
Isothermal amplification of specific sequences
Y. Tong, New England Biolabs, USA
Abstract:
This chapter focuses on the mechanisms of isothermal amplification of specific sequences. More than 15 different isothermal amplification technologies – including loop-mediated isothermal amplification (LAMP); strand displacement amplification (SDA); cross priming amplification (CPA); rolling circle amplification (RCA); helicase-dependent amplification (HDA); recombinase polymerase amplification (RPA); nucleic acid sequence-based amplification (NASBA) and transcription mediated amplification (TMA) – have been grouped for discussion and comparison. The following aspects are compared and presented for each platform: amplification scheme, primer design, major amplified products, detection methods, advantages and/or disadvantages. Furthermore, the impacts, clinical applications and future trends of isothermal amplification technologies are discussed. With the rapid development of modern biotechnologies, the mechanisms of nucleic acid amplification in vivo will be further discovered by scientists, and the related proteins will be manufactured with superior quality and at low cost in vitro. Therefore, the performance and application of isothermal amplification can be further exploited.
Key words
loop-mediated isothermal amplification; nucleic acid sequence-based amplification; transcription-mediated amplification; strand displacement amplification; helicase-dependent amplification; isothermal amplification
3.1 Introduction
A fast and sensitive diagnostic assay for infectious diseases provides information that can be utilized to determine treatment, infection control, and prevention measures. A desirable test should be rapid, accurate and cost-effective and have little need for operational skills. PCR-based molecular diagnostic assays provide rapid and accurate tests. However, the requirement for an expensive real-time thermal cycler limits their application in small hospitals, decentered laboratories, resource-limited settings and other point-of-care (POC) diagnostics.
Isothermal amplification can obviate the need for a thermal cycler. Variant isothermal amplification technologies use different mechanisms to reinitiate new rounds of DNA synthesis. The components of amplified products are also different. Although an initial heating step for template denaturation is still necessary for some technologies, an expensive thermocycler is not required for nucleic acid amplification. Instead, a cost-efficient heat block or a portable fluorescence reader (e.g. ESE-Quant tube scanner provided by Qiagen Inc. (Stockach, Germany)) can be used for amplification and detection. On the other hand, most of the isothermaltechnologies are complicated compared with PCR in terms of the components of the reaction mixture, primer design or amplified products. This limits their application as routine research tools.
The chapter is focused on the principles of isothermal amplification of a specific sequence. More than 15 platforms are grouped and discussed. Primer design is the key issue for all the technologies. Tm is one of the foundations of primer design. Therefore, Tm estimation will be discussed first and separately.
3.2 Melting temperature (Tm) estimation and categories of isothermal amplification technologies
Tm, the melting temperature of an oligonucleotide duplex, is defined as the temperature at which the oligonucleotide is 50% annealed to its complementary sequence. Accurate estimation of the Tm of a primer–template duplex is a critical factor of primer design for all the isothermal amplification technologies. The primer annealing temperature in a PCR assay can be adjusted and optimized based on different sequences. However, the annealing temperature is almost predetermined by the technology itself for an isothermal amplification technology. Therefore, the adjustment range is limited.
A primer Tm depends on several factors (Ahsen et al., 2001; Owczarzy et al., 2008). Generally, Tm is higher with higher concentration of primers, or higher GC content of primers, or longer length of the primers, or more salts in buffers. Several algorithms have been proposed, tested and used for Tm calculation. Some of the most frequently used equations and websites for Tm estimation are listed in Table 3.1. The equations are listed in order from simple to complicated. Tm is not only a property of an oligonucleotide, but also a property of an oligonucleotide under specific conditions. Because different isothermal amplification technologies operate under various conditions (buffers, proteins and temperature) which might be different from the theoretical calculation, the best fit of a Tm estimation needs to be determined experimentally for a particular technology.
Table 3.1
| Equation | Example website | |
| 1 | Wallace–Ikatura equation (basic): Tm = 2(A+T) + 4(G+C) A+T is the total number of A and T, G+C is the total number of G and C. |
http://www.promega.com/techserv/tools/biomath/calc11.htm |
| 2 | Marmur–Shildkraut–Doty equation (salt adjusted): Tm = 81.5 + 16.6(log10[Na+]) + 0.41(%GC) – b/n [Na+] is the molar salt concentration, b is between 500 and 700, n is the length of the oligonucleotide. |
http://www.promega.com/techserv/tools/biomath/calc11.htm |
| 3 | Nearest neighbor: Tm = ΔH°/(ΔS° + RInCt) ΔH° and ΔS° are associated with duplex formation calculated from nearest-neighbor thermodynamic parameters, R is the ideal gas constant (1.987 cal.K−1.mol−1), Ct is the molar concentration of oligonucleotide. |
http://www.basic.northwestern.edu/biotools/oligocalc.html |
| 4 | Adjusted nearest neighbor:  |
http://www.idtdna.com/analyzer/Applications/OligoAnalyzer |

Isothermal amplification technologies, which were invented in the past two decades, can be grouped or categorized based on reaction mechanisms, as illustrated in Table 3.2. They can be divided into two major groups based on the polymerase used in the platform. The technologies, depending on DNA polymerases, can be further grouped by other critical components (additional enzymes or specific structures). To achieve nucleic acid amplification at a constant temperature, strand separation is the crucial step. Isothermal amplification using DNA polymerase highly depends on the strong strand displacement activity of the polymerase. Strand displacement activity is defined as the ability of a protein to displace downstream DNA encountered during synthesis. Without further clarification, the DNA polymerases mentioned in this chapter only represent those with strong strand displacement activities, which are always the large polymerase fragments. IfBst DNA polymerase (from Bacillus stearothermophilus) is used, the amplification temperature is in the range of 52–65 °C. Primers should be designed for efficient annealing in this range. However, many isothermal amplification technologies have sophisticated reaction schemes and require a fine-tuned (adjusted) primer design compared with that of PCR. In most cases, the available tools for primer design are not sufficient. Therefore, it is always necessary to perform complicated experiments of primer screening and assay optimization to develop an isothermal amplification assay with good sensitivity, specificity and robustness.
