
Microfluidic devices for rapid identification and characterization of pathogens
H. Becker, T. Hansen-Hagge and C. Gärtner, microfluidic ChipShop GmbH, Germany
Abstract:
The scientific progress in microfluidics and the technical development of microfluidic-enabled devices for diagnostic applications have led to a significant growth in industrial interest in bringing such systems into the diagnostic market. In this review, we discuss the underlying technological challenges present in the development of microfluidic devices that integrate all steps of a molecular diagnostics workflow, for example. We present a variety of examples of how different types of pathogen analyses are carried out using microfluidic chips. Different chip-based methods such as polymerase chain reaction (PCR) or immunoassays are discussed in detail. An overview on existing commercial solutions complements these examples.
Key words
microfluidics; cartridge; chip; molecular diagnostics; immunoassay; polymerase chain reaction (PCR)
8.1 Introduction
Similarly to the microelectronic revolution 50 years ago, a comparable development can be seen with the introduction of miniaturization in the life sciences with the initial concept of the so-called ‘miniaturized total analysis system’ (μ-TAS), also often called ‘Lab-on-a-Chip’ (LoC) technology, which deals with the handling and manipulation of miniature amounts of liquid in analyses conducted within life sciences research and was introduced about 20 years ago (Manz et al., 1990; Vilkner et al., 2004; Lee and Lee, 2004; Haeberle and Zengerle, 2007; Becker, 2008). Recent years have witnessed an explosive growth of scientific activities in the Lab-on-a-Chip technology. While the number of scientific publications within the ‘microfluidics’ area has dramatically increased in the timeframe of 2000 to 2010, the progress in commercializing microfluidicsenabled products has been much slower than anticipated (Blow, 2009; Becker, 2009a), reflecting a ‘looking for a problem methodology’ rather than a widely used truly enabling technology. In the last few years, however, this situation has changed, as a critical mass of knowledge seems to have been achieved, which led to a significant increase in commercial activities making use of LoC technologies. Nowadays almost no product development in the field of diagnostics (especially molecular diagnostics) or the analytical sciences takes place that does not in one form or another involve elements with microfluidic functionality. Furthermore, the value of this technology has finally also been realized in the markets, as can be seen from the large number of acquisitions in the field. The technological as well as commercial challenges for microfluidic technology will be described in this paper using the example of microfluidic devices designed for rapid identification and characterization of pathogens.
8.2 Challenges and technical as well as commercial solutions
There is unquestionably a need for smart, robust, reliable as well as rapidly functioning, and portable devices able to detect an infection of harmful pathogens or a biological threat (Weile and Knabbe, 2009).
First very simple ‘dipsticks’ have been sold as ‘lateral-flow tests’ since the late 1980s (Mark et al., 2010). Examples that are still on the market today are test strips for pregnancy (Chard, 1992), drug abuse (Litman et al., 1983; Pacifici et al., 2001), cardiac markers (Siebenhaar et al., 2010) and also upcoming bio-warfare protection (Shyu et al., 2002). Apart from these very basic analysis systems, the number of appropriate microfluidic devices on the diagnostic market has only been slowly increasing. One explanation of this phenomenon is the presence of a significant number of still underestimated challenges and obstacles, which have to be overcome during device development (Becker et al., 2008; Mark et al., 2010 and 2012). A (semi-)quantitative analysis is always accompanied by a set of control or reference analyses performed in parallel, which accordingly increases the number of microfluidic channels, valves (Oh and Ahn, 2006), mixers (Nguyen and Wu, 2005) and control or monitoring elements. All these elements have to find their place on a cartridge with limited footprint and have to cooperate in a smooth manner.
Complexity as well as distinctive features of the analytical process (e.g. discrimination of different HPV sub-types (Anic and Giuliano, 2011; see below) via multiplex analysis or investigation of pathogens in grain, which is quite resistant to lysis, etc.) will strongly affect the choice of chip material (e.g. polymers with high glass transition temperature and low autofluorescence; material that is inert to organic solvents; structured glass, etc.), geometry (e.g. microtiter plate or microscopy-slide-sized chips, compact disc type cartridge, laminated cartridge layers, etc.), dimensions and fabrication mode of the cartridge (e.g. hot embossed, injection molding, etching, etc.). For disposable devices, which are required for any diagnostic test, polymers turn out to be the material of choice in most cases due to material cost and fabrication options.
An early but periodically repeated estimation of fabrication costs (COG, costs of goods) during the course of diagnostic device development is essential in order to achieve the acceptance of the future product by the market (Becker, 2009b). Here, it has to be kept in mind that the cartridge is not composed only of structured plastic but, rather, a composition of quite different materials and elements (e.g. membranes for filtration, magnetic beads for nucleic acid purification, gold arrays for electrochemical detection, etc.). Against this background, it is also very important to keep in mind that the assembly of different elements to a functional unit (back-end processing) will considerably affect the costs of production for the final integrated device. In analogy to fabrication experiences in the microelectronic world, these so-called back-end processes can account for up to 80% of the overall manufacturing cost, and therefore have to be thoroughly investigated during product development.
