
Nucleic acid sequencing for characterizing infectious and/or novel agents in complex samples
J.C. Detter and S.L. Johnson, Los Alamos National Laboratory, USA
K.A. Bishop-Lilly, Navy Medical Research Center-Frederick, USA
P.S. Chain, Los Alamos National Laboratory, USA
H.S. Gibbons, US Army Edgewood Chemical Biological Center, USA
T.D. Minogue, US Army Medical Research of Infectious Disease, USA
S. Sozhamannan, Critical Reagents Program, USA
E.J. Van Gieson, Defense Threat Reduction Agency, USA
I.G. Resnick, IGR Consulting, USA
Abstract:
Identification of microorganisms using nucleic acid sequencing has become a well-established field, with a variety of applications. With rapid changes in both hardware and software technologies over the last decade, the ability for a single microbial type to be identified in a complex sample is becoming easier and more robust with each turn. In this chapter we explore the history of sequencing and the upcoming challenges, and the importance of sequencing to public health and biodefense. We will also look at how sequencing fits with other orthogonal data types and the technology’s current abilities and limitations, as well as taking a forward look at sequencing overall.
Key words
sequencing; next-generation sequencing (NGS); public health; biodefense; detection; microbiology; metagenomics
1.1 Pathogen sequencing and applications in public health and biosecurity
The technology and infrastructure developed in support of the Human Genome Project opened up opportunities for advances in many unrelated areas of science and technical applications. Microbial sequencing and follow-on characterization has benefited greatly and opened up exciting new avenues of investigation for microbes, microbial communities and microbial activities. One area that has enjoyed significant advances is pathogen sequencing in support of public health and biosecurity.
1.1.1 The need for rapid detection and genetic characterization of pathogens as demonstrated by the 2001 anthrax attacks
In the fall of 2001, at least four envelopes containing Bacillus anthracis spores were mailed through the United States postal system. They were addressed to the New York Post, Tom Brokaw at NBC, and the Washington, DC offices of senators Daschle and Leahy. There were 22 suspected cases identified, divided evenly between inhalational and cutaneous cases. Of those, there were five fatalities (summarized in Rasko et al.1). The aftermath included a climate of fear and an unprecedented forensic challenge in attribution of the crime to the perpetrator; i.e. finding the source of the spores.
This unfortunate act of domestic bioterrorism taught us several things. First, it demonstrated the importance of rapid identification of an etiological agent as well as rapid identification of those who have been exposed to the agent. Case histories of the 11 inhalational anthrax patients clearly demonstrate that early administration of the appropriate antibiotic results in improved chances of survival2 (see Plate I in colour section between pages 256 and 257).
Second, in the forensic investigation that ensued, it became quite clear that existing genotyping methods, such as multi-locus variable-number tandem repeat analysis (MLVA) or single nucleotide polymorphism (SNP) typing systems, were insufficient to identify differences between isolates, as were initial efforts using Sanger sequencing. Therefore, whole genome sequencing (WGS) and analysis of various colony morphotypes were conducted. Additional findings that resulted from the forensic investigation are: evidentiary samples in a case such as this one need not be sequenced to ‘completion’ or ‘finished’, or closed to a single contig; following microbiological isolation of morphological variants, sequencing each morphotype to 9–12 × average depth of coverage was sufficient1; although several genome sequences were closed in the course of the investigation, no novel high-quality variations were identified in the closed genome sequences that were not present in the draft sequences; and, finally, it was noted that high-quality reference sequences are essential to render evidentiary draft genomes useful.1
In the case of the anthrax letter attacks, WGS was used to characterize the pathogen for the purpose of attribution, rather than as part of a rapid response scenario. However, given the short window in which the appropriate antimicrobial therapy or other countermeasures must be administered to save the victims, what would have been the outcome had the agent involved been a genetically engineered form of B. anthracis (e.g. ciprofloxacin resistant), and had the engineered phenotype not been discovered in the course of routine microbiological testing? In a mock rapid response exercise, Chen et al. demonstrated recently that WGS could have been employed upfront and operationally relevant information could have been obtained in time to improve survival rates.3
These lessons learned from the anthrax attacks are significant, but there are some caveats. B. anthracis is a highly monomorphic and new species, and spore formation supports its genomic stability. For instance, genome sequences of several Ames isolates were compared in the anthrax investigation and it was noted, that despite 21 years of laboratory growth, no distinguishing mutations were found, either in the chromosome or on the two plasmids. The mutations present in the Porton Down strain were attributed to the plasmid curing process. It is not clear whether a similar picture would emerge if the organism in question had not been such a monomorphic species – if, for instance, it had been an organism with as plastic a genome as Shigella sp. or Escherichia coli.
A further issue relates to scalability of the WGS sequencing approach for forensic genetic investigations. The combination of microbiological studies and individual WGS was costly and time-consuming. Is this approach feasible in a rapid response scenario when saving lives depends on the outcome of these genetic studies? The idea of direct metagenome sequencing of nucleic acid materials extracted from clinical specimens (ClinSeq) has gained momentum, and it could be a potential time-saver in a rapid response scenario. However, there are certain limitations of metagenomic sequencing, including the effects of matrix, depth and breadth of sequence coverage required and the bioinformatic challenges associated with sifting through mounds of data to identify the causal agent and causal genetic variations, if any (some of these aspects are treated in later sections). Finally, there is also the challenge of linking the potential agent to the disease, i.e. fulfilling Koch’s postulates, which may not be feasible within the time frame of real events.
1.1.2 Applications of WGS in a public health event caused by E. coli
Whereas, in the anthrax investigation, genomic characterization of the agent was conducted after rather than during the incident, in another more recent example WGS played a critical role in characterizing the agent of an outbreak in real time. This outbreak, involving a more virulent strain of E. coli than usual, occurred in May–June 2011 in Germany. The source of the bacterium was traced to fenugreek sprouts, but there was also significant secondary transmission (human to human and human to food). There were over 3800 total cases, including a higher than usual proportion of adults and an unusually high number of hemolytic uremic syndrome (HUS) cases (reviewed in Beutin et al.4).
This outbreak was notable in that it became the debut of so-called ‘open-source’ genomic analysis and served as a paradigm for future outbreaks/events. It was characterized by rapid, crowd-driven, round-the-clock analysis of Ion Torrent draft sequence by bioinformaticians worldwide and aggregation of the resulting data in a wiki. Notably, this rapid sequencing and analysis resulted in design of diagnostic primers 5 days after release of the draft genome.5
Independently, another team of researchers used third-generation PacBio sequence data to elucidate the reason for increased virulence. By running three sequencers in parallel for 5 h per isolate, these researchers rapidly achieved 75× average coverage per genome. All the bioinformatic analyses of the sequence data from various sources revealed some clues to the unusual virulence of the EAEC bacteria that is not normally associated with HUS syndrome and a possible model for the evolution of this pathogen itself.
In short, there were at least three genetic changes that occurred during the evolution of this pathogen: (1) acquisition by an enteroaggregative E. coli (EAggEC) strain of Stx2 genes (Shiga toxin genes found on a bacteriophage in enterohemorrhagic E. coli (EHEC)), (2) acquisition of a plasmid encoding a Type III aggregative adhesion fimbrial gene cluster (AAF/III) that has been postulated to enhance the virulence by aggregating the bacteria on the intestinal epithelium, and (3) acquisition of a plasmid that confers multiple antibiotic resistance. However, the key virulence factor is the ability of these bacteria to produce shiga toxin, and, notably, toxin production is enhanced in the presence of certain classes of antibiotics normally prescribed to combat bacterial diseases. Rasko et al. provided experimental evidence that exposure to ciprofloxacin did indeed result in increased expression of the shiga toxin gene by the German isolate.6 Thus, the German E. coli outbreak highlighted the power of the WGS approach to decipher relevant genetic characteristics of a pathogen from an outbreak scenario, and provided valuable diagnostic assays and possible treatment options.
A further instance in which time is limited and WGS data may be useful is when ruling out bioterrorism, such as in the recent case of a rapidly progressive, fatal, inhalational anthrax-like infection of a welder in Texas. In this case, the patient sought medical care just 2 h after onset of his illness. Within 10 h of his arrival, he exhibited signs of multi-organ system failure and he was started on antibiotic therapy. On day 3, B. cereus was identified from his cultures and ciprofloxacin was added to his regimen; nevertheless, he died later that day. PCR ruled out the possibility that the strain was a so-called ‘conventional’ strain of B. cereus. Given those PCR data and the rapid course of infection in the patient, his healthcare providers wondered whether the organism had acquired genes conferring increased virulence, and, if it had, whether it was a naturally emergent strain or genetically engineered. Therefore, to rule out the possibility that the pathogen had been manmade and to practice their institution’s emergency response preparedness, they undertook WGS using the Illumina GXII platform. Bioinformatic analysis indicated that the isolate was likely a natural strain of B. cereus with a pXO1-like plasmid and B. anthracis-like virulence factors,7 similar to the previous cases in welders involving a similar strain called G9241.8,9 Although in this case WGS was very useful for ruling out terrorism, it begs the question as to whether immediate metagenomic sequencing of the patient’s tracheal aspirate and/or bronchoalveolar lavage would have identified B. cereus faster and resulted in earlier administration of appropriate antibiotics.
Early administration of appropriate antibiotic is also important in the context of routine lower respiratory tract infections. In fact, the Centers for Medicare and Medicaid Services (CMS) require that antibiotics be administered within 6 h of the onset of symptoms. However, despite the fact that most cases are treatable if the etiologic agent is known, an etiologic identification is made in fewer than 10% of cases.10 Could a rapid metagenomic sequencing approach potentially increase the number of cases in which a causative agent can be identified in lower respiratory tract infections? Can MGS provide the same valuable information for other types of infections, and possibly decrease the time taken to identify sources of neonatal illness, such as the recent fatal case of Cronobacter sakazakii infection in a ten-day-old infant?11
1.1.3 Forensics and attribution
Genomics vaulted to prominence as a forensic tool during the investigation of the 2001 anthrax attacks on the United States postal system, and rapidly became established as a major tool in the emerging field of microbial forensics.12 The ultimate aim of using genomics as a forensic tool is to generate discriminatory (i.e. exclusive or inclusive) signatures that help narrow the range of potential suspects by establishing connections to the attack material and, equally important, eliminating those with no connection to the attack material. Genomic forensics has significant elements in common with the emerging discipline of genomic epidemiology and with genome-level studies of in vitro strain evolution, both of which rely on the ability to generate large numbers of high-quality signatures to enable the tracing of strain lineages.13–17 Because it must be defensible in court, information generated by genomic forensics will also be required to meet the standards of admissibility of scientific evidence known as the Daubert standard. Briefly summarized, these rules stipulate that scientific techniques be testable, be subject to peer review and publication, have known error rates, established standards, and be generally accepted by the scientific community. The increasing prevalence of WGS in the scientific community to address epidemiological questions and the proliferation of peer-reviewed papers in this field are promising, but to date no general laboratory standards or accreditation programs have been established for forensic genomics laboratories.
