
Transplantation of engineered cells and tissues
J. Mansbridge, Histogen, Inc., USA
Abstract:
It has long been known that transplantation of organs between different individuals leads to rejection. However, by contrast, implantation of tissue engineered products containing live cells has thus far shown no clinical evidence for immune rejection. Limited testing for specific mechanisms of rejection has shown no evidence of either humoral or cell-mediated reaction to the implants. This chapter discusses mechanisms of the immune response to grafts, the role of antigen-presenting cells and the reasons for this finding. A comparison is made between the autologous and allogeneic approaches to the manufacture of tissue engineered products. Methods are suggested for manufacturing vascularized organs to avoid rejection problems on implantation.
Key words
cellular tissue engineered implant persistence; antigen-presenting cells; mechanism of graft rejection; commercial tissue engineering; vascular tissue engineering
8.1 Introduction
For more than 50 years it has been clear that transplantation of organs from one animal to another of the same species causes, with very few exceptions, a vigorous immune response that would lead to the rejection of the transplanted organ (Medawar, 1957). The concept of the impossibility of organ transplantation, without the use of immunosuppressants or other means to control the immune system, has reached general acceptance and is regarded as dogma. However, in recent years, since the development of tissue constructs grown in vitro, it has been found that such structures, when implanted, cause little immunological reaction and have never been clinically rejected, even when allogeneic. A major reason for the absence of rejection is the lack of professional antigen-presenting cells (macrophages, B-lymphocytes, dendritic cells, endothelial cells), which provoke the rejection of transplanted organs, in the implanted constructs so far used.
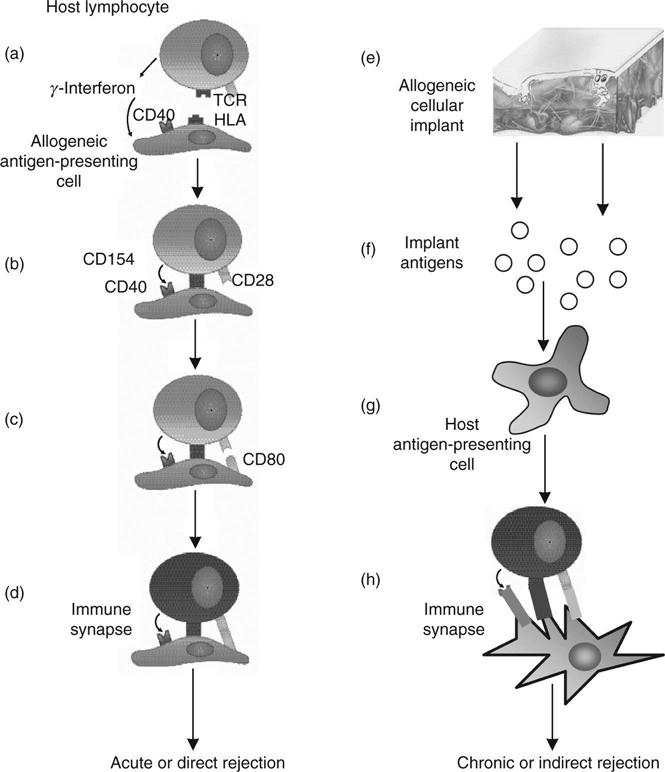
Two major pathways of organ rejection are considered in this chapter – the direct and the indirect (Fig. 8.1). Acute rejection by the direct pathway involves an initial interaction between a T-cell receptor (TCR) on a host lymphocyte and a surface molecule of the major histocompatibility complex (MHC) on a donor antigen-presenting cell (APC). This is usually a Class II molecule, such as HLADR (Fig. 8.1a). The lymphocyte may secrete γ-interferon which induces HLADR and CD40 in many cells. In the second reaction (Fig. 8.1b), CD40 on the donor cell binds with CD154 on the lymphocyte. This induces expression of CD80 and CD86 on the transplant that interacts with CD28 on the host lymphocyte. These costimulatory reactions, along with the specific initial reaction of the host TCR with the foreign MHC, comprise an ‘immune synapse’ that activates the lymphocyte (Fig. 8.1d). There are several such lymphocyte-activating pathways but this appears to be the most important in acute transplant rejection. In the chronic, or indirect, pathway, antigens shed by the allogeneic transplant (Fig. 8.1e) are ingested by antigen presenting cells, such as the macrophage (Fig. 8.1f), which then matures to a dendritic cell (Fig. 8.1g). The dendritic cell then exposes these peptides on MHC Class II molecules, such as HLADR. If these peptides are detected as foreign by a TCR on a lymphocyte, the same set of costimulatory reactions as occur in the acute pathway lead to activation of the lymphocyte. Activation of the lymphocytes, which are of the T-helper class, leads to both cell-mediated and humoral responses. The response develops more gradually than the acute pathway and persists as a chronic condition, as the release of antigens and the early parts of the pathway are slow processes. The direct pathway of organ rejection involves activation