After selection of cartridge material and mode of fabrication, it is recommended to define, produce and validate separately each individual function of the bioanalytical process protocol, such as sample processing, cell lysis, filtration, analyte purification, target sequence amplification, and detection by the use of dedicated microfluidic modules. Positively evaluated units can subsequently be compiled into an integrated platform. This process ideally culminates in a fully integrated sample-in answer-out cartridge.
A typical bioanalytical process encompassing sample collection and processing, analyte amplification, analyte purification, detection and the overall solution management is schematically illustrated in Fig. 8.1. The type of sample can be very different (e.g. biopsy, swab, sputum, blood, etc.) and, accordingly, the ‘world-to-chip’ interface (Schulte et al., 2000) has to be adapted to the respective type of sample. Although these interfaces are still an often overlooked but important item during product development, more and more existing standards from the targeted application area (e.g. Luer-Lock compatible interfaces in clinical diagnostics) are used, however, with disadvantages mainly in terms of size. For this reason, a similar press-fit interface with a reduced footprint (called ‘mini-Luer’) has been developed, allowing up to 32 fluidic ports on a device the size of a microscopy slide.

The next step – the various sample preparation processes, such as disintegration and/or dissolving of the sample, cell lysis, extraction of DNA/RNA, sample concentration, etc. – have so far typically been carried out off-chip due to their complexity and the different nature of the various samples. Moving these steps onto the device represents the biggest challenge (Kim et al., 2009), because several media (wash buffer, carrier buffer, beads, lysing agents, etc.) usually have to be handled sequentially, robustly and with a high precision in terms of volume, times or sequence. All processes require interfaces and plumbing in very restricted device areas.
As discussed later in more detail, the next process step usually is the amplification of the analyte if nucleic acids are used as analytical targets. This amplification step is then frequently followed by a process step for the separation of the analyte of interest. For this separation, various techniques exist, such as electrophoresis, chromatography (up to now not well developed on-chip) and the use of capture probes (e.g. DNA arrays) or other filtration mechanisms in order to isolate the desired component spatiotemporally or to remove unwanted components from the mixture.
The final analytical step comprises the detection of the analyte of interest. For many larger, lab-based systems, optical detection methods (Kuswandi et al., 2007) like laser-i nduced fluorescence still act as a benchmark with respect to sensitivity. For portable systems, electrochemical analysis methods or various other sensor methods (e.g. electrochemical sensors, surface acoustic waves (SAW), quartz crystal microbalance (QCM), thermal measurements) are becoming increasingly of interest. It should be noted that all the preceding process steps have to be matched to the selected detection method in order to generate the best results.
A minor but nevertheless important design step of an integrated device in diagnostics is the layout of a waste container system in order to retain all liquids used in the process on-chip. This is often necessary in order to avoid the risk of contaminating the instrument and to prevent carry-over from one measurement to the next. The required volume of such waste reservoirs can be critical, frequently stressing the limited real estate on the chip.
As mentioned above, the type of analyte, pathogen and sample source affect the strategy of resulting approaches for the development of microfluidic analytical devices. Normally, analytic targets are either nucleic acids (McCalla et al., 2011) or pathogen-indicating proteins (Kuswandi, 2007; Lee and Lee 2004; Lee et al., 2009, 2010). In some cases, such as HIV diagnostics, intact cells are the targets for an immunological analysis (Chin et al., 2011). Pathogens (Gordon et al., 2011; Loubiere and Moatti, 2010; Franz, et al., 1997) can belong to quite diverse classes of different biological kingdoms, such as eukaryotes (helminths, fungi, etc.), prokaryotes, and – very important – viruses. The latter encompass a very central class of pathogenic agents. As a first overview, a non-exhaustive list of commercial (available) devices is given in Chin et al. (2012).
8.3 Pathogens and analytes
One important advantage of the LoC technology (here we expand the notation from ‘lab on a chip’ also to lab on a cartridge/compact disc/cassette) (Janasek et al., 2006; Manz et al., 1990) is its POC (point of care, but here also used in the context of point of occurrence) applicability (Lewandrowski, 2009). To illustrate this, different scenarios are described in the following.
