6) [19] were found to be more potent inhibitors than the others. Similarly, a study on sialic acid‐conjugated nanogels [21] showed that 50‐nm‐sized SA conjugated nanogel with only 12% SA conjugation was much more potent, inhibiting 80% of the influenza A virus, than the 70‐nm‐sized nanogel with 80% SA, which inhibited only 60% of the virus from binding to the cells (Figure 9.4).
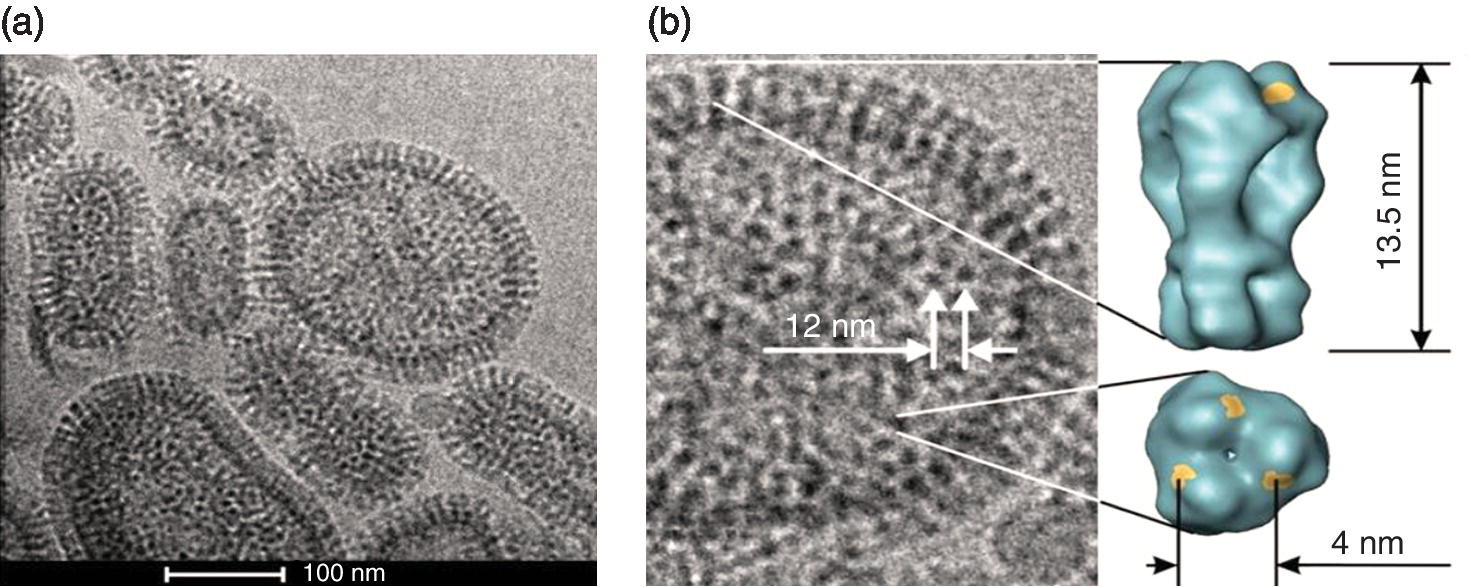
Figure 9.3 (a) The cryo‐transmission electron microscopy (TEM) image of influenza A virus (X31/H3N2) shows an abundant distribution of hemagglutinin (HA) glycoprotein trimers on the virus surface. (b) Inter trimeric distances and the length of the stem domain of HA3 are indicated as observed in the TEM image. Patches on the globular domain of HA trimers show the receptors for the sialic acid sugar residues.
Source: (a) Adapted from Ref. [9]. Reproduced with permission of American Chemical Society. (b) Adapted from Ref. [71]. Reproduced with permission of John Wiley and Sons.

Figure 9.4 Sialic acid‐functionalized polyglycerol nanogels consisting of a number of chemically crosslinked dendritic polyglycerol (dPG) units.
Source: http://pubs.rsc.org/en/content/articlehtml/2014/md/c4md00143e. Used under CC‐By 3.0 https://creativecommons.org/licenses/by‐nc/3.0/.
Ligands can be linked via optimized spacers to the carrier backbone not only to increase the local ligand density for rebinding but also to reach the less exposed receptors on the target surface. Furthermore, the inhibitory potency of the multivalent compound should be expressed in terms of ligand concentration to assess the impact of multivalent presentation. Zeta potential measurement of the target and the multivalent inhibitor provides further information about non‐specific electrostatic interaction at the interface. The pathogen‐cell binding inhibition by multivalent ligand architectures is the result of not only enhanced affinities of multivalent ligands but an overall effect of other additional effects like steric shielding, clustering, or statistical rebinding [22], and so on. Although the quantitative analysis of these effects is not straightforward, a careful comparison of binding constants and inhibitory concentrations of the multivalent inhibitor may unravel the contributions of other effects towards prevention of pathogen adhesion to cells. Depending on the size and properties of the targeted pathogen, different carrier backbones, for example, dendrimers, nanoparticles, linear or hyperbranched polymers, nanogels or microgels, and even bigger two‐dimensional (2D) adaptable platforms, can be selected. Studies by Haag and coworkers [23], Kiessling and coworkers [22], and Whitesides and coworkers [1,24] showed that the size, geometry, and also the inhibitor‐to‐pathogen size ratio play a crucial role in the binding mechanism. In the following sections, we will describe the different scaffold types with suitable examples from the literature to discuss the role of multivalency and other additional effects in pathogen–cell binding inhibition.
9.3 Multivalent Carbohydrate Ligands
Carbohydrate–protein interactions play an important role in the binding of pathogens to a cell surface. Monovalent carbohydrates are typically weakly bound to proteins with their dissociation constants ranging from mM to μM. When presented in a multivalent fashion, multiple simultaneous interactions of these carbohydrate ligands can lead to high affinities, which is useful for the binding of pathogens with cell surfaces. Various natural and unnatural glyconjugates (neoglycoconjugates) with different spatial arrangements have been explored for preventing pathogen–cell binding inhibition and thus infection [25]. Examples of such ligands include Pk saccharide for Shiga‐like toxins (SLT) [26], monosialotetrahexosyl ganglioside (GM1 oligosaccharides) for cholera toxin (CT) [27], α‐mannoside and galabiose for FimH [28] and P‐fimbriae [29], respectively, expressed on Escherichia coli (E. coli), sialic acid and silayl lactose for influenza, α‐D‐galactose and L‐fucose for cytototoxic lectin A and B, respectively, produced from Pseudomonas aeruginosa [30,31], and many more. Different multivalent carbohydrate‐conjugated ligands can be used as inhibitors of dendritic cell‐specific intercellular adhesion molecule (ICAM 3) grabbing non‐integrin (DC‐SIGN)‐mediated binding of different pathogens [32] like HIV‐1, HIV‐2, SIV, Ebola virus, hepatitis C virus, cytomegalo virus, and dengue virus, bacteria (Mycobacterium tuberculosis, Helicobacter pylori, and Klebsiella pneumonia), yeasts (Candida albicans), and parasites Leishmania and Schistosoma) to the dendritic cells on mucosal surfaces [3]. In the recent investigation by Bernardi and coworkers [33,34], a tetravalent dendron with four copies of linear trimannoside was synthesized. This tetravalent derivative inhibited the DC‐SIGN‐mediated trans HIV infection process of CD4 + T lymphocytes in the presence of elevated viral loads in cellular and cervical explants models (Figure 9.5a). The multivalent linear di‐ and trimannosides on the G3 Boltron‐type dendrimers are also known to strongly inhibit cell infection by Ebola pseudo‐typed viral particles by blocking DC‐SIGN [35] receptor in the nanomolar concentration range (Figure 9.5b). Virus‐like glycol‐dendri‐protein‐nanoparticles up to the size of 32 nm and bearing up to 1620 mannose residues based on a proteic scaffold Qβ phage were prepared [36]. These glycodendrinonanoparticles were capable of mimicking pathogens both in size and valency, blocked a model infection of T‐lymphocytes and human dendritic cells by Ebola virus in the picomolar range (Figure 9.5c).

