
Electrophoretic approaches to sample collection and preparation for nucleic acids analysis
C. Bradburne, Johns Hopkins University, USA
Abstract:
Gel electrophoresis has been an integral part of molecular biology labs for decades, finding utility in analysis, separation, molecular engineering and clean-up of nucleic acids. It continues to be refined, and emerging technologies are allowing fine control of DNA and RNA in a gel. One of these technologies, SCODAphoresis, even provides superior separation from common inhibitors such as soil humic acids, enabling DNA extraction and analysis from previously un-extractable sample matrices. The electrophoretic sample separation of nucleic acids will continue to be an important part of the sample-to-answer workflow, providing cleaner analytes for downstream assays like clinical diagnostics, polymerase chain reaction (PCR) and sequencing.
Key words
zone electrophoresis; SCODAphoresis; pulsed-field; isotachophoresis; dielectrophoresis; non-linear
13.1 Introduction
Sample preparation remains a vital, initial component of both laboratory analysis and field analytic detection systems for biomolecules. It is as critical as any decision about the assay platform, since it shapes the success of all downstream measurements. Technologies using electrophoresis have been used for years in specific applications of sample preparation, particularly for nucleic acid and protein processing, offering advantages in purity, analyte resolution, analysis and the potential for automation. Innovative methods based on electrophoresis have also begun to appear and find their way into novel applications. Here, we will examine the basis for current and emerging electrophoretic technologies for sample preparation, with a focus on nucleic acids. In particular, electrophoresis of DNA and RNA has progressed significantly from its historical use in genetic engineering, molecular biology and sequencing, and new applications are now being realized that offer simpler workflows, better purification, concentration and fine manipulation of individual species of DNA in a sample. Many of these applications can be automated and easily coupled with ‘lab-on-a-chip’-styled analytical platforms, and capillary sequencing or high-throughput analysis systems. Commercial platforms utilizing these technologies are enabling simpler workflows, standardization, better isolation and even high-resolution analysis of target nucleic acids. Of particular interest are difficult sample matrices such assoil, bone or tooth material, or organisms present at very low levels. Nucleic acid detection in many of these is now being enabled as techniques such as non-linear electrophoresis continue to advance.
In this chapter, we will examine properties and separation parameters for nucleic acids employing electrophoresis techniques. We will examine methods that use uniform direct current (DC) fields to drive sample separation from contaminants, such as pulsed-field electrophoresis and isotachophoresis. We will then discuss non-linear techniques that employ alternating current (AC) fields, such as dielectrophoresis and SCODAphoresis. Finally, we will compare sample preparation performance for several electrophoretic technologies, discussing such performance benchmarks as yield, purity, concentration and separation from contaminants.
13.2 Separation parameters for nucleic acids for use in sample preparation
Isolation of pure nucleic acids from their complex matrices is a vital component of any sequence detection strategy. For cellular applications, DNA and RNA are contained within some biological context: associated with a variety of storage, functional and regulatory molecules. Cell walls, phospholipids, lipopoly saccharides, proteins, and anabolic and metabolic organic compounds all need to be separated from nucleic acids prior to any downstream amplification and detection. The chemistries of these components are very different, and so purification methods tend to be complicated, or inefficient, such as nucleic acid-binding columns that only bind a small fraction of the DNA. Failures in sample prep can result in target loss, downstream assay inhibition such as inhibiting polymerase chain reaction (PCR), and false negatives. In addition, techniques that selectively over-represent a particular species relative to a target species can create the potential for false positives, and increase the ‘noise’ relative to candidate signals. Downstream nucleic acid biodetection is usually preferred because of its sensitivity and the robustness of the reagents to conditions, but it is also dependent on the application of comprehensive, non-biased and effective nucleic acid purification techniques.
There are a wide variety of applications that require nucleic acid purification, ranging from simple pure cultures to complex metagenomic environmental samples. Most purification techniques take advantage of some unique nucleic acid property or combination of properties as separation parameters to remove them from contaminating background. As with any biomolecular isolation method, useful separation characteristics include charge, size, density, length, the isoelectric point (pI), hydrophobicity and affinity. Depending on the desired assay, methodologies aim to exploit differences between the properties most likely to cause downstream results, separating the problematic species as much as possible. Examples for which robust commercial products exist include gel filtration products for size exclusion, charged silica beads for selective binding, anion exchange chromatography and electroelution from gel-like matrices. While most are effectivefor some types of mixtures, there remain many samples and applications in which the physicochemical associations between nucleic acids and the complex mixture of molecules in a sample can confound DNA and RNA separation.
