
Nylon-based polymer nanocomposites
A. Dasari, Nanyang Technological University (NTU), Singapore and Madrid Institute for Advanced Studies of Materials (IMDEA Materials Institute), Spain
Abstract:
This chapter highlights fundamental and recent major developments in the application of various nanoparticles in the design and performance of nylon-based materials. Specifically, it deals with techniques employed to prepare these nanocomposites, their physico-chemical characterization, mechanical and functional properties as well as applications and future prospects.
9.1 Introduction
Nylon, a family of synthetic polymers known generically as polyamide (or aliphatic polyamide), is one of the most commonly used engineering plastics. It is a semi-crystalline polymer, which was used to replace silk in military applications such as parachutes, flak vests, and many types of vehicle tire during World War II. It also replaced many mechanical and structural parts previously cast in metal, such as machine screws, gears, and other low- to medium-stress components. To further improve the mechanical properties of these materials, microscale particles like glass/carbon fibers, talc, and wollastonite are generally incorporated. But, due to the increasing emphasis on ‘multi-functionality,’ nanostructured materials have taken the limelight. Polymer nanocomposites are a good example of this class of nanostructured materials with the potential to exhibit unique combinations of mechanical, physical, optical, and thermal properties at relatively lower particle loadings than those in typical traditional composites. Moreover, many inherent properties of polymers, such as crystal structure, melt viscosity, and glass transition, are affected by the incorporation of nanoparticles. Hence, understanding the processing/structure/property relations in polymer nanocomposites is crucial for fully exploiting the fundamental characteristics of nanoparticles (large surface area and aspect ratio) in achieving combinatorial properties as well as improving the magnitude of property enhancements.
Over the last two decades, the study of these nanocomposite materials has been a mushrooming field of research yielding innovative advanced materials with high added value. This chapter highlights the major developments and applications of various nanoparticles in the design and performance of nylon-based materials.
The various techniques employed to prepare these nanocomposites, their physico-chemical characterization, mechanical and functional properties as well as future prospects will be outlined.
9.2 Types of nanoparticle and their modification
All nanoparticles have at least one dimension smaller than 100 nm and can be classified as one-, two-, or three-dimensional depending on their size and the number of dimensions at the nanoscale;1 that is, nanoplatelets are treated as one-dimensional, nanotubes two-dimensional, and equiaxed nanoparticles three-dimensional, where all three dimensions are less than 10 nm. Schematic representations along with representative examples are given in Table 9.1.
Table 9.1
Different classes of nanoparticles along with representative examples

Source: Adapted from Ruiz-Hitzky and Van Meerbeek (2006).1
In the development of polymer nanocomposites, the main challenge is to disperse the nanoparticles homogeneously and individually within the matrix because of their high tendency to agglomerate. Equally challenging is to control the interactions between these nanoparticles and the polymer matrix via physical interaction or chemical bonding. The extent of dispersion and interfacial interactions with the matrix will affect the inherent properties of the polymer and determine the magnitude of ultimate property improvement. So, the first step is to improve the dispersion of the nanoparticles in the matrix. Creating high shear forces during processing of polymer nanocomposites was considered as a way of breaking up the strongly bound aggregates of nanoparticles. But it was found that, if the matrix polymer lacks sufficient compatibility with the nanoparticles, stress alone cannot achieve fine dispersion of the nanoparticles.2 Therefore, depending on the matrix-filler combination, their compatibility has to be enhanced by functionalizing the fillers with organic surfactants (and other chemical agents). For this purpose, both physical and chemical methods have been used, such as the surface treatment of nanoparticles and the addition of coupling or grafting agents and compatibilizers. The following paragraphs will describe the various prevalent and potential approaches adopted in the surface treatment of nanoparticles, with representative examples.
9.2.1 Three-dimensional (3-D) nanoparticles
During the last decade, many three-dimensional (3-D) nanoparticles including Al2O3,3–5 zinc oxide (ZnO),6,7 calcium carbonate (CaCO3),8–12 and SiO2 13–18 have been incorporated into nylon matrices either as preformed nanoparticles or synthesized in situ by the sol–gel process. The former is common for nylon/CaCO3 nanocomposites, in which the nanoparticles are often surface coated with saturated or unsaturated fatty acids like stearic acid, aminocaproic acid, or amino acid.8–11 The basic idea is to reduce the surface energy of the CaCO3 nanoparticles and the particle–particle interaction, thus facilitating dispersion of the nanoparticles. But it should be noted that, in general, this surface treatment simultaneously weakens the particle–matrix interaction. However, by varying the number of reactive or functional groups in the fatty acid, the extent of the particle–matrix interfacial interaction can be tuned. For instance, amino acid and aminocaproic acid, which are more reactive compared with stearic acid, have a stronger interaction with the nylon matrix.11,12 In stearic acid, the carboxylic (−COOH) group at one end of the molecule forms a strong ionic bond with the calcium in the CaCO3, while the –CH3 non-polar group at the other end of the molecule interacts far less strongly with the nylon matrix, resulting in a much weaker particle–matrix interaction. In amino acid and aminocaproic acid, there are more functional groups to interact with CaCO3 and the nylon matrix (Fig. 9.1). In fact, even in the absence of any surface treatment, the interaction between CaCO3 nanoparticles and the nylon matrix is sufficient to provide a slight improvement in the mechanical properties. However, the agglomeration of untreated CaCO3 nanoparticles in the nylon matrix inevitably affects many other properties required of the composite.
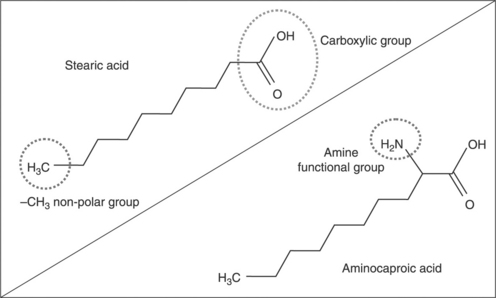
9.1 Chemical structure of stearic acid and aminocaproic acid with an additional amine functional group.
Surface treatment with organosilane (also termed silanization) is another popular strategy in the modification of nanoparticles. Organosilane is a silicon-based chemical compound with both organic- and inorganic-compatible functionalities within the same molecule. One end of the molecule bears a hydrolyzable group such as alkoxy or acetoxy while the other end of the molecule has an organo-functional group like epoxy or amine. The inorganic compatibility comes from the alkoxy groups and the organic compatibility stems from the organo-functional group. Silanization is commonly used in the surface modification of SiO2 nanoparticles13–16 and, during silanization, the alkoxy group on a silane molecule reacts with an hydroxyl group on the SiO2 nanoparticle surface, thus forming stable covalent –Si–O–Si– bonds. Typical silane-coupling agents that have been used in the preparation of nylon/SiO2 nanocomposites are γ-aminopropyl-triethoxysilane (APS),15 hexamethyl-disilazane (HMDS),15 and γ-glycidoxypropyl-trimethoxysilane (GPS).16 Silanization of silica nanoparticles can be carried out in situ13,15 or pretreated.14,16
In situ surface modification of SiO2 involves condensation polymerization, in which the hydrolysis product, sodium metasilicate, is used as a monomer and the organosilane is used as a chain terminator. First, sodium metasilicate (the monomer) is hydrolyzed to form silicic acid in the presence of hydrochloric acid. Condensation polymerization takes place by anhydration during hydrolysis of silicic acid. The Si and O atoms bond to each other to form three-dimensional tetrahedral network structures. Large numbers of hydroxyls remain on the surface of the silica nanoparticles. At this point, an organosilane molecule like APS is introduced and the silanol groups of APS generated by hydrolysis can react rapidly with the hydroxyl groups of SiO2 to form a modified layer on the silica surface. The organic chains substitute for a portion of the active groups on the SiO2 surface, thus resulting in steric hindrance. This prevents the SiO2 from continuously growing or agglomerating. By controlling the reaction conditions, nano-SiO2 particles capped with different organic compound(s) can then be obtained. For the surface treatment for preformed nano-SiO2 particles, the process is much easier. An organosilane solution, in ethanol14 or toluene,16 is first prepared, followed by the addition of SiO2 nanoparticles under rapid stirring. The treated nanoparticles are collected and dried for the subsequent preparation of nanocomposites.
9.2.2 Two-dimensional (2-D) nanoparticles
Examples of two-dimensional (2-D) nanoparticles are carbon nanotubes (CNTs), halloysite nanotubules, ZnO nanofibres, etc. In the following discussion, we will only consider CNTs as they are an ideal reinforcing agent for high-strength multifunctional polymer composites because of their exceptionally high Young’s modulus ~1 TPa,19 which is comparable to that of diamond ~1.2 TPa. Moreover, CNTs have unique structural and physical properties: electronically they can be either metallic or semi-conductive depending on their geometrical structure; good ballistic transport properties; extremely high thermal conductivity; and high optical polarizability. Depending on their structural configuration, CNTs are either single-walled carbon nanotubes (SWCNTs) or multi-walled carbon nanotubes (MWCNTs). SWCNTs are formed from only a single layer of a graphene sheet while MWNTs consist of several layers of coaxial graphene sheets.
As with other types of nanoparticle, a critical issue in taking advantage of the superior properties of CNTs is the ability to disaggregate and control their dispersion in the polymer as well as the molecular interaction with the matrix. As such, various modification strategies for CNTs have been established. One strategy involves the physical adsorption of surfactants, biomacromolecules, or polymers on the surface of the CNTs so that these functionalized CNTs can be dispersed easily in a wide variety of polar/non-polar solvents and polymer matrices. Anionic surfactants like sodium dodecyl sulfate (SDS)20 and sodium dodecylbenzene sulfonate (NaDDBS),21,22 cationic surfactants suchas hexadecyltrimethylammonium bromide (CTAB),23 and non-ionic surfactants like triton-X10024 have been used to decrease the aggregation of CNTs and improve solubility. In general, the interaction between the surfactants and the CNTs depends on the nature of the surfactants, which includes alkyl chain length, head-group size, and charge. This is clearly exemplified when comparing the surfactants SDS and NaDDBS, which has a benzene ring. The π-stacking interaction of the benzene rings on the surface of the CNTs increases the binding and surface coverage of NaDDBS surfactant molecules significantly.22 Besides using surfactants, non-covalent functionalization of CNTs has also been accomplished with the use of polymers, which physically wrap around the CNTs, forming supramolecular complexes.25–30 In these cases, the graphitic sidewalls of the CNTs provide the possibility for π-stacking interactions with organic polymers. CNTs can be wrapped or encapsulated with poly(p-phenylenevinylene-co-2,5-dioctyloxy-m-phenylenevinylene)26–28 and amphiphilic poly(styrene)-block-poly(acrylic acid)29 copolymers. Encapsulation significantly enhances the dispersion of modified CNTs in different solvents and polymer matrices because of the permanently fixed copolymer shell. However, with this type of non-covalent surface modification, the forces between the wrapping molecule and the CNTs might be weak and, when acting as a filler in a polymer composite, the efficiency of load transfer is low.