Figure 9.5 (a) Tetravalent linear trimannoside conjugate for HIV infection inhibition. (b) Trimannoside conjugates of G3 Boltorn‐type dendrimers for Ebola pseudotyped virus infection inhibition. (c) Mannose conjugated proteic scaffold Qβ‐(Man9)180 bearing 1620 mannose residues for Ebola pseudovirus infection inhibition, prepared by click reaction of propargylated Qβ phage with pentaerythritol‐based azide terminated glycodendron.
Source: (c) Adapted from Ref. [36]. Reproduced with permission of Nature Publishing Group.
Both Shiga and cholera toxin infections cause millions of deaths per year. These toxins belong to the family of bacterial AB5 toxins, where the enzymatically cleavable A subunit is connected to the B‐pentamer. The B‐pentamer contains an oligosaccharide recognizing domain which strongly binds to the receptors on the outer leaflet of many cell membranes. Several multivalent oligosaccharides have been explored that target the B5 subunit of both cholera and Shiga‐like toxins to block its cellular uptake. Excellent multivalent binders that match the topology of the receptor B5 protein structure were designed and explored. The ‘starfish’‐like inhibitors initially designed by Bundle and coworkers [37,38] with their planar and radially distributed binding units have shown their potential in blocking the cellular uptake of bacterial cholera and Shiga toxins [39,40]. This concept was further expanded from pentavalent to octa‐ and decavalent inhibitors of AB5 toxins (Figure 9.6). The higher avidity of the octavalent system compared with the pentavalent was explained for a situation where several statistical options for binding were available [8].

Figure 9.6 (a) Structure of decavalent starfish inhibitor of Shiga‐like toxins. (b) Multivalent (single and branched) galactose binders for cholera toxin. (c) Structure of an octavalent inhibitor of Shiga‐like toxins.
The multivalent version of the oligoscaccharide of ganglioside (GM1os) using glycodendrimers synthesized by Pieters and coworkers [40] strongly inhibits the cholera toxin B5‐subunit (CTB5). The oligovalent glycodendrimer, for example, was 380 000‐fold more potent with IC50 = 50 pM than the monovalent GM1‐mimic (Figure 9.7). The tetravalent and octavalent galactose with long spacer arms, prepared as simplified versions of GM1os, competed well with the natural GM1os [41].
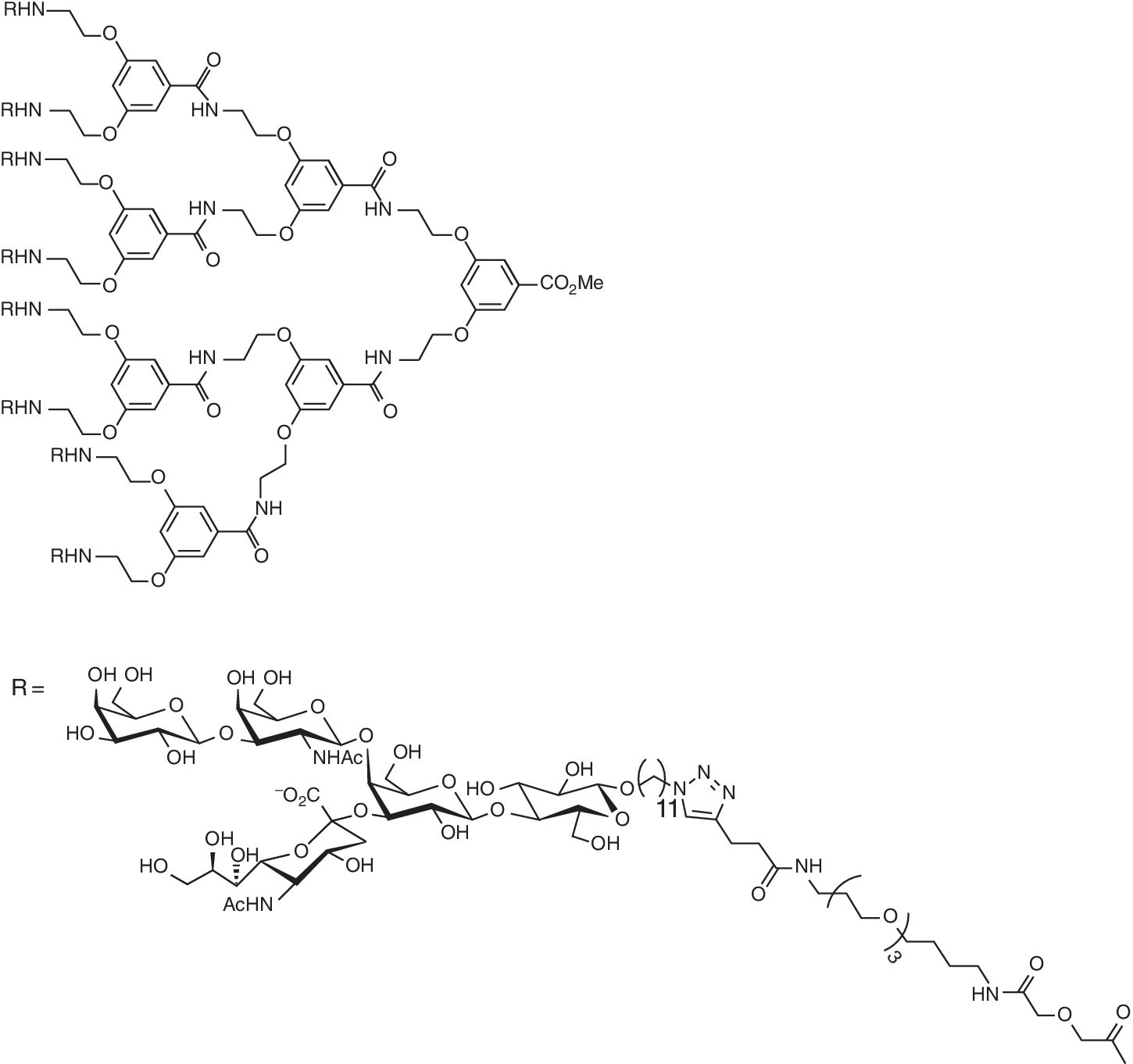
Figure 9.7 Octavalent GM1os inhibitor of the CTB5‐subunit.