Historically, the simplest way to perform DNA purification involved exploiting charge and hydrophobicity by implementing a solution-phase differential extraction between aqueous and organic solvents, specifically a mixture of phenol and chloroform. Proteins and other organic compounds were removed by separation into the phenol and chloroform or an insoluble interphase, while charged nucleic acids in the aqueous layer could then be further cleaned by selective solubility, precipitating with ethanol, in which other contaminants were soluble. The DNA was then resuspended in a polar solvent such as plain or Tris-EDTA-buffered water. While straightforward and removing the bulk of contaminants, this technique does not prevent the co-purification of charged organic molecules and metabolites with solubility properties similar to those of the nucleic acids, so they co-purify in polar solvents. Many of these compounds, such as the ubiquitous humic acids within soil, inhibit downstream enzymatic reactions such as PCR and sequencing, or create conditions that alter hybridization properties;1 the nucleic acid component must be purified further. Historically, methods that exploit size and density properties have been developed, but simpler and faster approaches are now available, typically using some type of column material that takes advantage of some combination of size, charge and hydrophobicity properties. While not very quantitative, most of these reagents provide sufficient yield and recovery for successful downstream processing, although any given sample may still require significant optimization to remove inhibitors and contaminants. The reagents still require a significant number of steps and therefore are not amenable to automation.
The majority of biological molecules are charged and have a unique shape. Since methods for recovering material from gels (electroelution and passive diffusion) are well established, many extensions to current electrophoretic platforms have been investigated. Specifically, by including excellent control parameters it is possible to better exploit the distinctive biophysical characteristics of nucleic acids. For example, systems are being built that allow the nanomanipulation of individual molecules based on the biophysical and electrostatic interactions of DNA/RNA with electric fields, solution components and physical gel matrices. Other applications include protocols that provide purification of a product from the reaction components, size selection, concentration of dilute samples, and separation from contaminants such as PCR inhibitors.
13.3 Electrophoresis using uniform electric fields for sample preparation and analysis
The best-known electrophoretic technique, used for many years for DNA/RNA analysis and size selection, is ‘zone’ electrophoresis. In zone electrophoresis, molecules are immersed in a solution that creates a common charge-to-mass ratio, allowing them to be separated into ‘zones’, or bands, based on the common physical characteristic of size (Fig. 13.1(a)), a proven technique familiar to all biologists. All components in the sample migrate through a gel matrix of either agarose or polyacrylamide, driven by a uniform electric field with a constant, DC current. Since each nucleotide contains a phosphate group with a negative charge, the charge-to-mass ratio is nearly equal for almost any DNA or RNA molecule, and so they separate based solely on their size due to physical interaction with the gelpolymer matrix. Resulting bands are generally chemically pure once the bandshave been excised and the DNA extracted from the gel, making this a useful technique for selection and purification of nucleic acids of small sizes (under 20 kb). Indeed, it has formed the basis of DNA manipulation for molecular biology and genetic engineering purposes for decades. When used within a sample preparation workflow, it is currently the standard for obtaining DNA libraries of suitable quality, purity and size distribution for Next-Generation Sequencing (NGS).
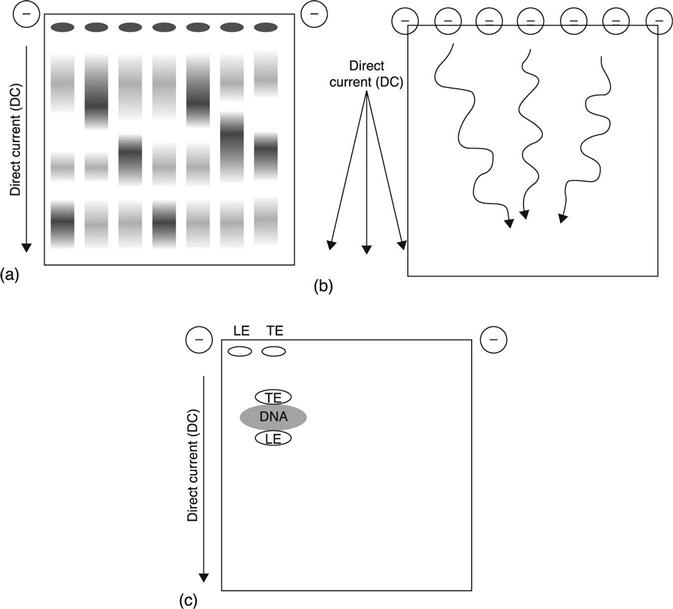
For purification of larger DNA fragments, variations of this technique can be employed. In pulsed-field electrophoresis, the direction of the electric field is alternated in three directions, with a net forward migration resulting from the combination of the three (Fig. 13.1(b)).2,3 This technique can be used for DNA of length up to 10 Mb,4 but it takes much longer due to the low mobility of the larger fragments. In addition, both basic and pulsed-field electrophoresis dilute the products when separating them, making the approach infeasible for samples with low starting mass. Additionally, limitations on the reproducible gel porosity, thermodynamic stability, and heat generation mean very large fragments (>2–10 Mb and above) cannot be efficiently resolved.