Robust, reliable, and cheap POC testing of HIV (Briggs et al., 2012) – especially, but not exclusively, in developing countries – is one very urgent need. This includes both first diagnosis of the disease as well as monitoring of the course of the disease under therapy. A second family of viruses, which should be controlled at the POC, is represented by the influenza virus (Mak et al., 2012). Here, LoC-based bioanalytical testing facilitates a comprehensive monitoring of the spreading of the infection from a local outbreak to an epidemic or even pandemic dissemination. In addition, LoC devices will allow a rapid discrimination between harmful and ‘harmless’ virus strains. And a rapid LoC-based influenza diagnosis in the doctor’s office by a general practitioner will allow an immediate therapeutic response to the infection and may help to prevent spreading of the influenza inside the doctor’s waiting room. As a third example, human papilloma viruses (HPV) have been intensively studied, and it has been found that certain HPV strains cause cervical cancer and others do not. Fortunately, an early therapy can erase the viral infection and the concomitant risk of cancer. Against this background, it is anticipated that POC diagnostics will permit rapid screening for those HPV subtypes causing cancer (Saxena et al., 2012). This will definitely increase the frequency of identified risky HPV infections, will decrease the time span between diagnosis and therapy, and, thereby, will help to save lives.
The battle against bacterial infections is still not won – despite antibiotics. This is due to the capability of bacterial pathogens to develop multiple resistances against antibiotic drugs in a surprisingly short period of time. Accordingly, increasing interest can be noted in the development of improved therapeutic as well as diagnostic tools (Mothershed and Whitney, 2006) for bacteria such as methicillin-resistant Staphylococcus aureus (MRSA; in fact, the name of this S. aureus strain could also be multi-resistant Staphylococcus aureus) (Lu et al., 2013). It is expected that a rapid and locally focused monitoring of MRSA – and other multi-resistant bacteria – will support containment of further outbreaks of the respective types of pathogens (Focke et al., 2010). Mycobacterium tuberculosis (Niemz et al., 2012) is also a pathogen able to gain multiple resistances against drugs. On top of multi-resistant Mycobacterium tuberculosis, totally drug-resistant TB was identified in 2002 in Italy. Other activities focus on the development of POC tests for lower respiratory tract infections such as community or respiratory-acquired pneumonia or tuberculosis (see, e.g., the European project RAPP-ID, http://www.imi.europa.eu/content/rapp-id). In summary, an adequate response to the ‘rearmament’ of pathogenic bacteria is urgently required and POC diagnostics will be one important weapon to win this ‘war against antibiotics resistance’. On top of this, bacteria are also misused as weapons by humans. This creates a strong need for POC diagnostics in the framework of CBRN (chemical, biological, radiological and nuclear) defense scenarios as well.
The amplification of target DNA sequences via PCR (Mullis et al., 1986; Saiki et al., 1988) is a very efficient and well-established technique for the detection of the respective target in the investigated sample. Accordingly, this methodology is well established in food, clinical, forensic and veterinary analysis and – to a slightly lesser extent – POC diagnostics (Zhang and Xing, 2007; Park et al., 2011). Unfortunately, the detection of pathogen-indicating DNA does not necessarily indicate the presence of an ongoing infection with the respective living and virulent pathogen. Due to the enormous sensitivity of this amplification technique, residual debris of destroyed pathogens within the sample could also give rise to a false (regarding biological threat) positive signal. One possible way out of the above dilemma is the examination of RNA instead of DNA (Mocharla et al., 1990). The RNA pool represents a collection of all expressed genes of a living organism. Thus, the turnover of the pool happens very rapidly. As a consequence, this phenomenon keeps the monitoring of expressed genes and, therewith, of the infection status up-to-date. As stated above, and as will be described in detail below, the development of a miniaturized PCR platform will have to address a large number of challenges before such a complex technology will be successfully implemented in a small device.
The conversion of the immunological ELISA (enzyme-linked immunosorbent assay) approach (Henares et al., 2008; Dutse and Yusof, 2011; Engvall and Perlmann, 1971) into a LOC device (Rossier and Girault, 2001) is less complicated than the PCR approach. As a consequence, this technique is realized in quite different lateral flow devices (Glad and Grubb, 1978; Martinez, 2011; Clerc and Greub, 2010). Moreover, the method is sensitive enough to offer an unequivocal diagnosis of pathogens in many cases (von Lode, 2005) and the antibody/antigen interaction can specifically monitor and quantify molecules such as drugs and metabolites (Ibrahim et al., 2009), large proteins (Minkstimiene et al., 2009), nucleic acids (Blazkova et al., 2009) and even whole pathogens (Rica et al., 2009). The quality of each individual analysis depends on a large variety of different parameters (density of the trapping antibody, temperature, pH, storage time of chip and ingredients until analysis, etc.). Accordingly, control experiments with internal standards are recommended in those cases where a simple ‘yes’ or ‘no’ is not sufficient. Such a sophisticated approach cannot be achieved by most lateral flow devices. In addition, the implementation of internal standards or multi-parameter analysis (Lee et al., 2009) is also desired because this allows semi-quantitative information to be obtained with respect to the number of infectious organisms in the investigated sample.