Table 3.2
Categories of isothermal amplification technologies
| Category: polymerase | Subcategory: other key components | Isothermal amplification technologies |
| Based on DNA polymerase | Based on multiple primers to form intermediate stem-loop products | LAMP (Notomi et al., 2000), SmartAmp (Mitani et al., 2007) |
| Based on additional enzymes (restriction or nicking enzymes) | SDA (Walker et al., 1992a and 1992b), NEAR (Maples et al., 2009), NEMA (http://bioustar.com/newSite/platforms/jspt_3.jsp), CPA (Fang et al., 2009) | |
| Based on additional enzymes (RNase H) | ICAN (Mukai et al., 2007), ICA (Jung et al., 2010) | |
| Based on padlock probe to form rolling circle structure | RCA (Fire and Xu, 1995) and related technologies, including RAM (Beals et al., 2010), PG-RCA (Murakami et al., 2009) | |
| Based on a group of proteins to maximally mimic the amplification in vivo | RPA (Piepenburg et al., 2006), HDA (An et al., 2005) | |
| Based on RNA polymerase | NASBA (Sooknanan and Malek, 1995), TMA (Kacian and Fultz, 1995), 3SR (Guatelli et al., 1990) |

Isothermal amplification is based on either complicated primers (more than two primer pairs) or complicated proteins (more than three proteins). The design details will be discussed in the following sections of the chapter, ranging from the most complicated platforms (more than three primer pairs) to simple platforms (just one pair of primers, similar to PCR). Most reviews or book chapters regarding isothermal amplification technologies refer to descriptive figures of the reaction mechanisms or a copy of the original publications. It is good to understand the reaction scheme from the original design. However, the common principles or core design ideas of isothermal amplification can be buried in the diversity of the presentation formats from different technologies. In this chapter, we try to present the principles in a more common or universal way. Therefore, the connection(similarities) or difference among the technologies can be presented and compared in a straightforward way. Figure 3.1 uses two of the technologies as an example to show the universal primer design scheme. Generally, one to three pairs of primers are designed for isothermal amplifications. One primer pair, defined as outer primers (Table 3.3), is used for strand displacement purpose for most technologies. Another primer pair, defined as inner primer pairs (Table 3.3), has the most diverse design purposes among different technologies. Some technologies use a third primer pair for additional purposes, e.g. speed acceleration or detection.
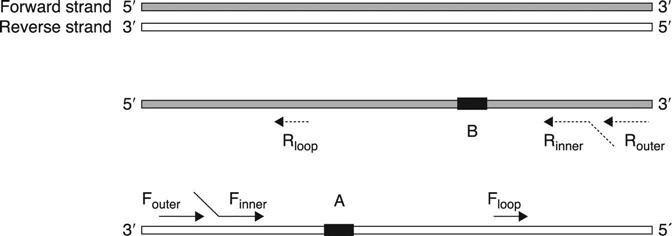
Table 3.3
| Terminology in this chapter | Definition | Terminology in LAMP | Terminology in SmartAmp |
| Finner | Inner forward primer. 3′ end sequence binds the reverse complement DNA strand, 5′ end sequence contains either the sequence of stem-loop forming region A (represented as the black rectangle in Fig. 3.1) or selffolding sequence. The 5′ end part is the important structure to form the stem-loop DNA structure. | FIP (forward internal primer) | FP (folding primer) |
| Fouter | The outermost forward primer, binding the reverse complement DNA strand. For most isothermal amplification technologies, it serves a strand displacement function together with DNA polymerase. | F3 (forward outer primer) | OP1 (outer primer) |
| Rinner | Inner backward primer. 3′ end sequence binds the forward DNA strand, 5′ end sequence contains either the sequence of stem-loop forming region B (represented as the black rectangle in Fig. 3.1) or self-folding sequence. The 5’ end part is the important structure to form the stem-loop DNA structure. | BIP (backward internal primer) | TP (turn back primer) |
| Router | The outermost reverse primer, binding the forward DNA strand. For most isothermal amplification technologies, it serves a strand displacement function together with DNA polymerase. | B3 (backward outer primer) | OP2 (outer primer) |
| Floop | Forward loop primer. Binding the reverse complement DNA strand, between the corresponding position of Rinner and stem-loop forming region B. It is an optional primer for both technologies to accelerate the reaction speed. | BLP (backward loop primer) | BP (Booster primer) |
| Rloop | Reverse loop primer. Binding the forward DNA strand, between the corresponding position of Finner and stem-loop forming region A. It is an optional primer for both technologies to accelerate the reaction speed. | FLP (forward loop primer) |

3.3 Isothermal amplification based on DNA polymerases
3.3.1 LAMP and SmartAmp
This group of isothermal amplification depends on DNA polymerase and complicated primer sets (four to six primers) to generate intermediate products (stem-loop DNA structures), which serve as templates for the following exponential amplification. The formation of the stem-loop DNA structure by the primer sets is the key point of the reaction. Once the intermediate products are formed, the autocyclic strand displacement reaction can progress rapidly at a constant temperature.