9.4 Scaffold Architecture
9.4.1 Linear and Dendritic Scaffolds
Different linear, branched, and globular carriers can be used as a scaffold to prepare multivalent ligand inhibitors of infection by different pathogens. The optimal choice of the scaffold for the preparation of a multivalent ligand architecture is difficult. Ligands are presented more rigidly and exposed on the dendritic carriers than the relatively flexible linear backbones, which determine the affinity of ligands. Therefore, the optimum ligand densities for a particular target might be different for the dendritic and linear carriers. Flexible ligands can attain different conformations to afford the maximum number of ligand–receptor pairs but, at the same time, high flexibility also incorporates high conformational entropy to the binding. The sugar‐conjugated, water swollen linear polymers can sterically stabilize the pathogen surface. For example, higher molecular weight linear polymers were extensively used in the past by Roy and coworkers (18,20), Whitesides and coworkers (1,42), and other groups (43,44). An additional steric shielding effect was proposed by Whitesides and coworkers [19] to account for the nanomolar inhibition of the influenza virus by the water swollen linear PAA sialosides. Chen and coworkers [45] have also recently reported zanamivir (ZA) conjugated linear poly‐L‐glutamine (PGN) polymers for inhibition of influenza viral fusion and release at subnanomolar concentrations of ZA [46]. These PGN‐ZA conjugates were also effective against monovalent ZA‐ and oseltamivir resistant influenza virus. Recently amphiphilic antiviral peptides that form high order supramolecular assemblies for the multivalent display of inhibitors have also gained a lot of importance for preventing binding of influenza virus to the host cells. The stearylated antiviral peptide B (PeBGF), for example, showed enhanced inhibitory effect against the serotypes of both human pathogenic influenza virus A/Aichi/2/1968 H3N2, and avian pathogenic A/FPV/Rostock/34 H7N1 in the hemagglutination inhibition assay. The unspecific binding to the cell lipid membranes was found a serious limitation of this type of self‐assembling antiviral peptide [47]. The large size of dendritic or globular architectures [48–52] can sterically shield some receptors at the target surface from binding. However, affording perfect dendrons with size greater than 10 nm is still challenging, for example, the diameter of the G8 polyamidoamine dendron is not greater than 8 nm. Synthesizing hyperbranched polymers [53,54] is less challenging, for example, 2–20 nm sized hyperbranched polyglycerol backbones can be afforded depending on the degree of polymerization. Even larger sized globular backbones may include nanogels or metallic nanoparticles.
Also, fullerenes have emerged as carbon‐based, globular carriers for the preparation of multivalent ligand architectures. The mannosylated fullerenes [55] with different linker lengths were investigated to inhibit the FimH of E. coli. The lipopolysaccharide (LPS) protects and stabilizes the bacterial membrane. The glycosylated fullerenes 1, 2, and 3 displaying the mannopyranose core structure of bacterial L,D‐heptosides have shown the inhibition of the LPS heptosyltransferase WaaC in the low micromolar range (IC50 = 11, 47, and 6.7 μM) and thus inhibit the bacterial LPS synthesis (Figure 9.8). The monomeric glycosides, on the other hand, displayed IC50 values above 400 mM [55]. Recently Muñoz et al. [56] reported the synthesis of the giant tridecafullerenes decorated with 120 peripheral carbohydrate (mannose or galactose) units. The mannosylated “superballs” have shown great potential in inhibiting cell infection by artificial Ebola virus particles with IC50 in the subnanomolar range.

Figure 9.8 Dodecavalent glycoclusters based on fullerene hexa‐adducts to inhibit the LPS heptosyl‐transferase WaaC with nanomolar affinity.
Dendritic or linear backbones also have different biological impacts. Heparin, for example, is a natural linear sulfated polysaccharide, which has been used in the last few decades as a viral entry inhibitor, as well as for prevention and treatment of thrombosis [57–61]. However, its therapeutic application is limited due to its anticoagulant effect [62]. Further drawbacks are associated with the preparation of heparin, since it has to be isolated from mammalian organs. This way of extraction always bears the potential risk of contamination by pathogens and leads to a highly polydisperse heparin [63].
On the basis of these disadvantages dendritic polyglycerol sulfate (dPGS) has been developed as a synthetic heparin mimetic, which has a similar activity profile but not showing the characteristic limitations of heparin (Figure 9.9). In contrast to heparin, dPGS can be fully synthesized with a straightforward protocol that results in a low polydisperse product, which shows only a low anticoagulant effect but is able to strongly interact with leukocytes and pathogens via electrostatic interactions [23,64]. Furthermore, dPGS is tunable in size, is highly biocompatible, and shows a high anti‐inflammatory effect in vivo [63,65,66].
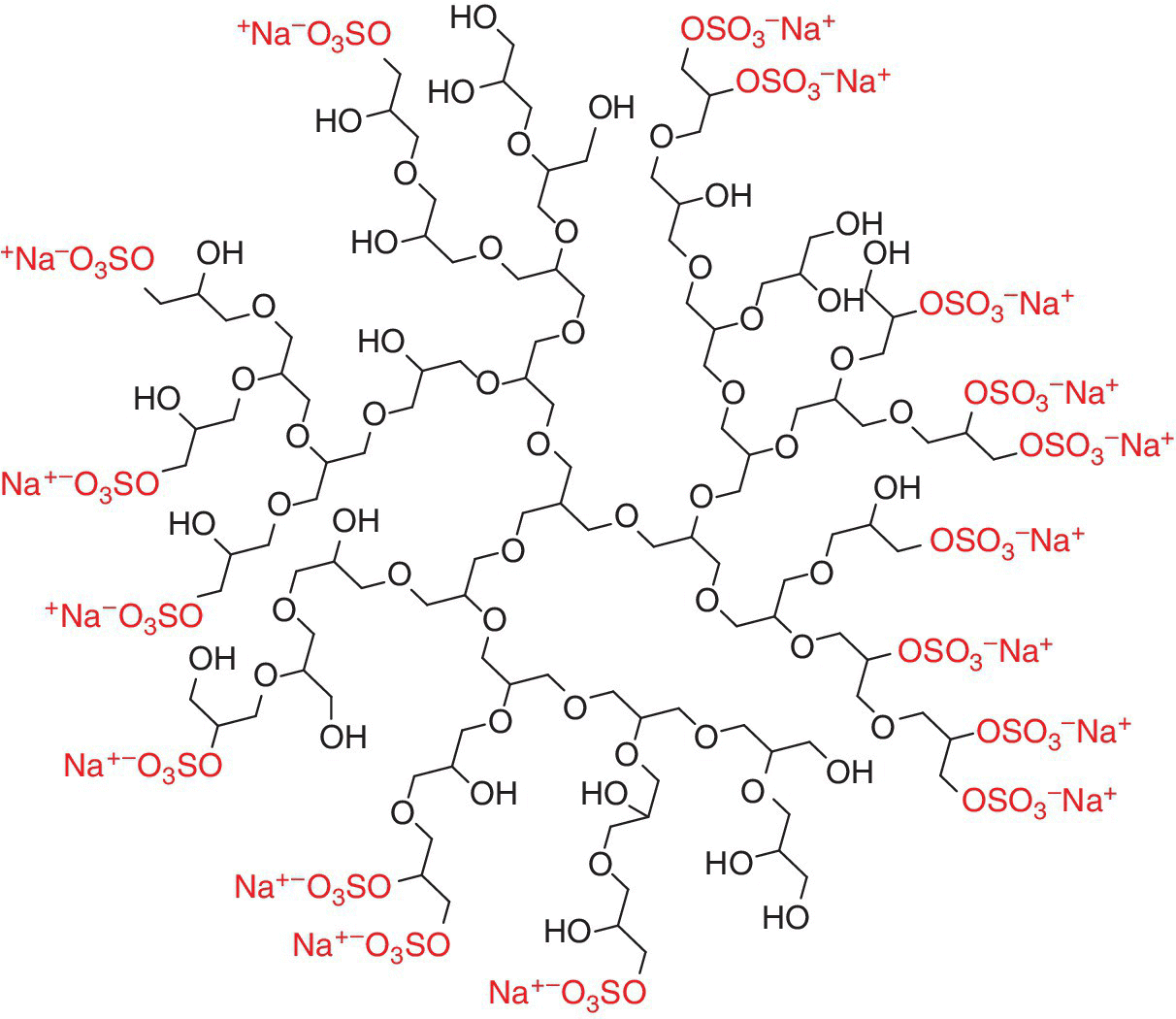
Figure 9.9 Schematic representation of an idealized dendritic polyglycerol sulfate structure. The negatively charged sulfate end groups and the corresponding counterions are shown in gray.