8.4 Chip-based analysis of protein-based analytes in microfluidic devices
The term ELISA describes a technique in which biomolecules are immobilized on an activated solid phase in an initial step (Fig. 8.2(a)). In order to become activated, the surface of chip-based cavities or channels must be, for instance, exposed to oxidizing plasma and subsequently incubated with an appropriate chemical surface modifier, which carries reactive chemical groups, such as the epoxy group. These chemical groups finally allow the immobilization of the trapping molecule (e.g. antigen; Fig. 8.2(b)) to the surface. This can be an antigen, an antibody or any other biomolecule that can be involved in an immunological binding reaction. The next step is the visualization of the immobilized complex of trapping and target molecule, as depicted in Fig. 8.2(b). In this example, a secondary antibody binds to an antigen as target and visualizes the complex with the help of a fluorescent dye.

As described earlier, the very basic design of lateral flow devices does not allow quantitative information to be generated at a high sensitivity level. In fact, lateral flow devices only allow a qualitative result of reduced sensitivity. Although sufficient in many cases, the potential of this still commercially very successful technique has reached its limit. Accordingly, cartridge-based ELISA tests have been developed in order to improve the reliability, sensitivity and robustness of analyses at a POC environment.
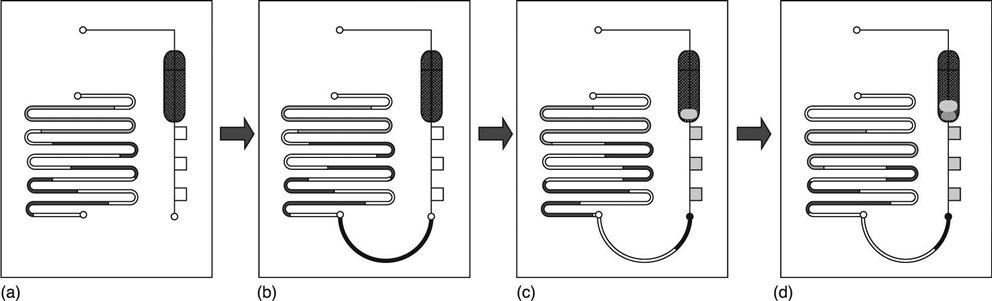
Similarly to the sample repertoire of lateral flow devices, the majority of cartridge-based ELISA tests are designed as blood analysis tools, where either blood plasma or serum is separated from blood cells by on-cartridge filtration or whole blood is directly analyzed, as is realized in the case of a device developed by the company Daktari Diagnostics. Alternatively, the blood sample is lysed onchip and transported in processed form over the sensor area (Table 8.1) (listed as a Claros Diagnostics technique, now a subsidiary of Opko Diagnostics) (Fig. 8.3 and 8.4). This approach is exemplified by a device from Opko Diagnostics (http://www.opko.com/products/point-of-care-diagnostics/) with a workflow shown in Fig. 8.3. As shown in Fig. 8.3(a), the cassette is composed of two independent fluidic systems, each being essentially a long channel with variable geometry. The system shown on the left is used for the storage of multiple liquid reagents in the form of plugs of liquid separated by gas segments. The geometry of the channels is optimized for stability of the air–gas interface upon storage. The system shown on the right is designed to perform the assay: from the inlet (bottom) the channel defines multiple detection zones, and then turns into a waste collection chamber. Each of the detection zones can be coated with a different capture probe, to perform multiplex assays. The illustration shows three zones, although actual designs have up to ten detection zones. At the time of manufacture, the cassette is loaded with appropriate reagents (in storage channels, and detection zones). All ports of the fluidic systems are sealed for transportation and storage. To operate the test, the user collects the specimen of whole blood in a sample collection device. The volume of 12 μ1 of blood necessary to run the assay can be obtained from a finger stick. The user connects the collection device to the cassette (Fig. 8.3(b)), effectively connecting the two fluidic systems into one long fluidic structure. The cartridge is inserted in the analyzer, which applies a source of vacuum at the outlet of the system (at the port located downstream of the waste chamber). The pressure gradient sustains flow of sample into the detection zones, and also starts moving the reagent plugs towards the waste collection chamber (Fig. 8.3(d)). The fluidic system is essentially a single conduct (Fig. 8.3(d)), and all fluids flow towards the vacuum source, sequentially traveling through the detection zones. The last fluid to flow through the detection zone is an amplification reagent, which creates an optically detectable signal.