of host T lymphocytes by direct stimulation by antigens in the transplant. The most important reaction involves interactions of Class II HLA (e.g. HLA-DR) on the cells of the transplant with T-cell receptors on the host lymphocytes. Such binding, together with costimulatory reactions between CD40 on the transplant and CD154 on the host lymphocyte and between CD28 on the lymphocyte and CD80 or CD86 on the transplanted cells, lead to lymphocyte activation and a cascade of events terminating in rejection of the transplant. The indirect pathway requires presentation of transplant antigens taken up by the host cells to the host lymphocytes, ultimately leading to gradual rejection. Most of this discussion will be concerned with the first mechanism. It has been observed that fibroblasts, used in scaffold-based, three-dimensional culture, lack costimulatory CD 80 and CD86. It has also been found that they respond selectively to γ-interferon, failing to induce HLA Class II molecules that are frequently involved in initiating rejection. Fibroblasts in this type of culture fail to induce either humoral or cell-mediated immunity and fail to activate lymphocytes in a mixed-cell reaction.
This discussion will centre on experience with Dermagraft. After a brief description of the product and clinical experience with it, evidence will be presented for its persistence in wounds. Dermagraft is made by seeding neonatal, human fibroblasts to a degradable, biocompatible scaffold made of lactate-glycolate copolymer. Under these conditions, the fibroblasts proliferate and lay down a large amount of extracellular matrix, much as the capsule made in vivo in response to a foreign body. This material is implanted into chronic wounds and aids their healing.
The consequences for the commercialization of tissue-engineered products of the use of allogeneic rather than autologous cells are large. While the testing required by the regulatory authorities for an allogeneic product may be more extensive than for autologous devices, the advantages in manufacturing, including bioreactor design, automation and scale up, more than outweigh the disadvantages. This is true even if some parts of the construct, such as the vascular system, have to be autologous. This topic is discussed further below (Section 8.5)
8.2 The immune response to tissue engineered products
Experience with tissue engineered skin products designed for treating chronic wounds has shown no evidence for rejection. To date, some hundreds of thousands pieces of both Dermagraft and Apligraf and smaller numbers of other allogeneic, live cell skin constructs have been implanted without a single report of immunological rejection. This provides the best evidence for lack of rejection and places an upper limit to the incidence of the phenomenon at less than 1 in about 100 000 implants. However, the question arises as to why this should be so.
As a Food and Drug Administration (FDA) requirement, in later clinical trials of Dermagraft, and for post-market surveillance, immunological responses to the allogeneic implant were specifically investigated. Neither humoral nor cell-mediated responses were detected. Briscoe et al., (1999) made a direct comparison between the responses of SCID-Hu mice to allogeneic human skin and to a bilayered human allogeneic tissue skin construct (Apligraf), which included both keratinocytes and fibroblasts. While they could quite clearly show rejection of the skin, the tissue engineered construct was not rejected.
In several xenogeneic implants using Dermagraft performed at Advanced Tissue Sciences in intact animals (pigs, dogs, rats), without immunosuppression, that lasted for 2–4 weeks no rejection was observed. These results all show that immune-based rejection has not been a clinical problem in the use of allogeneic implants for the treatment of wounds.