9.4.2 Multivalent Gold Nanoparticles
Competitive, multivalent nanoparticle binding inhibitors that are based on the rigid core, for example, gold nanoparticles [67], have been extensively used to prevent virus infections. The use of gold nanoparticles, moreover, gives high contrast “dark spots” in transmission electron microscopy, due to their high electron density, which is helpful for visualizing the multivalent attachment of ligand‐coated gold nanoparticles with pathogens (Figure 9.10). Furthermore, the possible controlled size preparation of these gold nanoparticles allows for a size dependent study of rigid multivalent nanoparticles. In the recent past, various gold nanoparticles functionalized [67] with multivalent ligands have been studied for the biological interactions and for biolabels. For example, mannose‐conjugated gold nanoparticles were used to visualize the position of individual FimH proteins on the type 1 pilus of E. coli with electron microscopy. The quantitative analysis [68] of multivalent interaction of different mannose‐conjugated gold nanoparticles with tetrameric plant lectin, concanavalin A (ConA), shows the dependence of affinity on nanoparticle size and linker of mannose ligands. Martínez‐Avila et al. [69] synthesized a series of gold nanoparticles with different spacers and densities of oligomannoside. Surface plasmon resonance‐based competitive assays were applied to evaluate selected mannose‐ conjugated gold nanoparticles as inhibitors of DC‐SIGN binding to HIV envelop glycoprotein gp120. The gp120 was direcctly immobilized on the sensor chip surface and binding measurements of fluid‐phase DC‐SIGN at a fixed concentration in the presence of mannose‐conjugated gold nanoparticles or free alkyl amino (oligo)mannosides were performed at varying stoichiometric ratios. The disaccharide Manα1‐2Manα containing gold nanoparticles was found to be the best inhibitor, showing nanomolar inhibitory concentrations (100% inhibition at 115 nM). This was 20 000 fold higher than the corresponding monomeric disaccharide (100% inhibition at 2.2 mM). Furthermore, these multivalent manno‐glyco nanoparticles [70] also inhibited DC‐SIGN‐mediated HIV‐1 trans‐infection of human T cells at nanomolar concentrations in experimental settings that mimicked the natural route of virus transmission. Papp et al. [71] chemically conjugated 2‐ and 14‐nm‐sized gold nanoparticles with sialic acid‐terminated dendron and investigated for binding with influenza virus. The larger sized gold nanoparticle inhibitors had high affinity for HA on the influenza virus surface as observed by electron microscopy techniques and biochemical analysis (Figure 9.10). This result pointed out the relevance of the contact area between an inhibitor and its target. Vonnemann et al. [72] studied different sized polysulfated gold nanoparticles for the vesicular stomatitic virus (VSV)–cell binding inhibition. The polysulfated nanoparticles smaller than the size of the VSV with diameters ≤50 nm inhibited VSV–cell binding only to a small extent, whereas the VSV‐sized nanoparticles (≥52 nm) formed VSV/nanoparticle clusters and effectively inhibited virus–cell binding and thus infection (Figure 9.11). For the virus‐sized nanoparticle inhibitors, the increased contact area between the virus and inhibitor enhanced the polyvalent effect and thus led to cluster formation. This effect was not observed for the nanoparticle inhibitors, which were much bigger than the virus itself.
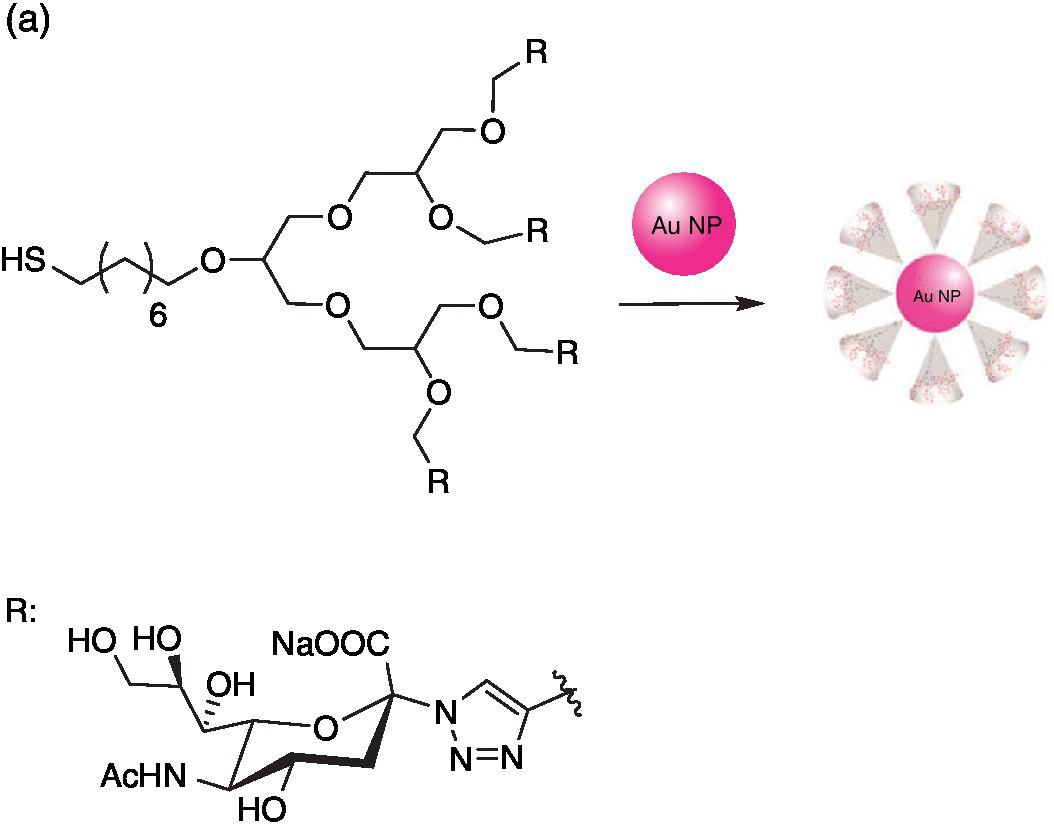
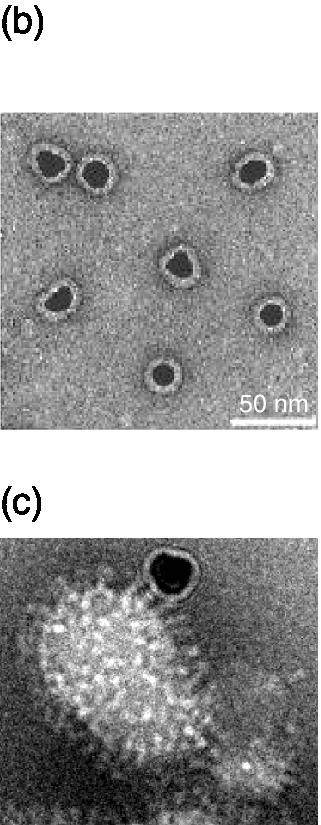
Figure 9.10 (a) Synthesis of sialic acid‐functionalized gold nanoparticles (Au NPs). (b) Electron microscopic visualization of 14‐nm‐sized gold nanoparticles with a sialic acid corona. (c) Preparation showing multiple binding of individually functionalized Au NPs to viral HAs.
Source: Adapted from Ref. [71]. Reproduced with permission of John Wiley and Sons.

