
Biodegradable polymer nanocomposites
Abstract:
Fossil carbon reserves are used to produce items whose durability was considered either to be an advantage or irrelevant depending on the application. Economic and environmental concerns are propagating interest in polymers that either are derived from alternative sustainable sources such as biomass or are biodegradable so that the risks of plastic residues in the environment are reduced or removed. Nanocomposite technology can engineer polymers to provide new materials, which is significant because of increasing legislative pressure and consumer demand for biodegradable high-performance materials, particularly in the packaging, biomedical and automotive sectors. This chapter introduces the most significant biodegradable polymers and reviews both methods of production and the physical properties of biodegradable nanocomposites.
13.1 Introduction
Increasing environmental awareness and concern over the disposal of plastic waste has led in recent years to a growing demand from consumers for biodegradable polymers. Many of these polymers are also derived from renewable resources, such as plant-based feedstocks or bacterial fermentation. Hence these materials meet the need to find replacements for fossil-fuel feedstocks as well as addressing growing concerns over end-of-life disposal issues and the necessity of finding alternatives to landfill.
As expected, the vast majority of applications for biodegradable plastics are in short-life, disposable products (Johnson et al., 2003). An important market is in food packaging, such as clear films, trays, clam-shell packaging and blister packs. Other items of packaging are compost sacks and bags. Biodegradable polymers are also used for convenience items such as disposable cutlery, plates and cups. In agriculture there are some obvious uses for compostable materials in plant pots, mulching film and yarn. In the medical sector, biodegradable polymers find use in drug delivery systems, sutures and disposable gloves.
A key barrier to the use of biodegradable polymers is their poor technical performance compared with many conventional polymers, particularly in terms of mechanical properties and barrier properties. For this reason nanocomposite technology has been applied to biodegradable polymers. This chapter reviews biodegradable nanocomposites, describing their methods of production and the effect of nanofillers on mechanical, thermal and barrier properties, as well as the effect on biodegradability.
13.1.1 Biopolymers, bio-derived polymers and biodegradable polymers
First it is necessary to define some of the terms used in this chapter and to distinguish between ‘biopolymers’, ‘bio-derived polymers’ and ‘biodegradable polymers’.
Biopolymers are polymers that are produced by the metabolic processes of living cells. This, therefore, includes carbohydrates such as cellulose and starch, and proteins such as keratin and enzymes. Also included in this group are poly(hydroxyalkanoate)s (PHAs), which are polyesters produced in vivo by bacteria such as Ralstonia eutropha. Another biopolymer of interest is chitin, which is extracted from crustacean shells.
Bio-derived polymers are polymers made from renewable, sustainable resources; a definition that excludes fossil carbon sources such as crude oil and coal. Hence this category of polymers includes biopolymers and it also includes two other groups of polymers, both of which are polymerised from monomers that are bio-derived. In the first of these ‘synthetic’ groups are polymers that retain the source material’s property of biodegradability, an important example being poly(lactic acid) or polylactide (PLA). In the production of PLA, lactic acid is first produced by fermentation of a carbohydrate feedstock such as maize sugar and the lactic acid is then extracted and polymerised to give PLA. Another group of bio-derived polymers that has more recently emerged are polymers that are identical to the current fossil-derived polymers, such as polyethylene and polyvinyl chloride, but produced from bio-derived ethylene. These polymers are not biodegradable but have the properties of their fossil-derived equivalents, with which they can be mixed and recycled.
Biodegradable polymers have the capability of being broken down into carbon dioxide and water by the action of microorganisms, such as bacteria and fungi. As discussed above, a large number of bio-derived polymers are biodegradable, but so also are a number of fossil-derived polymers. These include polyvinyl alcohol (PVA), polycaprolactone (PCL) and a number of other aliphatic and aromatic polyesters.
13.1.2 Degradable, biodegradable and compostable
Some definitions relating to the biodegradation of polymers are given below.
Degradation refers to a chemical process in which the long chains of polymer molecules are broken down into shorter lengths, for example by the action of water, oxygen, ultra-violet light or heat.
Biodegradation is a biological process that occurs only after the plastics have started to degrade. A biodegradable plastic is defined in EN ISO 472:2001 as a ‘degradable plastic in which degradation results in lower molecular weight fragments produced by the action of naturally occurring microorganisms such as bacteria, fungi and algae’. It is stated explicitly that the degradation is brought about by the action of living organisms rather than physical or chemical processes.
A compostable plastic is defined by the ASTM D 6400 standard as: ‘A plastic that undergoes degradation by biological processes during composting to yield CO2, water, inorganic compounds and biomass at a rate consistent with other known compostable materials and leaves no visible, distinguishable or toxic waste.’ The process of composting is defined in the British standard PAS 100:2005 ‘Specification for Composted Materials’ as a ‘process of controlled biological decomposition of biodegradable materials under managed conditions that are predominantly aerobic and that allow the development of thermophilic temperatures as a result of biologically produced heat’. Hence compostability is a very specific and commercially important form of biodegradability. To be described as compostable, a plastic must conform to international standards EN13432 and ASTM D6400 and biodegrade within 180 days.
It is often tacitly assumed that because a polymer can be described as ‘biodegradable’ then no further discussion as to its disposal is necessary and it must have a beneficial impact on the environment. However, this is not the case, and there will clearly be situations where the closed-loop recycling of biodegradable polymers is the preferred option.
Also, it is not helpful to simply describe a product as ‘biodegradable’ without being more specific. Conversion to carbon dioxide (CO2) is the most direct measurement of biodegradation. Narayan (2009) argues that the measurement of carbon dioxide evolved when a sample is incubated in soil or compost is the true measure of biodegradability, and that it is unacceptable to claim that a plastic will ‘eventually biodegrade’ without stipulating the disposal environment, time period and extent of biodegradation according to international standards.
13.1.3 Sustainability
Biosustainable polymers can be interpreted as those which are derived from renewable resources; a definition that excludes fossil carbon sources such as crude oil and coal. Inherent in this definition is the principle that supply will always meet demand, a situation that is unlikely ever to occur in the case of fossil carbon. In this category, the family of compounds known as poly(hydroxyalkanoate)s (PHA) have received much attention because they are easily produced in quantity through the fermentation of carbon-rich substrates by microorganisms, in particular bacteria. In this family is the polymers poly(beta-hydroxybutyrate) (P(3HB)).
The primary carbon source for biopolymers is carbon dioxide, CO2, which is ‘fixed’ by plants and other autotrophic (‘self-feeding’) organisms, such as bacteria, into simple sugars, which are then polymerised to give cellulose or starch, or converted into other biomolecules such as proteins (polypeptides) or lipids. For much of the biota, sunlight provides the energy to drive these processes through the mechanism of photosynthesis, although some autotrophs derive energy by facilitating the oxidation of reduced sulphur in minerals such as pyrites: even in the darkest cave or the deepest ocean trench, there can be found organisms that fix carbon dioxide. Notwithstanding current developments in emulating photosynthesis by artificial structures, plant and microbial biosyntheses are likely to remain the most significant source of carbon-based biopolymers for some time.
While plants and microbes are the most efficient source of fixed carbon precursors for polymer production, further energy is required to extract and process the polymers for use. In the production of poly(lactic acid), lactic acid is first produced by fermentation of a carbohydrate feedstock such as maize sugar (Huang, 2005). The lactic acid is then extracted and polymerised to give PLA. By contrast, poly(hydroxyalkanoate)s are produced in vivo by bacteria such as Ralstonia eutropha. The polymer is then extracted with solvents, which are then recovered. More recently, plants capable of PHA synthesis have been developed by genetic manipulation (Poirier et al., 1992).
These processes were subjected to a life cycle analysis (LCA) by workers at Dartmouth College and Monsanto (Gerngross and Slater, 2000). They found that the production of 1 kg of PHA would require three times the 29 MJ required to produce the same weight of polyethylene (PE) (Table 13.1). In terms of fossil-fuel requirements, 1 kg of PHA requires the power generated by 2.65 kg of a fossil fuel whereas the same amount of PE requires 2.2 kg, of which only 1.3 kg would be used for energy production. In their words, ‘What is gained by substituting the renewable resource for the finite one is lost in the additional requirement for energy.’ The result of this LCA contributed in part to Monsanto’s decision in late 1999 to cease developing PHA production systems. However, realising that energy production was the major hurdle in this exercise, these workers showed that by burning corn ‘stover’, i.e. waste stem and leaf matter, and using this biomass-derived energy as the power source, the production process became energetically viable. The fossil-fuel requirement of PLA is rather lower (Table 13.1) and Nature Works are currently marketing it on the basis that it requires 30% to 50% less fossil fuel than the equivalent weight of polymer derived from hydrocarbon feedstock (Leaversuch, 2002).
Table 13.1
Energy required to produce plant-derived and fossil-fuel-derived plastics

aNote: The polyamide type was not specified by the authors of this article.
Source: Gerngross and Slater, 2000.
In 2003 workers at Cargill Dow (Vink et al, 2003) published a comprehensive life cycle assessment of NatureWorks™ PLA production, comparing it with a range of thermoplastics on a weight basis, i.e. each parameter was expressed per kilogram of polymer produced. The polymers included: polyamide 6,6 (PA-6,6), polyamide 6 (PA-6), polycarbonate, ‘cellophane’, low-density polyethylene, poly(propylene) (PP) and poly(ethylene terephthalate) (PET). Two scenarios for PLA production were also considered, one in which the processing energy was derived from fossil carbon sources and one in which it was derived from biomass and wind power. Part of the analysis considered the fossil energy equivalent of the petroleum feedstock used to make the polymers in addition to that used to generate the required power. On this basis, production of all the other polymers consumed more fossil energy than PLA production, although, when the fossil feedstock element was discounted, PET consumed less energy than PLA. If the energy sources were biomass and wind power, however, the energy consumption of PLA production was greatly reduced. This alternative strategy led Cargill Dow to declare an objective to decrease fossil energy consumption from 54 MJ/kg PLA to 7 MJ/kg within 5 to 8 years from 2003, with a similar objective to reduce greenhouse gas emissions from + 1.8 to − 1.7 kg CO2 equivalents/kg PLA.