Achieving separation with these techniques is due to the mostly linear fashion in which sample components migrate through the gel: in general, the electrophoretic velocity (v) of a molecule is linearly proportional to the electric field (E) applied.5 Given that each molecule migrating through a gel has a particular electrophoretic mobility, or (μ), the following simple equation applies:
[13.1]
Most systems are set up such that μ is constant within the limits of the controllers, but newer devices are allowing manipulation of this mobility parameter, so that it can be used to enhance separation. Facilitation of this non-linear migration of nucleic acids through a gel, relative to the more linear migration of contaminants, represents a powerful new parameter to facilitate separation of one from the other.
Isotachophoresis is one such non-linear technique, widely used since the 1920s and with applications to biological molecules appearing in the 1940s.6 It uses electrostatic ‘stacking’ to sandwich and separate complex molecules, including nucleic acids (Fig. 13.1(c)). In this example the nucleic acids are mixed with two electrolytes: a ‘leading electrolyte’ (LE) and a ‘terminating electrolyte’ (TE); all three components have the same charge. During migration, the LE has a slightly higher electrophoretic mobility (μ) than the nucleic acid sample, which has a slightly higher μ than the TE. The result is that, during migration, the nucleic acid in the sample is ‘stacked’ in between the LE and the TE. This concentrates the migrating sample band, producing sharply defined leading and lagging edges. The technique therefore combines purification with concentration, in contrast to linear zone electrophoresis, a real advantage for dilute analytes. It is currently being developed and used in a capillary electrophoresis format as a component in an automated microfluidic workflow that is coupled to subsequent analysis platforms.4,7
13.4 Electrophoresis using non-uniform electric field gradients for sample preparation and analysis
Rather than use a uniform DC electric field, which neutralizes charge and limits how nucleic acids can be separated, modulating the electric field allows separation based on both structural and compositional properties. This opens a very wide range of possibilities for fine separation, focusing and trapping of nucleic acids in a sample, and much work has been done in this area over the past few decades. Important issues remain to be addressed for many, such as run-to-run reproducibility, and the establishment of reliable standards.
For non-linear electrophoresis, in which there is no net neutral charge-to-mass ratio, elements of the electrolytic and physical polymer characteristics of nucleic acids must be considered. Several articles review the detailed conceptual equations and quantitative biophysical properties for nucleic acids, gel matrix and buffer interactions,4,8,9 and so only a qualitative overview is presented here.
DNA and RNA behave as polyelectrolytes in solution; that is, they are polymers with repeating units of electrolyte groups, both of which properties can be considered during electrophoretic separations. The DNA polyelectrolyte contains two negative charges per base pair, resulting in an extremely high linear charge density. These charges along the polymer attract whatever small cations are present in the solution, such as Na+, resulting in a positively charged cloud around the polymer.9 Changes in cation concentration affect the behavior of the nucleic acids and are an important parameter in electrophoretic separations.