Figure 9.11 Schematic representation of the size‐dependent virus inhibition by ligand functionalized gold nanoparticles according to the TEM data. (a) Although smaller sized gold nanoparticles decorate virions, the inhibition of virus–cell binding turned out to be inefficient. (b) Larger virus‐sized gold nanoparticles induced the formation of virus‐inhibitor clusters, thus inhibiting the virus–cell binding more efficiently.
Source: Adapted from Ref. [72]. Reproduced with permission of Royal Society of Chemistry.
Vonnemann et al. [23] explored the quantitative impact of virus concentration, inhibitor size, steric shielding, and multivalency in the inhibition process by spherical inhibitors for both strong and weak ligand/receptor pairs. Different sized streptavidin‐coated silica nanoparticles were used as strong binding inhibitors for 10‐µm‐sized biotinylated silica particles. The dPG and dPGS‐coated gold nanoparticles for leukocyte‐selectin‐coated binders were taken as an example for weak ligand/receptor pairs. The impact of the varying sizes and ligand densities on the evaluation of IC50 was systematically investigated during inhibition. A modified version of the Cheng–Prusoff‐equation was adapted to account for the multivalent inhibitors covering the surface of the binder. In this equation, IC50 = kdmulti + 0.5P[B], the first term was the contribution to multivalency, and the prefactor of the binder concentration [B] accounted for the steric shielding contribution. Their analysis concluded that the optimal size of the globular inhibitor was smaller than the binder’s in most cases. The multivalent dissociation constant Kdmulti for large globular inhibitors exponentially depended on the contact area and therefore was much lower than the virus (pathogen) experimental/biological concentration, which shows the predominance of the steric shielding effect over multivalency in body fluids. The impact of steric shielding [23], furthermore, was only noticeable when all the inhibitors were bound to the pathogen.
Their analysis showed that the multivalent association constant for large globular inhibitors exponentially increased with inhibitor/virus contact area and ligand density. The particle and volume normalized IC50 value of an inhibitor predominantly depended on its multivalent association constant at a very low virus concentration. Compared with the multivalency effects, the contribution of steric shielding to the IC50 values of inhibitors was only minor, and its impact was only noticeable if all inhibitors were bound to the binder (virus).
Inorganic nanoparticles are usually conjugated with ligands via poly(ethylene glycol) (PEG) spacer to protect them from serum protein adsorption [73]. The attachment of a ligand to a PEG spacer can significantly impair its receptor affinity, which is counterbalanced by the nanoparticle’s multivalency. Therefore, affinity testing of monovalent ligand with PEG spacer before attaching to the nanoparticle is highly recommended. A recent study by Hennig et al. [74] showed that PEGylation of a model ligand EXP3174, which was a small molecule antagonist for the angiotensin II receptor type 1 (AT1R), decreased the receptor affinity by 58‐fold to the native ligand. After attachment of the PEGylated ligand to the nanoparticle, receptor affinity was regained in the low nanomolar range similar to the affinity range of the native ligand.
9.4.3 2D Platforms
Globular inhibitors decorated with multiple functionalities are able to bind several receptors at once but they are limited in their contact area due to their shape and size. Of course, the size of spherical particles is well tunable, but it can never provide the same contact area as its weight or size comparable sheet‐like analog. Since interactions of virus particles or bacteria are based on dynamic processes at the interfaces, a huge surface area with optimum ligand density is advantageous.
Furthermore, flexible 2D platforms coated with numerous multivalent ligands are not only able to bind several pathogen receptors at once but can ideally wrap the whole infectious particle. To satisfy all these expectations, a supramolecular host–guest construct was developed on the basis of adamantyl‐functionalized, thermally reduced graphene oxide (TRGO) decorated with multivalent sugar ligands (Figure 9.12a).
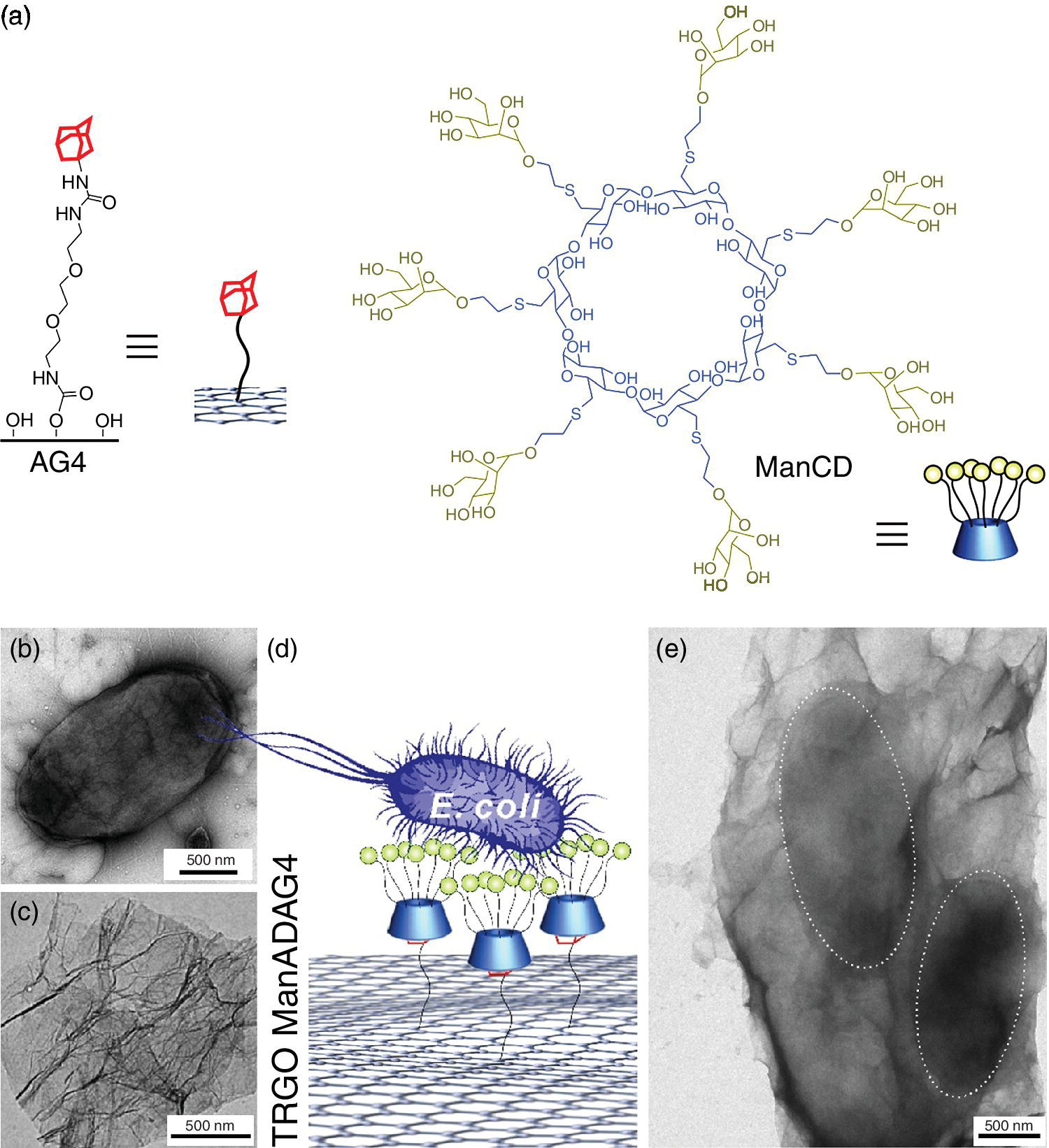
Figure 9.12 Schematic of (a) chemical structures of adamantyl‐functionalized graphene derivative (AG4) and heptamannosylated ß‐cyclodextrin (ManCD). TEM images of the (b) ORN178 E. coli and (c) ManCD‐AG4 hybrid. (d) Schematic representation for capturing E. coli by mannosylated cyclodextrin, adamantly‐functionalized (ManCD@AG4) graphene sheets. (e) E. coli agglutination incubated with ManCD@AG4. The dashed gray circles outline the captured bacteria.