Another strategy for CNT modification is the covalent grafting of polymers to surface-bound carboxylic acid groups (−COOH) or hydroxyl groups (−OH) on CNTs. Typically, these functional groups are created on CNT surfaces during oxidation by oxygen, air, concentrated sulfuric acid, nitric acid, aqueous hydrogen peroxide, or an acid mixture. Depending on the acid treatment temperature and time, oxidation procedures and oxidizing agents, the extent of the induced −COOH and −OH functionality differs.30,31 Their presence on a nanotube surface helps the attachment of organic or inorganic materials, which enhance the dispersion and solubility of functionalized CNTs in solvents and in polymer matrices. This is because these functional groups are very reactive and a number of chemical reactions can occur with these groups. Covalent functionalization takes place either through the attachment of as-prepared or commercially available polymer molecules onto the CNT surface via amidation, esterification, or radical coupling (‘grafting to’) or through the in situ polymerization of monomers onto the reactive surfaces of a CNT with the aid of active compounds (initiators or comonomers) via radical, cationic, anionic, ring-opening, and condensation polymerization (‘grafting from’).32 This approach has been used successfully to graft many polymers, including nylon, onto CNT surfaces.33,34 Grafting of CNTs with nylon-6 can be achieved in a two-step process.34 The first step is the formation of the acyl caprolactam initiator by the addition of ε-caprolactam to isocyanate-functionalized CNTs. The second step is the formation of polymer covalently functionalized CNTs in which the nanotube-bound acyl caprolactam initiates anionic ring-opening polymerization of ε-caprolactam in the presence of sodium caprolactamate as a catalyst. This approach is highly efficient and the resulting functionalized CNT surfaces are uniformly coated with nylon-6.
9.2.3 One-dimensional (1-D) nanoparticles
In contrast to the 3-D and 2-D nanofillers discussed earlier, plate-like or layer structured nanofillers are one-dimensional (1-D) with a thickness of, typically, a few nanometers while the other two dimensions are in the sub-micron range. But, as with other nanofillers, layered fillers are difficult to disperse individually in polymers due to the large contact area between the particles and the enhanced particle–particle interaction. As a result, most of the layered fillers (and in particular silicates) exist naturally as stacks and hundreds or thousands of these layers are stacked together by van der Waals forces (and, in some cases, electrostatic forces). Nevertheless, this configuration provides an excellent opportunity for fine-tuning their surface chemistry through ion exchange reactions with organic and inorganic cations. This in turn leads to many possibilities and prospects for tailoring various properties required for specific end applications, other than just merely dispersing the layers in a polymer matrix.
The clay minerals used for polymer nanocomposites can be classified into three groups, as schematically shown in Fig. 9.2:35 the 2:1 type, the 1:1 type, and layered silicic acids. Of these, the most commonly used for the preparation of polymer nanocomposites belongs to the family of 2:1 phyllosilicates and, in particular, smectite clays because of their swelling properties and high cation exchange capacities. For a 2:1 type clay, the crystal structure is made up of layers of two tetrahedrally coordinated silicon atoms fused to an edge-shared octahedral sheet of either aluminum or magnesium hydroxide. The layer has a thickness ~0.94 nm and lateral dimensions varying from 30 nm to several microns, depending on the particular layered silicate (for example, saponite ~50–60 nm, montmorillonite ~100–150 nm, and hectorite ~200–300 nm), thus providing a large surface area ~750 m2/g.36 These layers organize themselves into stacks interspaced with interlayer or intra-gallery space leading to a regular van der Waals gap between the layers. Isomorphic substitution within the layers (for instance, Al3 + replaced by Mg2 + or by Fe2 +, or Mg2 + replaced by Li+) generates a net negative charge. If the layers are negatively charged, this charge is generally counterbalanced by alkali or alkaline earth cations located in the interlayer, such as Na+ and K+. Therefore, this type of layered silicate is characterized by a moderate negative surface charge, termed the cation exchange capacity (CEC).
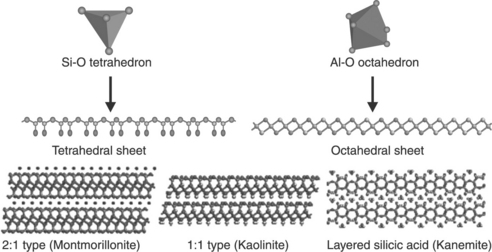
9.2 Classification of clay minerals and their structures. Reprinted with permission from Zeng et al.35 Copyright (2005) American Scientific Publishers.
The CEC of layered silicates is an important factor during the synthesis of polymer nanocomposites as it determines the amount of cationic surfactants that can be intercalated into the layered silicate intra-gallery through ion-exchange reactions to render the hydrophilic phyllosilicates more organophilic and compatible with organic polymers. Typical cationic surfactants for layered silicates include, but are not limited to, primary, secondary, tertiary, and quarternary alkylammonium or alkylphosphonium cations.37 These organic cations not only diminish the surface energy of the layered silicates and improve the wetting characteristics of the polymer matrix, but also increase the interlayer spacing of the layered silicates. The basal spacing of pristine clay layers depends on the type of clay; for example, pristine montmorillonite has a basal spacing of ~1.7 nm. Upon modification with organic cationic surfactants, the spacing (interlayer or intra-gallery) could be in the range ~1.9–4.0 nm, resulting in an organically modified clay (Fig. 9.3).38 However, the space between the silicate layers depends greatly on the length of the alkyl chain and the ratio of cross-sectional area to available area per cation.36,39,40

9.3 Cation-exchange reaction between silicate and an alkylammonium salt. Reprinted with permission from Zanetti et al.38 Copyright (2000) John Wiley & Sons Inc.
Apart from the above-mentioned alkylammonium-based surfactants, silanization was also adopted to improve the compatibility and the interfacial interaction between nylon-6 and layered silicates.41 Organosilanes with various functional groups were used by Kim et al.,41 i.e., APS, GPS, and 3-isocyanate-propyltriethoxysilane (IPS). The experimental results indicated that epoxy functional groups on GPS were more effective in terms of enhancing the nanoparticle-matrix interaction than amino groups on APS.
Further, with non–polar polymers, proper functionalization of the polymer matrix or the addition of compatibilizers is necessary to enhance compatibility with the organoclay. For example, with polypropylene (PP) or polyethylene (PE), maleic anhydride grafted polypropylene or polyethylene (PP-g-MA or PE-g-MA) is added, respectively, to improve the dispersion and swelling of the clay layers. The underlying reason is attributed to the possible interaction via hydrogen bonding between the oxygen group of the silicates and the grafted maleic anhydride group, which would assist the desired nanoscale dispersion of the organoclay in the polypropylene or polyethylene matrices.36,42–44 Figure 9.4 shows the differences in dispersion and exfoliation of organoclay in polypropylene without and with various contents of PP-g-MA.45

9.4 Transmission electron micrographs (TEMs) showing (a) poor compatibility of the organoclay layers with the polypropylene matrix resulting in agglomerates and good dispersion of the organoclay layers in a mixture of polypropylene and PP-g-MA with (b) 50 wt% and (c) 100 wt% PP-g-MA.
Several other approaches for the surface modification of nanoparticles have been established but are not discussed here. Although inherently different nanoparticle surface-modified technologies have been developed, they all address important issues that affect nanocomposite properties. The key to success for any nanoparticle surface-modification technology is to disperse the nanoparticles individually and homogeneously within the polymer matrix and to achieve an optimized chemical affinity with the surrounding polymer matrix so as to facilitate stress transfer from the ‘softer’ matrix to the ‘harder’ nanoparticles. An optimized interfacial interaction between the two components is crucial to ensure superior mechanical properties, particularly fracture toughness. If the interfacial interaction between the two components is too strong, the resulting nanocomposites may fail and undergo plastic deformation, while if it is too weak the interfacial interaction may lead to lower tensile strength and premature brittle fracture.
9.3 Current manufacturing techniques
A fundamental knowledge of the nature of the different manufacturing techniques is important in determining the ultimate performance of polymer nanocomposites. In this section, typical processing techniques for polymer nanocomposites and, in particular, nylon-based nanocomposites will be described. The three most common techniques to fabricate nanocomposites are solution processing, in situ polymerization, and melt processing.
9.3.1 Solution processing
The general protocol for all solution-processing methods consists of the dispersion of nanoparticles in a liquid medium by vigorous stirring or ultra-sonication, followed by mixing of the nanoparticle suspension with a polymer solution and, subsequently, controlled evaporation of the solvent with or without vacuum. For example, Vaia and coworke46 adopted this approach and compared the dispersion of clay layers with and without melt-processed nylon-6/quarternary alkylammonium-modified montmorillonite (organoclay) nanocomposite pellets. That is, they used film casting of solution-processed nylon-6 and organoclay using 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP) as a solvent and film casting of reconstituted, melt-processed nylon-6/organoclay nanocomposite pellets in HFIP. The former yielded an aggregated structure since the nylon-6 chains were not intercalated into the layered silicates. There was a small decrease in gallery height (from 1.85 nm to 1.7 nm), which might be due to the de-intercalation of excess ammonium modifier and dissolution within the nylon-6 matrix. In contrast, film casting from reconstituted, melt-processed nylon-6/organoclay nanocomposite pellets in HFIP resulted in a well-intercalated morphology. During melt processing, there is strong physico-absorption and hydrogen bonding between the nylon-6 chains and the organoclay layers. Specifically, amide and carbonyl groups on the polymer probably interacted with the oxygen plane of the layer surface as well as the weakly acidic SiOH and the strongly acidic bridging hydroxyl groups present at the layer edge. Due to these strong interfacial interactions in melt processing, the physico-absorbed nylon chains were not displaced on addition of HFIP during solution processing, and the HFIP effectively dispersed the individual layers by dissolution of the surface-absorbed nylon chains. This result underscores the importance of process history and the crucial balance of the interfacial interactions between the layered silicate and the surrounding medium.
Nylon-based nanocomposites are more commonly prepared by melt processing and in situ polymerization approaches rather than by solution processing. More often than not, these two approaches yield better quality nylon-based nanocomposites. Other drawbacks of solution processing are the need for suitable solvent or polymer solvent pairs and the high costs associated with the solvents, their disposal, and their negative impact on the environment.40
9.3.2 In situ polymerization
In situ polymerization has been demonstrated to be a successful approach for the preparation of nylon-based nanocomposites. Typically, in this process, the nanoparticle is firstly dispersed in the monomer or precursor and the mixture is subsequently polymerized by adding an appropriate catalyst or an organic initiator. A higher percentage of nanoparticles may be easily dispersed by this method and the nanoparticles can form strong interactions with the polymer matrices. Unlike solution or melt processing, in situ polymerization involves mixing the nanoparticles with short chain molecules instead of macromolecules. Thus, there is less possibility for nanoparticle agglomeration in the composites as there is less entanglement and steric hindrance between the short chain molecules. Indeed, in situ polymerization was first employed at the Toyota Central Research Laboratories to synthesize a nylon-6/clay nanocomposite.47–52 The researchers showed that the ε-caprolactam monomer was able to swell the ω-amino-acid-modified layered silicates and subsequently initiated ring-opening polymerization to obtain the nylon-6 layered silicate nanocomposite. Also, they showed that the carbon number of the amino acid should be greater than 8 for significant swelling of the modified layered silicates.50 A more detailed description of the process can be found in Usuki et al.52
The preparation of nylon-based nanocomposites with 3-D nanoparticles using in situ polymerization has also been reported.4,16,18,53 Some researchers even treated the nanoparticles with organosilane agents before in situ polymerization, so as to achieve a better dispersion of the nanoparticles in the matrix.4,16,53 By employing this approach, Ou and coworkers53 prepared nylon-6/silica nanocomposites in which the silica nanoparticles, pretreated with aminobutyric acid, were dispersed in ε-caproamide and aminocaproic acid at 90 °C, which were the monomer and initiator, respectively, followed by in situ polymerization at a high temperature under a nitrogen atmosphere. With this method, the modified silica nanoparticles, of diameter ~50 nm, were found to be well dispersed within the matrix (Fig. 9.5).53

9.5 Scanning electron micrographs (SEMs) of nylon 6/SiO2 nanocomposites with (a) 10 wt% unmodified and (b) modified SiO2 nanoparticles prepared by in situ polymerization. Reprinted with permission from Yang et al.53 Copyright (1998) John Wiley & Sons Inc.