8.2.1 Reasons for the lack of rejection of tissue engineered implants
One possible explanation for the lack of immune rejection of tissue engineered implants is that the cells do not survive. In the case of the bilayered construct, Apligraf, the keratinocyte layer is lost within a couple of weeks. This, however, cannot be taken as evidence for immune rejection as the keratinocytes would be expected to be lost in any case as they are terminally differentiated. During expansion culture to obtain sufficient cells from the initial biopsy to be commercially viable, stem cells are diluted or lose their phenotype and essentially all the keratinocytes in the implant are transit-amplifying cells that may be expected to proceed through their physiological differentiation process and cornify.
8.2.2 Persistence of implanted allogeneic fibroblasts
The persistence of fibroblasts in these systems has been studied by implanting the construct, which is usually derived from a male donor, into female patients and examining extracted DNA for the presence of sequences unique to the Y chromosome. This approach has been criticized on the grounds that it is insufficient to demonstrate the presence of cells (Epstein, 2003). However, it has been used in several diverse systems (Rooney et al., 1998; Tisdale et al., 1998; Shapero et al., 2000) and shows great sensitivity. In the case of Apligraf, used for treating venous ulcers, male DNA has been found within the first 4 weeks (Phillips et al., 2002), occasionally at 6 weeks (Griffiths et al., 2004) and, in the case of the treatment of epidermolysis bullosa, at 28 weeks (Fivenson et al., 2003).
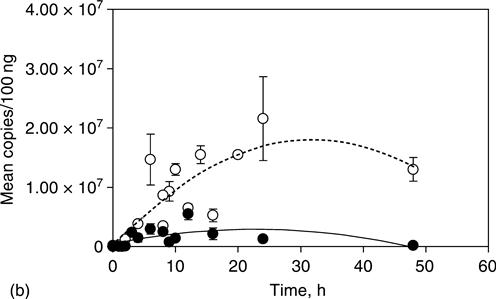
In the case of Dermagraft, studies were conducted in conjunction with a venous ulcer trial in which 10 female patients received a single implant. Biopsies were taken at 1–2 weeks and at 6 months or when the ulcer healed. DNA was extracted and the male-specific SRY sequence was amplified by a 2-stage nested technique (Fig. 8.2a). Samples were extracted from histological sections. Dewaxed paraffin sections, from which the epidermis was removed, and PBML were incubated for 3 h at 55 °C followed by 15 min at 99 °C in 50 mM tris pH 8.5, 0.045% Tween 20, 0.045% NP40, 1 mM EDTA, 1 mg/mL Proteinase K and stored at –20 °C (Leach, 1993 personal communication). Two sets of primers that can be nested were used for polymerase chain reaction (PCR) amplification of SRY, a male-specific marker. The use of nested primers permits very high PCR amplification without incurring problems of overamplification (appearance of artifactual bands) to the extent seen with comparable amplification with a single set of primers. Thus SRY sequences amplified with SRY1 and SRY2 (Calzolari et al., 1993) for 30 cycles followed by SRY4 and SRY5 (Petrovic et al., 1992) gave clean 290 bp bands without background with male DNA (Fig. 8.2b) and no band with female whereas 60 cycles using female DNA with either primer pair alone gave variable artifactual bands, some of which showed mobilities comparable with those obtained from male DNA.