Other important properties of polymers related to electrophoretic separations are stiffness/flexibility. For DNA, this is in part determined by the number and size of the individual chain units in the covalent polymer, the forces acting on those individual components, and the degree to which they form freely movable units. Stiffness and rigidity are represented for a polymer by the number of persistence lengths (lp), which typically ranges from 30–60 nm for DNA, depending on environmental factors like temperature and ionic strength, and varies between different locations on the same polymer. In polymer science, the lp can be represented as half the Kuhn length (l), or (lp = l/2), where (l) is the length of the individual chain units in the polymer, and each (l) can be acted upon by particular forces, resulting in a unique orientation. For electrophoresis applications, gel matrices typically have pore sizes that are larger than the Kuhn length (l), but smaller than the radius of gyration of the molecule, with much of the dynamic interaction between the molecule and the pore being driven by the randomness of Brownian forces.8 Persistence lengths for nucleic acids can be manipulated through ionic buffers, temperature and other factors that influence the transition between rigid and relaxed forms of the molecule.10 As persistence lengths decrease for very small fragments, properties may change and be very different from those of medium to larger-sized fragments, which can be important for purification and separation of small, non-coding RNAs and cDNAs in mixed samples. Theseconsiderations are important for designing effective protocols for non-linear electrophoresis, and separations using AC-generated fields.
One such separation concept is dielectrophoresis (DEP), which involves the idea that any molecule will become polarized when subjected to a non-uniform electric field, and exhibit some level of electrophoretic mobility (μ).11–15 Different molecules will have different mobilities, and therefore conditions can be adjusted for the selection of a particular molecule over another. For DNA molecules, a strong electric dipole moment can be induced in an electric field, and mobility induced in the presence of a high field gradient.16 Interestingly, low-frequency AC fields seem to provide a good separation force. DNA exhibits strong μ below 10 kHz, while most other biological molecules in solution are only weakly polarized and therefore exhibit little mobility. This is thought to occur because of a large difference in dielectric ‘relaxation times’, or (τ), between the two groups.16,17 Generally, as a field is generated, the charge carriers on the molecule complex rearrange themselves in response. The speed with which they ‘relax’, or return to equilibrium, is (τ). As (τ) becomes greater a lower frequency is required to create (μ), and any electrophoretic drift in the direction of the actual field itself is generally mitigated by the AC current (Fig. 13.2(a)). DEP has been used successfully to separate and collect both small and large DNA fragments, and electrodeless DEP has also been demonstrated to mitigate hydrolysis and damage to collected DNA resulting from physical contact with electrodes.18 Such ‘trapping’ can be done using an etched quartz plate, which creates large, local field gradients under DEP, allowing focusing and concentration of nucleic acids; alternatively, trapping can occur by using a quadrupole field (Fig. 13.2(b)).19,20 To date, much of the work in this field has been driven by anticipated applications in microfluidics and automated sample purification devices.
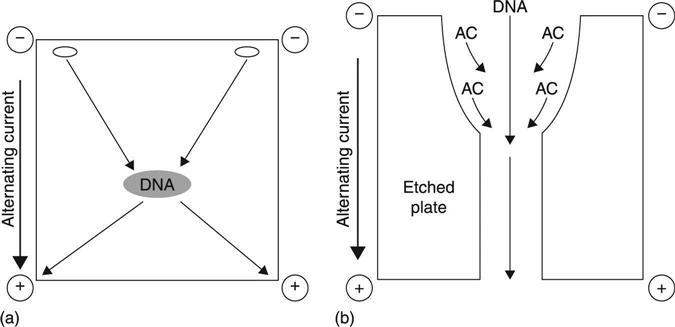
An interesting variation on dielectrophoresis is called Synchronous Coefficient of Drag Alteration (SCODA, or SCODAphoresis). The basic principle of SCODA is to force all molecules in a complex sample to undergo periodic motion, while synchronously altering the drag of the nucleic acid to create a net drift towards a central collection point (Fig. 13.3(a)). In SCODA, electrophoretic mobility is imposed in an AC field, as in standard dielectrophoresis, but electrophoretic drift is optimized to allow selection of long, linear, highly charged molecules, properties typical of DNA and RNA. An oscillating uniform field of a particular frequency (ω) is applied to a gel, using electrodes positioned at each of its four corners. The net force per time is zero, which causes molecules within the gel to orbit, acquiring a circular motion (both nucleic acid and contaminants). As materials orbit, a small quadrupole field is superimposed onto the first field and synchronously timed with its frequency oscillation. This field induces in the nucleic acids a net DC drift(described in more detail