Apart from directly mixing treated nanoparticles with a caprolactam monomer in a conventional in situ polymerization process, another route for achieving homogeneous dispersion of nanoparticles within a polymer matrix is through the integration of the well-established sol–gel process and in situ polymerization. The sol–gel process, as described earlier, is a wet-chemical technique primarily used for the fabrication of metal-oxide nanoparticles, starting from organic precursors such as metal alkoxides and metal chlorides, which eventually form a three-dimensional cross-linked inorganic network structure via various forms of hydrolysis-condensation reactions. Sengupta et al.17 added tetraethoxysilane (TEOS) to polyamide-66 (PA66) dissolved in formic acid under vigorous stirring with a magnetic stirrer bar at room temperature for 1 h. The solution was then kept at room temperature for 12 h to allow the hydrolysis and condensation reactions of TEOS. Finally, the solution was poured onto glass plates for the solvent to evaporate under a fume hood. This method permitted the synthesis of PA66/SiO2 nanocomposite with no aggregation of nanoparticles.
The preparation of nylon/CNT composites by in situ polymerization has also been very successful.54–58 Gao and coworkers54 functionalized MWCNTs using a solution of sulfuric acid and nitric acid to add –COOH groups to the nanotube surfaces. The MWCNTs/nylon-1010 composites were then prepared by polymerization of a nylon-1010 monomer salt in the presence of oxidized MWCNTs. A similar approach was used by Zhao et al.55 to prepare nylon-6/MWCNT composites using ε-caprolactam monomers. Xu et al.56 prepared nylon-6/functionalized-CNT composites from hexanolactam and 6-aminoaldehexose acid. Surprisingly, they found that their synthesized CNTs hindered the polymerization of nylon-6 and they later modified the sequence of the process by polymerizing hexanolactam and 6-aminoaldehexose acid for 2.5 h before adding CNTs. The underlying causes of the inability of the monomer to polymerize in their initial processing approach were investigated using Fourier transform infrared spectroscopy. The analysis revealed that the reason might be because the –COOH groups on the oxidized CNTs created a strong nylon-6/CNT interface linked by C–O–N chemical bonds. These chemical bonds consisted of more stable nylon-6/CNT molecule chains and further growth of nylon-6 molecules could be obstructed by the formation of these bonds.
9.3.3 Melt processing
Due to the fact that thermoplastic semi-crystalline polymers like nylon soften when heated above their melting point, the fabrication of nanocomposites based on these polymers by melt compounding has proved to be a very valuable technique. This processing technique is compatible with conventional industrial processes; additionally the process is solvent-free, thereby reducing the associated costs and effect on the environment. In general, this process involves the mixing of a polymer melt with nanoparticles above the softening point of the polymer, under shear (or even in the absence of shear, in some cases). The melt-blended materials can then be processed by several methodologies such as film extrusion, injection molding, or compression molding, depending on the final morphology and shape of the products required. Nonetheless, there are some limitations that should be taken into consideration when using melt processing to prepare nylon-based nanocomposites. First, the polymer matrix degrades during processing, in general, at high temperatures and shear stresses. Second, the success of this technique requires the polymer to be highly compatible with the nanoparticles.
The preparation of nylon-6 layered silicate nanocomposites by melt processing was first demonstrated by Liu et al.59 following work reported by Vaia et al.60 in the preparation of polystyrene (PS) layered silicate nanocomposites. Subsequently, melt processing has become the mainstream method in the manufacture of numerous polymer nanocomposites incorporating a wide range of nanoparticles, from spherical3,9–11 to plate-like,39,61 and fibrous nanoparticles.62–64 For nylon-based nanocomposites, much work has been spent on understanding the dispersion behavior of the nanoparticles in the resulting nanocomposites with respect to parameters such as processing conditions, the chemical nature of the matrix, the effect of nanoparticle surface modifications, the type of surface modifier, and melt rheology. Obviously, it is impossible to make an exhaustive list of works that have covered these topics in this chapter. Therefore, characteristic and important examples will be provided during the discussion of the structure–property relation in the next section.
9.4 Structures and properties
As mentioned previously, nanoparticles have the potential to exhibit a combination of different properties (physical, chemical, thermal, and mechanical). In addition, the exceptionally large surface-area/volume ratio of the nanoparticles, which is available for interaction with the polymer matrix, offers a chance to fine-tune the specific properties of nanocomposites required for end applications. An overview of some of these properties is given below, with particular emphasis on nylon-based matrices.
9.4.1 Mechanical properties
In general, the stiffness of a nanocomposite, even with agglomerated particles within the matrix, has a certain degree of improvement, though not to a large extent. This is because of its strong dependence on the orientation of anisotropic particles.65 Further, the effect of aspect ratio is evident by comparing the magnitude of the improvement in stiffness for various polymer nanocomposite systems. With high-aspect-ratio nanoparticles like CNTs and clays, when well dispersed in the polymers, significantly improved stiffness has been achieved compared with the neat polymers. For instance, in the presence of only 2 wt% of MWCNTs, the elastic modulus of the resulting melt-processed nylon-6 nanocomposite was substantially improved by about 214% from 396 MPa to 1242 MPa (Fig. 9.6).64 Moreover, the advantage of high-aspect-ratio nanoparticles over conventional reinforcements is evident in Fig. 9.7, which compares the tensile modulus of nylon-6 materials reinforced with organoclay and glass fibers.66–68 Even at very low loadings of filler, the nanoclay-reinforced nylon-6 shows a tremendous positive difference in the relative Young’s modulus (E/Em, where E and Em are the Young’s modulus of the composite and neat matrix, respectively). As explained earlier, the substantial improvement originates from the exceptionally high interfacial surface area, and it was also shown that, if the magnitude of intercalation is higher, greater improvements in elastic modulus and tensile strength can be achieved, as intercalation provides higher aspect ratios and optimizes the number of reinforcing elements.69

9.6 Variation of tensile properties for nylon-6 and its nanocomposites against MWCNT loading. Reprinted with permission from Liu et al.64 Copyright (2004) American Chemical Society.

9.7 Comparison of elastic modulus (relative to a polyamide-6 matrix polymer) for nanocomposites based on clay (montmorillonite) and glass fibers. Reprinted with permission from Fornes and Paul.66 Copyright (2003) Elsevier.
In some cases, however, enhancements in stiffness and strength were not realized even with the incorporation of high-aspect-ratio fillers like CNTs in the nylon matrix. This was seen in styrene maleic anhydride (SMA) copolymer encapsulated SWCNTs where a ~10% drop in tensile modulus was noted for nylon-6 composites with 1.5 wt% SMA-modified SWCNTs.70 No improvements in yield strength were noticed in the nanocomposites. This was attributed to the loose network-like structure of the SWCNTs or the existence of SWCNT agglomerates acting as defects. These unusual experimental results for the mechanical properties of nylon composites imply that further research in terms of processing methods and CNT functionalization, as well as a better understanding and control of the polymer–CNT interfacial interaction, is necessary for the design and tailoring of nylon–CNT composites with desirable and predictable performance.
Specifically with regard to the mechanical properties of polymer layered silicate nanocomposites, comparative results have been reported for intercalated and exfoliated structures. Some mathematical models have also been proposed to explain the reinforcement efficiency of layered silicates in polymers by considering factors such as aspect ratio and orientation.66,71 Much work has also been spent on understanding the influence of several other parameters like processing conditions,72 the effects of the origin of the clay, the surfactants used,39 the cation exchange capacity,73 and the properties of the polymer matrices.74 These aspects have been comprehensively reviewed in several publications36,37’40,75–77 and thus will not be reiterated here.
On the other hand, properties measured at larger deformation, notably tensile yield strength, as well as the corresponding deformation mechanisms depend strongly on the microstructure and interfacial interaction. This is shown in Table 9.2, which compares three types of MWCNTs with different surface functionalization, used in the preparation of nylon-6/MWCNT nanocomposites by melt processing.78 The first type of MWCNTs were synthesized in an ethanol flame, designated as F-MWCNTs; the second type are F-MWCNTs grafted with long-chain molecules of n-hexadecylamine (H-MWCNTs) and the third type are commercially available MWCNTs used as a benchmark, denoted as C-MWCNTs. The inherent defects in F-MWCNTs make them chemically active, and abundant functional groups such as carboxyl and hydroxyl groups are readily found on the surfaces of the F-MWCNTs. This facilitated further functionalization of the F-MWCNTs without oxidation or detriment to the primary structures of the nanotubes. Hence, the surface grafting of F-MWCNTs with long-chain molecules of n-hexadecylamine (H-MWCNTs) resulted in better nanoscopic dispersion and interfacial adhesion in the polyamide-6 (PA6) matrix, so that it had the highest tensile modulus of the three nanocomposites.
Table 9.2
Tensile properties of PA6 and its composites with various MWCNTS
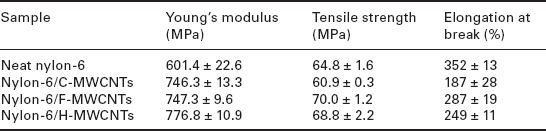
Source: Adapted from Liu et al.78
Based on the majority of studies, there is general agreement that the presence of layered silicates or CNTs results in either no improvement or even deterioration in fracture toughness and ductility of nylon-based nanocomposites74'79–82 Chen et al.80 investigated the embrittlement mechanisms of nylon-66 layered silicate nanocomposites and found that micro-sized and sub-micron voids developed around the layered silicates, which coalesced and formed premature cracks without any significant matrix plastic deformation, thus slashing the fracture toughness of the nanocomposites. He et al.81 also showed that, in nylon-6 with 2.5 wt% organoclay, crazes were initiated in the matrix adjacent to the interface between the clay platelets and polymer, which developed into micro-cracks on continued deformation to failure. This resulted in decreased KIC. At higher clay loadings, small-angle X-ray scattering results revealed a lower density of crazes; it was suggested that the large number of micro-cracks (due to the higher clay loading) had prevented the crazing mechanism from becoming fully operative before failure, thus leading to even lower toughness. We also reported similar observations for a nylon-6/organoclay layer nanocomposite in which only a limited number of micro-voids were found along the crack paths in the vicinity of an arrested crack tip, as shown in Fig. 9.8.82

9.8 TEM micrograph of polyamide-6/organoclay (95/5) nanocomposite showing voids along the path of an arrested crack tip. Black arrows indicate the direction of crack growth and the white arrow shows the delamination of the clay layers. Reprinted with permission from Lim et al.82 Copyright (2007) Elsevier.
It has also been shown that the presence of stiff layered silicates restricts the mobility of the surrounding chains, thus limiting their ability to undergo plastic deformation.83 Due to the strong tethering junctions between individual clay layers and the matrix (due to the ionic interaction between the ends of the polymer chains and the surface of the negatively charged clay layers), full-scale debonding at the polymer–clay interface was rarely observed under stress conditions, indicating that the constraint on the polymer adjacent to the clay was not relieved, limiting the ability of the polymer to undergo plastic deformation. In many studies with CNTs, the toughness of the composites fell. For instance, the presence of even 2 wt% CNTs reduced the toughness from 12.4 kJ/m2 to 10.0 kJ/m2.56 This reduction was attributed to the nucleation of cracks at the interface between encapsulated nylon-6 layers on CNT surfaces and the nylon-6 matrix.