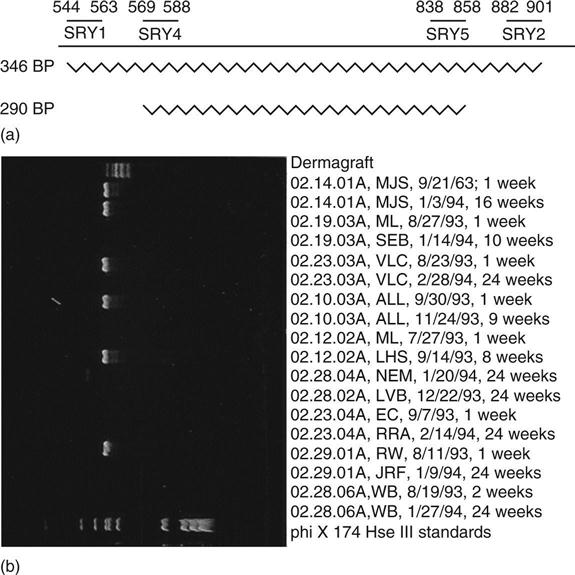
This techinique was capable of detecting single molecules, giving easily distinguishable positive or negative signals (Fig. 8.2b). DNA samples were split into aliquots predicted to contain one DNA molecule in every 10 reactions to allow counting of individual molecules. In this experiment, cells were invariably detected in the first biopsy from all the patients. However, Dermagraft DNA was also detected in six of nine of the later biopsies, which were taken at 2–6 months (Table 8.1). One problem with this very sensitive type of determination is the possibility of male cells circulating and persisting in the mother following a pregnancy with a male foetus (Bianchi et al., 1996). At least one of the patients who showed persistence of Y-chromosome-detectable sequences had not had a male child.
Table 8.1
Detection of male DNA in biopsies from sites implanted with Dermagraft
| Patient | Early point | Late point | ||
| Week | Allograft cell density, cells/mm3 | Week | Allograft cell density, cells/mm3 | |
| 1 | 1 | 22 | 9 | 51 |
| 2 | 1 | 109 | 8 | 54 |
| 3 | 1 | 53 | 16 | 168 |
| 4 | 1 | 156 | 20 | 0 |
| 5 | 1 | 200 | 24 | 68 |
| 6 | 1 | 47 | 24 | 0 |
| 7 | 24 | 2912 | ||
| 8 | 2 | 495 | 24 | 0 |
| 9 | 2 | 512 | 24 | 224 |
| 10 | 1 | 99 | 24 | 0 |
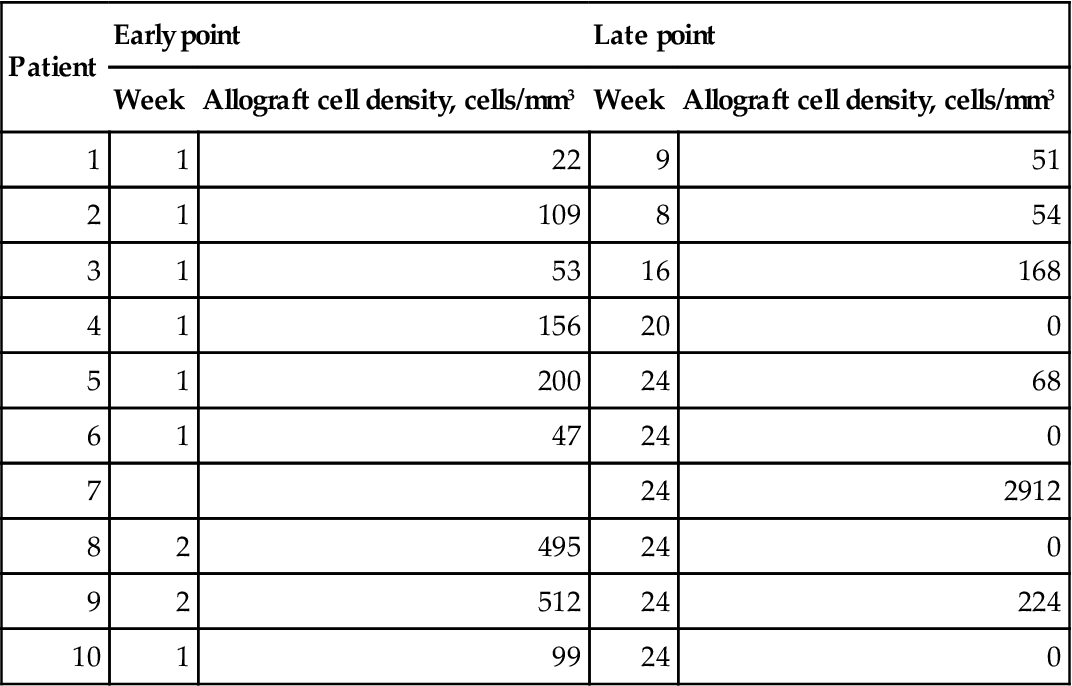
Thus, fibroblasts from tissue engineered skin implants colonize the ulcer bed and may persist for up to 6 months. The number of cells detected was very small in any individual biopsy (<10); however, this is not surprising considering the volume of the biopsy used (about 1 nL) and the frequency of apoptosis at late stages of normal wound healing (Greenhalgh, 1998).