Source: Adapted from Ref. [4]. Reproduced with permission of American Chemical Society.
The formation of host–guest inclusions on the carbon surface provides a versatile strategy, for not only increasing the intrinsic water solubility of graphene‐based materials, but also promote binding of the biofunctional groups with the sheet‐like architecture. The combination of the unique 2D surface area of the graphene and the specific binding ability of carbohydrates results in a flexible platform that is able to wrap selectively and agglutinate E. coli (Figure 9.12b–e). Through the powerful thermal IR‐absorption properties of the graphene‐based material, it is possible to kill the wrapped bacteria by short IR‐laser irradiation with high efficiency [4].
Functionalization, flexibility, and size are essential criteria for embedding pathogens into the carbon‐based 2D architecture. Additionally, implementing functional groups partially convert hybridized honeycomb‐like structures from sp2 to sp3 and thus results in increased flexibility of the sheet [75]. Due to these changes, the modified sheets can bend, extensively fold, and even wrap smaller particles. If the size of the 2D architecture is much smaller than that of the bacteria or the virus particle, several sheets will adhere on the pathogen and can fully cover the whole surface to isolate the infectious particle. Since the inhibition of cell–pathogen is still an urgent and unsolved problem, development of new multivalent 2D platforms is a new strategy for efficiently blocking pathogens.
9.5 Nano‐ and Microgels for Pathogen Inhibition
Hydrogels are highly hydrated three‐dimensional (3D) systems, which offer an excellent biocompatibility as well as an adjustable stiffness and swelling behavior [76]. Due to these facts, hydrogels are perfect candidates for various biomedical applications [77] such as targeted drug delivery [78–80], biosensing [81], or tissue engineering [82–84]. Furthermore, the gel size can be varied from 20 nm (nanogels) [21,85] up to 400 µm (microgels) [86] by different techniques. The commonly used techniques are miniemulsion, microfluidic templating, or inverse nanoprecipitation [87–89]. In the case of miniemulsion, the gel formation is based on a polymer crosslinking, for example, partially acrylated polyglycerol in a surfactant‐stabilized aqueous nanodroplet. This technique, however, is limited to nanometer‐sized gels on its own, but in combination with droplet‐based microfluidic and macro crosslinkers, for example, poly(ethylene glycol) diacrylate, the gel size can be augmented to microgels (Figure 9.13). By embedding cleavable bonds or linker into the polymer matrix, such as acetals, disulfides, ketals, or phosphate esters, a high biodegradation and an efficient degradation can be guaranteed [85,87].

Figure 9.13 (a) Schematic representation of a nano‐ or microgel formation via miniemulsion and microfluidic templating. The nanogel (on the left) consists of crosslinked acrylated polyglycerol, while the microgel (on the right) is the result of combining the above‐mentioned nanogel and a poly(ethylene glycol) diacrylate macro crosslinker. (b) TEM micrograph of nanogel particles. (c) Optical micrograph of water‐swollen microgel particles.
Source: Adapted from Ref. [86]. Reproduced with permission of Elsevier.
Furthermore, hydrogels can be post modified with different functionalities such as sulfates, amines, or specific glycan structures to build up a new type of multivalent pathogen inhibitor. One example for such an inhibitor is the polyglycerol‐based glycoarchitecture coated with multiple sialic acids for pathogen interaction (e.g., Figure 9.4). On the basis of influenza virus, Papp et al. [21] demonstrated the high inhibition potential of modified nanogels depending on the gel size and the degree of sialic acid functionalization.
Until now, nano‐ and microgels have been frequently used as transporters for encapsulation and targeted release of cells or pharmaceutical biomacromolecules triggered by external stimuli such as pH changes or reductive environments [88,89]. However, in the future, degradable hydrogels could be designed not only as a direct pathogen inhibitor but also to develop 3D pathogen traps. Additionally, microgels [90] are easily able to mimic the natural extracellular matrix, which is known to interact with various types of pathogens. Ideally, after adhesion of the infectious particle to the gel surface, diffusion into the gel network should take place so that the pathogen is trapped inside. There is still much work to be done for developing biocompatible or biodegradable 3D gel networks for efficient pathogen inhibition in biological systems.
9.6 Conclusion
Prevention of pathogen inhibition is one of the major goals for human health. Different multivalent ligand scaffolds have been synthesized and explored in the area of multivalent pathogen inhibition. The design of efficient multivalent inhibitors is a complex process and involves optimizing ligand density, flexibility, size, and charge. The larger sized inhibitor architectures such as 2D platforms and 3D gel networks can be extremely useful to combat pathogens like micrometer‐sized bacteria. However, the biocompatibility, biodegradability, and systemic clearance are inevitable for their application in biological systems. Many problems have still to be solved in this area. No reports are available yet on the fate of bound inhibitor–pathogen complexes in biological systems. Questions have also been raised on the applicability of pathogen binders for the late stages of infection. In spite of these challenges, the area of multivalent pathogen inhibitors has a great potential for future research.
Acknowledgments
We would like to thank the Deutsche Forschungsgemeinschaft (DFG) for financial support within the Collaborative Research Centre 765 as well as the European ITN “Multiapp” where different biophysical methods are being used to understand multivalency quantitatively at molecular level and thus the knowledge gained is applied to design novel multivalent interfaces. We are grateful to Dr Pamela Winchester for careful language polishing and Dr Wiebke Fischer for her support with the graphical design.
References
- 1 Mammen M, Choi SK, Whitesides, GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37(20):2754–94.
- 2 Fasting C, Schalley CA, Weber M, et al. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. 2012;51(42):10472–98.
- 3 Bhatia S, Dimde M, Haag R. Multivalent glycoconjugates as vaccines and potential drug candidates. Med. Chem. Commun. 2014;(5):862–78.
- 4 Qi Z, Bharate P, Lai CH, et al. Multivalency at interfaces: Supramolecular carbohydrate‐functionalized graphene derivatives for bacterial capture, release,.and Disinfection. Nano Lett. 2015;15(9):6051–7.
- 5 Jiang W., Kim BY, Rutka JT, Chan WC. Nanoparticle‐mediated cellular response is size‐dependent. Nat. Nanotechnol. 2008;3:145–50.
- 6 Moscona A. Medical management of influenza infection. Annu. Rev. Med. 2008;59:397–413.
- 7 Kane RS. Thermodynamics of multivalent interactions: Influence of the linker Langmuir 2010;26(11):8636–40.
- 8 Kitov PI, Bundle DR. On the nature of the multivalency effect: A thermodynamic model. J. Am. Chem. Soc. 2003;125(52):16271–84.
- 9 Bhatia S, Camacho LC, Haag R. Pathogen inhibition by multivalent ligand architectures. J. Am. Chem. Soc. 2016. DOI: 10.1021/jacs.5b12950.
- 10 Karlson KA. Meaning and therapeutic potential of microbial recognition of host glycoconjugates. Mol. Microbiol. 1998;29(1):1–11.
- 11 Landers JJ, Cao Z, Lee I, Piehler LT, et al. Prevention of influenza pneumonitis by sialic acid‐conjugated dendritic polymers. JID 2002;186(9):1222–30.
- 12 Kim Y, Cao Z, Tan W. Molecular assembly for high‐performance bivalent nucleic acid inhibitor. PNAS 2008;105(15):5664–9.