But the high flexibility of CNTs, in a few cases, was shown to deflect cracks via matrix crack bridging under loading and to the pull-out of the nanotubes. Bridging by the nanotubes prevents a crack from opening and thus restrains catastrophic crack propagation in the polymer matrix. An example of bridging of a matrix crack is shown in Fig. 9.9. This mechanism is believed to be the main energy absorbing mechanism in nylon-6/MWCNT composites.64 The pull-out mechanism, inspired by conventional polymer/fiber composites (including work done against sliding friction in pulling out the fiber), governs the extent of energy absorption. Hence, the very high interfacial areas in polymer/nanotube composites are expected to result in drastic improvements in the work of fracture due to nanotube pull-out. But recently, based on a scaling argumenti84 correlating the radius (r), fiber strength (σ), and interface strength (τ) with the energy absorbed per unit cross-sectional area by fiber pull-out (i.e., ![]() )), it has been shown that improvements in toughness in polymer/CNT nanocomposites cannot be attributed to the nanotube pull-out mechanism, as the pull-out energy significantly decreases when the fiber radius is scaled down to the nanoscale. Wichmann et al.85 argued that this conventional correlation is not valid for nanotubes by simply considering the Kelly–Tyson expression (critical length,
)), it has been shown that improvements in toughness in polymer/CNT nanocomposites cannot be attributed to the nanotube pull-out mechanism, as the pull-out energy significantly decreases when the fiber radius is scaled down to the nanoscale. Wichmann et al.85 argued that this conventional correlation is not valid for nanotubes by simply considering the Kelly–Tyson expression (critical length, ![]() ), that is, it is impossible to vary the fiber radius independently without changing other parameters. They further suggested that, if spatial or only local bonding existed between the nanotubes and the matrix, there would be partial debonding of the interface allowing crack bridging similar to conventional polymer/fiber composites, as shown and analyzed by Gao et al.86 two decades ago.
), that is, it is impossible to vary the fiber radius independently without changing other parameters. They further suggested that, if spatial or only local bonding existed between the nanotubes and the matrix, there would be partial debonding of the interface allowing crack bridging similar to conventional polymer/fiber composites, as shown and analyzed by Gao et al.86 two decades ago.

9.9 SEM image of a micro-crack linked by MWCNTs sheathed with polymer beads as indicated by the white arrows in a nylon-6 nanocomposite. Reprinted with permission from Liu et al.64 Copyright (2004) American Chemical Society.
Aside from these few cases where improvements in toughness and ductility have been reported, the majority of studies show reduced ductility and toughness when high-aspect-ratio nanoparticles are incorporated in nylon. Even in those few cases that report improvements, the properties of the nanocomposites were not consistent and the failure mechanisms were not well understood. Nevertheless, to achieve a balance between stiffness and toughness in nanocomposites, ternary nanocomposites (prepared by adding a soft elastomeric dispersed phase to binary polymer nanocomposite systems) have been developed.2,82,87–92 Though this is the best-known approach to date for counteracting the embrittlement of polymer nanocomposites,2 its associated disadvantages must also be considered. The final microstructures are generally complex and control of the location of the rigid fillers (either in the matrix or as elastomeric particles) is necessary for achieving enhanced properties.
With 3-D nanoparticles, the enhancement in stiffness and strength of the nanocomposites is not as significant. But, generally, a positive ductility and toughness response is noted with these materials. Li et al.16 observed a ~25% improvement in impact strength on incorporation of nano-SiO. (10 nm), treated with amine and epoxy-functionalized silanes, into a nylon-6 matrix compared with neat nylon-6. They also noted that with unmodified nano-SiO2 the impact strength was reduced by 50%. To increase the fracture toughness of a material, it is necessary to initiate several effective energy dissipation processes like crazing and shear yielding around the crack tip so that the total amount of plastic energy absorbed during fracture is high. Zhang and coworkers93 studied the toughness of nylon-66/TiO2 nanocomposites using the essential work of fracture (EWF). They found that the essential work of fracture (initiation) increased with increasing TiO2 content up to 3 vol% while the non-essential work of fracture (propagation) decreased. Fractographic analysis revealed that the nanoparticles acted as stress concentrators, triggering debonding and then inducing a large amount of local plastic deformation of the matrix. With an increasing number of TiO2 nanoparticles, the plastic zone became more and more constrained and thus the non-essential work of fracture decreased. The reduction of resistance to crack propagation was due to the inevitably agglomerated nanoparticles at higher nanoparticle loadings, leading to a high level of stress concentration, which favored catastrophic crack propagation. In another work, similar results for the toughness property and deformation mechanism were obtained for polyamide-66 with SiO2 and Al2O3 nanoparticles.94
9.4.2 Wear and scratch resistance
Wear generally originates from damage induced by rubbing bodies due to repeated applications of mechanical, impact, and other kinds of forces. Therefore, the surface loses mechanical cohesion and debris is formed from material dislodged from the contact zone.95–98 For reliable and effective functioning of materials, it is necessary to reduce material damage and material removal, or, in some cases, to control the extent of the material removed. Generally, the specific wear rate K (mm3 N–1 m–1) is determined by
where V (mm3) is the wear volume loss calculated from
Note that t (s) is the test duration, A (mm2) cross-sectional area of the wear track, L (N) normal load, R (mm) the wear track radius, and v (m/s) the sliding speed.
However, as polymers are highly sensitive to scratch and wear damage, they exhibit various modes of deformation, e.g., abrasive, adhesive, fatigue, corrosive, erosive, and delamination, even within a narrow range of contact variables.99–103 Moreover, there is always an overlap of different mechanisms in any particular contact process and any combination of the different mechanisms may represent the actual situation. This indicates the complexity of wear and scratch damage in polymers, and the difficulty in accurately quantifying and predicting the extent of damage and the damage modes, and thus limits their application. Nonetheless, for polymers, different wear mechanisms can be grouped into two main categories: cohesive and interfacial wear processes (schematically shown in Fig. 9.10).104 In cohesive wear processes, the frictional work is dissipated in relatively large volumes adjacent to the interface, either through the interaction of surface forces and the resultant traction stresses or simply via geometric interlocking of interpenetrating contacts. The extent of this surface zone is defined by the contact geometry and contact stresses generated in the surface. Cohesive wear processes are mainly controlled by the mechanical properties of the interacting bodies. Most mechanical wear processes can be grouped under this category, such as abrasive, fatigue, and fretting. In contrast, interfacial wear processes involve dissipation of frictional work in much thinner regions and at greater energy densities. This creates a large increase in local temperature. The chemistry of the surfaces and the forces emanating from them should be considered, instead of the mechanical properties of the interacting materials, to determine the extent of wear damage. Transfer wear and chemical or corrosive wear are interfacial wear processes.

9.10 Different wear mechanisms under cohesive and interfacial wear processes. Adapted from Briscoe and Sinha.104
Fortunately, from the tribological viewpoint, the major benefits of polymer nanocomposites relative to micro-sized particle composites are: (i) material removal is expected to be less as the nanoparticles have a similar size to the segments of the surrounding polymer chains; and (ii) the bonding between the nanoparticles and the matrix is expected to be better due to their large specific surface areas.105–107 However, there are many important factors affecting the tribological behavior of polymer nanocomposites, including particle size, aspect ratio, hardness, concentration, orientation, nature of the interface between the polymer matrix and the particles, tribochemical reactions, and transfer films that may arise due to the interaction of particles and counterfaces. Additionally, the indirect influence of nano-additives on polymeric materials should be considered when investigating their tribological response.
Many studies have described wear and scratch damage in polymers filled with different nanoparticles and the majority of them reported improved tribological properties. This was attributed to the presence of the nanoparticles themselves or to improved mechanical properties like the modulus and hardness or to the formation of transfer films on the slider contact surface. However, when dealing with polymer nanocomposites, it is not valid to assume that nanoparticles irrevocably improve wear and scratch (and friction) properties. Material properties like the modulus, hardness, fracture toughness, or the wear rate or scratch penetration depth, are not the sole indicators for comparing and ranking candidate materials.98,108,109 It was also found that nanoparticles by themselves, even if uniformly dispersed with good interfacial interaction with the matrix, do not irrevocably improve the wear (and friction) properties. Although it is important to consider these factors, it is necessary to thoroughly understand all nanostructural parameters and the relations between penetration depth and material deformation and sub-surface damage in the wear and scratch track. This knowledge is critical for understanding the surface integrity of these materials. The correct physical and spatial characterization of nanocomposites is required. For example, while the orientation and dispersion of clay layers are independent structural features, they must be simultaneously considered in determining the effective structural reinforcement in polymer/clay nanocomposites; otherwise, the results can be misleading if only dispersion or orientation of the structures is considered.109
Moreover, considering the complexities involved in evaluating the tribological response of polymer nanocomposites, only a limited effort has been made to model the stress fields induced by different slider geometries. Hence, to date, no explicit correlations between material parameters and wear and scratch damage, particularly for polymer nanocomposites at the nanoscale, are available. For a more in-depth review of the wear and scratch response of polymer nanocomposites, refer to Dasari et al.98
9.4.3 Barrier properties and permeability
Barrier properties are of prime importance in bottling, food packaging, packaging hydrocarbon solvents, and the protective coating industries. In comparison with other physical and mechanical properties of polymer/clay nanocomposites, which have mixed results, barrier properties are mostly positive, that is, dramatic decreases in permeability to different media can be achieved at relatively low loadings of nanoparticles (particularly with high-aspect-ratio fillers like layered silicates) in contrast to the neat polymers and polymer micro-composites. 110–116 This is due to the high aspect ratios of impermeable silicate platelets forcing the gas or liquid molecules to traverse a tortuous path in the polymer matrix surrounding these silicate particles. This obviously increases the effective path length for diffusion (Fig. 9.11).37,117 As is evident, aspect ratio, loading, orientation, and degree of dispersion have a significant effect on barrier properties. In addition, many factors, like the surface treatment of the nanoparticles, processing techniques, and the degree of crystallinity and cross-linking of the polymer, can have an indirect bearing on the barrier properties of these materials.

9.11 Comparison of tortuosity for the diffusion path in a polymer filled with micro-sized particles and polymer/clay nanocomposites. Reprinted with permission from Ray and Okamoto.37 Copyright (2003) Elsevier.
According to the tortuous-path model,117 the oxygen gas permeability of a film decreases to 1/5 and 1/2400, compared with the pure film, for 5 and 95 wt% of inorganic platelets, respectively. The tortuosity factor τ is calculated as117
where L/W is the aspect ratio of the layered filler and Vf is the volume fraction. This is for an ideal case where clay particles are completely exfoliated, uniformly dispersed, and oriented such that the direction of diffusion is normal to the direction of the sheets (at a preferred angle of 0°). This arrangement will obviously result in higher tortuosity and the model also suggests that platelet-shaped particles are more efficient at maximizing the path length than particles with other shapes. Recently, Bharadwaj, 118 based on geometric arguments, considered a range of orientations of the clay layers relative to the flow direction of the gas or liquid across the film and studied their effect on the relative permeability. As expected, the permeability is highest when the clay layers are aligned in the flow direction. He also concluded that dispersing longer sheets (L > 500 nm) than the normal length of clay platelets in a polymer matrix not only increased the tortuosity, but also reduced the dependence of the relative permeability on the orientation of the layers.
Further, the effect of tortuosity on the permeability can be expressed as
where PPCN and PP are the permeability coefficients of the nanocomposite and the pure polymer, respectively, and Vf is the volume fraction of clay.37,117
Yano et al.119 studied the effect of clay length on the relative permeability coefficients of polyimide with 2 wt% of organoclay. They compared experimental data and theoretical values predicted using Eq. 9.4 (Fig. 9.12) and found a good fit, that is, as the length of the clay increased, the relative permeability decreased drastically.

9.12 Theoretical predictions (line) and experimental data (symbols) showing the effect of clay-layer size on relative permeability for polyimide/clay nanocomposites. Reprinted with permission from Yano et al.119Copyright (1993)John Wiley & Sons Inc.