8.2.3 Humoral immune responses to Dermagraft
In a series of 50 sera from patients treated with at least three pieces of Dermagraft, the presence or absence of antibodies reactive with Dermagraft components was assessed by staining immunoblots of proteins from detergent-lysed Dermagraft. No increase in antibodies to human proteins was observed in comparing blots stained with sera taken after treatment compared with sera taken before. The only reaction consistently seen was to bovine serum albumin; however, no consistent change was seen in this protein following Dermagraft implantation.
8.2.4 Cellular immune response to Dermagraft
Immune rejection of transplants is largely through a cell-mediated T-lymphocyte response. The activation of a cell-mediated response to Dermagraft implantation was investigated using the secretion of cytokines by activated T cells. This was chosen in preference to determination of the blast response by tritiated thymidine incorporation because of the difficulty of estimating cellular tritium uptake in the heterogeneous lymphocyte/matrix/attached cell system. The secretion of γ-interferon by patient-derived lymphocytes in response to Dermagraft was determined by enzyme-linked immunosorbent assay (ELISA). Blood samples were taken before treatment and at the end of treatment, following at least three Dermagraft implants. The samples were transported to a research laboratory at the Mayo clinic within 24 h, lymphocytes isolated and incubated with Dermagraft for 3 days. The supernatant was harvested and stored at –70 °C until sufficient had been collected for economical analysis by ELISA.
In all patients, peripheral blood mononuclear lymphocytes isolated and tested in this way showed substantial stimulation by phytohemagglutinin, the positive control. However, the results showed no activation following Dermagraft treatment in any of the 25 patients. In all the data, a single case of lymphocyte activation was observed, but this occurred in a patient prior to treatment with Dermagraft. It thus appeared to be unrelated to Dermagraft and was interpreted as a non-specific result.
A similar study was performed using samples from 10 patients implanted intra-orally with a single piece of Dermagraft in conjunction with a clinical trial for the application of Dermagraft for gingival reconstruction. In this case, all positive controls showed reaction, but none responded to Dermagraft. Taking the results together, no case of implant-stimulated cellmediated immunological response was observed in 35 patients.
These results provide no evidence for an immunological response to Dermagraft, and, with the lack of clinical evidence for rejection, such a response does not occur, or is, at most, a very rare event. These data are consistent with numerous reports of variable survival of allograft cells in a host without immunosuppression (Hefton et al., 1983; Hull et al., 1983; Sher et al., 1983; Thivolet et al., 1986; Palmer et al., 1991; Otto et al., 1995).
8.2.5 Selective response to γ-interferon by fibroblasts in scaffold-based three-dimensional culture
While the lack of antigen-presenting cells in tissue engineered constructs may be the major factor in the lack of immune response to such implants, three-dimensional culture also modifies cells in a manner that may contribute to their survival. In the presence of γ-interferon, which is likely in a wound site, cells respond to the presence of γ-interferon by inducing MHC Class II molecules, such as HLADR, and costimulatory molecules, such as CD40. Such a reaction might be expected to precipitate an immune response. However, we have found that fibroblasts in scaffold-based three-dimensional culture respond selectively, the majority of cells failing to induce these molecules (Fig. 8.3). Gene expression analysis shows a large number of genes induced in fibroblasts by γ-interferon and a spectrum of differential responses between monolayer and three-dimensional cultures (Table 8.2). The set of genes markedly less induced in three-dimensional than monolayer culture include MHC Class II and molecules involved in the antigen-presenting pathway, while other genes, such as HLA Class I, are comparatively unaffected. Three-dimensional fibroblast cultures, therefore, show a selective response.