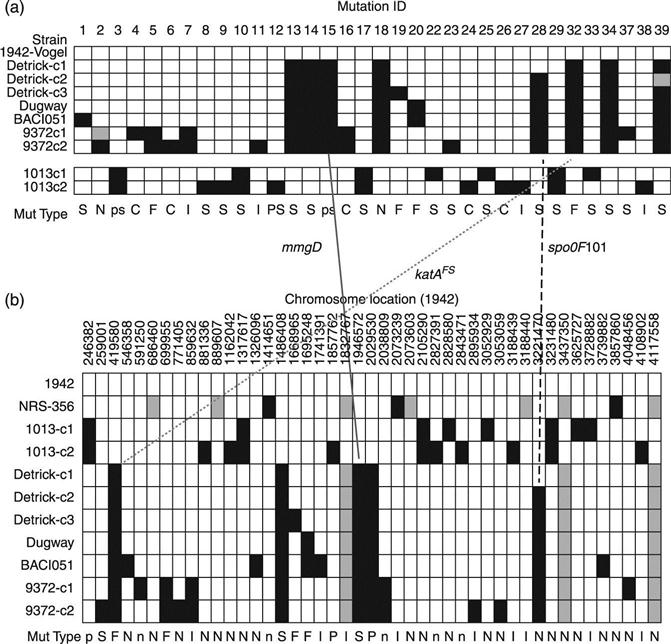
The forensic genomic investigation of the attack materials followed the discovery of colony morphology variants in the spore preparations derived from the letters; each variant was completely sequenced and the genetic variants characterized.18 The unique mutations present in each of five variants formed the basis for discriminatory PCR assays that were utilized to establish the origin of the spore preparations and exclude other potential sources.
Current methods
The genomics conducted for Amerithrax investigation followed then-conventional sequencing protocols based on capillary electrophoresis. Hence, the process was labor-intensive and very time-consuming, but produced what was then considered ‘gold standard’ information. Even with the long Sanger-based reads, one of the mutations underlying Morphotype A was not discovered until late in the process when the assemblies were examined for potential assembly conflicts, which revealed a short amplification.18
During the course of the investigation, the next generation of sequencing technologies emerged and found a place in laboratories across the world. In particular, rapid, short-read sequencing technologies could be brought to bear on the forensic samples. While the mutation that caused Morphotype A was almost missed by conventional sequencing, the duplication was very apparent as a spike in genome coverage (indicative of a structural variation or copy number variation) when next-generation (Sequencing by Oligonucleotide Ligation and Detection (SOLiD)) short-read sequencing was employed; furthermore, all of the other SNPs that had previously been characterized in the other morphotypes were readily identified in the SOLiD data sets.19 The rapidity and precision with which the newer technologies could operate were apparent, and subsequent work by members of our consortium has demonstrated the ability to perform such work from purified DNA in as little as 36 h using the 454 platform.3 Likewise, improvements in bioinformatics analysis tools make the identification of structural variations more automated.
Modern Methods and Approaches
More recently, our consortium conducted a retrospective genomic analysis of the Bacillus atrophaeus var. globigii (BG, a non-pathogenic surrogate for anthrax, vide infra) lineage that shared many of the characteristics of a forensic investigation,20 including differentiation based on non-genomic traits (e.g. colony morphology), source tracing, strain ‘matching’ and signature identification. In that study we retraced the ‘military’ lineage of BG using a combination of 454 and conventional sequencing and finishing, and, based on circumstantial open-source publications from the period,21 laboratory-verified phenotypes, and the propagation of genomic signatures over decades, were able to establish that the BW workers at Camp Detrick had deliberately selected a hypersporulating strain for their large-scale growths. Thus, our study not only established where the strains in use today had originated, but was able to assign the ‘intent’ behind the use of a given strain.
In the analysis of evidence, data with different levels of confidence can support different stages of the investigation, from lead generation through prosecution. Because the Amerithrax case was closed due to the death of the primary suspect, the data standards of the genomic investigation were not tested in court. Nevertheless, some basic principles can be stated that were derived from the Amerithrax work and from our own subsequent study.
1. Complement to other forensic techniques – Genomic analysis would not stand by itself in a microbial forensics investigation, but would be complemented by other techniques.22,23
2. Use case for genomic analysis – Like ballistics or fingerprint analysis, genomic analysis must provide information that can be used to match the evidentiary materials to a data base of reference materials.
3. Confirmation of motive – Incertain cases (e.g. the discovery of hypersporulating BG), the discovery of mutations can provide evidence that reinforces the attribution not only of source, but of the motive behind the selection of a particular variant.
4. Requirements for finishing – Fully finished sequences for all materials may not be required for the ‘lead generation’ stage. Draft sequence (or even raw data), provided that both reference and tester data sets exhibit high consensus, quality and coverage, can generate the discriminatory signatures that support assay development for inclusion and exclusion of evidentiary materials (Fig. 1.1). Given the speed at which WGS can now be conducted, it is expected that discriminatory signatures will be available much earlier in the course of an investigation, with subsequent finishing efforts intended to ‘clean up’ the data for use in court.
Future issues
It is important to note that, in the 2001 Amerithrax case, reference samples collected by the FBI from anthrax laboratories were screened by PCR for the presence of the discriminatory mutations. This process would be repeated today, but, given the likelihood that additional mutations and/or variants might be present in those samples, it is also conceivable that each sample would be subjected to WGS (and possibly deep sequencing; see Section 1.1.5) to provide additional possible signatures. A thorough understanding of known error rates, at both the read-level and the assembly level, is necessary to assign probability and likelihood to any identified variation. These rates are largely platform dependent; thus, orthogonal sequencing technologies should be applied.
In addition to the genomic evidence provided by the attack agent itself, other nucleic acids may be present in a forensic sample that may allow attribution of evidentiary material to sources. Human DNA from the suspect may be present in trace quantities, as might potential DNA associated with growth media or the source’s location. These may be present in low abundance, requiring amplification or enrichment from the major component. The ability to match the former through conventional forensic techniques is not guaranteed, and methods for ‘matching’ the latter to reference data bases are not established.
Most critically. for WGS to become a significant forensic technique, it must be proven to be able to survive scrutiny in a court of law. As such, information generated by genomic forensics will be required to meet the standards ofadmissibility of scientific evidence known as the Daubert standard described earlier. As the use of these techniques proliferates, a unified standard for accreditation of forensic genomics laboratories should be established.
1.1.4 Combined microbiology and next-generation sequencing (NGS) applications in bioforensics
NGS analysis was critical for the discovery and exploitation of the molecular targets used in the Amerithrax forensics application; however, just as importantwas the upfront microbiology responsible for elucidation of the independent morphotypes.1 Current trends toward greater reliance on sequencing and bioinformatic data are reaching fruition and providing insights that once were the purview of classic microbiology and biochemistry.24 As discussed later in this chapter, complete linkage of genotype to phenotype is a necessary aspect toward making this a full-blown reality. In the interim, and given the Amerithrax example, classic characterization methods remain relevant and can help frame the biological questions that sequencing-based technologies are uniquely situated to solve.
Sample testing
Regardless of the outcome from Amerithrax, substantial investment was made in the processing of samples both in the forensic aspect, analyzing sample for the correct combination of morphotypes,1 and in the less publicized direct detection of anthrax in samples from across the country. Indeed, in 2001, we received over 30 000 independent environmental, clinical and reference samples to be tested for the presence of Bacillus anthracis. This operation was a 65-person, 24 h a day, 7 days a week operation effort over the course of 8 months, employing real-time PCR, immunological detection and classic microbiology for pathogen detection. In this context, real-time PCR was the workhorse upfront detection vehicle that was used to determine whether the latter two methods were applied. Specifically, duplicate positive results from the same nucleic acid sample triggered a further evaluation and confirmation of anthrax; of the ~30 000, only 219 returned positive results. As sequencing technology moves further toward providing a solution in terms of multiplex and parallelism, response to biological events such as these could be reduced in scope while still providing a similar level of confidence in the identification at issue. Key aspects, some of which will be addressed later in this chapter, toward making sequencing a viable detection method include specificity, time-to-answer, reference standards and adequate analysis algorithms.
1.1.5 Deep sequencing to look at genomic variations in microbial populations
Genomic variations, both adaptive and neutral, underlie the evolution of all organisms. In microbes, several factors may contribute to rapid appearance of variants in populations: high population densities, rapid replication rates and high error rates of replication enzymes. However, these variants are present at relatively low frequencies. For instance, the observed frequency of any particular mutant in Escherichia coli is usually <10–5. In cases where the new phenotype can be selected for, even very infrequent spontaneous mutants can be detected. Often, however, it is not possible to select for a particular phenotype, and in suchcases bacteria must be screened for it. Microbial mutation frequencies vary by loci and may be higher than average in some hypervariable regions, in certain mutant backgrounds and in viral populations. To the best of our knowledge, direct population-based genotyping by WGS has not been performed for bacterial strains. However, due to their smaller genome size as compared with bacteria and eukaryotes, studies have been performed to detect variants in viral populations.
We and others have used NGS technologies, such as 454-based pyrosequencing, for genetic fingerprinting of ‘purified’ variants isolated from a population.3,20,25,26 From these experiments, we have gained some understanding of the power of NGS technologies to detect variations from draft sequences (see later section), frequencies of true positive/false positive variations and the limitations of the WGS approach for variation detection. It is rather easy to generate a unique genetic fingerprint of a ‘purified’ variant; however, this work has not been performed at the population level (population fingerprinting) to detect variants present as a minor component of a mixed bacterial population.
Genotyping viral populations for detection of rare variants
One of the best examples of rare variant detection in viral populations pertains to the detection of rare drug-resistant human immunodeficiency virus type 1 (HIV-1) mutants circulating in viral populations within patients.27,28 Direct sequencing after PCR amplification cannot detect poorly represented variants (<20%) in the heterogeneous virus population existing in a patient’s circulation. In a study aimed at determining the abilities of ten technologies to detect and quantify a common HIV-1 mutant (possessing a K103N substitution in the reverse transcriptase (RT) gene) using a blinded test panel containing mutant–wild-type mixtures ranging from 0.01% to 100% mutant, two technologies, allele-specific reverse transcriptase PCR and a Ty1HRT yeast system, could quantify the mutant down to 0.1–0.4%. Pyrosequencing and single genome sequencing (SGS) (45 sequences/mixture) had intermediate detection limits of 2% mutant but were not quantitative below 10% (Table 1 in Halvas et al.27).
Direct population sequencing and reverse hybridization (line probe assay (LiPA))-based methods are the most common methods for detecting hepatitis B virus (HBV) drug resistance mutations, although only mutations present in viral quasi-species with a prevalence of >20% can be detected by sequencing, and only known mutations present in ≥5% of the population can be detected by LiPA. Massively parallel ultra-deep pyrosequencing (UDPS; GS FLX platform) was used in a study to analyze HBV quasi-species in RT and hepatitis B S antigen (HBsAg), the results indicating that UDPS has a relative sensitivity much higher than both direct sequencing and LiPA and that UDPS results are quantitative, allowing establishment of the relative frequency of both known mutations and novel substitutions.29
Rare variant detection in pooled DNA samples
Targeted massively parallel sequencing (MPS of specific regions that are first PCR-amplified) has been evaluated for rare variant detection and allele frequency estimation in pooled DNA samples using Illumina’s sequencing technology. All expected alleles at a frequency of 1% and higher were reliably detected, plus the majority of singletons (0.6% allele frequency).30 Thus, variant detection by MPS has so far been demonstrated in viruses and localized genetic loci in human genomes. Coverage depth of a DNA sample at a locus in a genome is primarily determined by the size of the resequenced region and capacity of the sequencing instrument. For example, with a total sequence yield of 1 Gb, each position of a 10-kb region can be read on average approximately 100 000 times. Multiple genomic regions, and/or DNA from multiple individuals, might be pooled without losing statistical power to detect unique variants. For detection of rare alleles, e.g. in DNA from viruses, tumors or pooled DNA, 0.02-1% has been reported as minimal detectable allele frequency (Out et al.30 and references therein).