Nevertheless, huge permeability resistance to O2, He, CO2, water vapor, and many other solvents and gases has been reported with polymer/clay (montmorillonite) nanocomposites (even with intercalated structures). For example, in polyethylene terephthalate/clay nanocomposites, a twofold reduction in O2 permeability with only 1 wt% clay was noted. 120 Even for hydrogen and water vapor, the permeability coefficients of nylon-6 with only 0.74 vol% of montmorillonite were less than 70% of the equivalent coefficients for nylon-6. In polyimide nanocomposites, the permeability coefficient of water vapor showed a tenfold reduction with 2 wt% synthetic mica relative to pristine polyimide.121 Moreover, studies have indicated that a percolation threshold exists in polymer layered silicate nanocomposites, after which permeability does not decrease or has a negative effect.122,123 Similar behavior has also been observed for other physical properties of polymer nanocomposites.124,125 In this regard, Lu and Mai126 proposed a renormalization group (RG) model and identified the influence of aspect ratio, orientation, and extent of exfoliation of clay layers on the barrier properties of polymer/clay nanocomposites. The basic hypothesis of the RG approach is that the probability p that a cell acts as a barrier is the same at all orders. It was also concluded that the aspect ratio of clay layers is the controlling and determining factor of barrier properties.
But it is important to note that the tortuous path theory, and other proposed relations (e.g. the Cussler127 formula or the power law equation128), are based on the assumption that the presence of nanoparticles does not affect the diffusivity of the polymer matrix, which in reality is not generally true. Some liquids and vapors often have noticeable solubility in polymers, plasticizing them. Moreover, organophilic clay gives rise to superficial adsorption and to specific interactions with some of the solvents. Further, the presence of clay layers, as is well known, induces polymorphism, alters crystallization kinetics (temperature, rate, crystal fraction, and percentage crystallinity), and promotes the formation of small, irregular crystallites of polymers due to their heterogeneous nucleation effect. These increases of the surface area of crystals and the fraction changes in the crystalline and amorphous parts affect the permeability properties, as the crystalline regions are generally more impermeable to penetrant molecules than amorphous regions. Furthermore, as polymers usually have a wide range of relaxation times associated with the motion of the polymer segments, the barrier properties are affected by changes in temperature and the concentration of the permeant.
9.4.4 Thermal properties
Apart from the mechanical properties, thermal properties like dimensional stability, heat distortion temperature (HDT), and thermal stability dictate the widespread applicability of polymeric materials. For instance, the low thermal deformation tolerance of polylactic acid (PLA), a widely marketed biodegradable polymer, has restricted its application in electronics, microwavable packaging, etc. Therefore, it is important to understand and resolve these issues to widen the applicability of polymeric materials.
First, the dimensional stability of polymeric materials during molding is a key parameter, particularly in automobile applications, where polymers with essentially high coefficients of thermal expansion (CTEs) are integrated with metals, which have much lower CTEs.67 Moreover in batteries, a polymer electrolyte is employed as both electrolyte and separator. The heat generated during charging and discharging may cause deformation of the polymer electrolyte (temporary or even permanent dimensional changes), which may result in reduced efficiency or even a short circuit: good dimensional stability is essential. Layered silicate platelets, due to their shape and size, have proved to be useful in reducing the CTEs of polymers in two directions.129–131 That is, with changes in temperature, expansion or contraction of the matrix occurs but the fillers resist this process by inducing opposing stresses in the two phases. Yoon et al.130 measured the linear thermal expansion behavior in the flow direction (FD), the transverse direction (TD), and the normal direction (ND) in ~6.3 × 12.7 × 3.2 mm3 rectangular bars of nylon-6/clay nanocomposites. As shown in Fig. 9.13, the addition of clay reduced the thermal expansion coefficient in FD and TD but increased it in ND. They attributed this to the high constraint effect of nylon-6 in FD and TD, which resulted in a ‘squeezing’ effect in ND, thus increasing expansion in this direction. Hwang and Liu132 showed that the dimensional stability of polyacrylonitrile (PAN) electrolyte improves even with just 3 wt% clay. Within the temperature range 30–75 °C, the thermal expansion coefficient of the PAN/clay electrolyte was 778 × 10–6 mm/°C whereas for the neat PAN electrolyte it was 3400 × 10–6 mm/°C.

9.13 Linear thermal expansion coefficients (a) below and (b) above the nylon-6 glass transition temperature for annealed nylon-6/clay nanocomposites in three directions. Reprinted with permission from Yoon et al.130 Copyright (2002) Elsevier.
HDT is another thermal property and, as the name indicates, it quantifies heat resistance on external loading. An increase in HDT has been observed in clay-based nanocomposites for many polymer systems. The HDT of nylon-6 nanocomposites has been shown to increase from 65 °C (pristine nylon-6) to 150 °C with 5 wt% of clay. Such an increase in HDT is very difficult to achieve in conventional polymer composites reinforced by micro-particles. Ray et al.133 also concluded that the addition of clay layers to PLA increased its HDT; with a load of 0.98 MPa, HDT increased from 76 °C to 93 °C with 4 wt% of organoclay and it further increased to 115 °C with 10 wt% of organoclay. Even the incorporation of CaCO3 nanoparticles in a PP copolymer considerably increased its HDT along with the flexural modulus and impact strength.134
Further, it is important to understand the effect of additional functional additives and organic surfactants for modifying nanoparticles on the thermal stability of the polymeric matrix. For instance, despite the advantages of the organic modification of nanoparticles, it considerably degrades the polymer’s thermal stability. In particular, the commonly used long-chain alkylammonium surfactants have poor thermal (and photochemical) stability 135 and, normally, surfactants account for (~ 20–30% of the inorganic filler loading. Amine groups start to decompose usually from ~180 °C,136 which is below the processing temperatures of most engineering polymers, like nylon-6. Thus, the presence of such a large amount of low molecular weight bound or unbound surfactant may adversely affect thermal properties in addition to the mechanical and physical properties of the resultant nanocomposites.
Much effort has been directed towards developing alternative routes for producing nanocomposites without using the conventional alkylammonium surfactants. These include modifying the clay layers with thermally stable surfactants (usually aromatic compounds), water-assisted approaches, partially exchanged systems that decrease the amount of surfactant required,137 etc. Thermally stable surfactants (like phosphonium, pyridinium, iminium, and imidazolium salts) decompose at higher temperatures than conventional alkylammonium surfactants and are stable, particularly during processing. In particular, the treatment of clay with imidazolium salts (Fig. 9.14) via standard ion-exchange methods showed a tremendous improvement in thermal stability of the modified clays and ultimately polymer nanocomposites.61 Gilman et al. obtained an improvement of ~100 °C (in terms of peak decomposition temperature) by using 1-alkyl-2,3-dimethylimidazolium-treated clay in nitrogen in comparison to that of common alkylammonium clay.61 Further polyhedral oligomeric silsesquioxane (POSS)-treated clays were also used in polymers to obtain a positive effect on thermal stability, as POSS molecules are stable up to 300 °C.138–140 POSS molecules have hybrid (organic–inorganic) architecture and their structure contains a stable inorganic Si–O core, which is intermediate between silica and silicones. This core is covered externally by organic substituents which can be modified to yield a wide range of polarities and functionalities. This great variety gives a diversity of silsesquioxanes.

9.14 Structures of different imidazolium salts used to treat sodium montmorillonite from Gilman et al.61.
9.4.5 Other properties
There are many other properties of polymer nanocomposites, which have not been discussed, but are important in many commercial applications. These include biodegradability (the ability of functionalized nanoparticles to speed the process), optical properties, corrosion, photo-degradation resistance, etc. The flame-retardant properties of polymer nanocomposites are also worth mentioning. A significant amount of recent research has been on this topic. See, for example, various publications 141–143 and references therein. This is because, even at low loadings of nanoparticles and with no additional flame retardants in the system, the heat-release and mass-loss rates were greatly reduced, in addition to a huge delay in burning compared with the neat polymers. The reduction in the peak heat-release rate is an important parameter for fire safety, as it represents the point in a fire where heat is likely to propagate further (flame spread) or ignite adjacent objects. The improved flame retardancy was mainly attributed to the structural collapse of the nanocomposite during combustion and the formation of a multi-layered inorganic barrier on the polymer surface.
9.5 Applications of nylon-based polymers
Before discussing the various applications of polymer nanocomposites, it is important to realize that the scientific literature on these materials is immense and many research and development centers have studied these materials only in the past couple of decades. As with other major discoveries and inventions, it generally takes a few decades for them to have a large commercial effect, as economics is beyond the realm of scientific investigation and additional advances may be required to make the materials commercially viable. Moreover, biomedical applications require an in-depth understanding of the side effects of nanoparticles on humans, which may take even longer to establish. Nevertheless, many polymer-based nanocomposites are increasingly being used in applications including structural, transportation, biomedical, and sports.
There is wide speculation that the enhancements in tensile strength, stiffness, and the heat distortion temperature through the application of nanoparticle-filled engineering thermoplastics like nylon will challenge metal and glass applications in many new areas. The automotive and aerospace industries are leading the way in the development of nanocomposite applications. This is because of improved reinforcement, corrosion resistance, noise dampening, dimensional stability, scratch and wear resistance, and superior barrier properties. 144,145 An additional advantage of polymer nanocomposites is the ease of processing (extrusion and molding into different shapes as required). Some applications are shown in Fig. 9.15, with particular emphasis on layered silicates. The first application for polymer layered silicate nanocomposites was an automotive application by Toyota for a timing belt cover.37 It had good rigidity, excellent thermal stability, and no warpage. Soon after, Mitsubishi introduced nylon-6/clay nanocomposite-based engine covers on their gasoline direct injection (GDI) models and General Motors added an external thermoplastic polyolefin/clay nanocomposite step assist to its Safari/Chevrolet Astro vans.141

9.15 Commercial applications of polymer/clay nanocomposites. (a) A step-assist on a GMC Safari van that incorporates a thermoplastic olefin/clay nanocomposite. (b) GM’s 2005 Hummer H2 cargo bed uses ~7 lb of a molded-in-color thermoplastic olefin nanocomposite. (c) Noble Polymers’ Forte polypropylene/clay nanocomposite seat backs for the 2004 Acura TL. (d) Thermoplastic olefin nanocomposite in the body side molding of GM’s 2004 Chevrolet Impala. (e) Geoflow’s linear low-density polyethylene/clay nanocomposite drip emitter for irrigation tubing ensures the timed release of herbicide. (f) Putsch and Süd-Chemie jointly prepared Elan XP, a compound of polypropylene and polystyrene compatibilized by clay, which is used as an interior air vent for the Audi A4 and a Volkswagen van. (g) Nexans’ cable jacketing nanocomposite, the first such product for plenum cable used in office buildings due to the good flame retardancy of the polymer/clay materials. (h) Wilson’s double-core tennis balls have an inner core coated with a butyl-based rubber filled with nanoclay particles, which acts as a barrier and stops air escaping from the core. (i) Coors’ beer bottle, which has a layer of a polymer/clay nanocomposite because of the high barrier properties. Pictures are courtesy of the relevant company and technology websites.
Moreover, since nanocomposites are light, there is an average weight saving of 25% compared with highly filled-conventional polymers and 80% over steel, 144 which means large reductions in fuel costs. The savings are predicted to be 1.5 billion liters of gasoline over the life of one year’s production of vehicles with the added advantage of reduced CO2 emissions of more than 5 billion kilograms.35 Apart from the automotive industry, polymer/clay nanocomposites in molded parts are also used in construction (building sections and structural panels) and aerospace (flame-retardant panels and high-performance components) applications.