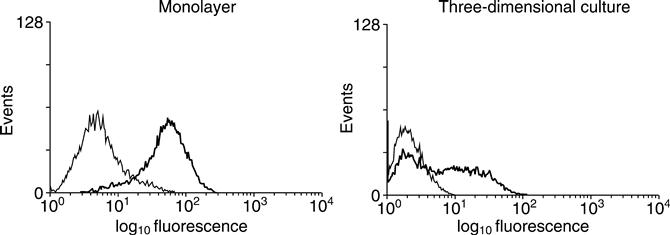
Table 8.2
Comparison between monolayer and scaffold-based three-dimensional culture of genes induced by γ-interferon
| Induced within 85% in monolayer and 3D cultures | Induced 3–6× more in monolayer than 3D culture | |
| HLA-class I, β2 microglobulin 9–27, 1–8U, C1 inhibitor | ||
| HLA Class II | HLA-DR, HLA-DMB, HLA-DP, IP-30, butyrophilin (BTF5), BiP | |
| Virus-associated | HPB, HPC, CMV associated, IDO, 2′-5′ polyA synthetase | |
| Transcription factors | IRF-9, IFP-35, IRF-1 |
Comparisons were made between median values from three independent determinations using Affymetrix U95A expression arrays, Genes are selected from a total of 13 genes showing similar induction in monolayer and three-dimensional culture and 43 that showed a difference similar to those discussed. Genes were selected on their relevance to the expression of MHC Class II genes and antiviral properties.
In an investigation of the reason for the selective response, we have examined parts of the signal transduction pathway from γ-interferon. In particular, we have determined the phosphorylation of signal transducer and activator of transcription-1 (STAT-1), which transmits a signal from the γ-interferon receptor to the nucleus, and the Class II transactivator (CIITA) which activates the promoter for HLA Class II genes such as HLADR. We observed that the initial γ-interferon receptor-mediated phosphorylation of STAT-1 on tyrosine 701 is very similar in monolayer and three-dimensional culture. Phosphorylated STAT movement into a detergent insoluble fraction, presumably the nucleus, although slightly slower in three-dimensional than in monolayer culture, is also little different (Fig. 8.4a). The difference in kinetics may be largely explained by differences in diffusion. However, expression of CTIIA, is markedly different. The CTIIA gene has a complex control region, comprising four promoters, some of which are important in particular cells, such as B-lymphocytes, and others are affected by agents such as γ-interferon (O’Keefe et al., 2001) (Fig. 8.4b). CIITA is much more highly induced by γ-interferon in monolayer than in three-dimensional culture. Other factors also involved in this pathway, such as Interferon Response Factor-1 (IRF-1), is also less induced in three-dimensional culture than monolayer. While this phenomenon does not appear to apply in collagen-based three-dimensional culture (Kern et al., 2001), it may be expected to occur as the cells migrate into the wound bed.
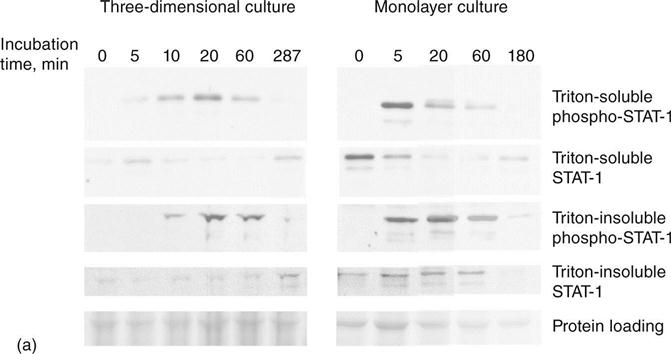
The general conclusion is that, in the absence of cells involved in antigen presentation, the cells generally used for tissue engineering may well not be immunogenic. The one immunogenic cell important for tissue engineering is the vascular endothelial cell. Barring a method to render them non-immunogenic, they will have to be obtained from an autologous source (Williams et al., 1989, 1994; Williams, 1995). It may be noted in passing that one organ that is regularly transplanted without major immune consequences in the majority of cases is the cornea, which contains no blood vessels.