An assessment of the contribution of biodegradable polymers to sustainable polymer production (Murphy and Bartle, 2004) listed several major findings: (1) that the LCAs published so far indicated that biopolymers have a particular advantage over petro-chemical-derived polymers where fossil energy consumption and global warming potential was concerned, (2) that the ‘eco-profiles’ of biopolymers were favourable on account of their plant biomass organic carbon content, (3) that disposal at the end of life was a significant element of the polymer life cycle for maximising the environmental benefit, and (4) that biopolymers offered the potential for adding value to waste and by-products from other industrial processes.
13.2 Biodegradable polymers
13.2.1 Poly(lactic acid)
Poly(lactic acid), Fig. 13.1, is an aliphatic ester of the poly(2-hydroxy) type, the most significant other example of which is poly(glycolic acid) (PGA). Although these two polymers were discovered in the mid-1950s, they were initially disregarded on account of their hydrothermal instability, which prevented their use in the injection moulding and extrusion processes. In the 1960s, the first use of PGA to make dissolving artificial sutures was reported (Schmitt and Polistina, 1969). In this application, sensitivity to water was an advantage and thus PGA provided the first practical alternative to denatured collagen. This property of in vivo degradability was also demonstrated for poly(L-lactic acid) and poly(DL- lactic acid) (Kulkarni et al., 1966). Consequently PLA/GA copolymers are widely used in biomedical applications because of their biocompatibility and range of properties obtainable by manipulation of the formulation conditions.

Lactic acid, unlike glycolic acid, contains an asymmetric carbon atom and therefore has two optically active enantiomers, D and L (Fig. 13.2). Lactic acid readily forms cyclic esters, or lactides, composed of two lactic acid units or dimers. Three optically isomeric lactides are therefore possible: L-lactide (a dimer of L-lactic acid), D-lactide (a dimer of D-lactic acid) and meso-lactide (a dimer of D- and L- lactic acid) (Fig. 13.3). A racemic mixture of D- and L-lactides may also be obtained. It may also be noted that the lactides contain two asymmetric carbon centres.

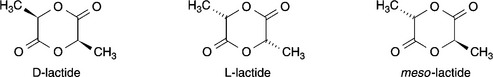
The standard production method, fermentation, gives L-lactic acid, which can be racemised to give DL-lactic acid if that product is desired. Either material can be converted to the lactide, which is then harvested by recrystallisation. Meso-lactide can be obtained from the DL-lactide recrystallisation filtrate (Vert et al, 1995).
Poly(lactic acid) may be produced by condensation polymerisation of the free acid or by ring-opening polymerisation of the lactide. In this respect there is an important interplay between the chirality of the monomer and the polymerisation method. Hence, if pure L- or D-lactic acid is polymerised by the condensation method, the product will be poly(D-lactic acid) or poly(L-lactic acid) respectively; condensation polymerisation of a racemic mixture will give a polymer with a random distribution of enantiomers in the chain. The important difference with ring-opening polymerisation is that the chain is built up by a pair addition mechanism, so that a pair of lactate subunits is added at a time. Consequently, polymerisation of L-lactide or D-lactide leads to the corresponding isotactic chain structures whereas polymerisation of meso-lactide leads to a non-random distribution of L-and D-enantiomers.
The polymer is produced by ring-opening polymerisation of the lactide in bulk or in organic solution: the former is preferred in industrial production. The process is subject to many factors, such as: initiator type and concentration; temperature; time; the presence of acids and water and the presence of alcohol initiators. In order to achieve high molecular weights, it is necessary to remove water in order to minimise hydrolysis of the polymer. Initiators in common use are generally metals such as tin (in the form of stannous octanoate) or zinc (as a powdered metal). The choice of initiator is partly dictated by the end-use of the polymer; for biomedical applications, biocompatible substances such as zinc are preferred (Vert et al., 1995). Other polymerisation catalysts such as lanthanide alkoxides have been reviewed.
While the crude polymer can be processed, it must be further purified before extrusion or injection moulding. Although PLA films can be formed by solventcasting methods, the high temperatures required to remove all traces of solvent can cause thermal degradation of the product. The optimal processing methods for PLA, therefore, are those that involve low temperatures, no solvents and the absence, or controlled presence, of water.
Free hydroxy acids can also be polymerised by vacuum distillation of water, in the presence or absence of a catalyst, to give oligomers up to 5000 daltons (Fukuzaki et al., 1989) with a polydispersity of 2. If the distillation temperature is raised above 180 °C in an attempt to obtain higher molecular weights, the product starts to discolour. However, by post condensation in an organic solution, molecular weights of 50 000 daltons are achievable (Buchholz and Entenmann, 1998).
The physical, mechanical and thermal properties of PLA along with those of PP, PET and PA-6.6 and another biodegradable polymer, poly(hydroxybutyrate) (PHB), for comparison, are shown in Table 13.2. The melting temperatures of PHB and PLA are near to those of PP, while those of PET and PA-6,6 are substantially higher. While the glass-transition temperature of PHB is comparable with that of PP, PLA has a relatively high Tg in the same region as PET and PA-6.6, which partly accounts for its low biodegradation rate and the higher temperatures required to compost it effectively. PLA has a very slow rate of crystallisation, which allows greater control over its degree of crystallinity (Wang et al., 2005). Its crystallinity is highly variable compared with the other polymers in the table and much lower values can be obtained. The degree of crystallinity of PLA is also highly dependent on the proportions of D- and L-lactic acid in the polymer, and higher crystallinities are obtained with the more optically pure polymers.

Comparing the mechanical properties in Table 13.2, it can be seen that the tensile moduli of PHB and PLA are considerably higher than those of the synthetic polymers, which makes these polymers suitable where stiffness is required as less material would be needed for a component. The tensile strength values of PLA and PHB are closer to that of PP than either PET or PA-6.6 and are therefore weaker. The brittleness of both biopolymers is reflected in the very low extension to break values (< 6%) compared with PP, PET and PA-6.6.
PLA is now an established polymer in biomedical applications by virtue of its biocompatibility and biodegradability. Both properties are due in part to its initial hydrolysis product being lactic acid, which is rapidly transformed into pyruvic acid and ultimately to carbon dioxide and water through common metabolic pathways. The realisation of the potential of PLA as a ‘green’ packaging polymer came somewhat later, when the focus of research switched from highly durable materials to those that would disappear in a convenient period of time.
As poly(lactic acid) is a polyester, it is unsurprising that the main mode of depolymerisation is hydrolysis, wherein the ester bond is cleaved to give a chain terminated with a hydroxyl group and another terminated with a carboxyl group. Although this process can occur in the presence of water alone, it is greatly accelerated by acid or alkaline conditions or the presence of catalysts such as enzymes. Of these, the esterases are the most significant and living cells produce a great number and variety.
It is known that the hydrolysis of the poly(lactic acid)/poly(glycolic acid) (PLA/GA) polymers is catalysed by carboxyl groups. As these are produced in increasing number as hydrolysis proceeds, the process is in effect autocatalytic (Pitt et al., 1981). It is not known if this is the case with pure PLA polymers. Another feature observed with the PLA/GA copolymers is the heterogeneous degradation of larger samples, where the interior degrades faster than the surface (Vert et al., 1995). In addition, larger samples degrade faster than very small or very thin samples (Vert et al., 1995) and this has important implications both in biomedical applications and in optimising the biodegradation properties of packaging materials.
Currently PLA is marketed as a ‘green’ plastic, most notably by the Cargill Dow consortium under the brand name NatureWorks. The main target application is food packaging and in 2006 the US retail chain Walmart announced that all clam-shell packaging used in its stores would be PLA. However, PLA does have other properties that make it attractive to the packaging industry, such as high optical clarity and high stiffness relative to cellophane, which would allow ‘downgauging’ of 25%, and, although its high glass-transition temperature is a hindrance to its compostability, it also makes it suitable for sealed package applications (Leaversuch 2001; 2002).
13.2.2 Poly(hydroxyalkanoate)s
In 1925, a microbiologist named Lemoigne working at the Pasteur Institute in Paris isolated a new polymer (Lemoigne, 1925) in a chloroform extract of a Bacillus megaterium culture and showed that it was a polyester of 3-hydroxybutyric acid (Lemoigne 1926; 1927), thereby giving one of the earliest accounts of a poly(hydroxyalkanoate).
The term poly(hydroxyalkanoate)s is generally used to describe polymers derived from 3-hydroxyalkanoic acids (beta-hydroxyalkanoic acids), distinguishing them from poly(lactic acid), which is an alpha-hydroxyalkanoic acid. PHA polymers have the generic structure given in Fig. 13.4.

As shown in Fig. 13.4, the length of the alkyl side chain may vary from 1 to 4 depending on the parent acid. For example, poly(3-hydroxybutyrate) gives a polymer in which the side chain is a methyl group. The polymer of 3-hydroxyvaleric acid has an ethyl side group. There is a tendency in the literature to use trivial names for the acids rather than the IUPAC systematic nomenclature; thus poly(3-hydroxybutyrate) is used in preference to poly(3-hydroxybutanoate) and poly(3-hydroxyvalerate) rather than poly(3-hydroxypentanoate).
There are two important structural differences between PLA and the poly(hydroxyalkanoate) polymers. The first is that the ester linkages in PHA polymers are separated by two carbon atoms as opposed to one in the case of PLA. The second is the possibility of extended alkyl side chains in the PHA polymers. These confer plasticity on the polymer and offer the possibility of tailoring the polymer properties by varying the side chain length. The molecular weight of microbial P(3HB) is in the range 10-106 daltons and the polymer is usually more than 50% crystalline with a well-defined melt temperature around 180 °C.
P(3HB) is produced and stored as granules of diameter 0.3 to 1.0 μm in the cytoplasm of cells of many prokaryotic microorganisms (bacteria and cyanobacteria) either as a carbon and energy reserve (when carbon is freely available but other nutrients are limited) or as a sink for reducing equivalents when oxygen is limited (Doi, 1990). Stored PHA may comprise between 30 to 80% of the dry weight of the cell (Doi, 1990). In the environment, microbes produce a range of copolyesters of 3-hydroxyalkanoic acids. These have been found in sewage sludge (Wallen and Rohwedder, 1974), marine sediments (Findlay and White, 1983) and both marine and freshwater cyanobacteria (Capon et al., 1983).