Table 8.1
List of companies offering microfluidic devices for pathogen analysis
| Company | Analyte* | Pathogen | Remarks | References | ||
| NA | Pro. | Cell | ||||
| Advanced Liquid Logic | X | X | X | HIV/MRSA In development | Sample prep. via ‘electrowetting’ | http://www.liquid-logic.com/technology |
| Alere | X | HIV | Via CD4 T-cell counting | http://www.alere.com/content/dam/aIere/docs/pressreleases/1107aIere14decpimacd4.pdf | ||
| Alere | X | HIV | Via HIV-specific antibodies | http://www.alere.com/content/dam/aIere/docs/pressreleases/1107aIere14decpimacd4.pdf | ||
| Arcxis | X | Infectious diseases | DNA and RNA preparation from blood | http://www.arcxis.com/ | ||
| Biosurfit | X | Viral infection In development | Compact disc and detection by surface plasmon resonance | http://www.biosurfit.com/ | ||
| Cepheid | X | MRSA/TB | Integrated microfluidic process from sample lysis to data read-out | http://www.cepheid.com/us/cepheid-solutions/systems/genexpert-systems/genexpert-iv/63?view=products | ||
| Opko Health Inc. | X | Infectious diseases | Chip-based ELISA | http://www.opko.com/products/point-of-care-diagnostics/hardwaredevice/ | ||
| Daktari Diagnostics | X | HIV | CD4+ T-cells are captured, lysed and the impedance of the lysate is monitored | http://www.daktaridx.com/ | ||
| Focus Dx (Quest) | X | Dengue viruses and other | Chip-based ELISA | http://www.focusdx.com/pdfs/pi/OUS/RT1500GM_G.pdf | ||
| Genefluidics | X | X | Non-specified pathogens | Cartridge for supply of electrochemical ELISA | http://www.genefluidics.com/index.php | |
| HandyLab(BD) | X | Bacteria | On-chip sample processing, PCR amplification and detection | http://www.bd.com/geneohm/english/handylab/ | ||
| Idaho Technologies (BioFire Diagnostics, Inc.) | X | Viruses/bacteria | In-pouch platform, from sample processing via PCR to fluorescence detection | http://www.biofiredx.com/ | ||
| IQuum | X | Viruses/bacteria | Lab-in-a-tube; from sample processing via PCR to fluorescence detection | http://www.iquum.com/ | ||
| Micronics (Sony) | X | Malaria/E coli | Immunological test via diffusion within a laminar flow of different solutions | http://www.micronics.net/products/research-and-development/active-labcards | ||
| PositivelD | X | MRSA, HPV and others | From sample processing to PCR | http://positiveidcorp.com/products_dragonfly.html | ||
| Rheonix | X | Infectious diseases | From sample processing via PCR to fluorescence detection In development | http://www.rheonix.com/ | ||
| Wave 80 Biosciences | X | X | HIV and other viruses | On cartridge amplification of lowconcentration analytes | http://www.wave80.com/technology.php | |
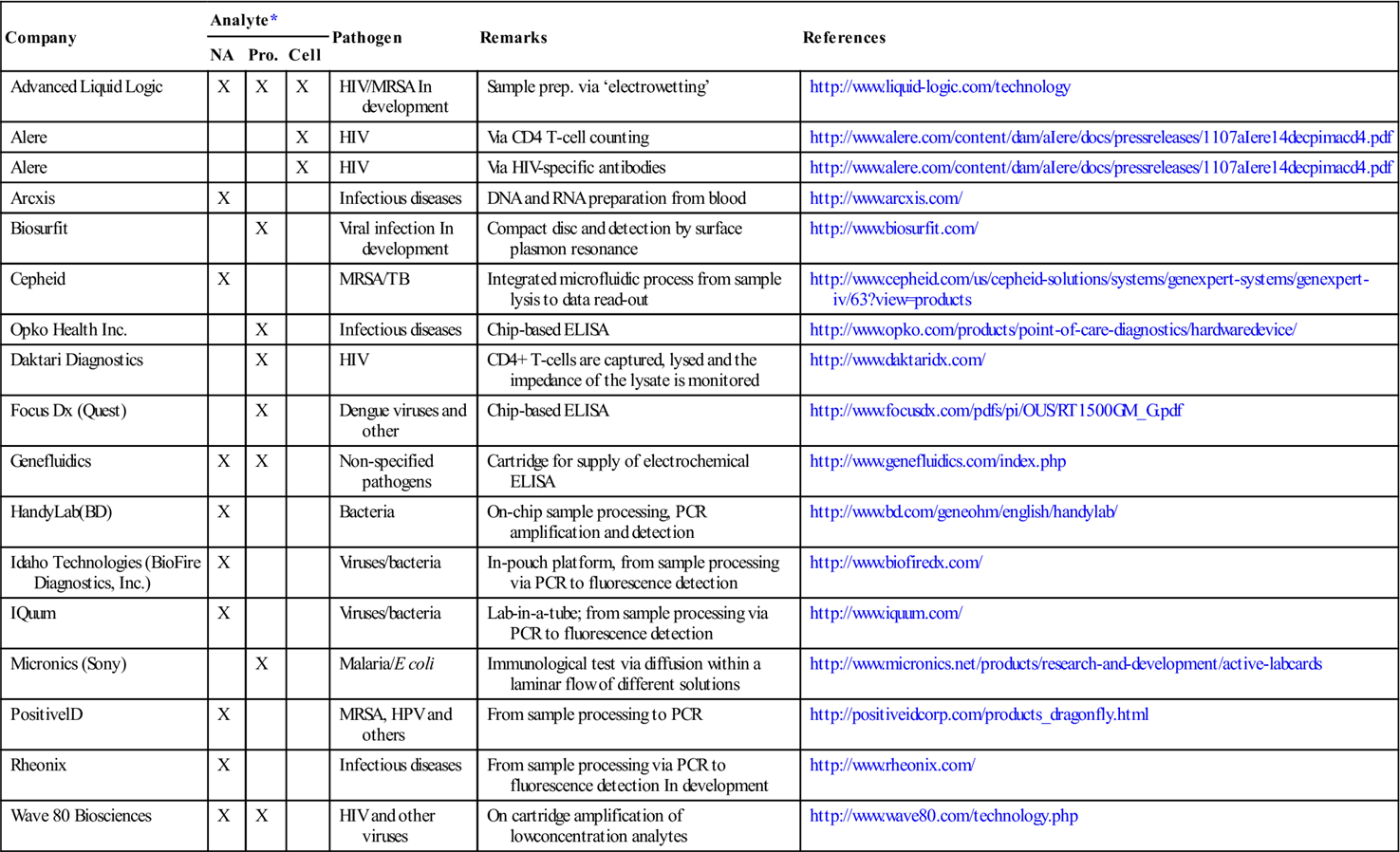
HIV=human immunodeficiency virus, HPV=human papilloma virus, MRSA=methicillin-resistant Staphylococcus aureus, TB = (multidrug- resistant) tuberculosis.
*NA=nucleic acids, Pro. = protein.