below), of particular drift velocity (dv), towards the center of the gel taking advantage of the unique (μ) described earlier. Most biological contaminants have quite different biophysical properties from nucleic acids: they continue to rotate at a constant radius, while nucleic acids migrate towards the center with each orbit. If the process is carried out long enough the nucleic acids can be collected in a central well, resulting in ‘electrodeless trapping’. In addition to this, DC-field ‘washing’ can be done to drive contaminants such as humic acids out of the gel while not affecting the motion or control of nucleic acids.21 This provides an extra level of separation which could not be achieved using standard DC electrophoresis. The resulting samples are of very high quality, since SCODAphoresis combines all the purification advantages of electroelution from agarose gels with sample concentration of up to 200×.21 In addition, its simplicity and reliance on non-moving parts are very desirable for automation, and devices employing microfluidics. Currently, the developers of this technology have introduced an autonomous, benchtop platform (initially offered in 2011) with the capacity to input up to 5 ml of sample and provide 60 μl of purified product. The developers and several users also report very good removal of contaminants such as humic acids that inhibit downstream reactions like enzymatic digestions.21,22
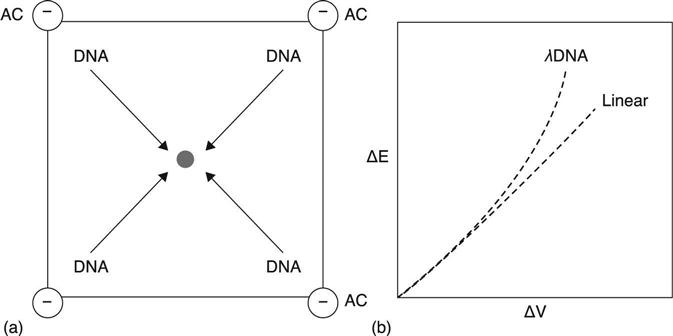
Since SCODAphoresis has so many desirable properties for concentration, purification and contaminant separation,21,22 its biophysical mechanisms and current utility are worth a more detailed look. The novelty of this technique lies in its exploitation of the non-linear dependence on the electric field of the velocity of a nucleic acid molecule through a gel medium. In a 2005 paper, Marziali et al.22 illustrated that the application of a reptating quadrupole field on λ phage DNA in a gel results in varying velocity of the DNA in that field. The observation was that as the field strength (E) increased the sample velocity (v) also increased, but in a slightly non-proportional manner (Fig. 3(b)).22 Discovering this non-linear relationship provided an important insight, giving access to a new parameter to manipulate, one that is relatively unique to long, charged polymers such as nucleic acids. The total electrophoretic mobility (μ) can be represented by the following equation:
[13.2]
where k represents the quadratic dependence on the field of the reptating DNA velocity, μ is the electrophoretic mobility as a function of the electric field (E) and μ0 is the field-independent mobility. In SCODAphoresis, a reptating field is provided at frequency ω, and there is a quadratic dependence of μ (E), which results in a doubling of the effect of this field to 2ω. This field is then heterodyned, a process in which a second AC field is introduced, which then combines in effect with the first to produce a new frequency that induces the electrophoretic DC drift. The heterodyning is done by providing a quadrupole field at 2ω, which results in a new average drift velocity proportional to the dipole and quadrupole fieldamplitudes. This drift velocity points to the center for all locations on the gel, and can therefore be used to focus and concentrate DNA.
As a fairly new approach to DNA purification, not many devices implement SCODAphoresis. SCODA platform advantages, such as high molecular weight DNA purification and minimal DNA shearing, are offset by physical limitations such as lengthy runs, a large ancillary chiller to prevent gel overheating, and an inability to directly purify anything with moderate salinity. Currently processing a single sample typically takes up to 4 h, exclusive of cell lysis, washing and solubilization steps. This limits its utility for biodefense and environmental monitoring applications, which may require constant sampling with immediate processing of an environmental matrix such as air, liquid or soil. However, it may be a good candidate to use in conjunction with a rapid test workflow, a method that provides adjudication for single positive samples, or confirmation of negatives for borderline calls. Yield in the system can also vary. A few reports have claimed that low yields for metagenomic applications can make library construction difficult,21,23 but another report claims 40–70% yield for challenging samples such as tar sands.23 For target detection in complex metagenomic samples such as soil, or clinical samples with large amounts of human background, yield may need to be monitored using spiked samples, to ensure that the downstream applications such as quantitative PCR or clinical diagnostic sequencing are not confounded.