Assuming the likelihood of detection of a rare variant by WGS follows a Poisson distribution, with 50× coverage of a genome the probability of detecting a rare variant present in 1/50, 1/100, 1/500, 1/5000 of a population is 0.63, 0.39, 0.095 and 0.010, respectively. Conversely, the required depth of coverage for the detection of a rare variant present in 1/50, 1/100, 1/500, 1/5000 of a population and at a detection probability of 90% is 115×, 230×, 1151×, and 11 513×, respectively. The average total sequence output obtained from a 454 Titanium pyrosequencing run is ~300–350 Mb, whereas a SOLiD v4hq run can produce up to 300 Gb and Illumina HiSeq v3 systems up to 600 Gb of total sequence data per run. For small genomes such as viruses or targeted genomic regions of small sizes (10 kb), the required depth of coverage is achievable with the 454 sequencer, but it is conceivable that SOLiD output size might be required for detecting bacterial variants (assuming an average genome size of 5 Mb and 60 Gb output/run, ~60 000× coverage is possible). However, these statistical predictions have not been tested empirically, and another factor that needs to be taken into account in these determinations is the error rate of the sequencing platform. Few published studies utilizing SOLiD sequencing to detect variations in human genome, and only one in B. anthracis, exist.19,31,32
1.1.6 Policy drivers for NGS
In light of the 2001 anthrax attacks, the United States government has worked to develop a robust whole-of-government approach to detecting and responding to biological outbreak events. The Departments of Defense, Homeland Security, and Health and Human Services have been tasked by a series of Presidential Directives to lead development and coordination of the government’s response capabilities. Neither policy documents from the Executive Office of the President (EOP) norany departments specifically call out nucleic acid sequencing as a requisite technology to enable the response capabilities; however, one can ascertain that this technology must play a larger and larger role in fulfilling the President’s directives to bolster the country’s response capabilities.
One of the more important directives to emerge from the EOP is Homeland Security Presidential Directive 21 (HSPD-21).33 This document calls for the US to plan and enable rapid response to a biological event, including the capacity to rapidly identify and characterize the threat. The document calls for the US to ‘strengthen laboratory diagnostic capabilities and capacity in order to recognize potential threats as early as possible’. In the case of engineered biological weapons, rationally designed detection methods (polymerase chain reaction-, immunological-, or array-based) will likely not be successful in fully identifying or characterizing a threat, pointing to a critical role for unbiased high-throughput sequencing. Furthermore, the mandate to ‘provide early warning and ongoing characterization of disease outbreaks in near real-time’ indicates that direct identification out of complex samples is far preferable to first culturing or propagating a threat prior to employing an identification technology. In support, former US Secretary of the Navy Richard Danzig has commented that future biological weapon attack samples are not likely to be as pristine as the 2001 anthrax samples34 Furthermore, rapid identification and attribution from a first wave of complex samples will be critical to preventing ‘reload’ or a second wave of attacks by perpetrators.
Based on the foundation laid by HSPD-21, the National Strategy for Countering Biothreats was released in 2009 by the National Security Council.35 Importantly, this document expanded upon HSPD-21 by calling for the promotion of global health security through building a ‘global capacity for disease surveillance, detection, diagnosis, and reporting’ for both natural disease outbreaks and biological weapon attacks. Specifically, the document states that ‘rapid detection and containment of, and response to, serious infectious disease outbreaks— whether of natural, accidental, or deliberate origin—advances both the health of populations and the security interests of States’. The following year, the 2010 National Security Strategy stated that timely and accurate insights on current and emerging biothreat risks were a key enabler to increasing national security36 Overall, the linking of public health and biodefense mission spaces has been a critical driver to whole-of-government approaches to rapid identification and characterization capabilities. In support, the Chairman of the Joint Chiefs of Staff called for the integration of biodefense capabilities. including that ‘the Department of Defense and Department of Homeland Security cooperate on … biological initiatives to maximize complementary research, development, test, and evaluation (RDT&E) and acquisition efforts and to minimize duplicative efforts and enhance technical cooperation’.37 While these documents do not explicitly call for a sequencing-based identification approach, the linking of public health and biodefense mission spaces greatly increases the number of targets which thenation must be prepared to identify, characterize, and respond. Unbiased high-throughput sequencing will likely play an increasingly predominant role in supporting the US government’s capacity to identify and characterize a diverse panel of natural or weaponized biological agents. The time-sensitive nature of response requires that this technology will need to be employed to identify biological agents directly from a variety of complex matrices.
Specific to biological weapon attacks is the necessity to attribute the attack to state or non-state actors, enabling an international military or diplomatic response. Attribution for the 2001 anthrax attacks took years, and, in fact, the case was not formally closed by the Department of Justice until 2010.23 The National Strategy for Countering Biothreats and the 2010 National Security Strategy both explicitly call for an expanded capability to attribute biological attacks to perpetrators, specifically by advancing the field of microbial forensics. In parallel, the National Research and Development Strategy for Microbial Forensics was released in 2009.38 This document specifically called out the need to ‘continue to support the development of rapid and cost-effective high-throughput sequencing and closure technologies that can be used to generate high confidence whole genome sequence data and genetic variation data for any known and unknown microorganism’. Specifically, high-throughput sequencing must be developed as an attributional tool to enable a much faster response capability than current state-of-the-art technologies.
1.2 Next-generation sequencing (NGS) technologies and the sequencing landscape
In the relatively short period of time since the first DNA sequencing occurred, sequencing technology has experienced dramatic increases in productivity coupled with cost decreases. While some of these advances are associated with automation and improvement of reagents, the greatest impacts have come from development of fundamentally different technical approaches to sequence determination. However, with diverse technical approaches come significant performance differences. The evolution of sequencing has not been linear.
1.2.1 Historical perspective of sequencing
First-generation sequencing technologies
In 1953 Watson and Crick first described the makeup of DNA,39 the language of genetics. However it was not until 1975 that a viable method of decoding that language became available.40 Other early methods, such as Maxam-Gilbert,41 were devised but were generally less robust and involved more hazardous chemical reagents. In the end, chain-termination methods42 won out and built the landscape of genomic sequencing. From this came the first complete genome sequence, that of the bacteriophage ϕX174.43
First described by Leroy Hood’s laboratory in 198644 and subsequently commercialized by Applied Biosystems in 1987, the first automated slab-gel chain-termination sequencer, the ABI 370, drastically changed how genomic sequencing was approached. The early 1990s found the United States National Institutes of Health (NIH) funding genome sequences of the first few cellular organisms (such as Mycoplasma capricolum, Caenorhabditis elegans, Escherichia coli and Saccharomyces cerevisiae) as well as J Craig Venter group’s first attempts to sequence expressed sequence tags from the human genome,45 at a cost of 75 cents per base.
In 1995 Applied Biosystems released the first commercially available capillary sequencing apparatus to automate the chain-termination chemistry, known as the ABI Prism 310 genetic analyzer. During the same year, The Institute for Genomic Research (TIGR) released the first complete genome of a free-living cellular organism generated by whole genome shotgun sequencing, the 1.8 Mb genome of Haemophilus influenzae.46 This breakthrough led to an explosion of microbial genomes as mapping to references became unnecessary.
Sequencing by ligation
The first of the ‘next-generation’ sequencing technologies, pyrosequencing, was described in the mid-to late1980s,47,48 but at the time was plagued with inefficiencies. The mentor–student team of Pål Nyrén and Mostafa Ronaghi overcame many of these issues in the late 1990s.49–51 This technological breakthrough resulted in the formation by Dr Jonathan Rothberg of 454 Corporation, originally a subsidiary of CuraGen Corporation but acquired by Roche Life Sciences in 2007. 454 Life Sciences began selling a parallelized pyrosequencing platform in 2005,52 beginning the rapid decline in sequencing costs.
The process works by releasing luciferase-bound pyrophosphate with the incorporation of each nucleotide, so that the light emitted is proportional to the number of bases added. This makes the process robust to substitution errors but sensitive to homopolymers (six or more of the same nucleotide in a row), as the light differences become difficult for the instrumentation to differentiate. The early FLX chemistry allowed 100 bp reads, the Titanium chemistry 200–400 bp reads, and now, with availability of the FLX+, reads of up to 1000 bp are possible.53 Of course, with longer reads genomic assembly becomes easier; however, the 454 technologies are the most expensive ‘next-generation’ sequences to produce and will no longer be commercially available after 2015, as the manufacturer has released plans to cease production.
Sequencing by synthesis – Illumina
The process first commercialized by Solexa in 2006, now known as Illumina, adds nucleotides in a controlled manner to distinct DNA templates and imaged after each incorporation.54 The process is dependent on reversible terminators55 and the data is produced in four distinct color channels so that one image is taken at the end of each cycle.56
Initial read lengths were 20–35 nt long; however, the current technologies offered routinely generate reads between 100 and 250 nt in length. Known for high read counts and low cost, recent releases in chemistry have reduced issues with sequencing through high GC areas and miscalls after a GGC motif. Many applications where high numbers of reads are important (SNP detection, metagenomic analysis, etc.) have benefited from this sequencing technology.
Sequencing by hybridization, SOLiD
The basic concept of hybridization sequencing is similar to that imposed with microarrays, and yields a technology that is highly efficient at sequencing short stretches of DNA (30–50 nt). Sequencing with microarrays has been around for a while,57 known to produce high-quality resequencing of known regions.58,59 Essentially, the aim is to locate regions of specific length through hybridization to synthesized oligonucleotide probes.54 The method is limited in read length, as longer probes have reduced ability to select for ‘perfect’ matches instead of single nucleotide discrepancies, quickly resulting in high error rates.54,60
Sequencing by hybridization is best known as the SOLiD system, available commercially since 2007 through Applied Biosystems. This platform has become highly popular in resequencing efforts where high accuracy is of the greatest importance.31,61–63 The SOLiD implementation utilizes a two-base encoding, resulting in 99.99% accuracy according to the manufacturer. The process is based on primers hybridizing to an adapter sequence within the library, and a set of four fluorescently labeled two-base probes vies to ligate onto the sequencing primer.