Production
A good example of a commercially produced PHA copolymer is Biopol, which is a copolymer of 3-hydroxybutyrate and 3-hydroxyvalerate, P(3HB-co-3HV). This is produced on an industrial scale by the bacterium Alcaligenes eutrophus growing on propionic acid and glucose in a controlled fermentation process developed by Holmes at ICI (Holmes et al., 1981). Since the initial patent in 1981, many variations of the process have been developed to produce alternative copolymers or to produce PHAs from alternative substrates. For example, one account (Skraly and Peoples, 2005) describes transgenic organisms capable of producing homopolymers of 3-hydroxypropionate or 3-hydroxyvalerate as well as their copolymers by the enzyme-catalysed conversion of diols such as 1,2-propanediol, 1,3-propanediol and, more interestingly, glycerol, which is a by-product of the biodiesel process. The PHA polymers are readily recovered and industrially useful as polymers or as starting materials for a range of chemical intermediates including 1,3-propanediol, 3-hydroxypropionaldehyde, acrylics, malonic acid, esters and amines (Skraly and Peoples, 2005). The genetic technologies available for improving PHA yields from fermentation processes were reviewed in 2003 by Reddy et al. (2003).
Another patented method involves first subjecting spent vegetable oil to a short anaerobic treatment to convert the oils to organic acids followed by separation of the acids from the sludge and fermenting them with a hydrogen bacterium to produce polyhydroxyalkanoic acids (Kazunari and Yoshito, 2000). This procedure is a means of disposing of a problematic waste by converting it to a useful product.
Very recently, researchers have reported the principle of producing styrene monomer by pyrolysis of waste polystyrene and using this as a substrate for poly(hydroxyalkanoate) production by Pseudomonasputida (Booth, 2006). They did not, however, offer any comparison with the simple alternative of using the regenerated styrene for further polystyrene production.
Limitations
The onset of thermal degradation of PHA is close to its melt temperature, which can impose limits on its commercial application. This has led investigators to study other poly(hydroxyalkanoate)s such as poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) in the search for a more thermally stable poly(hydroxyalkanoate). Another limitation is the slow crystallisation rate, which, for example, can make it difficult to form films.
Applications
A distinguishing feature of poly(hydroxyalkanoate)s is their biocompatibility, particularly of the short-chain polymers (3-HB) and PHBV (Gogolewski et al., 1993). This, coupled with their thermoplasticity, enables the manufacture of articles such as bone implants that do not cause inflammatory reactions with the associated risk of rejection (Saito et al., 1991).
Early commercial applications of PHAs included packaging films for bags and paper coatings as well as a variety of disposable items such as razors and hygiene products as well as cosmetic packaging. PHA is also a useful feedstock for producing pure R-3-hydroxybutyric acid, used in the production of the anti-glaucoma drug Trusopt (Reddy et al., 2003).
Biodegradability
PHAs are storage polymers that provide a rapidly available intracellular carbon source; thus they are readily hydrolysed by endo- and exoesterases. The main enzyme involved is PHB depolymerase, which is an extracellular endohydrolase identified in organisms such as Bacillus megaterium, an extremely common soil bacterium (Reddy et al., 2003). As these enzymes are produced extracellularly by many soil organisms, they are rapidly biodegraded in the environment, with up to 85% degradation in seven weeks being reported (Fletcher, 1993; Johnstone, 1990). A sample of PHA degraded within 254 days in a freshwater lake where the temperature remained less than 6 °C (Johnstone, 1990).
13.2.3 Starch
In the context of biodegradable nanocomposites, the role of starch is chiefly as a matrix polymer, whereas cellulose, in the form of fibrils or nanocrystals, plays the part of a reinforcing nanofiller. Starch is both a feedstock for biopolymer production and is itself a biopolymer, which can be processed to make nanocomposites.
In 2000 the world production of starch was estimated at 48.5 million tonnes, which was derived from maize (39.4 million tonnes), potatoes (2.6 million tonnes) and wheat (4.1 million tonnes) with other sources such as cassava and sago accounting for 2.5 million tonnes. Climate and geography dictate the principal source: in the US 24.6 out of 24.9 million tonnes were produced from maize, whereas in Europe 2.8 out of 8.4 million tonnes were derived from wheat.
Starch is a polymer of alpha-glucose, which exists in two forms: the linear amylose (Fig. 13.5) and the highly branched amylopectin (Fig. 13.6). In native starch, the proportions of the two molecules vary according to the plant of origin, with amylose comprising 15-30% and amylopectin 70–85% of the starch (BeMiller and Whistler, 2009). Amylose tends to exist in a tightly bound helical structure, which renders it largely insoluble in cold water, whereas the looser structure of amylopectin renders it more water soluble and digestible. The process of plasticisation opens up the helical amylose structure to give a material suitable for moulding and extrusion (Olkku and Rha, 1978). The molecular weight of amylose varies between 40 000 and 340 000 (250 to 2000 anhydroglucose units) while the molecular weight of the branched amylopectin may reach 80 000 000 (BeMiller and Whistler, 2009), which accounts for the high viscosity of plasticised starch. The proportions of amylose and amylopectin strongly influence the structure and physical characteristics of starch (Yoo and Jane, 2002) and in particular its behaviour in industrial processing and cooking. Consequently, much plant breeding and genetic research has been devoted to manipulating the relative amounts of these components. ‘Waxy rice’, for example, has little or no amylose, whereas the amylose content of maize can vary from 28% to 70% depending on the plant variety. The amylose content of wheat starch is typically 25% and that of potato is 20%. The production of processed food, such as potato crisps, generates a large amount of process water rich in so-called ‘grey starch’ and there is much interest in its potential as a starch feedstock for bio-derived materials. The variability in the structure and physical properties of the starch are so dependent on the plant from which it is obtained that the source should be considered the prime variable. Accordingly, the literature contains extensive reviews devoted to the properties of individual starches from different sources, such as maize (Gallant and Bouchet, 1986), wheat flour (Olkku and Rha, 1978) and peas (Ratnayake et al., 2002), while tuber-derived starches have been extensively reviewed as a group (Hoover, 2001).


13.2.4 Chitin
Chitin (Fig. 13.7) is the aminopolysaccharide equivalent of cellulose in terms of its natural abundance and role as structural biopolymer. It is a linear polymer of 2-acetamido-2-deoxy-β-D-glucose, although it is still frequently referred to by its trivial name, N-acetylglucosamine, and thus is an amino analogue of cellulose (Rinaudo, 2006). Purified chitin is an inelastic white material, which is insoluble in water. Deacetylation of chitin, by 5% sodium hydroxide for example, gives chitosan (Fig. 13.8), although complete deacetylation is difficult to achieve. Chitosan is soluble in acetic acid. Chitin itself is soluble in trichloroacetic acid (TCA) and other similar polar protic strong solvents, a property which provides a useful route to fibre spinning and further chemical modification.

The chemistry of chitin has been reviewed in detail (Pillai et al., 2009), as have its biomedical applications (Jayakumar et al., 2010; 2011), which arise from its adsorptive properties, non-toxicity, biocompatibility and biodegradability. Its adsorptive property also gives it the ability to form hydrogels with polymers such as poly(ethylene glycol) (PEG) and gelatin.
Chitin is a major component of invertebrate exoskeletons such as arthropod and crustacean shells as well as fungal cell walls. It is obtained mainly from marine sources as a by-product of harvesting organisms such as shrimp, prawn, lobster and other shellfish. The rate of production is such that in some areas it is a marine pollutant, for which reason its exploitation as a polymer feedstock has both economic and environmental benefits, if it is diverted from disposal to utilisation. The structure of the invertebrate exoskeleton is in itself an example of a naturally occurring nanocomposite and has been discussed in detail (Raabe et al, 2005).
Like cellulose, chitin is capable of acting as both the matrix polymer and the nanofiller in nanocomposites. For example, chitin-reinforced PCL has been explored as a route to a biodegradable nanocomposite (Morin and Dufresne, 2002) and chitin whiskers have been investigated as a nanofiller for natural rubber (Nair et al., 2003). Studies on chitin nanocomposites tend to focus on biomedical applications such as drug delivery from chitosan organically modified rectorite films (Wang et al, 2007).
13.2.5 Polycaprolactone (PCL)
Polycaprolactone is a semicrystalline linear polyester produced by ring-opening polymerisation of epsilon-caprolactone, which is commonly derived from fossil carbon. It has a much lower glass-transition temperature (Tg = − 60 °C) than other biodegradable polymers, which assists its biodegradability despite its high degree of crystallinity, typically 50%. The melting point (Tm = 60 °C) is also rather low. Like poly(butylene succinate) (PBS), PCL is often used in biodegradable polymer blends such as PCL/PHB (Lovera et al., 2007), PCL/starch (Averous et al., 2000) and PCL/PLA (Liu et al., 2000). As PCL and PLA are both synthesised by ring-opening polymerisation, PCL/PLA multiblock copolymers can readily be produced, to give biodegradable thermoplastic elastomers (Cohn and Hotovely Salomon, 2005).
13.2.6 Poly(butylene succinate) (PBS)
Of the synthetic biodegradable polymers available, poly(butylene succinate), see Fig. 13.9, has attracted much attention because it is comparable with poly(propylene) in terms of good thermal resistance and melt processability as well as its chemical resistance, whilst remaining biodegradable (Ray et al., 2005). Although it was originally derived from fossil carbon, there is considerable interest in developing it as a bio-sourced polymer, with the succinic acid moiety being produced by bacteria such as E. coli or commercially developed strains such as Basfi succiniproducens from glucose or glycerol feedstock. In Europe, the main producer is BASF, although another example of commercially available PBS is Bionolle, manufactured by Showa Highpolymer. PBS is suitable for film forming, sheet extrusion, injection moulding and moulded foam products, and the range of applications for which is it currently marketed include: mulching film, compost bags and household goods. It is also used in some civil engineering and construction applications. It has good fibre-forming properties and is therefore suitable for spun-fibre applications such as textiles. According to the manufacturers, PBS is degradable in a variety of media including compost, soil, fresh and salt water and activated sludge. Breakdown in as little as 12 weeks in suitable aerobic conditions has been reported (Mohanty et al., 2003a).

A related polymer is poly(butylene succinate)-co-adipate (PBSA). This is a random copolymer, which is synthesised by the polycondensation of butane-4-diol in the presence of adipic and succinic acids. PBSA has lower crystallinity than PBS and also greater flexibility in the polymer chains (Ray et al., 2008).