Loop-mediated isothermal amplification (LAMP) and smart amplification process (SmartAmp) belong to this group, utilizing the most complicated primer design strategy in all the isothermal technologies (Notomi et al., 2000; Mitani et al., 2007). To make the following discussion and comparison clearly understandable, the terminology is defined, as shown in Table 3.3. The terminology difference between LAMP and SmartAmp is also shown in this table. Figure 3.1 shows the design scheme of the technologies and the relative positions of the primers. Other isothermal amplification technologies use fewer primer pairs. However, the inner primer pairs are designed as complicated structures that are crucial for most isothermal amplification technologies. Generally, the inner primers can be designed in two to three parts. The 3′ end is always the hybridizationregion, while the 5′ end and the middle region have different design purposes. The detailed design structures of inner primer pairs are listed and compared separately in Fig. 3.2.
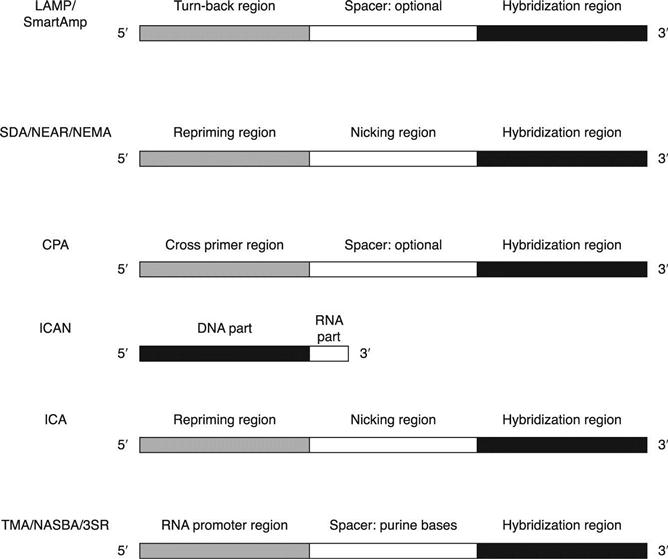
The amplification principles and primer design of LAMP and SmartAmp are similar. They both require a set of at least four specific primers which hybridize to six different specific regions on the nucleic acid template. The two outer primers have a role in strand displacement during the non-cyclic step only (initialamplification stage). The inner primers (also called turn-back primers or folding primers) have both sense and antisense sequence, in such a way as to help in the formation of stem-loop structure, a key aspect of the technology. In order to speed up the reaction, two additional primers (loop primers or stem primers), hybridizing another two different specific regions, can be designed and added to the reaction mixture (Nagamine et al., 2002; Tisi et al., 2010; Gandelman et al., 2011). Loop primers are designed in the region between the inner primer and stem-loop forming region (shown in Fig. 3.1). Stem primers are designed in the region between the two stem-loop forming regions with flexible orientation. LAMP uses a symmetric primer design. The two inner primers are like the turn-back primers in SmartAmp, which uses an asymmetric primer design with a turn-back primer and a folding primer. Compared with the LAMP technology, SmartAmp also employs additional proteins (MutS) to increase the amplification and detection discrimination for SNP (single-nucleotide polymorphism) identification. Some animations on the websites are helpful to understand the complicated principles, e.g. http://loopamp.eiken.co.jp/e/lamp/anim.html and http://www.dnaform.jp/smartamp/smartamp/movie/amp_only_e.html.
Primer selection and design are very challenging for this group of technologies. The following factors need to be considered: specificity of six to eight primer regions, GC content, secondary structure, primer Tm, primer length, primer distance and stability of end base pairs of primer (Parida et al., 2008). The recommended primer Tm from the outermost region (outer primer) to the innermost region (stem-loop forming region) spans from 57 °C to 67 °C. Obviously, this is a difficult task for human efforts alone, even with an advanced educational degree. Therefore, dedicated research work has been done to make both technologies feasible for researchers. The Eiken Chemical Co. (Tokyo, Japan) has developed software to aid the primer selection. The current version is 4. Other choices of software have also been developed, which are available online. The selection rules are specified clearly for each software program and in the review by Parida et al. (2008). The currently available tools are listed as follows.
• http://primerexplorer.jp/e/ (Eiken Chemical Co. Ltd)
• http://code.google.com/p/lava-dna/ (Torres et al., 2011)
• http://gerg.gsc.riken.jp/TP_optimization/ (Kimura et al., 2011)
Multiple detection methods have been developed for the amplified products of LAMP, listed in Table 3.4. The amplification products are stem-loop DNA structures with several inverted repeats of the target, also described as cauliflowerlike structures with multiple loops (Gill et al., 2011). Some of the detection methods are based on the high yield of final products, a unique feature of the technology. A large amount of DNA (10–30 μg/25 μl) can be synthesized in a short time (15–60 min) with high specificity and sensitivity in a typical LAMPreaction (Mori et al., 2001). The yield of final products is usually at least 20-fold that of PCR or other isothermal amplification technologies. Pyrophosphate ions are produced in large amounts to form a white precipitate of magnesium pyrophosphate (insoluble by-product). The turbidity of insoluble complex formed from the high-yield products is a unique feature of LAMP technology. This characteristic greatly facilitates design of the detection platform. The detection can be performed instrument-free (naked eye) or with portable devices, or with the same instruments as PCR. Some detection chemistries, like intercalating dyes or cationic polymers, have certain (minor to strong) inhibitions to LAMP. In the early research stage, addition of the detection reagent after amplification was suggested and applied for the assays. However, detection systems with open tubes always increase the chance of cross-contamination. Tao et al. have described a method of embedding the detection reagent in a wax capsule (Tao et al., 2011). This is a promising technology to solve common problems. In this method, a visualized LAMP method is established by the addition of a microcrystalline wax-dye capsule containing the detection dye. The wax capsule remains intact during amplification, and releases the dye to the reaction mixture only when the temperature is raised to the melting point (95 °C) following amplification. Thecolor difference between positive samples and negative samples is visualized by the naked eye, while the cooling wax turns into a solid barrier on top of the mixture to minimize the risk of aerosol contamination.