Single molecule sequencing – Pacific Biosciences
By sequencing single molecules, with read lengths limited by the lifetime of active polymerase, PacBio has attempted to change how we see genomic sequencing. The company was founded in 2004, based on work done at Cornell University that combined phototonics, semi-conductors and biotechnology.64,65 The process works by anchoring a single polymerase enzyme to the base of each nanopore, called a zero-mode waveguide due to the photonics involved, and video recording the incorporation of nucleotides based on the release of fluorescent dyes. To date, the instrument has only been available to a limited number of customers; however, with the extended read lengths, several publications have ensued in a short space of time.6,66–69
Single molecule sequencing – Helicos
Referred to by Helicos as the first true single molecule sequencing (tSMS) technology, this method utilizes a ‘virtual terminator’ to prevent the incorporation of more than a single nucleotide per cycle while imaging individual (singlestranded, poly-A-tagged) DNA molecules with known oligonucleotides and fluorescently labeled nucleotides.70 The company Helicos was founded in 200371 but faced challenges that delayed the platform release; however, they have pushed forward to release a series of promising peer-reviewed publications.72–74 This platform sequences single molecules, thereby eliminating amplification bias, and boasts a simplified, scalable library preparation. The reads are generally short, ~25 nt,73 but the sequencing is biased toward longer templates, which may be an issue with transcriptomic applications.74
Single molecule nanopore sequencing – Oxford
Oxford Nanopore Technologies (ONT) is expected to release both a full-sized, scalable sequencer (termed GridION) and a palm-sized sequencer (MiniION) utilizing alpha-hemolysin nanopores, expected to become commercially available late 2013/early 2014.75,76 The system will read the electrical disturbance of each base that passes through the pores – potentially allowing an experiment or to designate not the amount of run time but the amount of data needed, and stop sequencing at that point. The actual release date is still unspecified, but the company indicates that the GridION will be ‘less than other commercially available systems on an absolute level and that the cost per base will also be lower than other systems’, while the MiniION will cost less than $900 and run directly on a personal computer.
Semi-conductor sequencing – IonTorrent
The underlying theory behind the IonTorrent sequencer is simple and elegant: by detecting the intrinsic pH change (release of hydrogen ion) upon incorporation of nucleotides and only providing one species of nucleotide at a time, each polymerization event can be recorded in a digital format.77,78 Homopolymer repeats generate pH differences in exact proportion to the number of bases incorporated. In a semi-conductor chip of just under 1 cm2, microwells each contain a single ssDNA template molecule and a single DNA polymerase enzyme.78–80 Recent reports indicate an accuracy of 99.6% based on 50 nt reads and 100 Mb per run, with an accuracy for homopolymers of five repeats at 98%.81 Still, the platform is sensitive to long homopolymer stretches and may have difficulty determining the number of bases in a long repeat, similarly to 454 pyrosequencing.82
This technology, without modified nucleotides or optics, was licensed from DNA Electronics Ltd and developed by Ion Torrent Systems Inc. in 2010.78 For the semi-conductor sequencer sold through Applied Biosystems, the noted benefitsof ion semi-conductor sequencing are rapid sequencing speed, low upfront investment and low operating costs – with the cost reductions driven by the lack of optical measurements and modified nucleotides.80 The rate of sequencing is limited by the time it takes for substrate nucleotides to cycle through the system (described by the manufacturer as 4 s per cycle, so that each run takes approximately 1 h with 100–200 base reads generated).80 Two major drawbacks to use of this platform, particularly in the replacement of 454 pyrosequencing, are the shorter read lengths and the low throughput. The intention of the manufacturer is to increase the density of microwells on the chip and thereby increase throughput over time.78
Comparison of commercially available platforms
The cost of sequencing DNA has dropped dramatically over time; statistics kept by the National Human Genome Research Institute of the National Institutes of Health (NHGRI-NIH) show that the cost of sequencing a megabase of DNA is falling more quickly than predicted by Moore’s Law.83
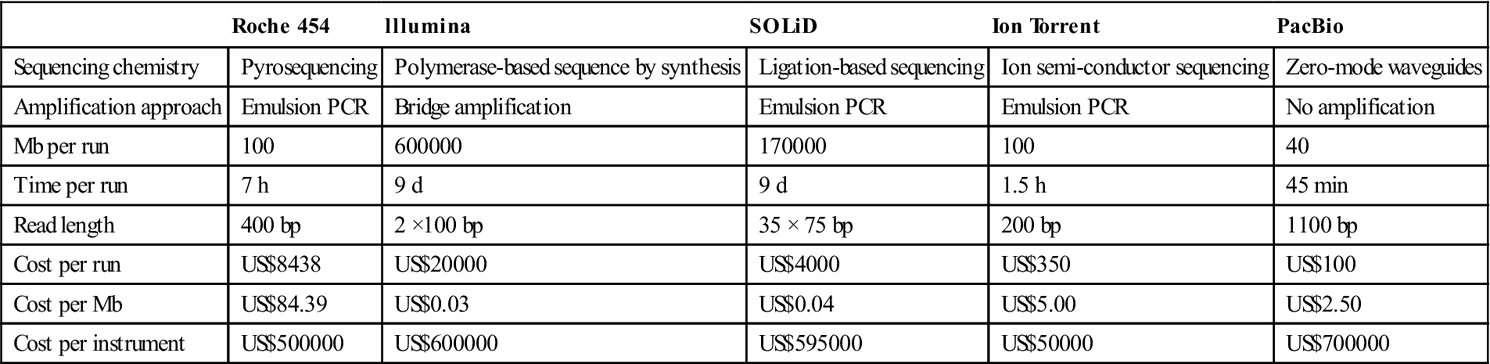
A comparison of commonly used commercially available second and third-generation sequencing platforms is shown in Table 1.1, and suggests that for the foreseeable future no single chemistry will be able to meet the needs of all applications; rather, a blended approach will be needed for WGS and individual platforms will likely outperform for singular uses.
Table 1.1
Comparing metrics and performance of next-generation DNA sequencers, data collected from Shendure and Ji84 as well as the respective manufacturer publications
| Roche 454 | lllumina | SOLiD | Ion Torrent | PacBio | |
| Sequencing chemistry | Pyrosequencing | Polymerase-based sequence by synthesis | Ligation-based sequencing | Ion semi-conductor sequencing | Zero-mode waveguides |
| Amplification approach | Emulsion PCR | Bridge amplification | Emulsion PCR | Emulsion PCR | No amplification |
| Mb per run | 100 | 600000 | 170000 | 100 | 40 |
| Time per run | 7 h | 9 d | 9 d | 1.5 h | 45 min |
| Read length | 400 bp | 2 ×100 bp | 35 × 75 bp | 200 bp | 1100 bp |
| Cost per run | US$8438 | US$20000 | US$4000 | US$350 | US$100 |
| Cost per Mb | US$84.39 | US$0.03 | US$0.04 | US$5.00 | US$2.50 |
| Cost per instrument | US$500000 | US$600000 | US$595000 | US$50000 | US$700000 |
1.2.2 Metagenomics
The term ‘metagenomics’, first coined by Handelsman et al. in 1998,84 refers to the study of community genomics, as opposed to traditional microbiology in which isolate genomes are sequenced and analyzed as distinct entities. The idea is to study a collection of genes sequenced from an environment. In other words, metagenomics is ‘the application of modern genomics techniques to the study of communities of microbial organisms directly in their natural environments, bypassing the need for isolation and lab cultivation of individual species’.85 These efforts have shown that the great majority of biodiversity in the microbial world was missed by traditional cultivation-based methods.86
Early efforts, led by the Pace group, utilized PCR of 16S ribosomal RNA sequences to explore microbial diversity outside the biases induced by culturing.86,87 As all bacterial and archaeal cells contain 16S ribosomal RNA, this highly conserved non-coding gene was ideal for identification of community members.86 Early on, before they were technologically possible, the Pace group proposed implementation of true metagenomic studies.87 Initial reports of cloning bulk DNA isolated from environmental samples were published in 1991,88 suggesting that interactions among environmental microbes are much more complex than previously noted among laboratory cultures, despite a focus on non-protein-coding, highly conserved genes. Comparisons of functional genes from grasses were soon after reported,89 as well as studies on the phylogeny of environmental microbial communities based on 16S.90 Needless to say, with a decade of existence under its belt, by the mid-1990s metagenomics had become an exploding field of research.
1.2.3 Technological innovations on the horizon for NGS
Anticipating the ‘next big thing’ in any technological field is always a risky business; however, there are several groups bidding to lead the next revolution in genomic sequencing. These endeavors include expanding on currently commercialized technologies (such as pyrosequencing, ligation and hybridization), exploring new methods for single molecule detection, often utilizing nanopores, and a few completely new ideas, such as electron microscopy and nanowires.
Pyrosequencing
Perhaps not advances in terms of increasing read length, read count or reducing run time, but definitely in terms of reducing the overall cost and thereby helping to make the small laboratory sequencer more readily available, are systems such as those put forward by Intelligent BioSystems (commercialized by AzcoBiotech) and LaserGen.91–93 Both companies are still in the pre-commercialization phase, but have issued press releases suggesting that beta-testing will begin by early 2014.
Another alternative, based on emulsion PCR, is that from GnuBio,94,95 which plans to produce a fully integrated sequencer, including genome region selection, amplification, sequencing and analysis – with a forecast price of US$50 000 – to be released by 2014 (personal communication). The system has been released to beta-testers and so appears to be on track for a 2014 commercial release. This sequencer and many like it are aimed at the clinical diagnostics market – attempting to break the barriers of cost and preparation to bring sequencing into routine human diagnostics.
Ligation
Made famous by Solexa (now Illumina), there are now other competitors offering similar chemistries. Most of those competitors appear to have gone out of business or underground, but one remains: the Polonator. Designed by the Church group at Harvard University, this system generates reads up to 30 bp long using emulsion PCR interrogated by sequential ligation steps.96–98 Recently the Polonator system, written all in open source and designed for use with non-vendor-specific reagents, became commercially available through Danaher.
Hybridization
Made possible by an increasingly large number of reference genomes, sequencing by hybridization is a fast, accurate and inexpensive way to resequence known organisms. It has long been dominated by well-known corporations such as Affymetrix, but newcomers such as Callida Genomics99,100 and NABsys are starting to take hold.101 Perhaps the most successful use of this sequencing type has been by Complete Genomics, a sequencing service dedicated to human resequencing and working to bring individual human sequence data into clinical practice.102
Microscopy
An idea that seems to be slowly taking off is one in which the atoms of DNA bases are stained and then the sequence read using microscopy. Halcyon Molecular is trying to read the code using electron microscopy,103 while ZA Genetics is working with electron microscopic methods of reading the sequence.104,105 Both companies are keeping details of their methodology quiet at this time.
Nanopores
Nanopores seem to have generated their own magic, with several companies rushing to come out with single molecule sequencing systems that take advantage of nanopores. Many of these technologies are being referred to as fourth-generation sequencing, although they all seem to incorporate various previous ideas into a nanopore framework, allowing an increase in parallelization of single molecule sequencing. Oxford Nanopore was discussed in the previous section, so here we will focus on other nanopore-based technologies.
BioNanomatrix, based out of Philadelphia, PA, quietly commercially released its instrument in late 2012, based on sequence motif labeling, using a restriction enzyme to nick a single strand at known recognition sites, and incorporating fluorescent dyes at those nicked sites. The DNA will be read into barcodes based on motifs defined by placement of and distance between those dyes. The imaging will utilize fluorescence microscopy as the DNA molecules are passed through long and narrow channels on a silicon wafer.