PBS is often blended with other polymers to give materials with improved properties such as impact strength. Examples of PBS blends with other biodegradable polymers include: PBS/PL A (Chen et al., 2005; Harada et al., 2007; Park and Im, 2002) and PBS/PHA (Qiu et al., 2003).
13.3 Methods of production of biodegradable polymer nanocomposites
13.3.1 Melt processing
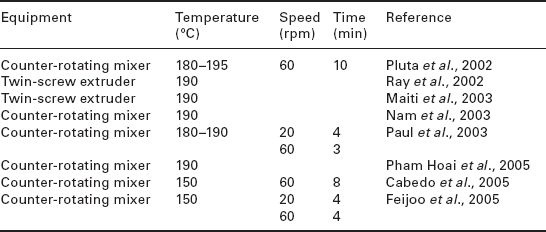
Although in situ polymerisation is a feasible technique (Paul et al., 2005b), the technique most favoured would appear to be melt extrusion (Ray et al., 2002a; 2003d; 2003b; Fujimoto et al., 2003b) or variations on melt compounding (Pluta et al., 2002; Nam et al., 2003; Cabedo et al., 2005; Paul et al, 2005b). The conditions used for melt compounding and extrusion of PLA/clay nanocomposites are summarised in Table 13.3. The favoured processing temperature is 180–190 °C, although a lower temperature of 150 °C has been used (Cabedo et al., 2005). In the melt-compounding process, the modal speed was 60 rpm, with some researchers favouring an initial phase at a slower speed, although reasons for this were not given. The total processing time ranged from three minutes to 10 minutes (Pluta et al, 2002).
Table 13.3
Processing conditions employed in the production of PLA/clay nanocomposites by melt intercalation
There is considerable variation in the PLA used in nanocomposite production, with five companies providing the entire stock (Table 13.4). Most researchers reported the quality of the PLA by noting its weight-average molecular weight, polydispersity and D-lactide content. The molecular weights of the PLA preparations ranged from 110 000 to 325 000; over a threefold variation. The polydispersity was more conservative, varying from 1.45 to 2.0. The purity was not reported by several workers, although it is known that the proportions of D- and L-isomers have a great influence on the physical properties of the polymer. However, from the known reports of purity it can be seen that the D-lactide content varied from zero to 12% (Paul et al., 2003; Cabedo et al, 2005).
Table 13.4
Grades of polylactide used on production and evaluation of PLA/clay nanocomposites

– indicates that data was not available.
aNotes: Calculated from Mn and the polydispersity.
bThe value is for the viscosity-average molecular weight.
The chemical nature of the organoclay also varied among the reports, although all the nanoclays were based on montmorillonite (MMT) with a cation exchange capacity in excess of 90 me/100 g. The counterion was nearly always a quaternary ammonium compound, i.e. a nitrogen atom is covalently bonded to four alkyl groups so that the ‘lone pair’ is also shared and the nitrogen centre acquires a positive charge. By using long-chain alkyl groups, the organoclay is more compatible with the polymer and so more easily wetted. The choice of alkylammonium compound is dictated partly by the commercial need for a low- cost compound. Thus, although Ray et al. (2002) concentrated on an octadecylammonium organoclay of high purity, most of the remaining researchers, as can be seen in Table 13.5, elected to use organoclays based on the cheaper hydrogenated tallow alkyls (HT), which are of animal origin. However, the purity of HT is somewhat variable, depending on its origin. A typical analysis for HT is: 65% C18; 30% C16 and 5% C14 (Chen and Yoon, 2005).

13.3.2 Solvent casting
Solvent casting is a suitable technique at the early stages of investigation of polymer nanocomposites because only small quantities of polymer need be used and the absence of heat treatment avoids thermal degradation of the polymer or nanofiller should they be susceptible. Chloroform remains the solvent of choice for many workers because it can be used equally successfully with PLA, PCL, PHA and PHBV and is also suitable for suspending cellulose microfibrils (Sanchez-Garcia et al., 2008). Biopolymer-layered silicate nanocomposites have also been made using chloroform both to dissolve PLA and to suspend the nanoclay (Krikorian and Pochan, 2003; McLauchlin and Thomas, 2009).
Others have favoured dimethylformamide (DMF) as a solvent for PHBV- cellulose nanocomposites (Ten et al., 2010). In all these studies, the dispersal method was sonication, with or without cooling. Ten et al. also reported a 17% reduction in the weight-average molecular weight of PHBV due to the sonication technique employed (Ten et al., 2010). It should be noted that, unlike PLA and PCL, PHB itself is not soluble in chloroform at room temperature and requires heating to 70 °C for effective dissolution (Yun et al., 2008).
While organic solvents are required for casting PLA films, some poly(hydroxyalkanoate) derivatives can be cast by alternative methods, such as casting from a latex of bacterial poly(β-hydroxyoctanoate) (PHO) obtained from Pseudomonas oleovorans, which can be cast into amorphous, thermoplastic elastomeric films (Dubief et al., 1999).
13.4 Properties of biodegradable polymer nanocomposites
13.4.1 Mechanical properties
Nanocomposites can be characterised either by tensile testing or by dynamic mechanical analysis. An example of the latter is given in Table 13.6. These materials were prepared by twin-screw extrusion (Ray et al, 2002a).
Table 13.6
Storage modulus of PLA-organo-montmorillonite exfoliated nanocomposites prepared by twin-screw extrusion

Source: Ray et al., 2002a.
At all temperatures, the storage modulus increased with increasing organoclay loading. The increases were more marked at temperatures below the Tg of the materials, so at 40 °C a clay loading of 7% increased the storage modulus from 1.60 GPa to 3.82 GPa, indicating that the polymer had become more elastic. The authors also noted increases in G′ above the glass-transition temperature, from 0.06 GPa to 0.19 GPa, and attributed this to the reinforcement effect of the clay particles, which restricted the movement of the polymer chains.
In another study, PLA/MMT nanocomposites were prepared by solvent casting from a chloroform suspension (Lee et al., 2003b). These samples also contained ammonium carbonate as a blowing agent and sodium chloride particles, which were leached out post-forming to produce scaffolds such as would be used in biomedical applications. Exfoliation of the layer silicate was confirmed by X-ray diffraction analysis and the disappearance of the peak at 26 = 4.56° due to interlayer spacing was observed. The authors reported a Tg of 63 °C for pristine PLA decreasing to 53 °C for the nanocomposite containing 5% organically modified montmorillonite (o-MMT), although the Tg of the 8% o-MMT nanocomposite was 58 °C. The recrystallisation temperature decreased from 111 °C to 90 °C and this was attributed to the clay platelets acting as nucleating sites and promoting crystallisation.
The mechanical properties were assessed by tensile testing and showed an increase in tensile modulus from 121 MPa for pristine PLA to 170 MPa for the 8% o-MMT nanocomposite. The tensile strength decreased from 48 MPa to 23 MPa. The increase in modulus was ascribed to mechanical reinforcement of the polymer by the clay platelets. In another study (Ogata et al., 1997), it was concluded that intercalation did not take place in solvent-cast composites although a twofold increase in Young’s modulus at 10% clay loading was observed. This increase was instead ascribed to a ‘superstructure’ forming in the blend film in the thickness direction.
The storage modulus of solvent cast nanocomposites has also been assessed elsewhere (Krikorian and Pochan, 2003). Here the conclusions were that exfoliated clay loading up to 15% gave a 61% increase in storage modulus at temperatures ‘around body temperature’, which is assumed to be from 36 °C to 38 °C.
PHA-cellulose nanocomposites based on a latex of bacterial poly(β-hydroxyoctanoate) obtained from Pseudomonas oleovorans have been cast into amorphous, thermoplastic elastomeric films (Dubief et al, 1999). Nanocomposite materials were prepared by stirring mixtures of the latex and colloidal suspensions of either hydrolysed starch or cellulose whiskers. After mixing, the preparations were either cast and evaporated or freeze-dried and moulded. The cellulose used was in the form of tunicin whiskers consisting of parallelepiped rods with the length ranging from 100 nm to several micrometres (average value around 1 μm) for widths of the order of 10–20 nm. The aspect ratio of these whiskers as estimated from transmission electron microscopy was around 67. The properties of the nanocomposites were assessed by determining the variation of storage modulus with temperature and were strongly related to the aspect ratio of the filler and to geometric and mechanical percolation effects. The authors also noted that specific polymer/filler interactions and geometrical constraints imposed by the particle size of the latex influenced the mechanical reinforcement effect of the cellulose whiskers.
In an accompanying publication (Dufresne et al., 1999), the same group showed that the high mechanical properties of the composite were the result of mechanical percolation created when the tunicin whiskers were present at 1% by volume or greater, the so-called ‘percolation threshold’. However, rather than through a direct interaction such as interparticular hydrogen bonding, the network was created through transcrystalline layers of polymer forming between the whiskers, the deleterious consequence of which was that the mechanical properties of the semicrystalline medium-chain-length PHA composite decreased disastrously when the melt temperature of the matrix was reached. The phenomena of transcrystallisation and mechanical percolation are therefore important considerations in the development of this type of nanocomposite.
This group also prepared a nanocomposite based on tunicin whiskers and plasticised starch using glycerol as plasticiser (Angles et al., 2000). The product contained four components (starch, cellulose, glycerol and water) and examination showed it to be a heterogeneous system containing glycerol- and amylopectin-rich domains with the plasticisers (water and glycerol) accumulating in the cellulose/ amylopectin interface. They observed that transcrystallisation of amylopectin had occurred, similar to that encountered with the PHA composites, and noted that this phenomenon increased with increasing water content. The mechanical properties, such as tensile modulus, were highly dependent on the water content and temperature, which was especially significant because at room temperature the glycerol-plasticised starch matrix was in its glass-rubber transition zone. This made the composite highly sensitive to fluctuations in temperature (Angles et al, 2000).
Although early work on biodegradable nanocomposites focused on PLA, PBS is attracting increasing attention due to its ease of processing (Ray et al, 2005; 2006a; 2006b; Guangxin and Yoon, 2005; Ray and Bousmina, 2006; Shih et al., 2007; 2008; Makhatha et al, 2008).