Capillary forces are the main moving power for lateral flow devices. In products more sophisticated than application of the lateral flow approach, fluid actuation is required, which can be realized in quite different ways. In the commercial devices listed in Table 8.1, for instance, capillary, pneumatic, centrifugal and, last but not least, gravitational forces have been applied. Pneumatic and centrifugal forces can be applied with the help of a syringe and a CD player-like device, respectively.
The implementation of sensors is as diverse as the mode of actuation. Detection can be by electrochemical (e.g. electrochemical impedance), fluorescent or colorimetric monitoring, or by adsorption. In order to improve the signal-to-noise ratio additionally, washing and dilution steps are included in the bioanalytical process of some of the devices. For this, the required liquids have to be provided, either from outside during the analysis or from reservoirs stored on board.
Many of the above products are still under development, and there are ongoing attempts to expand the portfolio of a system to additional analytes and, moreover, to simplify the technique in order to increase the acceptance of the user at the POC. Along these lines, mainly viruses, such as HIV, influenza or HPV, and bacteria, such as MRSA or Shigella toxin-expressing E. coli, are in the focus of portfolio expansion.
As a brief intermediate summary, the mode of actuation as well as control of flow direction (via valves, for instance) should be realized as simply as possible, in order to keep the configuration of the driving instrument manageable. The provision of liquids for reaction, washing steps and detection is an issue too. The storage of dissolved enzymes, for instance, will adversely affect the shelf life of the enzymes. Also, storage of dried ingredients next to water or buffer reservoirs may be problematic, because water vapor can penetrate into chambers which contain dried reagents, and thereby affect quality and shelf life of the biochemicals. Accordingly, the liquids should be stored separately in appropriate containers, which can be clicked onto the diagnostic cartridge just in time before use.
In an example of an ELISA analysis chip from our laboratory (Fig. 8.5), pathogenic Francisella tularensis bacteria have been used as test organisms and trapped onto the surface of ELISA test chips. The central part of this transparent cyclo-olefin polymer (COP) chip consists of six detection chambers, where the left four chambers are connected by one channel system. Cyclo-olefin polymer was chosen as cartridge material despite its comparatively high price because it has excellent optical properties, its material properties facilitate relatively easy structuring of microfluidic devices by injection moulding, and it can be obtained with a sufficiently high glass transition temperature.

All three independent sets of detection cavities can be separately addressed by the flanking turning valves as shown (Becker et al., 2012). The sample is introduced via a Luer adapter at the upper left side of the chip and the different solution can be applied to the chip by a set of three mini-Luers visible at the right side of the larger Luer adapter.