Table 3.4
Different detection methods of LAMP
| Detection principle | Detection device | Reference |
| Formation of insoluble by-product, magnesium pyrophosphate | Naked eye or real-time turbidimeter (Eiken Chemical Co., Ltd) | Mori et al., 2001 Mori et al., 2004 |
| Intercalating dye: ethidium bromide, SYBR green, pico green, SYTO-9, or propidium iodide | Naked eye ESE-Quant tube scanner, Smart cycler II or other real-time PCR equipment | Pham et al., 2005 Lucchi et al., 2010 Iwamoto et al., 2003 Chen and Ge, 2010 Hill et al., 2008 |
| Formation of an insoluble complex with DNA by cationic polymers (PEI) | Naked eye or microplate reader | Mori et al., 2006 |
| Metal ion indicator: calcein together with manganous ion, or hydroxy naphthol blue | Naked eye or microplate reader | Tomita et al., 2008 Goto et al., 2009 |
| BART: conversion PPi (inorganic pyrophosphate) to ATP by ATP sulfurylase | Research assembled system (real-time data collection) | Gandelman et al., 2010 |
| Probe hybridization: lateral flow dipstick | Open system or closed detection system (e.g. BESt™ cassette) | Kiatpathomchai et al., 2008 |
Note: BART, bioluminescent assay in real-time; PEI, polyethylenimine.
LAMP depends on a single protein, DNA polymerase, which is another important feature of this technology. Proteins are the components most sensitive to the inhibitors existing in clinical specimens. The lower complexity of proteins makes it possible to tolerate inhibition by clinical samples. The rate-limiting step is the formation of the intermediate stem-loop structure. Once it is formed, the remaining amplification is less sensitive to the variations of clinical sample input. This might be the reason for the reported robustness of the LAMP assay for clinical diagnostics (Francois et al., 2011; Kaneko et al., 2007).
Since Notomi and his colleagues developed LAMP in 2000, the technology has gained popularity in both the research field and the diagnostic field because of the user-friendly software, available reagents (enzymes, buffers, etc.), simple operation, and multiple choices of detection methods. Many researchers from different countries have worked on the basic research for different applications. As a result, there are more than 500 publications in PubMed so far (at the end of 2011). And the technology has been put into practice in detection of various pathogens, SNP typing and many other diagnostics. It has been demonstrated that, even though the primer design of LAMP is the most complicated system compared with PCR and the other isothermal amplification methods, it is feasible and applicable to both research and diagnostic use.
3.3.2 SDA, NEAR, NEMA, CPA
This group of technologies requires an additional enzyme (a restriction endonuclease or a nicking enzyme) or uses other strategies to simulate the effects of the enzyme.
Strand displacement amplification (SDA) was developed by Walker et al. in 1992 (Walker et al., 1992a, 1992b). This method uses two sets of primers, a DNA polymerase, a restriction endonuclease, and modified nucleotides. The restriction enzyme nicks one strand of double-stranded DNA (dsDNA), and the DNA polymerase extends the 3′ end from the nick. New strands extending from the 3′ ends will displace the downstream strands, which are dispatched from the dsDNA as amplified products. The displaced strands can also serve as new templates. However, because the restriction enzymes typically cut both strands of non-denatured and unmodified DNA, they are not suitable candidates for use in the SDA methods. To create a nick site in a single strand, non-standard nucleotides, such as α-thio-dNTP (dNTP[αS]), must be added to the reaction mixture to alter the enzymes’ action. Thiol-modified nucleotides are incorporated into the synthesized products to inhibit cleavage of the synthesized strand. This modification creates a nick site on the primer side of the strand, which can be extended by the polymerase. SDA can operate at 37–41 °C with Klenowexo-DNA polymerase and a restriction enzyme, such as HincII or Aval. SDA can also perform at around 52 °C with Bst DNA polymerase and a restriction enzyme, such as BsoBI (Little et al., 1999).
Two sets of forward and reverse primers (outer and inner primer pairs) are designed to achieve exponential amplification. The inner primer pairs (Finner and Rinner) are called amplification primers in SDA. The 3′ end of an inner primer, also called the target binding sequence, hybridizes at the target sequence (Fig. 3.2). It is about 10–25 nucleotides in length and confers hybridization specificity on the amplification primer. The inner primer also comprises a recognition site for a restriction endonuclease, 5′ to the target binding sequence. The recognition site is for a restriction endonuclease which will nick one strand of a DNA duplex when the recognition site is semi-modified. The 5′ tail of the inner primer is about 10–25 nucleotides, located at 5′ to the restriction endonuclease recognition site. It serves as a polymerase repriming site when the remainder of the inner primer is nicked and displaced during SDA. The repriming role of the 5′ tail nucleotides sustains the SDA reaction and allows the synthesis of multiple amplicons from a single target molecule. The sequence of the 5′ tail is not critical; it can be routinely selected and modified to obtain the desired Tm for hybridization (Walker et al., 1992b). The outer primer pairs (Fouter and Router) are also called bumper primers in SDA. The bumper primer anneals to a target sequence upstream of the amplification primer such that extension of the bumper primer displaces the downstream amplification primer and its extension product.
Little et al. developed a real-time detection method for SDA based on a dualdye labeled hairpin probe (Little et al., 1999). The region between these labels includes a stem-loop structure. The loop comprises a recognition sequence for the same restriction enzyme used in the assay. A target-specific sequence is at the 3′ end of the probe. Before SDA amplification, the two labels are proximal to each other, such that any excitation of the fluorescein (one label) leads to transfer of the emitted energy to the rhodamine label (the other label). The net effect is that very little emission from the excited fluorescein is detected. After SDA, the probes hybridize to the specific amplified sequences, and are cleaved by the restriction enzyme. This cleavage causes the physical separation of the two labels, such that no energy transfer from the excited fluorescein to rhodamine can occur. Therefore, the excited fluorescent signal is detectable, which is indicative of specific amplification of the target sequence.