The intercalation and exfoliation behaviour of PBSA-organoclay nanocomposites has also been studied in detail by combining melt rheology studies with scanning transmission electron microscopy (STEM) (Bandyopadhyay et al., 2010). It was found that increasing the organoclay loading up to 4% suppressed the viscous behaviour of pure PBSA, which became more gel-like at 6% clay content. At a clay content of 5% the behaviour was between the liquid and gel states. STEM studies indicated a highly delaminated nanocomposite microstructure at low clay contents, which became more flocculated with increasing clay content until a stacked–intercalated structure was produced at higher clay content. The authors reported a good agreement between the observed structures and the melt-state rheological behaviour. Other studies have reported similar trends and noted the shear-thinning behaviour of the nanocomposites (Eslami et al., 2010; Bhatia et al, 2009; 2010).
13.4.2 Thermal properties
The glass-transition temperature (Tg) of pure PLA is little affected by the incorporation of organoclays. For example, one study (Pluta et al., 2002) reported a Tg of 58.4 °C for annealed PLA and 58.2 °C for an annealed PLA/o-MMT nanocomposite. However, crystallising the samples isothermally at 120 °C both lowered Tg and produced greater differences between the materials, so that the Tg of crystallised PLA was 53 °C and that of the crystallised nanocomposite was measurably higher at 55 °C. They noted that this increase was for polystyrene nanocomposites and is ascribed to the restricted segmental motions at the interface of the organic and inorganic phases of the material. Other studies (Ray et al., 2003d) indicate that the Tg values of unfilled PLA and PLA containing 4% organoclay are effectively the same (60 °C). It is more likely that the difference in Tg recorded by these two authors is due to the different D-lactic acid contents, which were > 4% and < 2%, respectively. It has also been shown that the type of nanoclay had minimal effect on both T. and melting temperature (Paul et al, 2003). However, in this study, Tg was reduced from 55 °C to 15 °C by addition of 20 wt% PEG as plasticiser. It has also been reported that the melting point of PLA (177 °C) did not change with clay loading (Krikorian and Pochan, 2003).
It has been reported that the maximal thermal stability of PBS A/clay nanocomposites is obtained at 3% clay content (Bhatia et al., 2009). However, PBS/multi-walled carbon nanotube nanocomposites have a decomposition temperature 10 °C higher than PBS (Song and Qiu, 2009).
13.4.3 Barrier properties
It is in food packaging that the barrier properties of polymer thin films become highly relevant. In general, it has been found that incorporating up to 5% nanofiller by weight can greatly reduce the permeability to oxygen and water (Lange and Wyser, 2003). While it is generally accepted that the improvements in barrier properties arise because of the creation of a so-called ‘tortuous path’, which slows down the passage of gas or molecules, not all reported experiments show this. For example, melt-processed PBS-organoclay nanocomposites did not show greatly improved O2 barrier properties over the pure PBS (Ray et al., 2006a). The authors suggested that the high degree of crystallisation caused by the nucleating effect of the organoclay compromised the barrier property in this case. However, a 50% improvement in water barrier and a 48% improvement in oxygen barrier were reported for PLA-layered silica nanocomposites that were prepared by twin-screw extrusion and converted to blown films (Thellen et al., 2005).
As an example of the improvements in barrier properties achievable, the water permeability coefficient of PLA was reduced from 2.30 × 10− 14 to 1.03 × 10− 14by addition of 10% food grade mica by the solvent-casting technique, while the oxygen permeability coefficient was reduced from 2.77 × 10−18 to 1.09 × 10−18. The water vapour permeability coefficient of PHBV was reduced from 1.27 × 10− 14 to 0.60 × 10− 14 and the oxygen permeability increased from 1.44 × 10− 18 to 2.33 × 10− 18. The water vapour permeability coefficient of PCL was reduced from 3.39 × 10− 14 to 1.26 × 10− 14 and the oxygen permeability decreased from 7.06 × 10− 18 to 3.67 × 10− 18 (Sanchez-Garcia and Lagaron, 2010). Thus reductions of 50% or more in permeability are achievable in biodegradable nanocomposites.
13.4.4 Biodegradability
The biodegradation of PLA is sometimes compared to that of plasticised starch, which is also a candidate biopolymer. However, unlike starch, PLA requires a higher temperature than starch for biodegradation to occur and does not lend itself to home composting where lower temperatures frequently apply, compared with the more controlled conditions of municipal composting. However, a greatly enhanced biodegradation of PLA with 4% clay loading relative to neat PLA has been reported (Ray et al., 2003a). This was attributed to the greater amorphous nature of the PLA in the nanocomposite (40%), compared with the neat polymer (36%), as it is known that amorphous polymers are more readily degraded by microorganisms. A similar conclusion was reached with PBS/layer silicate nanocomposites where the increased biodegradation was ascribed to reduced crystallinity (Han et al, 2008). The degree of biodegradation appears to depend greatly on the organoclay used in the manufacture of the nanocomposite (Maiti et al., 2003).
A study of hydrolytic stability (Paul et al., 2005a) was carried out by immersing specimens in a pH 7.4 buffer at 37 °C for five and a half months. The authors do not stipulate that sterile conditions were used, although no infection of the samples was reported. A microcomposite prepared with Na.montmorillonite degraded within two months whereas neat PLA and a nanocomposite based on Cloisite 25A were still structurally unaltered after five months. The rapid degradation of the microcomposite was attributed to the hydrophilicity of the sodium clay used.
13.5 Applications of biodegradable polymer nanocomposites
13.5.1 Biomedical applications
Research in this field is very extensive, so this section offers a brief introduction and directs the reader to suitable reviews of the literature (Jayakumar et al., 2010; Katti, 2004). When originally discovered, PLA was largely ignored due to its biodegradability, as, at the time, the durable properties of other polymers were more popular. Interest in PLA resumed in the 1960s because of its biocompatibility (Kulkarni et al., 1966). As chitin also has this property (Rinaudo, 2006; Jayakumar et al, 2010), research into biomedical applications of these two polymers comprises the bulk of research carried out to date. PLA also has the property of being absorbed by living tissue, which makes it suitable as a temporary ‘scaffold’ for regeneration of tissue and bone. As with the food sector, legislation restricts the nanofillers that can be used in nanocomposites for biomedical applications, so that the main filler used is hydroxyapatite, which is itself biocompatible. Thus PLA/hydroxyapatite and collagen/hydroxyapatite nanocomposites are used in many orthopaedic applications. Another extensive research field is that of using PLA-based nanocomposites as controlled drug release agents (Chen et al, 2007).
The salient properties of biodegradable nanocomposites for biomedical applications are the extent and speed of absorption by the living tissue (Gogolewski et al., 1993; Katti, 2004; Li et al, 2004; Weir et al, 2004; Cui et al., 1998) and their mechanical properties, particularly at body temperature (Katti, 2004; Li et al., 2004; Lee et al, 2005a).
13.5.2 Packaging
Nanocomposite technology affords a practical solution to the problems often associated with biodegradable and bio-derived polymers for food use, namely weak mechanical properties and poor barrier properties (Sanchez-Garcia and Lagaron, 2010; Sorrentino et al., 2007; Lagaron et al., 2005; Ray et al, 2003c). The alternative, polymer blending, is limited to blending only with other biodegradable polymers if biodegradability is to be maintained. Biodegradable nanocomposites often have better water and oxygen barrier properties as well as greater solvent resistance and improved mechanical properties (Ray et al., 2002a; Sanchez-Garcia et al., 2008; Makhatha et al., 2008; Sanchez-Garcia and Lagaron, 2010; Rhim and Ng, 2007).
While these aspects deal with the shelf-life of the food product, nanocomposite technology can play a role in the end of life of the packaging, particularly where composting or anaerobic digestion is involved, and, in doing so, demonstrates clear advantages over polymer blends. Whereas blending with non-biodegradable polymers can compromise the biodegradability of the whole composite, using nanofillers can result in controlled and enhanced biodegradability (Ray et al., 2002a; 2003c; Fujimoto et al, 2003a). Post-consumer food packaging is usually contaminated with food waste, either remnants of the food that it protected or commingled with uneaten or discarded food. Although recycling is a possible option, the principle of disposing food waste and food packaging by either composting or anaerobic digestion is increasingly seen as a viable method, particularly if energy recovery in the form of biogas can be achieved at the same time (WRAP, 2009).
It has been pointed out that many of the additives, fillers and organic clay modifiers used in nanocomposite manufacture are not approved for food contact (Sorrentino et al, 2007), although one viewpoint is that there is no reason to believe that there is any immediate risk by using substances in approved lists (Lagaron et al., 2005). A comprehensive review of the legislation on food technology in 2005 did not reveal any legislation regarding nanocomposites or nanoparticles (Arvanitoyannis et al, 2005). However, the impacts and implications of nanotechnology in food applications were reviewed in 2009 (Bouwmeester et al, 2009). The potential for consumer exposure to and ingestion of nanoparticles was assessed with respect to the applications (barrier, strength and anti-microbial). It was concluded that, as the nanoparticles were to be directly incorporated into the polymer, there was potential for exposure to the food but not directly to the consumer. Various reports on the subject of nanoparticles in packaging call for new protocols for evaluating the migration of nanoparticles (Dainelli et al., 2008). The safety requirements for ‘active and intelligent packaging’, which includes biodegradable packaging, are stated in EU regulations 1935/2004/EC and 450/2009/EC.
In summary, this area is still very much under development and, for the time being, the ‘precautionary principle’ still very much applies in biodegradable nanocomposite research.
13.5.3 Automotive applications
The steadily increasing price of oil has an impact on all industries, not least the automotive industry, which has responded by seeking ways to make vehicles more fuel-efficient by substituting non-structural metal body parts with polymer nanocomposite materials (Presting and König, 2003; Hussain et al., 2006; Salonitis et al., 2010; Mohanty et al., 2003b; Moore, 2003; Leaversuch, 2001). In this application, the concern is not so much for biodegradability of the material, but for its sustainability in the context of rising and fluctuating oil prices. The manufacturer Toyota was one of the ‘early adopters’ of the new technology, although many other manufacturers are following suit and any list of names would rapidly be out of date.
Research in automotive applications has tended to focus on PLA, PBS and cellulose esters as the matrix polymer (Moore, 2003; Harris and Lee, 2008; Lee et al., 2009), while the nanofillers are either organoclays (Ray et al., 2006a) or plant fibres (Baldwin et al., 2003; Lanzillotta et al, 2002; Oksman et al, 2003; Mohanty et al, 2005) such as kenaf (Lee et al, 2009; Huda et al., 2008), hemp and cellulose fibres (Huda et al, 2004a; 2004b; Samir et al., 2004; Eichhorn et al., 2001).