In addition, there are two upstream cavities in front of the two singular detection zones. These cavities support the long-term storage of dried/lyophilized reagents, which are necessary for the simultaneous performance of reference and control experiments. Finally, the lower left side of the chip shown is occupied by a large waste reservoir with an integrated venting hole covered by a hydrophobic membrane.
A corresponding detection test chip (Fig. 8.6–8.8), solely encompassing two detection channels, has been generated by injection moulding of transparent, black., or white COP depending on the requirements of the read-out mode. Figure 8.7 shows fluorescent (a) and colorimetric (b) monitoring of pathogenic Francisella tularensis bacteria, which have been trapped onto the surface of a chip-based cavity at the indicated position. The colorimetric test was mediated by the reaction of an enzyme (horseradish peroxidise; HRP), giving rise to the violet precipitates shown. The positive control consisted of a trapping molecule, which was directly labelled with the dye or HRP. Irrelevant targets (e.g. Yersinia pestis bacteria) were used as negative controls. As illustrated by Fig. 8.8, fewer than 10 000 bacteria per experiment can unequivocally be identified in this platform.




8.5 Chip-based analysis of nucleic acid-based analytes in microfluidic devices
For nucleic acids-based molecular diagnostics, the sample (blood, sputum, saliva, etc.) has usually to be processed in order to extract the nucleic acids. Moreover, the initial concentration of analyte in most analysis scenarios is too low for direct investigation. Accordingly, target sequence multiplications via isothermal amplification, such as nucleic acid sequence-based amplification (NASBA) (Compton, 1991), helicase dependent amplification (HDA) (An et al., 2005), strand displacement amplification (SDA) (Walker et al., 1992), rolling circle amplification (RCA) (Lizardi et al., 1998), or PCR are essential tools that additionally have to be implemented in the cascade of the on-board bioanalytical process chain.
Considering solely the temperature management, isothermal nucleic acid amplification is superior to PCR. Here, however, the utilized enzymes and chemicals are more sensitive to fluctuations in different environmental conditions (pH, ionic strength and composition, temperature stability) than the thermophilic polymerase and, in addition, it is a delicate task to coordinate the activity of a whole orchestra of different enzymes, which have to act in concert during isothermal amplification.
PCR is more robust in a biological sense and, reflecting this robustness, it is possible to amplify target sequences directly out of whole blood (Chas, 2009); the respective inhibitor-resistant amplification is based, however, on the usage of specialized enzymes and reaction mixtures, which are exclusively provided by corresponding enzyme manufacturers. Apart from this special situation, PCR still requires at least two, normally three or more, temperature regimes that have to be applied to the chip for more than 20–30 PCR cycles.
If the volume of the PCR mixture is small (nanoliters) and the walls of the reaction cavities are thin, the PCR can be performed stationary in a chamber. Here, the increase and, even more important, decrease of the incubation temperature can be performed rapidly enough (<30 min – a period of time that is believed to be acceptable to a user). Alternatively, the fluid plug (and not the temperature profile) is mobile and is moved over two or three different temperature zones, which cover all needed incubation temperatures in a constant manner. This so-called ‘continuous-flow PCR’ (Kopp et al., 1996; Köhler et al., 1998; Gärtner et al., 2007) is uniquely suited for chip-based nucleic-acid amplification. As a consequence, the fluid actuation has to be adapted to the required mobility of the plug. Using this approach, very rapid amplifications (<10 min for the amplification of single copy genes out of genomic DNA of eukaryotes) can be achieved.
Moreover, techniques for mechanical, ultrasonic, thermal or chemical release of the target sequence have to be realized on board a chip, cartridge or cassette as well. In this line, illustrative examples from our work are given in the following. In most cases, pathogenic organisms can be efficiently disintegrated (lysed) and, as an additional advantage, the nucleic acid-dissolving buffer can also act as a selective transfer medium for nucleic acids to a functionalized bead surface. Here, the nucleic acid is stored intermediately until all contaminants are washed away. In Fig. 8.9(b), a chip is schematically shown carrying such functionalized beads. Additionally, these beads hold magnetic cores, enabling the controlled actuation of the beads with the help of an external magnet. The magnet, as well as a heating system for incubation at elevated temperatures, is supplied by an instrument (Fig. 8.9(a)), which controls each step of the nucleic acid purification and isolation.

In the next step, the pure nucleic acid has to be amplified (Fig. 8.10 and 8.11). In order to streamline the duration of analysis, a continuous-flow PCR with a mobile liquid phase migrating over different stationary temperature phases is favoured (Wang et al., 2009). One realization of this type of approach is given in Fig. 8.10. Here, the PCR solution is pushed as a slowly but evenly moving plug through a meandering channel with the help of a syringe pump. Thereby, the plug passes two or three (shown in this example) different temperature zones. The geometry of the channel and the number of meanders define the number of PCR cycles and the length of each step within each cycle.