A similar technology has been proposed by PathoGenetix of Woburn, MA (utilizing restriction enzymes to nick ssDNA) but with a shorter probe. The focus of this platform is speed, for clinical applications to move from sample to identification in approximately 3 h.106
Finally, NobelGen is developing a method to adhere fluorescently tagged probes to single DNA molecules, passing them through nanopores on a 96-well plate.107 The plate design will make it more compatible with current robotics, and each well is said to contain sufficient nanopores to sequence a full human genome. The described sample preparation takes approximately 5 h, but the run time is only 30 min.
Single molecule detection
Current commercial single molecule sequencing technologies include only the PacBio RS; however, even outside the nanopore platforms, more appear to be on their way. Base4Innovation appears to be close to releasing a commercial instrument, although the chemistry behind their platform is closely guarded. Cracker, a small start-up in Hsinchu, Taiwan, appears to be working to produce ‘SMRT on a chip’, a single molecule sequencing method with the polymerase affixed to a nanowell and fluorescent dye released with each incorporated base.108
Summary
There are a multitude of companies with either sequencing technologies or services anticipated on the market in the near future. Their approaches include furthering chain-termination (Sanger) sequencing, pyrosequencing, sequencing by hybridization, sequencing by ligation, etc. No single platform can provide for all the needs of the scientific community: some are more costly on a per-read or per-base basis, others require long preparation or run times, and some (especially those based on single molecule rather than consensus sequencing) have intrinsically high error rates. Often when commercial suppliers estimate costs they include only the reagents required for sample preparation, with little to no consideration of the labor hours involved to complete those steps. Many groups have decided that the best way to utilize these differing sequencing technologies is to use a blended approach – trying to maximize depth of coverage, read length (or read span in terms of long insert paired-end libraries) and cost. This blended approach does have its drawbacks, largely the upfront cost of purchasing and maintaining multiple sequencing platforms and the computational ability to merge the data sets together in a logical and useful fashion.
1.2.4 The data analysis challenge
A major challenge with utilizing sequencing as either a clinical diagnostic tool or a means of deriving information from pathogens in a rapid response situation in the biodefense context is the ability to process and handle the vast quantities of data generated by each of the sequencing platforms and to turn that information into results that can be acted upon by either a practicing clinician or a policy maker. In the case of bioterrorism, the objective will be to generate investigational leads as rapidly as possible to allow the process of attribution to occur, with the hope that any suspects might be apprehended quickly. In the case of a true biological warfare scenario, attribution would help determine how a nation should respond. For a clinician, the goal would be to diagnose the disease, describe the pathogen, and determine how and with what to treat an infected patient. In all three cases, robust data handling, management and analysis procedures are required to provide the information needed. The process should be automated to run efficiently and effectively; also, the reports generated should be easily understandable. Wherever possible, analytical procedures should highlight how the identified organism differs from previously characterized strains. Ideally, centers performing in such rapid response scenarios should have information flows pipelined, such that as much analysis as possible is performed automatically with outputs readily available to the analyst. At a minimum, a pipeline for rapid response should include the following modules:
1. Automated data quality assessment and assembly.
2. Identification of nearest neighbor to the strain level – BLAST of reads, contigs or using k-mer based approaches.109
3. Variant profiling – should include SNPs, indels. This process is usually based on a reference-mediated assembly using read-alignment algorithms,110,111 although other reference-independent methods are available.109
4. Identification of ‘accessory’ DNA elements previously unobserved for those strains. These would include deliberately introduced or naturally acquired genetic elements. Examples would be the pESBL plasmid from the E. coli O104:H4 outbreaks or a genetically engineered Yersinia pestis strain containing an antibiotic resistance cassette in the pCD1 virulence plasmid.112,113
5. Inventory of virulence factors and drug resistance determinants. This functionality requires the existence of data bases containing the nucleotide/protein sequences of the respective elements. Several such data bases exist, although variable in definitions and curation status.114–117 The Chinese VFDB is the most complete, best annotated and best curated117 of current virulence factor databases.
All of these steps require high-quality reference sequences and/or well-curated, annotated data bases with functional descriptions of each of the various genes. Conformity in annotation between each reference data base entry, particularly harmonization of annotations across reference sequences in GenBank, would be highly desired (see Section 1.4.5 on annotation).
1.3 Characterization of known pathogens
1.3.1 Traditional methods of characterizing known pathogens
NGS and sequencing-based analyses have dramatically transformed the landscape of everything from diagnostics to evolutionary biology.118 The need to characterize pathogens and to define the questions NGS can help solve remains. Currently, classic methods for nucleic acid characterization have been applied to guide more recent sequence-based efforts. However, as sequencing costs continue to decline the trend may reverse sooner than later. Overall, these classic methods for strain/speciesdifferentiation have their route in both genotypic and phenotypic characterization. Current phenotypic characterization tools evolved from classic microbiological techniques for bacterial/viral identification. Methods such as metabolic profiling, Gram staining, morphology and fatty acid typing evolved from classic biochemistry analytics to the current Omnilog, VITEK, electron microscopy and MIDI instruments used today. Similarly, the advent of Sanger sequencing allowed discrimination and identification based on nucleic acid signatures or genotype.119
Bacterial nucleic acid characterization
Nucleic acid characterization/sequencing remains at the core of our current understanding of bacterial evolution and strain differentiation.119 Numerous sequence-based techniques have evolved since the advent of Sanger sequencing; among the most relevant for characterization and bacterial identification/phylogeny are pulse field gel electrophoresis (PFGE), 16S, multi-locus sequence typing (MLST) and multi-locus variable nucleotide tandem repeat (VNTR) analysis (MLVA).120 These techniques arose as methods to better understand phenotypic patterns observed on the macroscopic scale. One of the first molecular techniques developed to better characterize strain-specific divergence is PFGE.
PFGE is not actually a sequence-based method in that no direct sequencing takes place. Instead, the method capitalizes on nucleotide variation at random restriction endonuclease sites within a query genome to develop differential patterns of genomic DNA fragments when run on gel electrophoresis.121 This method was initially and continues to be implemented in strain differentiation and is the upfront basis for investigation in the genomic arena.122 However, this method is prohibitive for identification of novel or emerging bacterial pathogens, as direct knowledge of the query organism is required for comparison with reference standards. One method that seeks to mitigate this issue and increase the resolution of strain differentiation is optical mapping. This method, while similar in nature to PFGE, allows generation of a genome-wide restriction pattern, and thereby interrogates the entire query genome as opposed to select fragments. Comparison of this method with a reference database might allow the identification of an unknown etiologic agent. The current limitation with optimized enzymes required to generate the optical map again necessitates some a priori knowledge of the infectious agent and its nucleic acid before application.
In contrast to PFGE, characterization of 16S RNA is a direct sequencing application that has been recognized as a useful target for phylogenetic studies since the 1960s.123–125 This gene is a structural part of the ribosomal complex, and as such has selective pressure to conserve sequences in certain functional areas while remaining selectively neutral in others.126,127 This dichotomy in selective pressure allows species-specific characterization, monitoring the changes and/or similarities in nucleotide composition between microorganisms at this genetic locus. One can infer the relatedness of those organisms, or phylogeny, based on similarity or dissimilarity across variable 16S regions.128 Given the unique phylogenetic qualities of this gene, it can also be used for direct identification of the bacterium. Current methods for analyzing this genetic locus span from Sanger sequencing to mass spectrometry of 16S amplicons using consensus primers.129–131
MLST is similar to 16S as a sequence-based typing method; however, the complexity of the analysis is increased in that several distinct genetic loci are interrogated.132 Initially described in Neisseria by Maiden et al.,133 this method characterizes sequence data for several housekeeping genes within the queried genome. This is accomplished through PCR amplification of the desired genes with subsequent sequencing of the resulting amplicons. Cladistics and phylogenetic relationships are inferred across all queried organisms through sequence similarity across the interrogated loci. This method is ideal for characterization of diverse/non-clonal organisms, as housekeeping genes are typically more recalcitrant to polymorphisms and therefore are a good target for parsing phylogeny of evolutionarily distal organisms. For more clonal bacterial organisms, evaluation of highly variable, low-selective pressure sites are required to parse genetically similar isolates within a bacterial population.
Toward this end, MLVA was developed and initially described for the characterization of Bacillus anthracis ecology and epidemiology.134 MLVA exploits nucleotide repeats inherent to organisms’ genomes that fluctuate in copy number based on individual strains, VNTRs. Specifically, VNTR loci are amplified via consensus PCR with subsequent sequencing used to reveal the number of tandem repeats representative of the query. Because of the lack of selective pressure and inherent variability of these regions, these molecular markers are ideal for characterizing clonal bacteria such as many of the bio-warfare and recently emerged infectious disease agents.135,136
Viral nucleic acid characterization
Classic virology, similarly to bacteriology, relies on morphology and host range to characterize and differentiate individual species. Typical characterization efforts entail isolating the virus through tissue culture followed by morphological evaluation (electron microscopy) and sequencing portions of the viral genome. The advent of Sanger sequencing and PCR allowed a more detailed phylogeny and characterization resolution of viruses.137,138 Indeed, sequencing specific loci within viral genomes for SNP discovery and strain differentiation has been applied widely across the many viral groups.139,140 Similarly to bacterial sequence-based techniques, this type of characterization was dramatically influenced by the evolution of NGS with parallelism and multiplex capabilities allowing increasedthroughput. This NGS effect is even more acute for viruses compared with bacteria due to the relative size of the viral genome, most <100 Kb, and the ease with which NGS has allowed an enhanced throughput of full viral genome sequencing. While this has profoundly impacted viral phylogenetic architecture, a notable lack of standards has made cross-comparability of data sets problematic.
1.3.2 Genomic standards and viral characterization
Language and vernacular are one of the critical aspects of communication, and, in the genomics context, shared vocabulary results in an understanding of data set quality and completeness. Classification terms of ‘finished’ or ‘draft’141 were adequate in the Sanger era, when genomic data were prevalent but not overwhelming. The need for these classic standards to be redefined arose as data generation became more expeditious than the downstream analysis and the term ‘draft’ spanned a large spectrum of genomic coverage and completeness. Chain et al.141 made the first attempt at tackling this issue and breaking down the ‘draft’ and ‘finished’ statuses for bacterial and eukaryotic genomes into bins-based metrics of coverage and post-sequencing processes applied to the data set. Draft nomenclature was broken into ‘standard draft’, ‘high-quality draft’, ‘improved high-quality draft’ and ‘annotation-directed improvement’, the major segregation being a coverage criterion of >90% between standard and high-quality drafts and remaining terms defined by application of specific bioinformatics processes. The parsing of ‘finished’ into ‘non-contiguous finished’ and ‘finished’ reflects more the necessity to say ‘good enough’ for some eukaryotic genomes for which resolution of highly repetitive and/or intractable gaps may not be resolved by conventional means. These terms are accepted as standards for genome submission; however, one set of organisms is ostensibly absent, viruses.
Defining common genomic standards is a critical issue moving forward in the characterization of viral nucleic acids; however, this issue is mired in complexity not found in bacteria or eukaryotes. While viruses are generally less complex than bacteria in terms of genomic content/architecture,142 the notion of quasi-species and application to viral populations and RNA viruses143,144 creates a situation that does not lend itself to standardization. Specifically, the idea that within any given sample a spectrum of different genotypes exists does not allow similar criteria to those defined above to be applied. In fact, the existence of quasispecies within a viral population suggests that a new lexicon must be developed, specific to viral genomes, before the redefinition of the bacterial and eukaryotic standards can be resolved. Should terms such as consensus sequence or criteria such as population representation be instituted to describe viral genomes? That is beyond the scope of this chapter; however, this issue needs to addressed, and in short order.