However, several drawbacks have hindered the general application of SDA. First, SDA requires a heat denaturation step prior to isothermal amplification. SDA enzymes must be added stepwise to the reaction after heat denaturation of target DNA, so the workflow is complicated. The multiple-step workflow necessitates opening the reaction vessel and exposing the sample to potential contamination. Second, the use of modified nucleotides will not only increase the manufactory cost, but also make the reaction mixture complicated. The added restriction enzymes are much less efficient in nicking the modified substrates, thereby leading to a slower amplification rate and lower product yield, while requiring a much higher amount of the requisite enzyme.
With recent research and discovery of nicking enzymes, the technologies of nicking enzyme amplification reaction (NEAR) and nicking enzyme mediated amplification (NEMA) have been developed (http://bioustar.com/newSite/platforms/jspt_3.jsp; Maples et al., 2009). Both technologies use nicking enzymes instead of restriction enzymes. Therefore, modified nucleotides are no longer required. NEAR only uses the inner primer pairs, while NEMA uses two sets of primers similarly to SDA. The recognition site for a restriction endonuclease of the inner primers is replaced by the corresponding site for a nicking enzyme. The reaction is performed at 54–60 °C with Bst DNA polymerase (Maples et al., 2009).
However, there are some risks of template-independent nucleic acid amplification (background noise) when employing DNA polymerase together with restriction endonucleases or nicking enzymes for in vitro nucleic acid amplification (Liang et al., 2004; Zyrina et al., 2007; Liang et al., 2006). Sometimes the non-specific amplification can affect the sensitivity of the assay.
Cross priming amplification (CPA) can perform strand displacement amplification without nicking activity from additional enzymes (restriction or nicking enzymes) (Fang et al., 2009). It uses two sets of primers to simulate the effects of the additional enzyme. The outer primer pairs are similar to those of SDA. The inner primer pairs contain no recognition sites for any enzymes (Fig. 3.2). Instead, the inner primers, also called cross primers, are designed as follows. The 3′ end of the Finner sequence hybridizes to the amplification target. The 5′ end of Finner is identical to the 3′ end sequence of Rinner, and vice versa (Fang et al., 2009). The displaced strand contains newly introduced priming sites on both ends. Thus it can serve as a template with priming sites for both cross primers on its 3′ end. A new priming site is introduced after each round of extension/displacement, resulting in multiple primer binding sites which accelerate the amplification process. The cross primer can also be used as a displacement primer in the following amplification. Overall, the primary purpose of the 5′ end sequence of the inner primer pairs in CPA is to introduce additional priming sites at both ends of the target. The DNA extension mainly relies on the annealing of multiple primers to multiple priming sites of both strands to drive the synthesis of new DNA (You, 2011). Additional primers may be designed for detection purposes. The final products are a mixture with different lengths and multiple forms of secondary structures. Although this technology employs multiple primer binding sites to increase the efficiency, the yield of final product is still far away from that of the LAMP method (similar to other isothermal amplification technologies). The amplification time is also in the range of 1 h.
Selection and design of the inner primer pairs are essential to the speed, specificity and sensitivity of the above technologies. Generally, the concentration of outer primers is lower than that of inner primers, which is similar to LAMP technology. However, there is no available software for primer design for the above technologies.
3.3.3 ICAN, ICA
This group of isothermal amplification technologies is highly dependent on an additional enzyme, RNAse H (Ribonuclease H). The enzyme can specifically degrade the RNA portion of DNA/RNA hybrids (Tadokoro and Kanaya, 2009). Prokaryotic RNase H, which is involved in DNA replication, repair and transcription, has been classified into RNases HI, HII and HIII based on the difference in their amino acid sequences. RNase HI represents type 1 RNase H, and RNases HII and HIII represent type 2 RNase H. Both types of enzymes are useful for nucleic acid amplification and detection, with more recent reports on Type 2 enzymes (Gašparič et al., 2008; Dobosy et al., 2011). Type 1 RNase H requires multiple RNA bases in the substrate for full activity. A DNA/RNA/DNA oligonucleotide with only one or two RNA bases is not cleaved by this type of enzyme when hybridized to a DNA oligonucleotide, while Type 2 RNase H can cleave a single ribonucleotide embedded within a DNA sequence when hybridized to a DNA oligonucleotide (Eder et al., 1993). The cleavage by both types of enzymes occurs on the 5′ side of the RNA residue, leaving a DNA oligonucleotide with a 3′-hydroxyl that is competent to serve as a primer. This unique feature of cleavage is crucial for this group of isothermal amplification technologies. The inner primer pairs are designed with one or more RNA bases, which can be cleaved by the RNase H enzymes to generate nick sites for isothermal amplification. Isothermal and chimeric primer-initiated amplification of nucleic acid (ICAN) and isothermal chain amplification (ICA) belong to this group.
ICAN was proposed in 2002 (Shimada et al., 2002; Mukai et al., 2007). It uses only one pair of primers (inner primer pairs) together with RNase H and DNA polymerase. In Mukai’s paper, Thermococcus litoralis RNaseH and BcaBEST DNA polymerase (Takara Bio Inc., Japan) were used for amplification at 55 °C. The inner primers of ICAN, also described as chimeric primers, are designed as 5′-DNA-RNA-3′, with a few RNA residues (one to six) at the 3′ end (Fig. 3.2). RNase H can cleave the RNA portion of the extended strand and introduce a nick site at the 5′-RNA/DNA-3′ junction of an extended strand synthesized from the chimeric primer. The yield of final products can be increased by adding more primers. Three amplification products are formed, which can be differentiated by gel electrophoresis. The long one contains both primer sequences (forward and reverse), the middle one contains only one primer sequence (either forward or reverse), and the short one contains no primer sequence. Two unique mechanisms, multi-priming and template-switching, have been proposed to explain the phenomenon (Uemori et al., 2007).