An alternative to the above flow-through-PCR is represented by the so-called Boyle-Mariotte-PCR (Fig. 8.11) (Brunklaus et al., 2012). Here, the PCR solution is pushed against the trapped air in a dead end channel system. The main advantage and disadvantage of this approach in comparison to the flow-through-PCR are the reduced space consumption and the higher efforts for flow regulation, respectively.
The final step of the process cascade from sample in to analysis result out is the detection of the pathogen-indicating sequence, for which one option is schematically depicted in Fig. 8.12. In this case, the biomolecules, here target-specific oligonucleotides, are immobilized on the surface of a cavity in an appropriate chip. The immobilized sequence specifically catches the matching reverse complementary hybridization partner out of the solution of single-stranded PCR product. Due to the fluorescent label on one sequence of the PCR primer set, the hybridization complex can be optically identified.

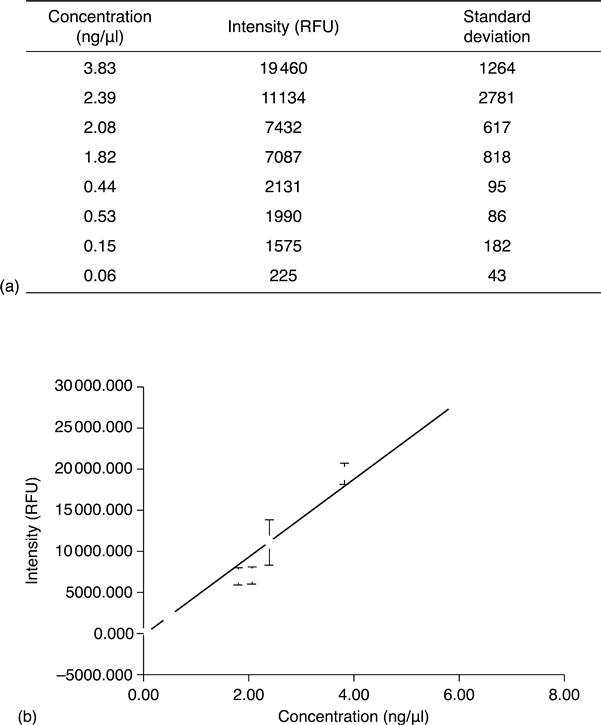
Results of such a hybridization experiment are given in Fig. 8.13. The data indicate that less than 100 pg/μl target PCR product can be detected by this approach.
8.6 Future trends
There is no discussion about the urgent need for simple, robust, reliable and rapid POC devices. The intention of this chapter is to give a tour d’horizon about the state of the art of microfluidic pathogen identification and analysis in quite different scenarios, such as putative threats from severe contaminations and infections as well as bio-weapons or immediate in situ health care. As also explained, the type of target molecules – DNA, RNA or antigenic protein – strongly influences the configuration of the LoC device. The type of organism – virus, bacteria or eukaryote – also has some effects on the design of the cartridge and the driving instrument. The complexity can additionally be increased, if the number of analytes, reference measurements or points of a standard row, which have to be included in the analysis, is high.
Reflecting this initial situation, quite different answers have been developed or will be commercialized soon. It is no surprise that currently most LoC devices on the market are directed towards antigenic proteins, which can relatively easily be implemented into a cartridge-based ELISA technology and which can be adapted to the lateral flow approach.
It is the intention of the more sophisticated LoC devices, however, to exhibit a better performance, a higher sensitivity as well as specificity, and a higher information density (e.g. inclusion of standard tests or testing of a panel of strains) than the simpler lateral flow devices. The technical challenge here, however, is that the ‘comforts’ of the lateral flow approach, such as low price per test or the portability of the device, must be retained. Regarding these points, there doubtlessly still remains significant developmental work to do, although technical progress in the area is impressive, as the given examples of commercialized devices illustrate.
We tried to illustrate, by examples of PCR- and fluorescence detection-based simultaneous investigation of multiple bacterial pathogens, that there is an impressive cohort of problems confronting us as well as currently available solutions to these challenges. PCR requires much space on the cartridge and driving instrument because a precise management of reaction temperature and volume (avoidance of evaporation at elevated temperature) is essential. Apart from this, the special requirements of the bioanalytical protocol and also the need for simultaneous investigation of different test samples imply an ambitious flow pattern of different liquids inside the cartridge. Moreover, the provision of these different liquids, as well as the management of actuation, motivates a still-ongoing progression of technical skills.
Currently, the need for smart POC LoCs meets a continuously growing repertoire of microfluidic tools, which enable scientists and engineers in this field to respond to the above prerequisites in a technically as well as commercially reasonable manner. The near future will certainly see a whole variety of such smart tools and devices.
8.7 Acknowledgements
Part of the work was carried out under the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 261810.