Excessive heating and salinity are important limitations on SCODAphoresis. As described earlier, salinity can influence net charge on a nucleic acid molecule, and heating can influence the Kuhn length and persistence lengths, thus affecting stiffness and rigidity. Heating is an inherent by-product of generating an electric field; this heat degrades the efficiency of the system and can cause chemical damage to the nucleic acids themselves. The current commercial system is sold with a separate chiller to mitigate the effect. The level of heating is related to the ionic strength and conductivity of the sample. Thus, there is a need to pre-treat some samples in order to desalinate them: samples typically are not run with salinity over 100–300 μS/cm. This is not a problem affecting a minority of samples: blood plasma contains significant amounts of electrolytes, with typical values of 6,670 μS/cm.24 Depending on how desalination is carried out, the sample yield or quality may decrease, so pre-treatments must be considered carefully. A microfluidics system that feeds into SCODAphoresis might have to consider a dialysis component or a Peltier platform, keeping in mind their deleterious effects on yield.
13.5 Comparison of electrophoretic techniques for sample preparation and contaminant rejection
The utility and selection of an electrophoretic technique must be considered as part of an overall workflow, and selection of the optimal solution can change depending on the application and downstream analysis. For many applications, such as low-complexity samples, simple bead or column-based techniques may be sufficient; for soil metagenomic samples, common problems include the presence of inhibitors and low concentrations of DNA from the lowest-abundance organisms, either of which may lead to false negatives. In particular, humic acids are abundant in soil and chemically inhibit many enzymes as well as interacting with DNA to interfere with probe and enzyme binding.25 For these samples, purification, retention and concentration for low-abundance targets are critical. Fragment sizes are also a consideration in method selection. For example, most protocols require cell extraction techniques that can shear the DNA, such as bead beating, vortexing and excessive pipetting. This may not be acceptable for some downstream applications, where the isolation of high-molecular weight DNA is a requirement for eukaryotic genome reconstruction, or for the construction of high-quality, metagenomic libraries for single molecule sequencers.26
A few studies have considered soil metagenomic library preparation comparing commonly employed laboratory extraction techniques side by side with linear and non-linear electrophoresis. Sample preparation typically involves chemical, physical or ultrasonic probe-induced cell breakage and separation of DNA and RNA, followed by a chemically selective extraction technology such as gel filtration or anion exchange on beads. An example of an optimization scheme can be found in Engel et al.,23 who compared four extraction/purification workflows on three diverse soil types: a MoBIO Powersoil extraction and purification; an extraction with cetyltrimethylammonium bromide (CTAB) and ammonium acetate, coupled with a Promega Wizard anion exchange purification; a CTAB extraction coupled with SCODAphoresis; and a CTAB extraction coupled with DC electrophoresis and size-exclusion chromatography. The authors found that MoBio and CTAB/Wizard workflows generally were fastest and provided DNA of high purity, but the MoBio preps highly sheared the DNA due to the bead-beating step. Since the targeted application was high-throughput sequencing, the optimal workflows were the CTAB/SCODA and MoBio kits, with preference given to the CTAB/SCODA method since it resulted in high-molecular weight DNA and low levels of inhibitors.23 Further combinations of extraction technique with the appropriate purification technology will depend on the soil components and biological context.
Environmental aerosol and water collection may be more straightforward in scope than soil metagenomic applications. Typical workflows involve aerosol collection and immobilization onto a functionalized surface such as a Whatman FTA filter, and direct processing of sample.27 Little comparative data exists on the effectiveness of applying non-linear electrophoresis techniques for environmental sample preparation, but it is likely that they will be useful. PCR-inhibiting compounds can still be found in the particulate matter collected with aerosols, and so the application of an electrophoresis technique would be desirable. Additionally, an ability to concentrate dilute samples is desirable, since many bioaerosol collection strategies tend to vastly underestimate the true concentrations (according to some reports, by anywhere from 10 to 24 times28). Therefore it isenvisioned that this could be a part of an automated sample collection workflow utilizing microfluidics.