1.3.3 Changing landscape of bacterial genetics: whole genome sequencing (WGS) and linking phenotype to genotype
Characterization of known pathogens encompasses defining the pathogen’s virulence and other adaptive features and establishing a link between the phenotype and the genotype. Bacterial genetics, the study of heredity and variation in bacteria, entails the understanding of how the genetic information stored (encoded in DNA) is expressed and regulated during the interplay of gene functions; i.e. in bacterial metabolism, growth, reproduction and faithful transmission of genetic material from generation to generation. To state it simply, it is the understanding of how the bacterium’s phenotype is determined by its genotype. The study of bacterial genetics involves three steps: (1) isolation of mutants with defined changes in phenotypes, (2) genetic/physical mapping of mutations and identification of the gene/locus of interest, and (3) verification of the genetic link to the phenotype by complementation of the mutant with a wild-type copy of the gene, typically expressed on a plasmid. All of these steps are time-consuming, taking anywhere from months to years, and various strategies to aid in these steps and minimize the time frame have been developed over the years, especially in model, genetically malleable organisms. Isolation of mutants and mapping of mutations have been the most arduous of these steps, especially in organisms in which genetic manipulations are arduous or are nonexistent.
Chemical and UV mutagenesis have been used to induce various types of mutations, but mapping these mutations without linked selectable markers is difficult, especially if the mutation gives rise to a non-selectable phenotype. To overcome this problem, transposons carrying selectable markers have been used to link mutations or create mutants. Transposon insertions allow facile mapping, cloning and identification of the gene of interest using the antibiotic marker encoded by the transposon. Generally, transposon insertions lead to gene inactivation, gene product loss and inactivation of essential genes, which may be lethal to the bacterium. Transposon insertions often result in phenotypes caused by polar effects on downstream genes, thus creating problems in identifying the true causal variation for a given phenotype. Often interesting mutants are those with altered functions due to protein non-synonymous changes rather than loss of function. Thus, mapping unmarked mutations poses a challenge.
Mutations
Genetic mapping is usually carried out by one of the three gene transfer modes: conjugation, transduction and transformation. Many of the mutagenesis and mapping methodologies were developed using Escherichia coli and its bacteriophages (in particular T4 and λ) as model organisms, and hence they havebeen the bacterial genetics ‘work-horses’ for decades. However, the luxury of well-developed genetic systems such as transposon mutagenesis and mutation mapping is not available in many other bacteria, and development of such systems has been slow in coming. A Tn5 transposon-based in vitro mutagenesis system has been shown to work in a broader variety of bacterial genera and species, but may not work in all (Epicentre Biotechnologies).
The advent of NextGen sequencing technologies is filling this void and is already changing the landscape of bacterial genetics. Rapid and low-cost WGS technologies have resulted in mutations mapped directly without resorting to traditional genetic approaches. WGS approach allows mapping of any type of mutation (point mutation or structural variations such as insertion or deletion or copy number variation) in any bacterium, without a need for the presence of closely linked selectable markers, by whole genome comparison of the parent with the mutant.
One mutation mapping bottleneck is the lack of a single bioinformatic tool to sift through volumes of sequence data produced by the various WGS platforms (anywhere from 350 Mb to 600 Gb), which is necessary to identify meaningful variations from among hundreds of putative variations that result from the relatively high error rates of some of the NextGen sequencing technologies. Bacterial geneticists can, however, tap into the wealth of experience in distinguishing true vs. false positive variations and linking causal variations to specific phenotypes that eukaryotic molecular biologists have accumulated through genome wide association studies (GWAS) of various genetic disorders. Fortunately, there are hardly any phenotypes linked to quantitative trait loci in bacterial systems, the so-called QTL phenotypes; i.e. most of the phenotypes are encoded by single loci, and thus verification of the link between the phenotype and the genotype is feasible without the need for statistical assertions.
Several articles have been published demonstrating the utility of the WGS approach for mutation mapping in bacteria and other organisms.25,26,145,181 Our own study using Roche-454-based whole genome pyrosequencing technology has identified a number of causal variations leading to specific phenotypic changes such as phage resistance and ciprofloxacin resistance in Bacillus anthracis.3 In this study, using Roche/454 sequence data, we have uncovered a heretofore unknown gene involved in conferring high-level ciprofloxacin resistance in B. anthracis. Interestingly, taking a similar approach, Serizawa et al. identified the same gene using short-read sequence data generated by the Illumina Genome Analyzer II platform.146 In a second example, we identified a gene, csaB, as being responsible for spontaneous resistance to AP50 phage infection. This gene has a demonstrated role in cell surface anchoring of various proteins, including the B. anthracis cell surface-associated S-layer protein, SAP,147,148 which has been the postulated receptor of AP50 phage (unpublished data). In these two examples, we have confirmed the roles of these genes in their respective phenotypes, by complementing the mutants with the wild-type copies of the respective genes.182 Thus, we have fulfilled some of the molecular Koch’s postulates, thereby lending credence to the WGS-based approach for linking phenotype to genotype.149
1.4 Discovery of novel agents
1.4.1 Examples of metagenome sequencing to determine etiologic agents
Identification of disease causal agents has historically relied heavily on the ability to culture the organism in the laboratory and/or use of organism-specific antibodies or sequence-based probes. These methods can be very limiting. For instance, many microorganisms are recalcitrant to laboratory culture. In addition to this, some etiologic agents considered ‘culturable’ have a false negative rate, such as endocarditis caused by staphylococci and streptococci.150 Serological assays are typically limited to identifying known or closely related organisms, and antigenic drift and shift can result in false negatives. Even highly sensitive PCR-based assays must be continually updated due to signature degradation.151 All of these reasons lead to a need for assays immune to these limitations and/or biases. Prior to the advent of high-throughput sequencing (HTS), high-density oligonucleotide microarrays were used to determine the presence of microorganisms. Syndrome-specific panels showed success in diagnosis of infectious disease.152 However, any sequence features sufficiently different from the array probe cannot hybridize, and result in false negatives. HTS represents a relatively unbiased approach to detection of causal agents of infectious disease.
The primary example is a study of Honeybee Colony Collapse Disorder (CCD) published in 2007.153 In this study, a metagenomic ‘survey’ was conducted, in which numerous healthy colony samples and diseased colony samples were characterized by 454 sequencing, and Israeli acute paralysis virus was identified as being correlated with CCD. Then, in early 2008, a study was published in which HTS was used to identify a novel arenavirus in three transplant patients who had died of an unexplained febrile illness.154 In this case, all available microbiological, serological, PCR and microarray assays had failed to identify a causal agent of the febrile illness. This report highlighted the power of HTS to detect a novel agent from a limited amount of sequence information. Indeed, of over 100 000 sequence reads, only 14 corresponded to the new virus. The sequences were the basis for design of multiple detection assays, including a RT-PCR assay to detect the virus in tissue samples. Using the same experimental approach, a second novel arenavirus was identified from clinical specimens originating from a highly fatal outbreak in South Africa.155
Published examples of metagenomic sequencing to detect a causal agent in diseases of animals and humans are already too numerous to be summarized here, despite the recent development of the application. In some cases, as in the case of a novel filovirus that caused an outbreak in Uganda in 2007, metagenomicsequencing was used to follow up when results from traditional diagnostic assays indicated a novel agent. The Ugandan filovirus had produced a positive result in a broadly cross-reactive IgM capture assay followed by mixed results in RT-PCR assays for known filoviruses, so metagenomic sequencing was employed to characterize the novel virus’s genome.156 In another example, more traditional diagnostic assays and metagenomic sequencing both played a role in detection and identification of human metapneumovirus causing fatal infection of wild Rwandan gorillas.157 The opposing scenario, in which traditional diagnostic assays completely fail and metagenomic sequencing plays more than just a supporting role, would include a more recent report of astrovirus encephalitis in an immune-compromised teenage boy.158
1.4.2 Some limitations of MGS (metagenome sequencing) as of today
Detection of pathogens
The earlier a pathogen can be detected, the less risk of morbidity and mortality to individuals, communities and economies. Therefore the interest is high, among many parties, in developing techniques for pathogen detection prior to human contact, also known as biosurveillance. Outside highly targeted approaches using PCR, this type of analysis was not readily feasible before NGS techniques, particularly high-throughput methods such as Illumina, became mainstream. The ability to generate billions of bases of sequence information from various environments has made plausible detecting pathogens from mixed communities of bacteria, potentially before they jump to human populations. This sequencing capability is also potentially important for bioforensics, the study of determining the source of an infectious agent after the agent has become active in humans. Difficulties include the types of tools available for metagenome analysis; the lack of appropriate reference genomes for adequate detection of ‘novel’ pathogens; difficulties in sample preparation, including matrix effects and biases introduced from extraction; and the need for sufficient depth and breadth of coverage to detect pathogens at potentially very low levels in a given sample. Also, as this is a young field, there are no determined standards, such as the properties that would lead to ‘identification’ of a pathogen in a sample, be it 1, 2, 10 or 1 million copies of a genome within the sample, and what proportion of the genome must be detected for the organism to be classified as present.
Assembly vs. read-based analysis
There are very few ready-to-use tools for metagenomic analysis. Several recent attempts have been made to adapt tools designed for assembly and analysis of single bacterial genomes for use in metagenomic assembly (IDBA, MetaVelvet, MetAmos, MetaRay),159–162 as well as adaptation of current web-basedtools for genomic analysis, IMG and RAST, to allow annotation and limited analysis of metagenomic samples online.163,164 However, there are only a few functions these tools are capable of performing. Assembly of metagenomes using current tools can require immense resources, computational time or both, and are likely to generate incorrect, chimeric or highly error-prone contigs. Assembled contigs can be analyzed for gene content, phylogenetic markers or both, using IMG-M, MG-RAST, MEGAN or other analysis tools. Even the best assemblies have at times only incorporated <1% of all reads generated from sequencing platforms, indicating that diversity is so great that >99+% of the diversity is not sampled by current sequencing techniques or represented in reference databases.
Read-based analysis can allow use of all reads in a sample. While not alleviating the lack of coverage depth and breadth, it makes it possible to generate preliminary information on the gene content of a sample, as well as preliminary phylogenetic/taxonomic distributions of dominant species. These read-based analyses are limited and highly error-prone, due to the short sequence lengths, potential sampling biases and a general lack of sufficient reference genomes for adequate analysis. Read mapping-based analyses can be useful, however, for targeted analysis (e.g. polling a sample for presence/absence/abundance of virulence genes, etc.). These analyses require a very specific data base of expected genes or genomes for detection and are therefore exclusive of other genes of interest.
There is no ideal, perfect tool available for detection of unknown pathogens from mixed samples. Tools that appear best suited for this task require immense resources, limiting the number and effectiveness of possible analyses.