- 13 Yamaguchi M, Danev R, Nishiyama K, et al. Zernike phase contrast electron microscopy of ice‐embedded influenza A virus. J. Struct. Biol. 2008;162(2):271–6.
- 14 Ruigrok RWH, Calder LJ, Wharton SA. Electron microscopy of the influenza virus submembranal structure. Virol. 1989;173(1):311–16.
- 15 Li S, Sieben C, Ludwig K, et al. pH‐Controlled two‐step uncoating of influenza virus. Biophys. J. 2014;106(7):1447–56.
- 16 Boettcher C, Ludwig K, Herrmann A, et al. Structure of influenza haemagglutinin at neutral and at fusogenic pH by electron cryo‐microscopy. FEBS 1999;463(3):255–9.
- 17 Weis W, Brown JH, Cusack S, et al. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1998;333(6172):426–31.
- 18 Roy R, Pon RA, Tropper FD, Andersson FO. Michael addition of poly‐L‐lysine to N‐acryloylated sialosides. Syntheses of influenza A virus haemagglutinin inhibitor and group B meningococcal polysaccharide vaccines. J. Chem. Soc. Chem. Commun. 1993;(3):264–65.
- 19 Lees WJ, Spaltenstein A, Kingery‐Wood JE, Whitesides GM. Polyacrylamides bearing pendant alpha‐sialoside groups strongly inhibit agglutination of erythrocytes by influenza A virus: Multivalency and steric stabilization of particulate biological systems. J. Med. Chem. 1994;37(20):3419–33.
- 20 Roy R, Andersson FO,.Harms G, et al. Synthesis of esterase‐resistant 9‐O‐acetylated polysialoside as inhibitor of influenza C virus hemagglutinin. Angew. Chem. Int. Ed. Engl. 1992;31(11):1478–81.
- 21 Papp I, Sieben C, Sisson AL, et al. Inhibition of influenza virus activity by multivalent glycoarchitectures with matched sizes. ChemBioChem 2011;12(6):887–95.
- 22 Gestwicki JE, Cairo CW, Strong LE, et al. Influencing receptor − ligand binding mechanisms with multivalent ligand architecture. J. Am. Chem. Soc. 2002;124(50):14922–33.
- 23 Vonnemann J, Liese S, Kuehne C, et al. Size dependence of steric shielding and multivalency effects for globular binding inhibitors. J. Am. Chem. Soc. 2015;137(7):2572–9.
- 24 Choi SK, Mammen M, Whitesides GM. Monomeric inhibitors of influenza neuraminidase enhance the hemagglutination inhibition activities of polyacrylamides presenting multiple C‐sialoside groups. Chem. Biol. 1996;3(2):97–104.
- 25 Bernardi A, Jimenez‐Barbero J, Casnati A, et al. Multivalent glycoconjugates as anti‐pathogenic agents. Chem. Soc. Rev. 2013;42:4709–27.
- 26 Ling H, Boodhoo A, Hazes B, et al. Structure of the shiga‐like toxin I B‐pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998;37(7):1777–88.
- 27 Fan E, Merritt EA, Verlinde CLMJ, Hol WGJ. AB5 toxins: structures and inhibitor design. Curr. Opin. Struct. Biol. 2000;10(6):680–6.
- 28 Heidecke CD, Lindhorst TK. Iterative synthesis of spacered glycodendrons as oligomannoside mimetics and evaluation of their antiadhesive properties. Chem. Eur. J. 2007;13(32):9056–67.
- 29 Lane MC, Mobley HLT. Role of P‐fimbrial‐mediated adherence in pyelonephritis and persistence of uropathogenic Escherichia coli (UPEC) in the mammalian kidney. Kidney Int. 2007;72(1):19–25.
- 30 Gilboa‐Garber N. Pseudomonas aeruginosa lectins. Methods Enzymol. 1982;83:378–85.
- 31 Imberty A, Wimmerova M, Mitchell EP, Gilboa‐Garber N. Structures of the lectins from Pseudomonas aeruginosa: insights into the molecular basis for host glycan recognition. Microbes Infect. 2004;6(2):221–8.
- 32 van Kooyk Y, Geijtenbeek TB. DC‐SIGN: escape mechanism for pathogens. Nat. Rev. Immunol. 2003;3(9):697–709.
- 33 Sattin S, Daghetti A, Thepaut M, et al. Inhibition of DC‐SIGN‐mediated HIV infection by a linear trimannoside mimic in a tetravalent presentation. ACS Chem. Biol. 2010;5(3):301–12.
- 34 Berzi A, Reina JJ, Ottria R, et al. A glycomimetic compound inhibits DC‐SIGN‐mediated HIV infection in cellular and cervical explant models. AIDS 2012;26(2):127–37.
- 35 Luczkowiak J, Sattin S, Sutkeviciute I, et al. Pseudosaccharide functionalized dendrimers as potent inhibitors of DC‐SIGN dependent Ebola pseudotyped viral infection. Bioconjug. Chem. 2011;22(7):1354–65.
- 36 Ribeiro‐Viana R, Sanchez‐Navarro M, Luczkowiak J, et al. Virus‐like glycodendrinanoparticles displaying quasi‐equivalent nested polyvalency upon glycoprotein platforms potently block viral infection. Nat. Commun. 2012;3:1303.
- 37 Kitov PI, Sadowska JM, Mulvey G, et al. Shiga‐like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 2000;403(6670):669–72.
- 38 Jacobson JM, Yin J, Kitov PI, et al. The crystal structure of shiga toxin type 2 with bound disaccharide guides the design of a heterobifunctional toxin inhibitor. J. Biol. Chem. 2014;289(2):885–94.
- 39 Tsutsuki K, Watanabe‐Takahashi M, Takenaka Y, et al. Identification of a peptide‐based neutralizer that potently inhibits both shiga toxins 1 and 2 by targeting specific receptor‐binding regions. Infect. Immunol. 2013;81(6):2133–8.
- 40 Pukin AV, Branderhorst HM, Sisu C, et al. Strong inhibition of cholera toxin by multivalent GM1 derivatives. ChemBioChem 2007;8(13):1500–3.
- 41 Branderhorst HM, Liskamp RM, Visser GM, Pieters RJ. Strong inhibition of cholera toxin binding by galactose dendrimers. Chem. Commun. (Camb.) 2007;(47):5043–5.
- 42 Sigal GB, Mammen M, Dahmann G, Whitesides GM. Polyacrylamides bearing pendant α‐sialoside groups strongly inhibit agglutination of erythrocytes by influenza virus: The strong inhibition reflects enhanced binding through cooperative polyvalent interactions. J. Am. Chem. Soc. 1996;118(16):3789–800.
- 43 Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell‐surface interactions. Curr. Opin. Chem. Biol. 2000;4(6):696–703.
- 44 Kanai M, Mortell KH, Kiessling LL. Varying the size of multivalent ligands: The dependence of Concanavalin A binding on neoglycopolymer length. J. Am. Chem. Soc. 1997;119(41):9931–2.
- 45 Lee CM, Weight AK, Haldar J, et al. Polymer‐attached zanamivir inhibits synergistically both early and late stages of influenza virus infection. PNAS 2012;109(50):20385–90.
- 46 Weight AK, Haldar J, Alvarez de Cienfuegos L, et al. Attaching zanamivir to a polymer markedly enhances its activity against drug‐resistant strains of influenza a virus. J. Pharm. Sci. 2011;100(3):831–5.
- 47 Lauster D, Pawolski D, Storm J, et al. Potential of acylated peptides to target the influenza A virus. Beilstein J. Org. Chem. 2015;11:589–95.