ICA is a new technology published in 2010 (Jung et al., 2010). It uses two pairs of primers (outer and inner primer pairs) together with RNase H and DNA polymerase. Hybridase thermostable RNase H (Epicentre Biotechnologies, Madison, WI, USA) and Bst DNA polymerase were used for the amplification at 60 °C. The outer primer pairs are similar to those of SDA. The inner primer pairsare also similar to those of SDA, as shown in Fig. 3.2. The only difference is that a stretch of RNA residues is designed in the nicking region for RNase H cleavage. The major products are double-stranded nucleic acids, including either the forward inner primer or the reverse inner primer. Besides the main amplification products, two other double-stranded nucleic acids are also expected to form, one that includes both the forward and the reverse inner primers and another that is composed of the region inside both the forward and reverse inner primers. However, this technology easily forms non-specific products (primer dimers) from negative samples and low copy number of template samples, based on the published information (Jung et al., 2010).
Cycling probe technology (CPT, dual-labeled chimeric DNA-RNA-DNA probe) is a sequence-specific real-time detection method (Duck et al., 1990). It can be applied for both ICAN and ICA technologies. In the real-time ICAN/ICA, RNase H has two roles. It introduces a nick to chimeric primers to drive the amplification reaction, as well as cleaving a hybridized probe to separate the fluorescence quencher from the reporter, resulting in increased fluorescence.
3.3.4 RCA and related technologies
Rolling circle amplification (RCA) of circulable oligonucleotides (‘padlock’ probes) is a method for the detection and amplification of short DNA sequences. The padlock probe consists of a single-stranded oligonucleotide whose 5′ and 3′ ends hybridize to a target of interest and are then ligated to create a single-stranded DNA (ssDNA) circle, which is then a substrate for RCA (Fire and Xu, 1995). However, RCA is a linear amplification process with only one primer, whose sequence is complementary to the ssDNA. It can take several hours to obtain a detectable signal. In order to improve the amplification efficiency, different formats of exponential RCA have been proposed, such as ramification amplification (Zhang et al., 1998), hyperbranched RCA (Lizardi et al., 1998), cascade RCA (Thomas et al., 1999) and exponential RCA (Alsmadi et al., 2003; Beals et al., 2010). All of the above exponential RCA technologies use one additional primer (second primer), with a sequence identical to a part of the ssDNA, to generate tandem repeat products. The second primer binds at every tandem sequence synthesized by the first primer. The second primers are extended by the polymerase to displace downstream growing strands, which in turn contain a binding site for the first primer. Therefore, exponential amplification is achieved by the two primers, continually initiating new rounds of strand displacement synthesis to provide new binding sites for the primers.
The amplified products are different from linear RCA and exponential RCA. Linear RCA generates a population of long single-stranded products, also called linear concatenated DNA molecules. Each product contains repeats complementary to the individual padlock template. It looks like a broad smear of high molecular weight DNA filaments by gel electrophoresis. The tandem repeatsgenerated by exponential RCA are shown as a double-stranded DNA ladder by gel electrophoresis.
Primer generation RCA (PG-RCA) is a special kind of exponential RCA (Murakami et al., 2009). It uses only ssDNA, nicking enzyme and DNA polymerase. No exogenous primers are required. The circular probe carries not only the hybridization sequence to the target but also the complementary sequence of the nicking enzyme. The reaction starts from ssDNA hybridization to a template DNA, and is followed by a cascade reaction of linear RCA and nicking reaction. The nicking reaction creates multiple primers for the circular probe from the linear RCA product. The smear and ladder DNA are observed on gel electrophoresis. However, background noise (template-independent DNA ladder products) is a crucial concern for this technology to be further applied. This is similar to some other isothermal technologies using a nicking/restriction enzyme together with a DNA polymerase.
RCA can use several real-time detection methods for product detection, such as molecular beacon (Nilsson et al., 2002), PNA probe (Smolina et al., 2004; Kuhn et al., 2001) and molecular zipper (Yi et al., 2006). Padlock probes are powerful reagents capable of distinguishing single nucleotide changes in DNA samples. However, it is difficult to conduct circularization of padlock probes and amplification in one tube simultaneously. Therefore, multiple steps are required. This limits its application in the field of rapid diagnostics. So far there are no commercial kits available for in vitro diagnostics.
3.3.5 RPA and HDA
Recombinase polymerase amplification (PRA) (Piepenburg et al., 2006) and helicase-dependent amplification (HDA) (An et al., 2005) both depend on synchronization of multiple proteins to amplify nucleic acid in vitro. Compared with previously discussed isothermal technologies, these two methods utilize just one pair of primers, which is similar to PCR. The core idea of the technologies is to maximally mimic the natural process in vitro. RPA mimics the process of recombination, while HDA mimics the process of the mismatch repair pathway.
PRA is mediated by the coordinated activities of four proteins: T4 UvsX (recombinase, RecA homologue, ATP-dependent ssDNA binding protein), T4 UvsY (recombinase loading factor), T4 gp32 (ssDNA binding protein) and Bsu DNA polymerase (from Bacillus subtilis). The recombinase forms a complex with DNA primers (pre-synaptic filament) and scans the duplex template for homologous sequences. When the homologous sequence is found, primers hybridize to the target sequence through a strand exchange mechanism and displace the parent strand. Primers are extended by DNA polymerases in both directions. UvsY stimulates the reaction in two ways: displacing the gp32 protein from ssDNA and interacting with UvsX to stabilize its interaction with the primers. Gp32 can bind the parent strand when the strand is displaced duringsynthesis. A low reaction temperature (37–42 °C) and fast reaction speed (15–30 min) represent the main advantages of RPA.