Lack of reference genomes
The most powerful tool in detecting pathogens is a data base of available sequenced genomes of known pathogens, to allow similarity searching of either contigs or individual reads. This is the basis for the identification of close relatives of previously identified pathogens, as well as potential annotation of genes present in a sample that may be associated with virulence properties. While most pathogen classes have been identified, isolated, sequenced and finished, there is typically more than one strain of individual pathogen with few or no finished genome representatives. This lack of knowledge of strain variations makes downstream analysis difficult. As sequencing capacity becomes less expensive, many genomes are being generated for these variants; however, very high-quality draft or finished genomes, coupled with annotation, are necessary to build a data base that will allow sensitive and accurate identification.
Depth and breadth of coverage of individual organisms in a sample
To detect any organism in a mixed sample, both the relative abundance of the organism and the required coverage of that organism to be able to definitively call it present within the sample must be considered. In detection of an unknown organism, it is possible that neither variable will be known. This can lead to several options, all dependent on the importance of speed of analysis, budgetary concerns, availability of sample and computational resources. Briefly, sequencing can be pre-determined (e.g. targeting approximately 200 billion bases from a sample) or determined by an algorithm, by first sequencing a smaller number of bases, and using estimation techniques, such as rarefaction curves or k-mer frequency prediction methods, to determine whether and how much additional sequencing will be required to achieve a reasonable level of certainty of presence/absence of an individual organism. This determination is dependent on the two factors mentioned above: the minimum acceptable limit of detection (i.e. the lower limit of detection) and the required genome coverage for a positive result.
Sample preparation
Despite this technique’s inherent potential for relatively unbiased detection, significant challenges are involved. Influencing data output and downstream analysis are: sample matrix, type and absolute numbers of organism(s), the method of nucleic acid extraction, sequencing platform type, data base limitations and lack of standards.
The biological matrix greatly impacts quality and quantity of recovered nucleic acid. For instance, whole blood contains multiple nucleases and PCR inhibitors (i.e. heme), tissue samples will necessarily contain large amounts of host genetic material, and archived frozen samples suffer from nucleic acid degradation. Sputum and nasal swabs often contain precious few of the organism(s) in question. No one extraction method is suitable for all sample types; however, since many novel pathogens implicated in infectious disease are RNA viruses, TriZol® (also called Tri-Reagent) is often the reagent of choice for extraction of RNA from multiple sample types, as it inactivates pathogens and stabilizes RNA, but must be added in excess of the native sample (3:1 ratio is common). Incomplete sample homogenization, failure to remove large amounts of cellular material (i.e. red blood cells) and carryover of organic phase material will result in impurities. RNA present in low amounts is often lost during processing, thereby necessitating the addition of a carrier, such as glycogen, which may interfere with downstream enzymatic reactions. Depending on the original sample volume, TriZol may additionally result in splitting samples into a cumbersome number of tubes, increasing the chance for clerical errors and requiring downstream sample pooling. Often two or more extraction techniques are combined to assure high-quality nucleic acid that is acceptable for NGS. Although great strides have been made in sample preparation and many application-specific kits are or are becoming commercially available, it is crucial that new and innovative techniques be developed.
To increase the likelihood of detecting a novel agent, it is critical to maximize the output of the metagenome breadth and coverage sequencing depth. This is difficult to accurately estimate a priori, as metagenomic samples yield uneven sequence coverage and with differing depth. At present, the community has no agreed-upon adequate genome coverage and depth standards for species identification. It remains formally possible, while currently unpublished, to identify a novel agent from a single novel microbial read in a complex metagenomic sample. Identification of novel agents has been reported with as few as 14 reads out of >100 000.154 Whereas identification of an agent may require detection of only one or more reads, the crucial next step, characterization, is absolutely dependent on complete (100%) representation of the agent’s entire genome at adequate depth of coverage, especially in the case of RNA viruses or other microorganisms likely to exhibit functionally relevant minority populations or quasi-species (discussed in Section 1.1.5). This makes follow-on experiments necessary to more fully characterize the microbial genome, at times using primer-based Sanger sequencing of the novel fragment(s). It would be optimal if some of the original sample was available for such experiments; however, in many cases the original sample may be precious or limited in terms of volume. This challenge is more pronounced when identifying viral agents as opposed to bacterial agents, as viral genomes are orders of magnitude smaller (~1 × 104−1 × 105 bp) than those of an average bacterial agent (~3−4 × 106 bp) and the overall amount of viral nucleic acid may be in the picogram range. This leads to two technical obstacles: 1) viral nucleic acid is outcompeted during amplification by other nucleic acids in the matrix, such as host ribosomal RNA if the matrix is tissue, or 2) if the overall amount of nucleic acids in the metagenome sample itself is low then library preparation of the sample may fail, as successive losses of genetic material occur in each step.
Just as there is currently no agreed-upon standard to indicate what breadth or depth of coverage would be required to make an ‘identification’, there is currently a paucity of knowledge regarding the actual limit of detection (LoD) for each sequencing platform and protocol. While finding of even one or several pathogen-specific reads in a clinical sample that is not found in control samples is likely to be interpreted as a positive result, in the absence of LoD data it is difficult to conclude with any confidence that a pathogen is not present simply because no pathogen-specific reads are detected. Recently, Moore et al. conducted a LoD study whereby serially diluted viral RNA was spiked into a colorectal biopsy sample and sequenced using the Illumina platform. In this study, although the proportion of viral reads detected was less than expected, potentially due to issues with quantitating the RNA, virus-specific reads were detected from samples spiked with sub-picogram RNA quantities.165 In another study, Cheval et al. spiked plasma and cerebrospinal fluid with known concentrations of 11 different viruses and assessed the level of detection by Roche-454 pyrosequencing as compared with Illumina, and found that the higher output (number of reads) produced by the Illumina platform resulted in better detection of the virusesper run. The authors report detection of viruses by 454 sequencing at titres of ≥103pfu ml−1 and viral genomes by Illumina present at ≥102.4 genome copies/ml.166 Other studies have suggested that the LoD by 454 sequencing for an RNA virus with a genome of ~10–11 kb lies between 102 and 103 pfu ml–1. This LoD is likely to be very protocol specific, as modifications introduced at each step of the procedure can have drastic effects on overall sequence read output and quality (Frey et al, unpublished data).
Conclusions
While there is no definitive way to determine whether a pathogen may be present in a sample, HTS techniques allow rapid, sensitive and modular analysis of a mixed community sample to identify potential pathogens at low abundance. Many current bottlenecks may be solved by development of better pathogen signatures detection heuristics and computational resources HTS data analysis. Large amounts of sequence data will likely always be required to generate a statistically valid detection of unknown pathogens within a sample. This is also important to reduce the number of false negatives in tests.
1.4.3 Problems with annotation data base(s) and some potential solutions
A fundamental and widely recognized issue plaguing the expanding genomic information data bases is the creation and propagation of erroneous and misleading annotations. This is not as problematic for well-characterized core metabolic genes. Often annotations are fully automated, relying entirely on comparison with previous annotations: for example, the lpxO gene of Salmonella enterica serovar Typhimurium, whose function was discovered and characterized in 2000 and was subsequently characterized in depth enzymatically to unambiguously identify its function. A cursory analysis of representative annotations of this gene product available in even the relatively well-curated RefSeq database yields the results shown in Table 1.2.
Table 1.2
Representative annotation of lpxQ gene products in the RefSeq database
| Date* | Accession # | Organism/genome |
| 2000 | AAF87784 | Salmonella enterica subsp. enterica serovar Typhimurium |
| 2001 | NC_003197.1 | Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 |
| 2004 | YP_219152; NC_006905 | Salmonella enterica subsp. enterica serovar Choleraesuis str. SC-B67 |
| 2008 | ZP_02346018 | Salmonella enterica subsp. enterica serovar Saintpaul str. SARA29 |
| 2008 | ZP_03162104.1 | Salmonella enterica subsp. enterica serovar Saintpaul str. SARA23 |
| 2012 | EHP21538 | Salmonella enterica subsp. enterica serovar Montevideo str. IA_2010008286 |
*Date of original submission from the GenBank entry
The protein homologues in question are >99.5% identical to the original entry. Most notably, the same gene product is found annotated differently in genomes submitted by the same group in the same study! If anything, it can be argued that the quality of automated annotation is actually decaying as more entries are deposited. Definitive functional evidence for any given gene is likely to come from only a very limited number of sources. Similar confusing annotations can be found with the extremely well-characterized spo0F gene of B. subtilis and related species (see Table 1.3).
Table 1.3
Representative annotation of spo0F gene products in the RefSeq database
| Date* | Accession # | Organism/genome | Gene name | Functional annotation† | Depositor |
| 1993 | AAA22787 | Bacillus subtilis | spoOF | SpoOF protein | Trach et al.171 |
| 2004 | YP 081038 | Bacillus Hcheniformis | spoOF | Two-component response regulator | |
| 2007 | YP_001422989 | Bacillus amyloliquefaciens | spoOF | Sporulation initiation phosphotransferase F | |
| 2010 | ZP 03593522.1 | Bacillus subtilis str. 168 | – | Two-component response regulator | |
| 2011 | YP_003975155.1 | Bacillus atrophaeus 1942 | – | Unnamed protein product, two-component response regulator | Gibbons et al.20 |
| 2012 | NP_391594 | Bacillus subtilis str. 168 | spoOF | Two-component response regulator‡ |
*Date of deposition into GenBank (original publication dates from 1985).
†Most specific functional data available in the RefSeq entry.
‡Entry does include references to experimental data (PMIDs).
The lack of standards in annotation and genetic nomenclature results in considerable loss of valuable time for researchers, particularly during function assignment to given mutations, as researchers must often comb through manydifferent entries for a given gene product. The ‘annotation confusion’ is the combined legacy of decades of idiosyncratic bacterial gene naming and renaming by individual investigators and the proliferation of mis-annotations perpetuated by automated annotation algorithms. Resolution of this issue may come through widespread crowd-sourcing (e.g. Wikigenes) and/or adoption of universal standards for functional annotation combined with effective methods for propagating literature-supported revisions to gene annotations throughout the expanding database of genomic information.
1.4.4 Pathogen discovery process
Classic pathogen discovery strategies typically employ a tiered diagnostic approach for etiologic agent identification in the event of an infection of unknown origin. Samples are screened for the presence or absence of pathogens endemic in the region using relatively inexpensive assays. These assays would include immunoassays, such as antigen-capture or IgM capture ELISAs, as well as realtime PCR-based diagnostics specific to a pathogen group (e.g. a pan-filovirus assay), or may be pathogen-specific. Classical microbiological methods, such as bacterial and viral culture, are also be used for pathogen identification. If these commonly used assays fail to identify the pathogen, a second tier of diagnostic assays will be implemented.
This second tier of assays or nucleic acid characterization tools is more expensive than the initial tier but provides broader detection abilities and/or does not require a priori knowledge of the pathogen. This includes microarray-based screenings, such as the ViroChip172–175 or the GreeneChip.152,154 The ViroChip is a microarray that contains probes from all of the known viruses found in GenBank. The GreeneChip is arrayed with oligonucleotide probes designed against known vertebrate viruses, bacteria, parasites and fungi. When these more traditional assays fail to identify the pathogen, NGS may be applied. A sequencing approach is unbiased and generates a significant amount of data. Using this approach, though, can be difficult, since disease causation would still have to be demonstrated. As the cost and time related to NGS decrease, use of microarrays for pathogen detection is becoming less common.