The three sectors, food packaging, automotive applications and biomedical, are all sectors which have a risk-averse approach to new product development. Consequently it is to be expected that progress in these applications is likely to be slow but steady (Hussain et al., 2006).
13.6 References
Angles, M.N., Vignon, M.R., Dufresne, A., EMBRAPA, UNESP, USP, Processing and characterisation of plasticised starch 1 tunicin whiskers nanocomposite materialsMattoso L.H.C., Leao A., Frollini E., eds. Natural Polymers and Composites. 2000:206. [14–17 May].
Arvanitoyannis, I.S., Choreftaki, S., Tserkezou, P. An update of EU legislation (directives and regulations) on food-related issues (safety, hygiene, packaging, technology, GMOs, additives, radiation, labelling): presentation and comments. International journal of Food Science and Technology. 2005; vol. 40(no. 10):1021–1112.
Averous, L., Moro, L., Dole, P., Fringant, C. Properties of thermoplastic blends: Starch-polycaprolactone. Polymer. 2000; vol. 41(no. 11):4157–4167.
Baldwin, K.A., Lee, E.C., Flanigan, C.M., SPE, E.D., Use of natural fibre reinforced composites in the automotive industryGPEC 2003: Plastics Impact on the Environment. Brookfield, CT: SPE, 2003. [26–27 February., pp. 87.].
Bandyopadhyay, J., Maity, A., Khatua, B.B., Ray, S.S. Thermal and rheological properties of biodegradable poly(butylene succinate)-co-adipate nanocomposites. Journal of Nanoscience andNanotechnology. 2010; vol. 10(no. 7):4184–4195.
BeMiller, J., Whistler, R.L. Starch: Chemistry and Technology, 3rd edn. London: Academic Press; 2009.
Bhatia, A., Gupta, R.K., Bhattacharya, S.N., Choi, H.J. An investigation of melt rheology and thermal stability of poly(lactic acid)/poly(butylene succinate) nanocomposites. Journal of Applied Polymer Science. 2009; vol. 114(no. 5):2837–2847.
Bhatia, A., Gupta, R.K., Bhattacharya, S.N., Choi, H.J. Effect of clay on thermal, mechanical and gas barrier properties of biodegradable poly(lactic acid)/poly(butylene succinate)(PLA/PBS) nanocomposites. International Polymer Processing. 2010; vol. 25(no. 1):5–14.
Booth, B. Polystyrene to Biodegradable PHA Plastics. Environmental Science and Technology. 2006; vol. 40(no. 7):2074–2075.
Bouwmeester, H., Dekkers, S., Noordam, M.Y., Hagens, W.I., Bulder, A.S., et al. Review of health safety aspects of nanotechnologies in food production. Regulatory Toxicology and Pharmacology. 2009; vol. 53(no. 1):52–62.
Buchholz, B., Entenmann, G. Semi-SolidMixtures of Amorphous Oligomers and Crystalline Polymers Based on Lactic Acid, 1998. [VS 5725881 A].
Cabedo, L., Feijoo, J.L., Villanueva, M.P., Lagaron, J.M., Saura, J.J., et al. Comparacion entre nanocompuestos biodegradables de poli(acido lactico) (PLA) amorfo con arcillas de distinta naturaleza. Revista De Plasticos Modernos. 2005; vol. 584:177–183.
Capon, R.J., Dunlop, R.W., Ghisalberti, E.L., Jeffries, P.R. Poly-3- hydroxyalkanoates from marine and freshwater cyanobacteria. Phytochemistry. 1983; vol. 22(no. 5):1181–1184.
Chen, G.X., Yoon, J.S. Clay functionalization and organization for delamination of the silicate tactoids in poly (l-lactide) matrix. Macromolecular Rapid Communications. 2005; vol. 26(no. 11):899–904.
Chen, G.X., Kim, H.S., Kim, E.S., Yoon, J.S. Compatibilization-like effect of reactive organoclay on the poly (l-lactide)/poly (butylene succinate) blends. Polymer. 2005; vol. 46(no. 25):11829–11836.
Chen, C., Lv, G., Pan, C., Song, M., Wu, C., et al. Poly (lactic acid)(PLA) based nanocomposites - A novel way of drug-releasing. Biomedical Materials. 2007; vol. 2:L1.
Cohn, D., Hotovely Salomon, A. Designing biodegradable multiblock PCL/PLA thermoplastic elastomers. Biomaterials. 2005; vol. 26(no. 15):2297–2305.
Cui, F.Z., Du, C., Feng, Q.L., Zhu, X.D., De Groot, K. Tissue response to nano- hydroxyapatite/collagen composite implants in marrow cavity. Journal of Biomedical Materials Research. 1998; vol. 42(no. 4):540–548.
Dainelli, D., Gontard, N., Spyropoulos, D., Zondervan-van den Beuken, E., Tobback, P. Active and intelligent food packaging: legal aspects and safety concerns. Trends in Food Science and Technology. 2008; vol. 19(no. 1):S103–S112.
Doi, Y. MicrobialPolyesters. US: VCH Publishers, Inc; 1990.
Dubief, D., Samain, E., Dufresne, E. Polysaccharide microcrystals reinforced amorphous poly(beta-hydroxyoctanoate) nanocomposite materials. Macromolecules. 1999; vol. 32(no. 18):5765–5771.
Dufresne, A., Kellerhals, M.B., Witholt, B. Transcrystallization in Mcl-PHAs/ cellulose whiskers composites. Macromolecules. 1999; vol. 32(no. 22):7396–7401.
Eichhorn, S.J., Baillie, C.A., Zafeiropoulos, N., Mwaikambo, L.Y., Ansell, M.P., et al. Current international research into cellulosic fibres and composites. Journal of Materials Science (USA). 2001; vol. 36(no. 9):2107–2131.
Eslami, H., Grmela, M., Buosmina, M. Linear and nonlinear rheology of polymer/layered silica nanocomposites. J. Rheology. 2010; vol. 54(no. 3):539–562.
Feijoo, J.L., Cabedo, L., Gimenez, E., Lagaron, J.M., Saura, J.J. Development of amorphous PLA-montmorillonite nanocomposites. Journal of Materials Science. 2005; vol. 40(no. 7):1785–1788.
Findlay, R.H., White, D.C. Polymeric beta-hydroxyalkanoates from environmental samples and Bacillus megaterium. Appl. Environ. Microbiol.. 1983; vol. 45:71–78.
Fletcher, A. PHA as natural, biodegradable polyesters. In: Plastics from Bacteria and for Bacteria. New York: Springer-Verlag; 1993:77–93.
Fujimoto, Y., Ray, S.S., Okamoto, M., Ogami, A., Yamada, K., et al. Well-controlled biodegradable nanocomposite foams. from microcellular to nanocellular. Macromolecular Rapid Communications. 2003; vol. 24(no. 7):457–461.
Fujimoto, Y., Ogami, A., Okamoto, M., Ray, S.S., Ueda, K., et al. Newpolylactide/ layered silicate nanocomposites. 5. Designing of materials with desired properties. Polymer. 2003; vol. 44(no. 21):6633–6646.
Fukuzaki, H., Aiba, Y., Yoshida, M., Asano, M., Kumakura, M. Direct copolymerisation of lactic acid with butyrolactone in the absence of catalysts. Makromolekulare Chemie. 1989; vol. 190(no. 7):1553–1559.
Gallant, D., Bouchet, B. Ultrastructure of maize starch granules. A review. Food Microstructure. 1986; vol. 5(no. 1):141–155.
Gerngross, T.U., Slater, S.C. How green are green plastics? Scientific American. 2000; vol. 283(no. 2):24–29.
Gogolewski, S., Jovanovic, M., Perren, S.M., Dillon, J.G., Hughes, M.K. Tissue response and in vivo degradation of selected polyhydroxyacids: Polylactides (PLA), poly(3-hydroxybutyrate) (PHB) and poly(3-hydroxybutyrate-co-3- hydroxyvalerate) (PHB/VA). Journal of Biomedical Materials Research. 1993; vol. 27(no. 9):1135–1148.
Guangxin, C., Yoon, J.-S. Nanocomposites of poly((butylene succinate)-co- (butylene adipate)) (PBSA) and twice functionalized organoclay. Polymer International. 2005; vol. 54(no. 6):939–945.
Han, S.I., Lim, J.S., Kim, D.K., Kim, M.N., Im, S.S. In situ polymerized poly(butylene succinate)/silica nanocomposites: Physical properties and biodegradation. Polymer Degradation and Stability. 2008; vol. 93(no. 5):889–895.
Harada, M., Ohya, T., Iida, K., Hayashi, H., Hirano, K., et al. Increased impact strength of biodegradable poly (lactic acid)/poly (butylene succinate) blend composites by using isocyanate as a reactive processing agent. Journal of Applied Polymer Science. 2007; vol. 106(no. 3):1813–1820.
Harris, A.M., Lee, E.C. Improving mechanical performance of injection molded PLA by controlling crystallinity. Journal of Applied Polymer Science. 2008; vol. 107(no. 4):2246–2255.
Holmes, P.A., Wright, L.F., Collins, S.H. Beta-hydroxybutyrate Polymers 1981; [EP0052459].
Hoover, R. Composition, molecular structure, and physicochemical properties of tuber and root starches: A review. Carbohydrate Polymers. 2001; vol. 45(no. 3):253–267.
Huang, S.J. Poly(lactic acid) and copolyesters. In: Bastioli C., ed. Handbook of Biodegradable Polymers. Shrewsbury: Rapra Technology Limited, Shawbury; 2005:287–301.
Huda, M.S., Mohanty, A.K., Drzal, L.T., Misra, M., Schut, E., SPE. Physico- mechanical properties of ‘green’ composites from polylactic acid (PLA) and cellulose fibers. In: GPEC 2004: Plastics - Helping Grow A Greener Environment. Brookfield, Ct: SPE; 2004:12. [2004 18–19 February].