Other promising applications of polymer nanocomposites are due to their outstanding barrier, permeability (particularly with high-aspect-ratio nanoparticles), and optical transparency properties. They are currently used in packaging for processed meats, cheese, confectionery, cereals, boil-in-the-bag foods, and extrusion-coating applications with paperboard for fruit juice and dairy products, and with co-extrusion processes in the manufacture of bottles for beer and carbonated drinks. Commercial grades of packaging films based on nylon-6 nanocomposites are available from Ube Industries and from Bayer AG.
Nanofibers are another type of nylon-based material that are used in applications. The outstanding properties of nanofibers (large surface area to volume ratio, flexibility in surface functionalities, and superior mechanical performance) make them suitable candidates for applications such as filtration, protective clothing, biomedical (including wound dressing), and as supports for enzymes and catalysts. A number of processing techniques 146 such as drawing, template synthesis, phase separation, self-assembly, and electrospinning have been used to prepare polymer nanofibers. Of these, electrospinning and electrostatic spinning have many advantages. For an extensive comparison of these techniques, the fundamentals behind them, their advantages and disadvantages, refer to the literature.147–150
While nano-layer silicates add muscle to nylon, CNTs impart electrical and thermal conductivity. Electro-conductive polymer nanocomposites are the preferred replacement for traditional steel in fuel delivery lines because they prevent the build-up of static electricity. They have also been developed for electro-paintable exterior body panel applications. In summary, though the main applications for nanocomposites are currently in packaging and transportation, their application will increase as R&D progresses.
9.6 Future trends
The critical challenge in the development of polymer nanocomposites with remarkable improvements in thermo-mechanical, electrical, and optical properties lies in the dispersion of the nanoparticles in the polymer matrix. In addition to the various surface-modified technologies in the preparation of polymer nanocomposites, there are still opportunities and challenges for improving dispersion and the interfacial properties of these materials. Thus, further research and development from these perspectives is required. Frequently, the poor characterization of these materials and inadequate quantitative descriptions of observed phenomena have led to contradictions and misleading impressions for these nanocomposites. Thus, much more work is needed to develop analytical and modeling tools to predict and verify the behavior of these types of nanocomposite.
9.7 Acknowledgements
The permission from various publishers to reproduce the figures in this chapter is much appreciated.
9.8 References
1. Ruiz-Hitzky, E., Van Meerbeek, A. Clay mineral and organoclay-polymer nanocomposite. In: Bergaya, F., Theng, B.K.G., Lagaly, G., eds. Handbook of Clay Science – Developments in Clay Science, Vol. 1. The Netherlands: Elsevier Ltd; 2006:583.
2. Dasari, A., Lim, S.H., Yu, Z.Z., Mai, Y.W. Fracture properties and mechanisms of polyamide/clay nanocomposites. In: Karger-Koscis J., Fakirov S., eds. Nano- and micromechanics of polymer blends and composites. Munich: Hanser; 2009:377.
3. Tabuani, D., Ceccia, S., Camino, G. Nylon-6 nanocomposites, study of the influence of the nanofiller nature on morphology and material properties. J Polym Sci B Polym Phys. 2009; 47(19):1935–1948.
4. Zheng, L.Y., Lau, K.T., Zhao, L.X., Zhang, Y.Q., Hui, D. Mechanical and thermal properties of nano-Al2O3/nylon 6 composites. Chem Eng Comm. 2010; 197(3):343–351.
5. Chen, C.H., Li, H.Y., Chien, C.Y., Yen, F.S., Chen, H.Y., Lin, J.M. Preparation and characterization of alpha-Al2O3/nylon 6 nanocomposite masterbatches. J Appl Polym Sci. 2009; 112(2):1063–1069.
6. Wang, S.B., Ge, S.R., Zhang, D.K. Comparison of tribological behavior of nylon composites filled with zinc oxide particles and whiskers. Wear. 2009; 266(1–2):248–254.
7. Zheng, J.R., Siegel, R.W., Gregory Toney, C. Polymer crystalline structure and morphology changes in nylon 6/ZnO nanocomposites. J Polym Sci Part B-Polym Phys. 2003; 41(10):1033–1050.
8. Moussa, M.A., Ghoneim, A.M., Hakim, A.A.A., Turhy, G.M. Electrical and thermal properties of nylon 6/calcium carbonate composites. Adv Polym Techn. 2009; 28(4):257–266.
9. Avella, M., Carfagna, C., Cerruti, P., Errico, M.E., Gentile, G. Nylon based nanocomposites: Influence of calcium carbonate nanoparticles on the thermal stability. Macromol Symposia. 2006; 234:163–169.
10. Avella, M., Errico, M.E., Gentile, G. Nylon 6/calcium carbonate nanocomposites: Characterization and properties. Macromol Symposia. 2006; 234:170–175.
11. Cayer-Barrioz, J., Ferry, L., Frihi, D., Cavalier, K., Suguela, R., Vigier, G. Micro structure and mechanical behavior of polyamide 66-precipitated calcium carbonate composites: Influence of the particle surface treatment. J Appl Polym Sci. 2006; 100(2):989–999.
12. Tomlinson, W.J., Coulson, C.N. Strength of surface-modified calcite nylon-6 composites. J Mater Sci Lett. 1992; 11(9):531–534.
13. Fang, X.W., Li, X.H., Yu, L.G., Zhang, Z.J. Effect of in situ surface-modified nano-SiO2 on the thermal and mechanical properties and crystallization behavior of nylon 1010. J Appl Polym Sci. 2010; 115(6):3339–3347.
14. Mahfuz, H., Hasan, M., Dhanak, V., Beamson, G., Stewart, J., et al. Reinforcement of nylon 6 with functionalized silica nanoparticles for enhanced tensile strength and modulus. Nanotechnol. 2008; 19(44):445702.
15. Xu, X.M., Li, B.J., Lu, H.M., Zhang, Z.J., Wang, H.G. The effect of the interface structure of different surface-modified nano-SiO2 on the mechanical properties of nylon 66 composites. J Appl Polym Sci. 2008; 107(3):2007–2014.
16. Li, Y., Yu, J., Guo, Z.X. The influence of silane treatment on nylon 6/nano-SiO2 in situ polymerization. J Appl Polym Sci. 2002; 84(4):827–834.
17. Sengupta, R., Bandyopadhyay, A., Sabharwal, S., Chaki, T.K., Bhowmick, A.K. Polyamide-6,6/in situ silica hybrid nanocomposites by sol-gel technique: synthesis, characterization and properties. Polym. 2005; 46(10):3343–3354.
18. Reynaud, E., Jouen, T., Gauthier, C., Vigier, G., Varlet, J. Nanofillers in polymeric matrix: a study on silica reinforced PA6. Polym. 2001; 42(21):8759–8768.
19. Treacy, M.M.J., Ebbesen, T.W., Gibson, J.M. Exceptionally high Young’s modulus observed for individual carbon nanotubes. Nature. 1996; 381(6584):678–680.
20. Duesberg, G.S., Burghard, M., Muster, J., Philipp, G., Roth, S. Separation of carbon nanotubes by size exclusion chromatography. Chem Comm. 1998; 3:425–436.
21. Moore, V.C., Strano, M.S., Haroz, E.H., Hauge, R.H., Smalley, R.E., et al. Individually suspended single-walled carbon nanotubes in various surfactants. Nano Lett. 2003; 3(10):1379–1382.
22. Islam, M.F., Rojas, E., Bergey, D.M., Johnson, A.T., Yodh, A.G. High weight fraction surfactant solubilization of single-wall carbon nanotubes in water. Nano Lett. 2003; 3(2):269–273.
23. Das, D., Das, P.K. Superior activity of structurally deprived enzymecarbon nanotube hybrids in cationic reverse micelles. Langmuir. 2009; 25(8):4421–4428.
24. Wang, H., Zhou, W., Ho, D.L., Winey, K.I., Fischer, J.E., et al. Dispersing single-walled carbon nanotubes with surfactants: A small angle neutron scattering study. Nano Lett. 2004; 4(9):1789–1793.
25. Cheng, F., Imin, P., Maunders, C., Botton, G., Adronov, A. Soluble, discrete supramolecular complexes of single-walled carbon nanotubes with fluorene-based conjugated polymers. Macromol. 2008; 41(7):2304–2308.
26. Curran, S.A., Ajayan, P.M., Blau, W.J., Carroll, D.L., Coleman, J.N., et al. A compositefrom poly(m-phenylenevinylene-co-2,5-dioctoxy-p-phenylenevinylene) and carbon nanotubes: A novel material for molecular optoelectronics. Adv Mater. 1998; 10(14):1091–1093.
27. Coleman, J.N., Dalton, A.B., Curran, S., Rubio, A., Davey, A.P., et al. Phase separation of carbon nanotubes and turbostratic graphite using a functional organic polyme. Adv Mater. 2000; 12(3):213–216.
28. Dalton, A.B., Stephan, C., Coleman, J.N., McCarthy, B., Ajayan, P.M., et al. Selective interaction of a semiconjugated organic polymer with single-wall nanotubes. J Phys Chem B. 2000; 104(43):10012–10016.
29. Kang, Y.J., Taton, T.A. Micelle-encapsulated carbon nanotubes: A route to nanotube composites. J Am Chem Soc. 2003; 125(19):5650–5651.
30. Georgakilas, V., Kordatos, K., Prato, M., Guldi, D.M., Holzingger, M., Hirsch, A. Organic functionalization of carbon nanotubes. J Am Chem Soc. 2002; 124(5):760–761.
31. Datsyuk, V., Kalyva, M., Papagelis, K., Parthenios, J., Tasis, D., et al. Chemical oxidation of multiwalled carbon nanotubes. Carbon. 2008; 46(6):833–840.
32. Sahoo, N.G., Rana, S., Cho, J.W., Li, L., Chan, S.H. Polymer nanocomposites based on functionalized carbon nanotubes. Prog Polym Sci. 2010; 35(7):837–867.
33. Qu, L.W., Veca, L.M., Lin, Y., Kitaygorodskiy, A., Chen, B.L., et al. Soluble nylon-functionalized carbon nanotubes from anionic ring-opening polymerization from nanotube surface. Macromol. 2005; 38(24):10328–10331.
34. Yang, M., Gao, Y., Li, H.M., Adronov, A. Functionalization of multiwalled carbon nanotubes with polyamide 6 by anionic ring-opening polymerization. Carbon. 2007; 45(12):2327–2333.
35. Zeng, Q.H., Yu, A.B., Lu, G.Q., Paul, D.R. Clay-based polymer nanocomposites: research and commercial development. J Nanosci Nanotechnol. 2005; 5(10):1574–1592.
36. Tjong, S.C. Structural and mechanical properties of polymer nanocomposites. Mater Sci Eng R: Reports. 2006; 53(3–4):73–197.
37. Ray, S.S., Okamoto, M. Polymer/layered silicate nanocomposites: A review from preparation to processing. Prog Polym Sci. 2003; 28(11):1539–1641.
38. Zanetti, M., Lomakin, S., Camino, G. Polymer layered silicate nanocomposites. Macromol Mater Eng. 2000; 279(6):1–9.
39. Fornes, T.D., Yoon, P.J., Hunter, D.L., Keskkula, H., Paul, D.R. Effect of organoclay structure on nylon 6 nanocomposite morphology and properties. Polym. 2002; 43(22):5915–5933.
40. Nguyen, Q.T., Baird, D.G. Preparation of polymer-clay nanocomposites and their properties. Advances in Polym Technol. 2006; 25(4):270–285.
41. Kim, E.S., Shim, J.H., Woo, J.Y., Yoo, K.S., Yoon, J.S. Effect of the silane modification of clay on the tensile properties of nylon 6/clay nanocomposites. J Appl Polym Sci. 2010; 117(2):809–816.
42. Wang, K.H., Choi, M.H., Koo, C.M., Choi, Y.S., Chung, I.J. Synthesis and characterization of maleated polyethylene/clay nanocomposites. Polym. 2001; 42(24):9819–9826.