There are multiple examples in the literature of using this tiered approach for novel pathogen identification.154,172,173,175–177
One example involves the identification of a novel adenovirus as the causative agent in a pneumonia outbreak at a US primate center.172 This outbreak was highly pathogenic, resulting in >80% (19/23) mortality among New World titi monkeys, and at least one researcher and a family member became acutely ill with a respiratory infection. Classical microbiological assays, including bacterial, fungal and mycoplasma culture, and viral respiratory panel assay, did not identify the pathogen. RNA isolated from clinical samples identified adenovirus as the potential pathogen with the ViroChip. Following PCR confirmation, WGS and phylogenetic analysis determined that this new titi monkey adenovirus (TMAdV) is highly diverse from other human and simian adenoviruses.
Another example of this tiered approach is the identification of a novel arenavirus from recent organ transplant recipients.4 Three individuals died not long after receipt of organ transplants from a single donor, presenting with febrile illness. Classical microbiological assays, as well as a panmicrobial oligonucleotide microarray analysis,152 failed to identify the pathogen. NGS and analysis identified an Old World arenavirus related to lymphocytic choriomeningitis virus (LCMV) and identification was confirmed by classical microbial techniques.
1.5 Future trends
1.5.1 Field-able sequencing
Current efforts to place sequencing ‘in the field’ involve the installation and setup of technologies in non-traditional laboratory spaces. Difficulties include the high energy consumption, large computing power required to decipher the data, and the training and equipment required to properly prepare and run samples through the system. While each hurdle is substantial, none are insurmountable.
Current platforms, such as the MiSeq and Torrent PGM, are small enough to fit in a mobile space. In fact, Life Technologies has showcased the portability of its PGM platform by placing it on a bus that travels around the United States (Figure 1.2(a)) and in the trunk of a Mini (Figure 1.2(b)). These mobile laboratories are able to provide enough power to the sequencer during the run for successful completion and then transfer the data generated to cloud-based storage. Of course, an open laboratory, such as in the trunk of a car, is highly susceptible to contamination and so may not be ideal for most applications.

More applicable is the installation of sequencing technologies in non-traditional laboratories, allowing the setup of all ancillary equipment and full training of staff involved. These laboratories can be designed to include electrical power conditioners, allowing for intermittent power access, as well as restricted Internet access and on-site analysis of the data generated by commercial off-the-shelf (COTS) bioinformatics tools.
1.5.2 Sequencing as diagnostic tool
To achieve the next step in disease diagnostics, sequencing-based applications present unique opportunities on the path to broad-spectrum, ‘assay free’ diagnostics. Even in present FDA-approved systems for multiplexed diagnostics, sequence-based recognition is necessary for assay design and function. Molecular identification depends on specifically designed reagents, whereas future systems will likely generate sequence data that will ultimately be compared against reference information in silico to make identification. The common thread between the present and future diagnostic systems is a guiding data base for either assay development or instrument result correlation with a known set of disease signatures.
Such a robust, validated data base does not exist in the public sector, and the FDA-cleared technologies that have been created based on known sequences (i.e. PCR systems, arrays or others) have often relied upon a proprietary data base (or internally verified versions of error-rich public data bases) to drive assay design containing the molecular comparator. In the case of present-day and historical systems, the data base was not large, because a limited number of assay targets were sought and the proprietary data bases could be more easily validated for a very specific set of applications. In the future, a data base driving sequencing-based system diagnosis will likely be much more significant in size, perhaps even approaching GenBank in size. Yet this data base will need to far exceed the presentstate of GenBank in terms of data quality (as do the current, proprietary data bases for diagnostics), as it will serve the needs of agnostic sequencing-for-diagnostics systems and continue to drive developments for assay-based systems.
In today’s molecular comparator systems, probe molecules must be synthesized or generated from a natural system, are fixed after generation and cannot be easily changed. Once incorporated in a multiplex system, addition of new elements is difficult and requires revalidation experiments. In future systems that might rely upon vast (much larger than today’s proprietary diagnostic, sequence-based data bases) high-quality, curated sequence databases, entries would be added (following a pre-defined quality metric) when required. This would enable reconfiguration by changing data base entries, leading to widely available target opportunities for Dx systems of the future. The fact that such a data base would be public also adds to the accessibility of this information for device design, and would therefore enable a potentially greater variety of technical approaches, as more developers could utilize this information.
The data in the present-day GenBank data base serve a very useful function in driving unprecedented scientific discovery, but do not have the consistent quality standards across all entries to serve as a diagnostic data base. The utility of GenBank in the basic research environment shows the power of a vast and centralized genomic repository. Making a high-quality reference data base available for diagnostics would fuel advancement in medical treatment rivaling the advancements in scientific discovery that were fueled by NCBI and GenBank.
Defining the next standards for sequence-based references
The definition of nucleic acid sequence reference standards is particularly challenging, given that the current ‘gold standard’ (e.g. Sanger sequencing) is far too expensive to be practical, and the many competing ‘next-generation’ sequencing technologies and platforms are all climbing learning curves in terms of error rates, read lengths and cost/base. Some new platforms are designed for resequencing human genomes and may be applicable for pathogen sequencing only under specific circumstances (e.g. high degrees of multiplexing samples). Error rates vary widely between technologies and platforms and are affected by changes in chemistry and software, in addition to physical components. This means that error rates are not static per platform, and standardized methods for determining error rates for each configuration also need to be developed. Methods to establish whether a particular sequence submission meets the standards of the reference data base also need to be developed.
For these sequence-based reference data bases to function appropriately as diagnostic tools, they will have to be coupled with the appropriate comparative analysis tools that can take a sequencing result from an unknown sample (likely metagenomic) and provide positive/negative predictive value (PPV/NPV) that exceeds current diagnostic systems. Also, the ‘price of entry’ for submitting a datapackage to these data bases will need to be clearly defined. The standards for data quality should be defined such that they can provide a predictable level of PPV and NPV, given the known error rate of a challenge sequence presented to the data base. For regulatory approval, the error rate of the ‘comparator’ needs to be a known quantity, and, now that the comparator is at a digital level, error rates related to the uncertainty of the query sequence and the quality of the data base sequence can be used to predict an overall PPV and NPV. Setting the desired PPV and NPV for a given error rate in the query sequences will allow us to define the desired quality of the data base sequence.
1.5.3 Evolving the cultural paradigm and legal framework for collaboration and exploitation of emerging technologies
NGS is truly a disruptive technological innovation. Both the extent and scope of fields of practice potentially impacted by NGS are great. By its nature, having the capability to cost-effectively and readily generate genomic information provides foundational information about organisms and communities of organisms. Hence, information generated by NGS has impacts throughout biology and the myriad fields of practice impacted by biology; and, by extension, the political and religious structures of society. A brief overview of the non-technical challenges facing application of NGS is provided below, since a comprehensive review is beyond the scope of this chapter. While significant and appropriate focus is occurring to address aspects of these challenges (i.e. impacts of NGS on medicine178),other areas of potential impacts are receiving less attention (e.g. intellectual property management179). Unforeseen impacts may also occur, as was the case with NGS WGS of strains of Vibrio cholerae providing the foundation for a lawsuit by Haitians against the UN humanitarian effort.180
The non-technical challenges of NGS can be divided into (a) sample acquisition and management, (b) information generation and (c) information management. Illustrative fields of practice that are utilizing NGS include genomic medicine, the broad area of research and development involving biological systems (e.g. wildlife biology, therapeutics, biofuels), criminology and national security. Each of these fields of practice has overlapping and unique challenges to confront in effectively utilizing NGS. Of equal or greater challenge is managing sample acquisition, information generation and information management across fields of practice.
A hypothetical example, employing previously envisioned activities/events, tracking a single sample of a human genome through scenarios of sample acquisition, sequencing and information management is illustrative. Imagine a time in the not too distant future when at birth an infant’s genome is sequenced by public health regulation, as is currently the case for diagnosis of such genetic anomalies as PKU, or voluntarily due to parental interest. The cost of this sequence is greater in the US because many human genes have been patented and royaltiesmust be paid for WGS and follow-on analysis. Of course, this cost may be avoided if the sequencing is performed in a country that does not recognize intellectual property ownership for natural genes and the information is transmitted over the Internet to the physician and the baby’s parents. The information is provided on some digital storage media for future use by the child’s parents as well as the physician, the hospital, and local and national health authorities for some set of specified uses. A professional from the burgeoning field of genomic counseling provides guidance to the parents as to how the child should be raised in order to maximize predicted phenotypic potential while minimizing adverse health effects associated with presence of deleterious alleles. Of course, this counseling does not occur until the parents have taken a basic course in medical genomics so that they do not take any rash action when presented with negative information.
Our hypothetical baby grows and matures, benefiting from the insights provided by predictive genomic analysis. The school authorities have additional data for making class assignments, and prophylactic medical care, diet and treatments for childhood illnesses are tailored to the child’s unique genotype. All goes well with effective management of the child’s genomic information, with the exception of addressing a paternity concern raised by the father, until an adolescent indiscretion brings the teenager to the attention of the authorities. At this point the ‘medical’ genomic information is requested by the law enforcement community. The nascent area of law that manages the interface between medical and law enforcement genomic information management is asked to rule on the request. After some deliberation, a redacted copy of the teenager’s genome sequence is provided to law enforcement and the matter is resolved. This matter is closed until a bodily fluid sample found at the scene of an alleged terrorist event yields a match to the redacted genome held in the national criminal genomic bank. Of course, it is not a complete match, since the entire genome is not available to law enforcement and the conclusions are probabilistic in nature. At this point a third community of practice, the national security community, enters into the picture. Since this is a potential act of terrorism having grave potential impacts for national security, greater latitude is afforded the investigating officials and the entire genome is provided. This transfer of the entire genome to the national security apparatus is not without controversy, since advocacy support groups decry the breach of privacy and racial profiling. Fortunately, the complete genome exonerates our twenty-something case study. But all is not well, since the love of his life he met on Facebook is now asking for a copy of his genome to run a compatibility profile with her own. Further, she is asking him to submit to a metagenomic analysis of his oral cavity and gut before she will share bodily fluids with him, since she has had her high school Better Health through Genomics course.
It is hard to imagine that the legal, ethical and cultural frameworks needed to realize most aspects of the above hypothetical case could be established in a time frame commensurate with availability of the technical capabilities. Aspects of these challenges are explored in the science fiction movie GATTACA (1997); stimulatingand perhaps prejudicing the lay public’s interest and views on application of the technology. Communities of practice are beginning to explore these challenges, such as the World Health Organization’s Ethical, Legal and Social Implications (ELSI) of human genomics component of the Human Genetics Programme focusing on human health. However, rationalization of guidelines and regulations across communities of practice (e.g. health, law enforcement, national security) will add a great additional level of challenge. Therefore, it is anticipated that lack of needed legal and ethical consensus will result in ‘back pressure’ on utilization of NGS technology. NGS is redefining the concept of disruptive technology.
1.6 Acknowledgments
We would like to acknowledge Arya Akmal for the statistical calculations described in Section 1.1.5. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of JPEO-CBD-MCS-CRP, the U.S. Department of Defense or the U.S. Government.