Clinical diagnostic samples have challenging parameters, and can vary in their preparation workflows, depending on the use-case, sample source and downstream target-application. Likewise, PCR inhibition can vary depending on the target and the source of the sample.29 The simplest, most straightforward sample for consideration would be a culture grown from a pure isolate, which would contain minimal contaminant material and is the most desirable for sequencing. Metagenomic analysis of clinical samples is more complex. Inhibitors to PCR and sequencing can be found in nearly all clinical sample sources, such as blood (heme), urine (urea), fecal (bile salts; polysaccharides), and tissue samples (collagen; myoglobin), so inhibitor rejection is an important consideration in sample preparation workflow. For targeted detection of microbial or virus pathogen nucleic acids in a complex clinical sample, even overly abundant human genomic background can inhibit PCR and sequencing reactions, so various strategies have been tried to enrich microbial target while rejecting human and other complex background. Interestingly, different clinical sample sources carry different considerations. Blood may contain human background and infected agent, while nasal and sputum samples will also contain genomic material from other agents present in the environment. Fecal samples are probably the most complex, containing genomic material from human, gut microfloral, and food sources, while also containing large concentrations of PCR inhibitors such as bile salts and complex polysaccharides.30,31 Electrophoretic technologies are well suited for use on clinical samples, although certain issues such as salinity (described earlier) need to be considered. It is envisioned that, as electrophoretic technologies find their way into more miniaturized and automated workflows, their use will grow for all of these use-cases.
13.6 Future trends
In the ‘sample-to-answer’ workflow, the sample preparation of nucleic acids can influence all downstream answers, and so electrophoresis techniques offer solutions to help limit false negatives and false positives, and to provide the most representative sample for analysis. DNA sequencing technologies and their workflows have advanced at an astounding rate over the last 5 years, with the price of sequencing per base pair greatly beating Moore’s law of computing.32 This is resulting in the exponential availability of throughput, data and capacity to sequence at a reduced cost. As sequencers become more and more automated and have simpler workflows, electrophoresis will provide a straightforward approach to automate sample preparation and sample handling in the ‘sample-to-answer’ workflow, and offers possibilities for the further automation of sequencing. As of late 2012, next-generation and third-generation single molecule sequencers are providing the capability to sequence on semi-conductor chips, with at least one platform (Ion-Torrent) having a size and appearance similar to a flash drive that can plug into a USB port.
A similar anticipated application is ‘Lab-on-a-chip’, which is the long-awaited moniker for individual microfluidic platforms that can multiplex sample handling, amplification, assay and analysis in an automated and miniaturized fashion.33 These platforms will need to be coupled with robust, accurate sample preparation technologies, and non-linear electrophoretic techniques such as DEP and SCODA offer this capability. While drawbacks such as conductivity, throughput and yield will need to be addressed, DEP and SCODA do offer the potential for fine control and focusing of DNA fragments, and even separation based on size or secondary modifications. Further utilization of DEP and SCODA are also finding their way into separation of other biomolecules besides DNA/RNA, and so combinatory approaches may be realized in which DEP and SCODA could separate multiple types of biomolecules from the same sample in a single automated device. Ultimately, the promise of Lab-on-a-chip devices will depend on the degree of integration33 of multiple biochemical analytical capabilities with these simple, straightforward and more universally applicable sample preparation techniques.
13.7 Sources of further information and advice
There are several good books, reviews and online groups which provide more information on electrophoretic technologies for nucleic acid sample preparation. For overviews of research articles, the journal Electrophoresis, published by Wiley and available online, publishes a yearly ‘Reviews’ issue in January (issue #1), which provides timely and leading edge overviews on electrophoresis of biomolecules. A recent book resource can also be found in Westermeier.4 Reviews describing nucleic acid dynamics, including the biophysical considerations and equations for the properties of nucleic acids in an electrophoresis matrix, are reviewed in Frank-Kamenetskii.9 An exhaustive overview of physical mechanisms of DNA during electrophoresis is given in Viovy.8 The wide and venerable efforts in the field of dielectrophoresis have only been touched on in this review. Several good review articles on dielectrophoresis can be found in Gascoigne and Vykoukal,13 Wang et al.15 and Pethig.20 Currently there are few review articles available for SCODAphoresis; however, an excellent description of the biophysical parameters can be found in Pel et al.,21 and a technical brief on the automated instrument design can be found in Broemeling et al.34
Online resources include a LinkedIn group searchable as ‘Sample Prep’, which contains dialogue and topics for DNA/RNA sample prep and electrophoresis. An annual meeting is also dedicated to this topic: ‘Integrating Sample Preparation. Techniques and Applications’, the sixth of which was held in Baltimore, MD in October 2012. It can only be expected that, as electrophoretic sample prep technologies become more and more integrated with microfluidics and automated approaches to DNA analysis, detection and sequencing, more resources for their use will become available.
13.8 Acknowledgments
The author thanks Jennifer Weller of the College of Computing and Informatics at the University of North Carolina Charlotte for detailed and critical review of this manuscript.