43. Kato, M., Okamoto, H., Hasegawa, N., Tsukigase, A., Usuki, A. Preparation and properties of polyethylene-clay hybrids. Polym Eng Sci. 2003; 43(6):1312–1316.
44. Ristolainen, N., Vainio, U., Paavola, S., Torkkeli, M., Serimaa, R., Seppala, J. Polypropylene/organoclay nanocomposites compatibilized with hydroxyl-functional polypropylenes. J Polym Sci B-Polym Phys. 2005; 43(14):1892–1903.
45. Lim SH, Dasari A, Yu ZZ and Mai YW, unpublished work.
46. Fong, H., Liu, W.D., Wang, C.S., Vaia, R.A. Generation of electrospun fibers ofnylon 6 and nylon 6-montmorillonite nanocomposite. Polym. 2002; 43(3):775–780.
47. Kojima, Y., Usuki, A., Kawasumi, M., Okada, A., Kurauchi, T., Kamigaito, O. Synthesis of nylon-6-clay hybrid by montmorillonite intercalated with ε-caprolactam. J Polym Sci Part A: Polym Chem. 1993; 31(4):983–986.
48. Kojima, Y., Usuki, A., Kawasumi, M., Okada, A., Fukushima, Y., et al. Mechanical properties of nylon-6-clay hybrid. J Mater Res. 1993; 8(5):1185–1189.
49. Kojima, Y., Usuki, A., Kawasumi, M., Okada, A., Kurauchi, T., Kamigaito, O. One-pot synthesis of nylon-6 clay hybrid. J Polym Sci Part A: Polym Chem. 1993; 31(7):1755–1758.
50. Usuki, A., Kawasumi, M., Kojima, Y., Okada, A., Kurauchi, T., Kamigaito, O. Swelling behavior of montmorillonite cation exchanged for omega-amino acids by ε-caprolactam. J Mater Res. 1993; 8(5):1174–1178.
51. Fukushima, Y., Inagaki, S. Synthesis of an intercalated compound of montmorillonite and 6-polyamide. J Inclusion Phenom. 1987; 5(4):473–482.
52. Usuki, A., Kojima, Y., Kawasumi, M., Okada, A., Fukushima, Y., et al. Synthesis of nylon 6-clay hybrid. J Mater Res. 1993; 8(5):1179–1184.
53. Yang, F., Ou, Y.C., Yu, Z.Z. Polyamide 6 silica nanocomposites prepared by in situ polymerization. J Appl Polym Sci. 1998; 69(2):355–361.
54. Zeng, H.L., Gao, C., Wang, Y.P., Watts, P.C.P., Kong, H., et al. In situ polymerization approach to multiwalled carbon nanotubes-reinforced nylon 1010 composites: Mechanical properties and crystallization behavior. Polym. 2006; 47(1):113–122.
55. Zhao, C.G., Hu, G.J., Justice, R., Schaefer, D.W., Zhang, S.M., et al. Synthesis and characterization of multi-walled carbon nanotubes reinforced polyamide 6 via in situ polymerization. Polym. 2005; 46(14):5125–5132.
56. Xu, C., Jia, Z., Wu, D., Han, Q., Meek, T. Fabrication of nylon-6/carbon nanotube composites. J Electronic Mater. 2006; 35(5):954–957.
57. Kang, M., Myung, S.J., JinH, J. Nylon 610 and carbon nanotube composite by insitu interfacial polymerization. Polym. 2006; 47(11):3961–3966.
58. Moniruzzaman, M., Chattopadhyay, J., Billups, W.E., Winey, K.I. Tuning the mechanical properties of SWNT/Nylon 6, 10 composites with flexible spacers at the interface. Nano Lett. 2007; 7(5):1178–1185.
59. Liu, L.M., Qi, Z.N., Zhu, X.G. Studies on nylon 6 clay nanocomposites by melt-intercalation process. J Appl Polym Sci. 1999; 71(7):1133–1138.
60. Vaia, R.A., Ishii, H., Giannelis, E.P. Synthesis and properties of 2-dimensional nanostructures by direct intercalation of polymer melts in layered silicate. Chem Mater. 1993; 5(12):1694–1696.
61. Gilman, J.W., Awad, W.H., Davis, R.D., Shields, J., Harris, R.H., et al. Polymer/layered silicate nanocomposites from thermally stable triakylimidazolium-treated montmorillonite. Chem Mater. 2002; 14(9):3776–3785.
62. Radhakrishnan, V.K., Davis, E.W., Davis, V.A. Influence of initial mixing methods on melt-extruded single-walled carbon nanotube-polypropylene nanocomposites. Polym Eng Sci. 2010; 50(9):1831–1842.
63. Shofner, M.L., Khabashesku, V.N., Barrera, E.V. Processing and mechanical properties of fluorinated single-walled carbon nanotube-polyethylene composites. Chem Mater. 2006; 18(4):906–913.
64. Liu, T.X., Phang, I.Y., Shen, L., Chow, S.Y., Zhang, W.D. Morphology and mechanical properties of multiwalled carbon nanotubes reinforced nylon-6 composites. Macromol. 2004; 37(19):7214–7222.
65. Kiss, A., Fekete, E., Pukanszky, B. Aggregation of CaCO3 particles in PP composites: Effect of surface coating. Composites Sci Tech. 2007; 67(7–8):1574–1583.
66. Fornes, T.D., Paul, D.R. Modeling properties of nylon 6/clay nanocomposites using composite theories. Polym. 2003; 44(17):4993–5013.
67. Paul, D.R., Robeson, L.M. Polymer nanotechnology: Nanocomposites. Polym. 2008; 49(15):3187–3204.
68. Lee, H.S., Fasulo, P.D., Rodgers, W.R., Paul, D.R. TPO based nanocomposites. Part 1. Morphology and mechanical properties. Polym. 2005; 46(25):11673–11689.
69. Fornes, T.D., Hunter, D.L., Paul, D.R. Nylon-6 nanocomposites from alkylammonium-modified clay: The role of alkyl tails on exfoliation. Macromol. 2004; 37(5):1793–1798.
70. Bhattacharyya, A.R., Potschke, P., Haussler, L., Fischer, D. Reactive compatibilization of melt mixed PA6/SWNT composites: Mechanical properties and morphology. Macromol Chem Phys. 2005; 206(20):2084–2095.
71. Weon, J.I., Sue, H.J. Effects of clay orientation and aspect ratio on mechanical behavior of nylon-6 nanocomposites. Polym. 2005; 46(17):6325–6334.
72. Dennis, H.R., Hunter, D.L., Chang, D., Kim, S., White, J.L., et al. Effect of melt processing conditions on the extent of exfoliation in organoclay-based nanocomposites. Polym. 2001; 42(23):9513–9522.
73. Chavarria, F., Nairn, K., White, P., Hill, A.J., Hunter, D.L., Paul, D.R. Morphology and properties of nanocomposites from organoclays with reduced cation exchange capacity. J Appl Polym Sci. 2007; 105(5):2910–2924.
74. Fornes, T.D., Yoon, P.J., Keskkula, H., Paul, D.R. Nylon 6 nanocomposites: The effect of matrix molecular weight. Polym. 2001; 42(25):9929–9940.
75. Alexandre, M., Dubois, P. Polymer-layered silicate nanocomposites: Preparation, properties and uses of a new class of materials. Mater Sci Eng R: Reports. 2000; 28(1–2):1–63.
76. LeBaron, P.C., Wang, Z., Pinnavaia, T.J. Polymer-layered silicate nanocomposites: An overview. Appl Clay Sci. 1999; 15(1–2):11–29.
77. Chen, B., Evans, J.R.G., Greenwell, H.C., Boulet, P., Coveney, P.V., et al. A critical appraisal of polymer-clay nanocomposites. Chem Soc Reviews. 2008; 37(3):568–594.
78. Liu, H.M., Wang, X., Fang, P.F., Wang, S.J., Qi, X., et al. Functionalization of multi-walled carbon nanotubes grafted with self-generated functional groups and their polyamide 6 composites. Carbon. 2010; 48(3):721–729.
79. Yu, Z.Z., Yan, C., Yang, M.S., Mai, Y.W. Mechanical and dynamic mechanical properties of nylon 66/montmorillonite nanocomposites fabricated by melt compounding. Polym International. 2004; 53(8):1093–1098.
80. Chen, L., Phang, I.Y., Wong, S.C., Lv, P.F., Liu, T.X. Embrittlement mechanisms of nylon 66/organoclay nanocomposites prepared by melt-compounding process. Mater Manufacturing Processes. 2006; 21(2):153–158.
81. He, C.B., Liu, T.X., Tjiu, W.C., Sue, H.J., Yee, A.F. Microdeformation and fracture mechanisms in polyamide-6/organoclay nanocomposites. Macromol. 2008; 41:193–202.
82. Lim, S.H., Dasari, A., Yu, Z.Z., Mai, Y.W., Liu, S.L., Yong, M.S. Fracture toughness of nylon 6/organoclay/elastomer nanocomposites. Compos Sci Technol. 2007; 67(14):2914–2923.
83. Dasari, A., Yu, Z.Z., Mai, Y.W. Transcrystalline regions in the vicinity of nanofillers in polyamide-6. Macromol. 2007; 40(1):123–130.
84. Windle, A.H. Two defining moments: A personal view by Prof Alan H Windle. Compos Sci Technol. 2007; 67(5):929–930.
85. Wichmann, M.H.G., Schulte, K., Wagner, H.D. On nanocomposite toughness. Compos Sci Technol. 2008; 68(1):329–331.
86. Gao, Y.C., Mai, Y.W., Cotterell, B. Fracture of fiber-reinforced materials. Zeitschrift Fur Angewandte Mathematik Und Physik. 1988; 39(4):550–572.
87. Dasari, A., Yu, Z.Z., Mai, Y.W. Effect of blending sequence on microstructure of ternary nanocomposites. Polym. 2005; 46(16):5986–5991.
88. Sue, H.J., Gam, K.T., Bestaoui, N., Clearfield, A., Miyamoto, M., Miyatake, N. Fracture behavior of α-zirconium phosphate-based epoxy nanocomposites. Acta Materialia. 2004; 52(8):2239–2250.
89. Liu, W.P., Hoa, S.V., Pugh, M. Morphology and performance of epoxy nanocomposites modified with organoclay and rubber. Polym Eng Sci. 2003; 44(6):1178–1186.
90. González, I., Eguiazábal, J.I., Nazábal, J. Rubber-toughened polyamide 6/clay nanocomposites. Compos Sci Technol. 2006; 66(11–12):1833–1843.
91. Tjong, S.C., Meng, Y.Z. Impact-modified polypropylene/vermiculite nanocomposites. J Polym Sci B-Polym Phys. 2003; 41(19):2332–2341.
92. Khatua, B.B., Lee, D.J., Kim, H.Y., Kim, J.K. Effect of organoclay platelets on morphology of nylon-6 and poly(ethylene-ran-propylene) rubber blends. Macromol. 2004; 37(7):2454–2459.
93. Yang, J.L., Zhang, Z., Zhang, H. The essential work of fracture of polyamide 66 filled with TiO2 nanoparticles. Compos Sci Technol. 2005; 65(15–16):2374–2379.
94. Zhang, H., Zhang, Z., Yang, J.L., Friedrich, K. Temperature dependence of crack initiation fracture toughness of various nanoparticles filled polyamide 66. Polym. 2006; 47(2):679–689.
95. Briscoe, B.J., Sinha, S.K. Scratch resistance and localised damage characteristics of polymer surfaces – a review. Materialwissenschaft Und Werkstofftechnik. 2003; 34(10–11):989–1002.