- 48 Mansfield ML, Klushin LI. Intrinsic viscosity of model starburst dendrimers. J. Phys. Chem. 1992;96(10):3994–8.
- 49 Murat M, Grest GS, Molecular dynamics study of dendrimer molecules in solvents of varying quality. Macromolecules 1996; 29(4):1278–85.
- 50 Stechemesser S, Eimer W. Solvent‐dependent swelling of poly(amido amine) starburst dendrimers. Macromolecules 1997;30(7):2204–6.
- 51 Tomalia DA. Architecturally driven properties based on the dendritic state. High Perform. Polym. 2001;13(2):S1–S10.
- 52 Newkome GR, Shreiner CD. Poly(amidoamine), polypropylenimine, and related dendrimers and dendrons possessing different 1 → 2 branching motifs: An overview of the divergent procedures. Polymer 2008;49(1):1–173.
- 53 Staedtler AM, Hellmund M, Sheikhi Mehrabadi F, et al. Optimized effective charge density and size of polyglycerol amines leads to strong knockdown efficacy in vivo. J. Mater. Chem. B 2015;3(46):8993–9000.
- 54 Kainthan RK, Muliawan EB, Hatzikiriakos SG, Brooks DE. Synthesis, characterization, and viscoelastic properties of high molecular weight hyperbranched polyglycerols. Macromolecules 2006;39(22):7708–17.
- 55 Durka M, Buffet K, Iehl J, et al. The functional valency of dodecamannosylated fullerenes with Escherichia coli FimH‐towards novel bacterial antiadhesives. Chem. Commun. (Camb.) 2011;47(4):1321–3.
- 56 Munoz A, Sigwalt D, Illescas BM, et al. Synthesis of giant globular multivalent glycofullerenes as potent inhibitors in a model of Ebola virus infection. Nat. Chem. 2016;8(1):50–7.
- 57 Bengali Z, Satheshkumar PS, Moss B. Orthopoxvirus species and strain differences in cell entry. Virol. 2012;433(2):506–12.
- 58 Bengali Z, Townsley AC, Moss B. Vaccinia virus strain differences in cell attachment and entry. Virol. 2009;389(1–2):132–40.
- 59 Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 2005;86(Pt 5):1279–90.
- 60 Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 1998;72(2):1577–85.
- 61 Whitbeck JC, Foo CH, Ponce de Leon M, et al. Vaccinia virus exhibits cell‐type‐dependent entry characteristics. Virol. 2009;385(2):383–91.
- 62 Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat. Prod. Rep. 2002;19(3):312–331.
- 63 Turk H, Haag R, Alban S. Dendritic polyglycerol sulfates as new heparin analogues and potent inhibitors of the complement system. Bioconjug. Chem. 2004;15(1):162–7.
- 64 Calderon M, Quadir MA, Sharma SK, Haag R. Dendritic polyglycerols for biomedical applications. Adv. Mater. 2010;22(2):190–218.
- 65 Kainthan RK, Janzen J, Levin E, et al. Biocompatibility testing of branched and linear polyglycidol. Biomacromolecules 2006;7(3):703–9.
- 66 Dernedde J, Rausch A, Weinhart M, et al. Dendritic polyglycerol sulfates as multivalent inhibitors of inflammation. PNAS 2010;107(46):19679–84.
- 67 Niemeyer CM. Nanoparticles, proteins, and nucleic acids: Biotechnology meets materials science. Angew. Chem. Int. Ed. 2001;40(22):4128–58.
- 68 Lin CC, Yeh YC, Yang CY, et al. Quantitative analysis of multivalent interactions of carbohydrate‐encapsulated gold nanoparticles with concanavalin A. Chem. Commun. 2003;(23):2920–1.
- 69 Martinez‐Avila O, Hijazi K, Marradi M, et al. Gold manno‐glyconanoparticles: Multivalent systems to block HIV‐1 gp120 binding to the lectin DC‐SIGN. Chem. Eur. J. 2009;15(38):9874–88.
- 70 Martinez‐Avila O, Bedoya LM, Marradi M, et al. Multivalent manno‐glyconanoparticles inhibit DC‐SIGN‐mediated HIV‐1 trans‐infection of human T cells. ChemBioChem 2009;10(11):1806–9.
- 71 Papp I, Sieben C, Ludwig K, et al. Inhibition of influenza virus infection by multivalent sialic‐acid‐functionalized gold nanoparticles. Small 2010;6(24):2900–6.
- 72 Vonnemann J, Sieben C, Wolff C, et al. Virus inhibition induced by polyvalent nanoparticles of different sizes. Nanoscale 2014;6(4):2353–60.
- 73 Khan S, Gupta A, Verma NC, Nandi CK. Kinetics of protein adsorption on gold nanoparticle with variable protein structure and nanoparticle size. J. Chem. Phys. 2015;143(16):164709.
- 74 Hennig R, Pollinger K, Veser A, et al. Nanoparticle multivalency counterbalances the ligand affinity loss upon PEGylation. J. Control. Release 2014;194:20–7.
- 75 Stankovich S, Dikin DA, Dommett GH, et al. Graphene‐based composite materials. Nature 2006;442(7100):282–6.
- 76 Richter M, Steinhilber D, Haag R, von Klitzing R. Visualization of real‐time degradation of pH‐responsive polyglycerol nanogels via atomic force microscopy. Macromol. Rapid Commun. 2014;35(23):2018–22.
- 77 Seliktar D. Designing cell‐compatible hydrogels for biomedical applications. Science 2012;336(6085):1124–8.
- 78 Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008;33(4):448–77.
- 79 Sisson AL, Steinhilber D, Rossow T, et al. Biocompatible functionalized polyglycerol microgels with cell penetrating properties. Angew. Chem. Int. Ed. 2009;48(41):7540.
- 80 Hamidi M, Azadi A, Rafiei P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008;60(15):1638–49.
- 81 Richter A, Paschew G, Klatt S, et al. Review on hydrogel‐based pH sensors and microsensors. Sensors 2008;8(1):561–81.
- 82 Nguyen MK, Lee DS. Injectable biodegradable hydrogels. Macromol. Biosci. 2010;10(6):563–79.
- 83 Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials 2003;24(24):4337–51.
- 84 Fedorovich NE, Alblas J, de Wijn JR, et al. Hydrogels as extracellular matrices for skeletal tissue engineering: state‐of‐the‐art and novel application in organ printing. Tissue Eng. 2007;13(8):1905–25.
- 85 Sisson AL, Haag R. Polyglycerol nanogels: highly functional scaffolds for biomedical applications. Soft Matter 2010;6(20):4968–75.
- 86 Steinhilber D, Seiffert S, Heyman JA, et al. Hyperbranched polyglycerols on the nanometer and micrometer scale. Biomaterials 2011;32(5):1311–6.
- 87 Steinhilber D, Witting M, Zhang X, et al. Surfactant free preparation of biodegradable dendritic polyglycerol nanogels by inverse nanoprecipitation for encapsulation and release of pharmaceutical biomacromolecules. J. Control. Release 2013;169(3):289–95.
- 88 Fleige E, Quadir MA, Haag R. Stimuli‐responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv. Drug Deliv. Rev. 2012;64(9):866–84.
- 89 Steinhilber D, Sisson AL, Mangoldt D, et al. Synthesis, reductive cleavage, and cellular interaction studies of biodegradable, polyglycerol nanogels. Adv. Funct. Mater. 2010;20(23):4133–8.
- 90 Rossow T, Heyman JA, Ehrlicher AJ, et al. Controlled synthesis of cell‐laden microgels by radical‐free gelation in droplet microfluidics. J. Am. Chem. Soc. 2012;134(10):4983–9.