Huda, M.S., Mohanty, A.K., Misra, M., Drzal, L.T., Schut, E., SPE, Effect of processing conditions on the physico-mechanical properties of cellulose fiber reinforced poly (lactic acid). ANTEC 2004. Proceedings of the 62nd SPE Annual conference in Chicago, IL, 16–20 May. 2004:1614.
Huda, M.S., Drzal, L.T., Mohanty, A.K., Misra, M. Effect of fiber surface- treatments on the properties of laminated biocomposites from poly(lactic acid) (PLA) and kenaf fibers. Composites Science and Technology. 2008; vol. 68(no. 2):424–432.
Hussain, F., Hojjati, M., Okamoto, M., Gorga, R.E. Review article: polymermatrix nanocomposites, processing, manufacturing, and application: an overview. Journal of Composite Materials. 2006; vol. 40(no. 17):1511.
Jayakumar, R., Menon, D., Manzoor, K., Nair, S.V., Tamura, H. Biomedical applications of chitin and chitosan based nanomaterials - A short review. Carbohydrate Polymers. 2010; vol. 82(no. 2):227–232.
Jayakumar, R., Prabaharan, M., Sudheesh Kumar, P.T., Nair, S.V., Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnology Advances. 2011; vol. 29(no. 3):322–337.
Johnson, R.M., Mwaikambo, L.Y., Tucker, N. Biopolymers. London: Rapra Technology Ltd; 2003.
Johnstone, B. A throw away answer. Far Eastern Econ. Rev.. 1990; vol. 147(no. 6):62–63.
Katti, K.S. Biomaterials in total joint replacement. Colloids and Surfaces B: Biointerfaces. 2004; vol. 39(no. 3):133–142.
Kazunari, M., Yoshito, Y. Production of Biodegradable Plasticfrom Vegetable Oil Waste 2000; [JP2000189183].
Krikorian, V., Pochan, D.J. Poly (l-lactic acid)/layered silicate nanocomposite: fabrication, characterization, and properties. Chemistry of Materials. 2003; vol. 15(no. 22):4317–4324.
Kulkarni, R.K., Pani, K.C., Neuman, C., Leonard, F. PolylacticAcid for Surgical Implants, United States 1966; [AD636716].
Lagaron, J.M., Cabedo, L., Cava, D., Feijoo, J.L., Gavara, R., et al. Improving packaged food quality and safety. Part 2: Nanocomposites. Food Additives and Contaminants. 2005; vol. 22(no. 10):994–998.
Lange, J., Wyser, Y. Recent innovations in barrier technologies for plastic packaging - A review. Packaging Technology and Science. 2003; vol. 16(no. 4):149–158.
Lanzillotta, C., Pipino, A., Lips, D., SPE, New functional biopolymer natural fiber composites from agricultural resources. ANTEC 2002. Proceedings of the 60th SPE Annual Technical Conference in San Francisco, CA. 2002:5. [5–9 May].
Leaversuch, R. Nanocomposites broaden roles in automotive, barrier packaging. Plastics Technology. 2001; vol. 47(no. 10):64–69.
Leaversuch, R. Renewable PLA polymer gets ‘green light’ for packaging uses. Plastics Technology. 2002; vol. 48(no. 3):50–55.
Lee, C.H., Hyun, Y.H., Lim, S.T., Choi, H.J., Jhon, M.S. Fabrication and viscoelastic properties of biodegradable polymer /organophilic clay nanocomposites. Journal of Materials Science Letters. 2003; vol. 22(no. 1):53–55.
Lee, J.H., Park, T.G., Park, H.S., Lee, D.S., Lee, Y.K., et al. Thermal and mechanical characteristics of polylactic acid nanocomposite scaffold. Biomaterials. 2003; vol. 24(no. 16):2773–2778.
Lee, L.J., Zeng, C., Cao, X., Han, X., Shen, J., et al. Polymer nanocomposite foams. Composites Science and Technology. 2005; vol. 65(no. 15–16):2344–2363.
Lee, Y.H., Lee, J.H., An, I.G., Kim, C., Lee, D.S., et al. Electrospun dual-porosity structure and biodegradation morphology of montmorillonite reinforced PLLA nanocomposite scaffolds. Biomaterials. 2005; vol. 26(no. 16):3165–3172.
Lee, B., Kim, H., Lee, S., Kim, H., Dorgan, J.R. Bio-composites of kenaf fibers in polylactide: Role of improved interfacial adhesion in the carding process. Composites Science and Technology. 2009; vol. 69(no. 15–16):2573–2579.
Lemoigne, M. Ann. Inst. Pasteur (Paris). 1925; vol. 39:144–173.
Lemoigne, M. Bull. Soc. Chim. Biol. 1926; vol. 8:770–782.
Lemoigne, M. Ann. Inst. Pasteur (Paris). 1927; vol. 41:148–165.
Li, H.Y., Chen, Y.F., Xie, Y.S. Nanocomposites of cross-linking polyanhydrides and hydroxyapatite needles: Mechanical and degradable properties. Materials Letters. 2004; vol. 58(no. 22–23):2819–2823.
Liu, L., Li, S., Garreau, H., Vert, M. Selective enzymatic degradations of poly (L-lactide) and poly (e-capro lactone) blend films. Biomacromolecules. 2000; vol. 1(no. 3):350–359.
Lovera, D., Marquez, L., Balsamo, V., Taddei, A., Castelli, C., et al. Crystallization, morphology, and enzymatic degradation of polyhydroxybutyrate/polycaprolactone (PHB/PCL) blends. Macromolecular Chemistry and Physics. 2007; vol. 208(no. 9):924–937.
Maiti, P., Batt, C.A., Giannelis, E.P. Biodegradable polyester/layered silicate nanocomposites. In: Nanomaterials for Structural Applications. Warrendale, PA, 15086, USA, MA: Materials Research Society, 506 Keystone Drive; 2003. [2–6 December 2002].
Makhatha, M.E., Ray, S.S., Hato, J., Luy, A.S. Thermal and thermomechanical properties of poly(butylene succinate) nanocomposites. Journal of Nanoscience and Nanotechnology. 2008; vol. 8(no. 4):1679–1689.
McLauchlin, A.R., Thomas, N.L. Preparation and thermal characterisation of poly(lactic acid) nanocomposites prepared from organoclays based on an amphoteric surfactant. Polymer Degradation and Stability. 2009; vol. 94(no. 5):868–872.
Mohanty, A.K., Drzal, L.T., Misra, M., Nano reinforcements of bio-based polymers - The hope and the reality. Polymeric Materials Science and Engineering Spring 2003 Meeting; New Orleans, LA, USA. American Chemical Society, Washington, DC, 2003:60–61. [email protected] [23–27 March 1155 16th St, NW, 20036, USA].
Mohanty, A.K., Misra, M., Drzal, L.T., SPE, Emerging green nanocomposites: striving for sustainability in automotive applicationsGPEC 2003: Plastics Impact on the Environment. Brookfield, CT: SPE, 2003. [26–27 February, pp. 69].
Mohanty, A.K., Misra, M., Drzal, L.T. Natural Fibers, Biopolymers, and Biocomposites. Boca Raton, FL, USA: CRC Press; 2005.
Moore, S. Polylactic acid: Autos au naturale. www.plasticstoday.com/articles/autos-au-naturale, 2003. [Plastics Today. Online:].
Morin, A., Dufresne, A. Nanocomposites of Chitin Whiskersfrom Riftia Tubes andPoly(caprolactone). Macromolecules. 2002; vol. 35(no. 9):2190–2199.
Murphy, R.J., Bartle, I.Summary Report, Biodegradable Polymers and Sustainability: Insight from Life Cycle Assessment. UK: National Non-Food Crops Centre, 2004.
Nair, K.G., Dufresne, A., Gandini, A., Belgacem, M.N. Crab Shell Chitin Whisker Reinforced Natural Rubber Nanocomposites. 2. Mechanical Behavior. Biomacromolecules. 2003; vol. 4(no. 6):1835–1842.
Nam, J.Y., Ray, S.S., Okamoto, M. Crystallization behavior and morphology of biodegradable polylactide/layered silicate nanocomposite. Macromolecules. 2003; vol. 36(no. 19):7126–7131.
Narayan, R., Fundamental principles and concepts of biodegradability - Sorting through the facts, hypes and claims of biodegradable plastics in the marketplace. Bioplastics Magazine. 2009; vol. 4(no. 1). www.ciras.iastate.edu/bioindustry/biobasedproductsZNarayan_biodegradable_plastics.pdf [Online:].
Ogata, N., Jiminez, G., Kawai, H., Ogihara, T. Structure and thermal/mechanical properties of poly(l-lactide)-clay blend. Journal of Polymer Science: Polymer Physics Edition. 1997; vol. 35(no. 2):389–396.
Oksman, K., Skrifvars, M., Selin, J.-F. Natural fibres as reinforcement in polylactic acid (PLA) composites. Composites Science and Technology. 2003; vol. 63(no. 9):1317–1324.
Olkku, J., Rha, C.K. Gelatinisation of starch and wheat flour starch - A review. Food Chemistry. 1978; vol. 3(no. 4):293–317.
Park, J.W., Im, S.S. Phase behavior and morphology in blends of poly (L-lactic acid) and poly (butylene succinate). Journal of Applied Polymer Science. 2002; vol. 86(no. 3):647–655.
Paul, M.-A., Alexandre, M., Degee, P., Henrist, C., Rulmont, A., et al. Newnanocomposite materials based on plasticized poly(l-lactide) and organo-modified montmorillonites: Thermal and morphological study. Polymer. 2003; vol. 44(no. 2):443–450.
Paul, M.-A., Delcourt, C., Alexandre, M., Degee, P., Monteverde, F., et al. Polylactide/montmorillonite nanocomposites. Study of the hydrolytic degradation. Polymer Degradation and Stability. 2005; vol. 87(no. 3):535–542.
Paul, M.-A., Delcourt, C., Alexandre, M., Degee, P., Monteverde, F., et al. (Plasticized) polylactide/(organo-)clay nanocomposites by in situ intercalative polymerization. Macromolecular Chemistry and Physics. 2005; vol. 206(no. 4):484–498.
Pham Hoai, N., Kaneko, M., Ninomiya, N., Fujimori, A., Masuko, T. Melt intercalation of poly(l-lactide) chains into clay galleries. Polymer. 2005; vol. 46(no. 18):7403–7409.