96. Lim, S.C., Ashby, M.F. Wear-mechanism maps. Acta Metallurgica. 1987; 35(1):1–24.
97. Sayles, R.S. Basic principles of rough surface contact analysis using numerical methods. Tribology International. 1996; 29(8):639–650.
98. Dasari, A., Yu, Z.Z., Mai, Y.W. Fundamental aspects and recent progress on wear/scratch damage in polymer nanocomposites. Mater Sci Eng R: Reports. 2009; 63(2):31–80.
99. Bahadur, S. The development of transfer layers and their role in polymer tribology. Wear. 2000; 245(1–2):92–99.
100. Fischer, T.E. Tribochemistry. Annual Review of Mater Sci. 1988; 18:303–323.
101. Zhang, M.Q., Rong, M.Z., Yu, S.L., Wetzel, B., Friedrich, K. Effect of particle surface treatment on the tribological performance of epoxy based nanocomposites. Wear. 2002; 253(9–10):1086–1093.
102. Friedrich, K. Wear models for multi-phase materials and synergistic effects in polymeric hybrid composites. In: Friedrich K., ed. Advances in Composite Tribology. Amsterdam: Elsevier Science Publishers B V; 1993:209.
103. Dasari, A., Yu, Z.Z., Mai, Y.W. Wear and scratch damage in polymer nanocomposites. In: Friedrich, K., Alois, K.S., eds. Tribology of Polymeric Nanocomposites, Tribology and Interface Engineering Series, Vol 55. Elsevier; 2008:374.
104. Briscoe, B.J., Sinha, S.K. Wear of polymers. Proceedings of the Institution of Mechanical Engineers J: J Eng Tribology. 2002; 216(J6):401–413.
105. Chang, L., Zhang, Z., Breidt, C., Friedrich, K. Tribological properties of epoxy nanocomposites – I. Enhancement of the wear resistance by nano-TiO2 particles. Wear. 2005; 258(1–4):141–148.
106. Kietzke, T., Neher, D., Landfester, K., Montenegro, R., Guntner, R., Scherf, U. Novel approaches to polymer blends based on polymer nanoparticles. Nature Mater. 2003; 2(6):408-U7.
107. Xing, X.S., Li, R.K.Y. Wear behavior of epoxy matrix composites filled with uniform sized sub-micron spherical silica particles. Wear. 2004; 256(1–2):21–26.
108. Dasari, A., Yu, Z.Z., Mai, Y.W. Nanoscratching of nylon 66-based ternary nanocomposites. Acta Materialia. 2007; 55(2):635–646.
109. Dasari, A., Yu, Z.Z., Mai, Y.W., Kim, J.K. Orientation and the extent of exfoliation of clay on scratch damage in polyamide 6 nanocomposites. Nanotechnol. 2008; 19(5):055708.
110. Messersmith, P.B., Giannelis, E.P. Synthesis and barrier properties of poly(ε-caprolactone)-layered silicate nanocomposites. J Polym Sci A: Polym Chem. 1995; 33(7):1047–1057.
111. Osman, M.A., Mittal, V., Morbidelli, M., Suter, U.W. Polyurethane adhesive nanocomposites as gas permeation barrier. Macromol. 2003; 36(26):9851–9858.
112. Gorrasi, G., Tortora, M., Vittoria, V., Galli, G., Chiellini, E. Transport and mechanical properties of blends of poly(ε-caprolactone) and a modified montmorillonite-poly(ε-caprolactone) nanocomposite. J Polym Sci B: Polym Phys. 2002; 40(11):1118–1124.
113. Doppers, L.M., Breen, C., Sammon, C. Diffusion of water and acetone into poly(vinyl alcohol)-clay nanocomposites using ATR-FTIR. Vibrational Spectroscopy. 2004; 35(1–2):27–32.
114. Xu, B., Zheng, Q., Song, Y.H., Shangguan, Y. Calculating barrier properties of polymer/clay nanocomposites: Effects of clay layers. Polym. 2006; 47(8):2904–2910.
115. Frounchi, M., Dadbin, S., Salehpour, Z., Noferesti, M. Gas barrier properties of PP/EPDM blend nanocomposites. J Membrane Sci. 2006; 282(1–2):142–148.
116. Nazarenko, S., Meneghetti, P., Julmon, P., Olson, B.G., Qutubuddin, S. Gas barrier of polystyrene montmorillonite clay nanocomposites: Effect of mineral layer aggregation. J Polym Sci B: Polym Phys. 2007; 45(13):1733–1753. [2007].
117. Nielsen, L.E. Platelet particles enhance barrier of polymers by forming tortuous path. J Macromol Sci: Chem. 1967; A1(5):929–942.
118. Bharadwaj, R.K. Modeling the barrier properties of polymer-layered silicate nanocomposites. Macromol. 2001; 34(26):9189–9192.
119. Yano, K., Usuki, A., Okada, A., Kurauchi, T., Kamigaito, O. Synthesis and properties of polyimide clay hybrid. J Polym Sci A: Polym Chem. 1993; 31(10):2493–2498.
120. Kim, S.H., Kim, S.C. Synthesis and properties of poly(ethylene terephthalate)/clay nanocomposites by in situ polymerization. J Appl Polym Sci. 2007; 103(2):1262–1271.
121. Yano, K., Usuki, A., Okada, A. Synthesis and properties of polyimide-clay hybrid films. J Polym Sci A: Polym Chem. 1997; 35(11):2289–2294.
122. Choi, W.J., Kim, H.J., Yoon, K.H., Kwon, O.H., Hwang, C.I. Preparation and barrier property of poly(ethylene terephthalate)/clay nanocomposite using clay-supported catalyst. J Appl Polym Sci. 2006; 100(6):4875–4879.
123. Ray, S.S., Okamoto, K., Okamoto, M. Structure-property relationship in biodegradable poly(butylene succinate)/layered silicate nanocomposites. Macromol. 2003; 36(7):2355–2367.
124. Koo, C.M., Kim, S.O., Chung, I.J. Study on morphology evolution, orientational behavior, and anisotropic phase formation of highly filled polymer-layered silicate nanocomposites. Macromol. 2003; 36(8):2748–2757.
125. Bharadwaj, R.K., Mehrabi, A.R., Hamilton, C., Trujillo, C., Murga, M., et al. Structure-property relationships in cross-linked polyester-clay nanocomposites. Polym. 2002; 43(13):3699–3705.
126. Lu, C.S., Mai, Y.W. Influence of aspect ratio on barrier properties of polymer-clay nanocomposites. Phys Rev Lett. 2005; 95(8):088303.
127. Cussler, E.L., Hughes, S.E., Ward, W.J., Aris, R. Barrier membranes. J Membrane Sci. 1988; 38(2):161–174.
128. Fukuda, M., Kuwajima, S. Molecular-dynamics simulation of moisture diffusion in polyethylene beyond 10 ns duration. J Chem Phys. 1997; 107(6):2149–2159.
129. Kim, D.H., Fasulo, P.D., Rodgers, W.R., Paul, D.R. Structure and properties of polypropylene-based nanocomposites: Effect of PP-g-MA to organoclay ratio. Polym. 2007; 48(18):5308–53323.
130. Yoon, P.J., Fornes, T.D., Paul, D.R. Thermal expansion behavior of nylon 6 nanocomposites. Polym. 2002; 43(25):6727–6741.
131. Chow, T.S. Effect of particle shape at finite concentration on thermal expansion of filled polymers. J Polym Sci B: Polym Phys. 1978; 16(6):967–970.
132. Hwang, J.J., Liu, H.J. Influence of organophilic clay on the morphology, plasticizer-maintaining ability, dimensional stability, and electrochemical properties of gel polyacrylonitrile (PAN) nanocomposite electrolytes. Macromol. 2002; 35(19):7314–7319.
133. Ray, S.S., Yamada, K., Okamoto, M., Ueda, K. New polylactide-layered silicate nanocomposites. 2. Concurrent improvements of material properties, biodegradability and melt rheology. Polym. 2003; 44(3):857–866.
134. Zhu, W.P., Zhang, G.P., Yu, J.Y., Dai, G. Crystallization behavior and mechanical properties of polypropylene copolymer by in situ copolymerization with a nucleating agent and/or nano-calcium carbonate. J Appl Polym Sci. 2004; 91(1):431–438.
135. Yu, Z.Z., Hu, G.H., Varlet, J., Dasari, A., Mai, Y.W. Water-assisted melt compounding of nylon-6/pristine montmorillonite nanocomposites. J Polym Sci B: Polym Phys. 2005; 43(9):1100–1112.
136. Xie, W., Gao, Z.M., Pan, W.P., Hunter, D., Singh, A., Vaia, R. Thermal degradation chemistry of alkyl quaternary ammonium montmorillonite. Chem Mater. 2001; 13(9):2979–2990.
137. Lan, T., Kaviratna, D., Pinnavaia, T.J. Epoxy self-polymerization in smectite clays. J Phys Chem Solids. 1996; 57(6–8):1005–1010.
138. Zhang, Y., Lee, S., Yoonessi, M., Liang, K., Pittman, C.U. Phenolic resintrisilanolphenyl polyhedral oligomeric silsesquioxane (POSS) hybrid nanocomposites: Structure and properties. Polym. 2006; 47(9):2984–2996.
139. Jash, P., Wilkie, C.A. Effects of surfactants on the thermal and fire properties of poly(methyl methacrylate)/clay nanocomposites. Polym Degradation and Stability. 2005; 88(3):401–416.
140. Fina, A., Tabuani, D., Carniato, F., Frache, A., Boccaleri, E., Camino, G. Polyhedral oligomeric silsesquioxanes (POSS) thermal degradation i. Thermochimica Acta. 2005; 440(1):36–42.
141. Laoutid, F., Bonnaud, L., Alexandre, M., Lopez-Cuesta, J.M., Dubois, P. New prospects in flame retardant polymer materials: From fundamentals to nanocomposites. Mater Sci Eng R. 2009; 63(3):100–125.
142. Kiliaris, P., Papaspyrides, C.D. Polymer/layered silicate (clay) nanocomposites: An overview of flame retardancy. Prog Polym Sci. 2010; 35(7):902–958.
143. Dasari, A., Cai, G., Yu, Z.Z., Mai, Y.-W. Flame retardancy of polymer-clay nanocomposites. In: Tjong S.C., Mai Y.-W., eds. Physical properties of polymer nanocomposites. Cambridge: Woodhead Publishing Ltd; 2010:347.
144. Garces, J.M., Moll, D.J., Bicerano, J., Fibiger, R., McLeod, D.G. Polymeric nanocomposites for automotive applications. Adv Mater. 2000; 12(23):1835–1839.
145. Njuguna, B., Pielichowski, K. Polymer nanocomposites for aerospace applications: Properties. Adv Eng Mater. 2003; 5(11):769–778.
146. Huang, Z.M., Zhang, Y.Z., Kotaki, M., Ramakrishna, S. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos Sci Technol. 2003; 63(15):2223–2253.
147. Reneker, D.H., Chun, I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnol. 1996; 7(3):216–223.
148. Doshi, J., Reneker, D.H., Electrospinning process and applications of electrospun fibers. Special Technical Session on Electrostatics in Polymer Processing and Charge Monitoring. IEEE Industry Applications Society 28th Annual Meeting. 2–8 October, 1993:1698–1703. [Toronto, Canada].
149. Li, D., Xia, Y.N. Electrospinning of nanofibers: Reinventing the wheel? Adv Mater. 2004; 16(14):1151–1170.
150. Reneker, D.H., Yarin, A.L., Fong, H., Koombhongse, S. Bending instability of electrically charged liquid jets of polymer solutions in electrospinning. J Appl Phys. 2000; 87(9):4531–4537.