However, the precise rules of primer design are not yet known. Trial-and-error method is applied for primer design and selection. Usually primers are selected as 30–35 nt in length, longer than those used in PCR. As a matter of fact, longer primer sequences increase the chances of non-specific amplification at low temperature. Primer dimers (non-specific amplification) are typically seen in non-template control and very low copy number of target amplification (manufacturer’s package insert, TwistDx Limited, Babraham, Cambridge, UK). In order to resolve the issue, target-specific probes, such as exo probe, fpg probe or LF probe, are applied in real-time and end-point detection (manufacturer’s package insert). However, it is not a fundamental solution. The sensitivity loss by dimer formation is not solved. Furthermore, there is still a certain distance from the PCR assay in terms of robustness and repeatability of the real-time data, based on the published information. All the above issues have limited its broad application in the research and diagnostic field.
HDA is mediated by the coordinated activities of three proteins: Tte UvrD (helicase, unwinding duplex), ET SSB (thermostable ssDNA binding protein) and Bst DNA polymerase. It employs a similar reaction mechanism to PCR, with the exception that HDA uses a helicase enzyme rather than heat to separate doublestranded DNA. Like PCR, the simple reaction scheme requires a pair of primers, a protein mix and buffer. This is the simplest platform in all the isothermal technologies. The similarity between HDA and PCR and the simplicity of the HDA scheme makes it easy to apply variant real-time PCR detection chemistries to HDA (Tong et al., 2008; Tong et al., 2012). The primer design and selection are aided by Primer 3 software, http://bioinfo.ut.ee/primer3-0.4.0/primer3/ (manufacturer’s package insert, BioHelix Corp., Beverly, MA, USA). However, the trial-and-error method is still important for assay optimization. This technology is more attractive with recent improvements in speed, sensitivity and robustness (Tong et al., 2011). Moreover, it can also be adapted to an instrument-free detection format, the BESt™ analyzer, for point-of-care diagnostics (Chow et al., 2008).
3.4 Isothermal amplification based on RNA polymerases
Nucleic acid sequence-based amplification (NASBA), transcription mediated amplification (TMA) and self-sustained sequence replication (3SR) all depend on RNA polymerase instead of DNA polymerase (Sooknanan and Malek, 1995; Kacian and Fultz, 1995; Guatelli et al., 1990). They are highly similar to each other. The techniques utilize a reverse transcriptase to produce DNA from the RNA templates, and an RNA polymerase to make RNA from a promoter engineered in the primer region. Since reverse transcriptase has RNase H activity, additional RNase H is an optional enzyme to remove the RNA from cDNA without heat denaturation. One pair of primers (inner primers) is required for the amplification. One of them can bea regular target-specific sequence. However, at least one of the primers is designed as a ‘promoter-primer’, which contains a highly conserved 5′ promoter sequence recognized by T7 RNA polymerase at the 5′end (Fig. 3.2). Double-stranded DNA intermediate products are formed by the amplification of two primers, and then an RNA polymerase recognizes the promoter sequence in the DNA template and initiates transcription. The amplification occurs at around 41 °C. ‘Beacon Designer’ from PREMIER Biosoft provides assistance with primer design. Generally, the promoter region is designed as 5′-AATTCTAATACGACTCACTATAGGG-3′. The first ten nucleotides following the promoter sequence should be a purine-rich sequence. Otherwise, extra purine residues (e.g. AGA, AGG) can be inserted immediately after the promoter region to prevent abortive transcription.
NASBA has been commercialized by bioMérieux Inc. (Durham, NC, USA), while TMA has been manufactured by Gen-Probe Inc. (San Diego, CA, USA). The primary products of amplification are single-stranded RNA (with length in the range of 120–250 nucleotides in most cases) and thus can be applied to detection formats by using probe hybridization without any denaturation step. Molecular beacon and hybridization protection assay (HPA) are implemented for specific product detection (Leone et al., 1998; Arnold et al., 1989; Jonas et al., 1993). This group of technologies is naturally suitable for RNA amplification and detection. However, a recent study by Deiman et al. showed that the efficiency of DNA amplification can be significantly improved by incorporation of restriction enzyme digestion prior to amplification (Deiman et al., 2008). Since RNA products are more labile outside the reaction tube than DNA products, the risk of laboratory contamination is thus substantially reduced. This is one of the advantages compared with other isothermal amplification technologies.
3.5 Future prospects
Since the early 1990s, the isothermal amplification approach has been developed into a simple, rapid and cost-effective tool by several distinct technologies. Some technologies depend on a simple protein mixture (like PCR) but multiple primers, while some technologies depend on a simple primer design (like PCR) but a complicated protein mixture. In the past two decades, some of the technologies have been successful in the transition from the research bench to the clinic. Several FDA-approved or CE-marked diagnostic kits have become available for use: for example, illumigene® C. difficile from Meridian Bioscience Inc. (based on LAMP), IsoAmp® HSV from BioHelix Corp. (based on HDA), ProbeTec Herpes Simplex Viruses Qx Amplified DNA Assays from Becton Dickinson (based on SDA), APTIMA Combo 2 Assay (CT/NG) from Gen-Probe Inc. (based on TMA) and NucliSENS EasyQ® HPV from bioMérieux Inc. (based on NASBA). Table 3.5 is a summary of the clinical applications of isothermal amplification technologies. It has fully demonstrated the feasibility and robustness of using the technologies for clinical application.