Pillai, C.K.S., Paul, W., Sharma, C.P. Chitin and chitosan polymers: Chemistry, solubility and fiber formation. Progress in Polymer Science. 2009; vol. 34(no. 7):641–678.
Pitt, C.G., Chasalow, F.I., Hibionada, Y.M., Klimas, D.M., Schindler, A. Aliphatic Polyesters. 1. Degradation of poly(epsilon-caprolactone) in vivo. Journal of Applied Polymer Science. 1981; vol. 26(no. 11):3779–3787.
Pluta, M., Galeski, A. Crystalline and supermolecular structure of polylactide in relation to the crystallization method. Journal of Applied Polymer Science. 2002; vol. 86(no. 6):1386–1395.
Pluta, M., Galeski, A., Alexandre, M., Paul, M.-A., Dubois, P. Polylactide/ montmorillonite nanocomposites and microcomposites prepared by melt blending: Structure and some physical properties. Journal of Applied Polymer Science. 2002; vol. 86(no. 6):1497–1506.
Poirier, Y., Dennis, D.E., Klomparens, K., Somerville, C. Polyhydroxybutyrate, a biodegradable thermoplastic, produced in transgenic plants. Science (Washington). 1992; vol. 256(no. 5056):520–523.
Presting, H., König, U. Future nanotechnology developments for automotive applications. MaterialsScience andEngineering: C. 2003; vol. 23(no. 6–8):737–741.
Qiu, Z., Ikehara, T., Nishi, T. Poly (hydroxybutyrate)/poly (butylene succinate) blends: Miscibility and nonisothermal crystallization. Polymer. 2003; vol. 44(no. 8):2503–2508.
Raabe, D., Sachs, C., Romano, P. The crustacean exoskeleton as an example of a structurally and mechanically graded biological nanocomposite material. Acta Materialia. 2005; vol. 53(no. 15):4281–4292.
Ratnayake, W.S., Hoover, R., Warkentin, T. Pea starch: Composition, structure and properties - A review. Starch-Stärke. 2002; vol. 54(no. 6):217–234.
Ray, S.S., Bousmina, M. Crystallization behavior of poly ((butylene succinate )- co-adipate) nanocomposite. Macromolecular Chemistry and Physics. 2006; vol. 207(no. 14):1207–1219.
Ray, S.S., Maiti, P., Okamoto, M., Yamada, K., Ueda, K. New polylactide/ layered silicate nan-ocomposites. 1. Preparation, characterisation and properties. Macromol. 2002; vol. 35:3104–3110.
Ray, S.S., Yamada, K., Ogami, A., Okamoto, M., Ueda, K. New polylactide/ layered silicate nanocomposite: Nanoscale control over multiple properties. Macromolecular Rapid Communications. 2002; vol. 23(no. 16):943–947.
Ray, S.S., Yamada, K., Okamoto, M., Ogami, A., Ueda, K. New polylactide/ layered silicate nanocomposites. 3. High-performance biodegradable materials. Chemistry of Materials. 2003; vol. 15(no. 7):1456–1465.
Ray, S.S., Yamada, K., Okamoto, M., Ueda, K. Biodegradable polylactide/ montmorillonite nanocomposites. Journal of Nanoscience and Nanotechnology. 2003; vol. 3(no. 6):503–510.
Ray, S.S., Yamada, K., Okamoto, M., Ueda, K. Control of biodegradability of polylactide via nanocomposite technology. Macromolecular Materials and Engineering. 2003; vol. 288(no. 4):203–208.
Ray, S.S., Yamada, K., Okamoto, M., Ueda, K. New polylactide-layered silicate nanocomposites 2. Concurrent improvements of material properties, biodegradability and melt rheology. Polymer. 2003; vol. 44(no. 3):857–866.
Ray, S.S., Bousmina, M., Okamoto, K. Structureandpropertiesofnanocomposites based on poly(butylene succinate-co-adipate) and organically modified montmorillonite. Macromolecular Materials and Engineering. 2005; vol. 290(no. 8):759–768.
Ray, S.S., Okamoto, K., Okamoto, M. Structure and properties of nanocomposites based on poly(butylene succinate) and organically modified montmorillonite. Journal of Applied Polymer Science. 2006; vol. 102(no. 1):777–785.
Ray, S.S., Vaudreuil, S., Maazouz, A., Bousmina, M. Dispersion of multi- walled carbon nanotubes in biodegradable poly(butylene succinate) matrix. Journal of Nanoscience andNanotechnology. 2006; vol. 6(no. 7):2191–2195.
Ray, S., Bandyopadhyay, J., Bousmina, M. Influence ofdegree ofintercalation on the crystal growth kinetics of poly(butylene succinate)-co-adipate nanocomposites. European Polymer Journal. 2008; vol. 44(no. 10):3133–3145.
Reddy, C.S.K., Ghai, R., Rashmi, R., Kalia, V.C. Polyhydroxyalkanoates: An overview. Bioresource Technology. 2003; vol. 87:137–146.
Rhim, J.W., Ng, P.K.W. Natural biopolymer-based nanocomposite films for packaging applications. Critical Reviews in Food Science and Nutrition. 2007; vol. 47(no. 4):411–433.
Rinaudo, M. Chitin and Chitosan: Properties and Applications. Progress in Polymer Science. 2006; vol. 31(no. 7):603–632.
Saito, T., Tomita, K., Juni, K., Ooba, K. In vivo and m vitro degradation of poly(3-hydroxybutyrate) in rat. Biomaterials. 1991; vol. 12(no. 3):309–312.
Salonitis, K., Stavropoulos, P., Chryssolouris, G. Nanotechnology for the needs of the automotive industry. International Journal of Nanomanufacturing. 2010; vol. 6(no. 1):99–110.
Samir, M.A.S.A., Alloin, F., Paillet, M., Dufresne, A. Tangling effect infibrillated cellulose reinforced nanocomposites. Macromolecules. 2004; vol. 37(no. 11):4313–4316.
Sanchez-Garcia, M.D., Lagaron, J.M. Novel clay-based nanobiocomposites of biopolyesters with synergistic barrier to UV light, gas, and vapour. Journal of Applied Polymer Science. 2010; vol. 118(no. 1):188–199.
Sanchez-Garcia, M.D., Gimenez, E., Lagaron, J.M. Morphology and barrier properties of solvent cast composites of thermoplastic biopolymers and purified cellulose fibers. Carbohydrate Polymers. 2008; vol. 71(no. 2):235–244.
Schmitt, E.E., Polistina, R.A., Polyglycolic acidprosthetic devices. 1969. [A61L17/12; A61L27/18; C08G63/06; D01F6/62B edn, A61B17/04, USA].
Shih, Y.F., Wang, T.Y., Jeng, R.J., Wu, J.Y., Teng, C.C. Biodegradable nanocomposites based on poly(butylene succinate)/organoclay. Journal of Polymers and the Environment. 2007; vol. 15(no. 2):151–158.
Shih, Y.F., Chen, L.S., Jeng, R.J. Preparation and properties of biodegradable PBS/multi-walled carbon nanotube nanocomposites. Polymer. 2008; vol. 49(no. 21):4602–4611.
Skraly, F.A., Peoples, O. Polyhydroxyalkanoate Productionfrom Polyols. . 2005.
Song, L., Qiu, Z. Crystallization behavior and thermal property of biodegradable poly(butylene succinate)/functional multi-walled carbon nanotubes nanocomposite. Polymer Degradation and Stability. 2009; vol. 94(no. 4):632–637.
Sorrentino, A., Gorrasi, G., Vittoria, V. Potential perspectives of bio- nanocomposites for food packaging applications. Trends in Food Science and Technology. 2007; vol. 18(no. 2):84–95.
Suyatma, N.E., Copinet, A., Tighzert, L., Coma, V. Mechanical and barrier properties of biodegradable films made from chitosan and poly(lactic acid) blends. Journal of Polymers and the Environment. 2004; vol. 12(no. 1):1–6.
Ten, E., Turtle, J., Bahr, D., Jiang, L., Wolcott, M. Thermal and mechanical properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Polymer. 2010; vol. 51(no. 12):2652–2660.
Thellen, C., Orroth, C., Froio, D., Ziegler, D., Lucciarini, J., et al. Influence of montmorillonite layered silicate on plasticized poly(l-lactide) blown films. Polymer. 2005; vol. 46(no. 25):11716–11727.
Vert, M., Schwarch, G., Coudane, J. Present and future of PLA polymers. In: Albertsson A., Huang S., eds. Degradable Polymers, Recycling, and Plastics Waste Management. 1st edn. NY: Marcel Dekker Inc; 1995:195–204.
Vink, E.T.H., Rabago, K.R., Glassner, D.A., Gruber, P.R. Applications of life cycle assessment to Nature Works™ polylactide (PLA) production. Polymer Degradation and Stability. 2003; vol. 80(no. 3):403–419.
Wallen, L.L., Rohwedder, W.K. Poly-beta-hydroxyalkanoate from activated- sludge. Environ. Sci. Technol.. 1974; vol. 8:576–579.
Wang, X., Du, Y., Luo, J., Lin, B., Kennedy, J.F. Chitosan/organic rectorite nanocomposite films: Structure, characteristic and drug delivery behaviour. Carbohydrate Polymers. 2007; vol. 69(no. 1):41–49.
Wang, Y., Ribelles, J.L.G., Sanchez, M.S., Mano, J.F. Morphological contributions to glass transition in poly(l-lactic acid). Macromolecules. 2005; vol. 38(no. 11):4712–4718.
Weir, N.A., Buchanan, F.J., Orr, J.F., Dickson, G.R. Degradation of poly-L- lactide. Part 1: In vitro and in vivo physiological temperature degradation. Proceedings of the Institution of Mechanical Engineers H, Journal of Engineering in Medicine. 2004; vol. 218(no. 5):307–319.
WRAP. Research Report: Biopolymer Packaging in UK Grocery Market. Banbury, UK: WRAP; 2009.
Yoo, S., Jane, J. Structural and physical characteristics of waxy and other wheat starches. Carbohydrate Polymers. 2002; vol. 49(no. 3):297–305.
Yun, S., Gadd, G., Latella, B., Lo, V., Russell, R., et al. Mechanical properties of biodegradable polyhydroxyalkanoates/single wall carbon nanotube nanocomposite films. PolymerBulletin. 2008; vol. 61(no. 2):267–275.