Table 3.5
Clinical applications of isothermal amplification technologies
| Technology | Manufacturer | Website | Multiplex capabilitya | Examples of clinical applications (CE marked or FDA cleared IVD products)b |
| LAMP | Meridian | http://www.meridianbioscience.com/illumigene/ | N | illumigene® group B streptococcus, illumigene®.C difficile |
| SDA | BD | http://www.bd.com | Y | BD ProbeTec™ ET CT (Chlamydia trachomatis), BD ProbeTec™ ET GC (Neisseria gonorrhoeae), BD ProbeTec™ ET CT/GC, BD ProbeTec™ Qx HSV (typing) |
| CPA | Ustar | http://www.bioustar.com/en | NA | Mycobacterium tuberculosis (TB), Chlamydia trachomatis (CT) |
| HDA | Biohelix | http://www.biohelix.com | Y | IsoAmp® HSV (non-typing) |
| TMA | Gen-probe | http://www.gen-probe.com | Y | APTIMA CT, APTIMA GC, APTIMA COMBO 2, APTIMA HCV, APTIMA HPV, APTIMA HIV-1 PROCLEIX HIV-1/HCV Assay, PROCLEIX WNV Assay (West Nile Virus) |
| NASBA | bioMérieux | http://www.biomerieux-diagnostics.com | Y | NucliSENS EasyQ® HSV 1/2, NucliSENS EasyQH®HIV-1, NucliSENS EasyQ® HPV, NucliSENS EasyQ® Enterovirus, NucliSENS EasyQ® hMPV |
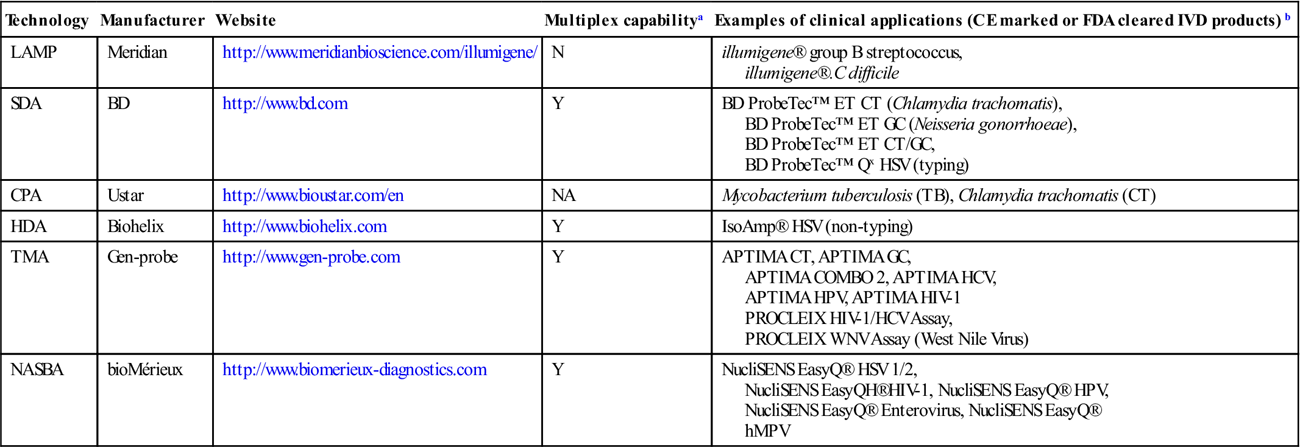
FDA, Food and Drug Administration; IVD, in vitro diagnostics.
Notes:
aDefined as the ability to amplify simultaneously at least two different targets (or one target and an internal control): yes (Y), no (N) or data not available (NA).
bThe commercial products available from each manufacturer are not limited to those listed in the table.
Although PCR is still the predominant technique of nucleic acid amplification for research and molecular diagnostics, the need for temperature cycling not only increases the cost of PCR-based devices, but also complicates the use of PCR in point-of-care or resource-limited settings. Isothermal amplification technologies are more suitable for these situations because they require less complicated thermal control than PCR. Without the need for thermal cycling, an isothermal system can be designed as a simple and low-energy consuming device. Therefore, it may outperform PCR in a portable, electricity-free amplification and detection system in the future. In recent years, active research work has been focused on the inexpensive, disposable, integrated diagnostic unit. In the innovated unit, sample lysis and nucleic acid extraction, amplification and detection are incorporated into a single unit. There are several advantages of the miniature unit, for example, less hands-on time, fewer steps, easy operation, prevention of cross-contamination and less use of expensive reagents. The integrated instrument can overcome the factors that have limited the practical application of POC devices for patient care.
There have been several reports on the mini-integrated devices developed from the technologies discussed in this chapter. Lutz et al. (2010) and Shen et al. (2011) reported, respectively, on using a fully automatic lab-on-a-foil system and a digital slipchip based on RPA. Mahalanabis et al. (2010) demonstrated an integrated microfluidic chip that performed HDA. Dimov et al. (2008) developed a microfluidic chip using NASBA. Sato et al. (2010) developed a microbead-based RCA system for DNA detection. Several microsystems have also been developed based on LAMP (Fang et al., 2010; Huang et al., 2011; Wang et al., 2011; Liu et al., 2011; Lee et al., 2008). However, most of the systems are in the research and design phase and have not reached the manufacturing stage. Some data presented in the research paper are short of amplification repeatability and precision, according to reviews by molecular diagnostic scientists. In order to develop the system further for clinical application, collaboration among experts in mechanical engineering, software engineering and molecular diagnostics is necessary.
For more details of individual isothermal amplification technologies, the original publications and some review literature have provided valuable information. For example, the review by Niemz et al. (2011) provides clear figures and summary tables of different technologies. The review by Gill and Ghaemi (2008) provides clear information on the amplification mechanisms. And several reviews provide a good reference source for the microchip system (Asiello and Baeumner, 2011).