8.3 Generality of the resistance of tissue engineered products to immune rejection
The scope of immunity from rejection among the allogeneic cells used in tissue engineering has not been determined. It is true for fibroblasts, almost certainly for keratinocytes and smooth muscle cells but certainly not for endothelial cells and hemopoietic cells. Reports indicate that cartilage cells are minimally immunogenic (Huey et al., 2012) and that conditions that promote functional tissue reduce their immunogenicity (Yuan et al., 2011). These data are consistent with Matzinger’s view that the immune system is activated by products of the cellular stress response (heat shock proteins) through toll-like receptors, leading to immune rejection (Matzinger, 2007). Thus, one approach may be to induce physiological differentiation and to minimize the stress response.
The low immunogenicity of bone marrow-derived mesenchymal stem cells has long been known (Aggarwal and Pittenger, 2005). This appears to be a property of many types of tissue-derived stem cells (Hardin-Young and Parenteau, 2004; Barry et al., 2005; Wolbank et al., 2007; Guo et al., 2009; Kode et al., 2009; Montjovent et al., 2009; Siepe et al., 2009; Wada et al., 2009, Davies et al., 2011). In the case of stem cells, it may be true that undifferentiated cells are non-immunogenic but that they may become immunogenic if they differentiate into antigen-presenting cells such as macrophages, dendritic cells and endothelial cells. The use of allogeneic cells has many advantages for manufacturing (see below), even if it can be applied only to part of the final construct. Determination of the range of cells showing minimal immunogenicity would be a valuable contribution to tissue engineering.
In unpublished experiments performed at Advanced Tissue Sciences, small diameter vascular implants were tested in dogs. These constructs comprised a smooth muscle tube lined with endothelial cells. Endothelial cells are antigen-presenting cells and immunogenic, so autologous cells were used (Williams et al., 1994). However, the smooth muscle cells were of allogeneic canine origin. The constructs were implanted for up to 12 weeks without any evidence for immunological rejection. These experiments need to be extended; however, it is likely that cells other than fibroblasts are tolerated, provided that antigen presenting cells, such as endothelial cells, B-lymphocytes, macrophages and dendritic cells are carefully excluded.
8.4 Testing and regulatory consequences
At this stage, regulatory authorities are requesting testing of new allogeneic products for immunogenicity. This places an appreciable resource burden on companies developing products, both in terms of collection of samples, effort and money, so the numbers requested have not been large. The results demonstrate the general lack of humoral or cell-mediated response to the allogeneic implants, but cannot exclude rare or idiosyncratic events.
8.5 Comparison between autologous and allogeneic tissue engineering
The contrast between autologous and allogeneic approaches to tissue engineering is striking. Autologous tissue engineering is a service industry, requiring heavy investment in plant (individual tissue culture suites, or burdensome cleaning, are required for each patient) and is usually performed by expensive, skilled and highly trained personnel. Economies of scale are not possible. However, safety testing is much reduced. In allogeneic approaches, the burden of testing is much greater. In recent experience, the cost of a new master cell bank was placed at about a million dollars overall. However, with judicious use, such a cell bank may last 15–20 years, so the cost is averaged over a considerable time. The advantage of allogeneic approaches is that manufacturing is much simplified, can be performed by less expensive, although trained, personnel and shows economies of scale. This approach is more suitable for large-scale production and can incorporate more easily specialized bioreactor design, automation and so forth. The cost of the product is much reduced. While these advantages are more difficult to achieve with autologous systems, ingenious design, such as development of completely automatic machines that will accept a fresh biopsy and produce the final product, all within disposable containers and without human intervention, is possible.
Although it may be feasible to make the bulk of a tissue-engineered construct from allogeneic cells, certain components, and particularly the vascular system, will have to be autologous because endothelial cells are antigen-presenting cells. At this stage, few tissue engineered vascular organs have been implanted, as problems associated with the development of a practical vascular system have not been solved. Thus far, implants, such as skin constructs, which do become vascularized, have been vascularized directly from the host following implantation, an approach that may well have limited their size.
One case in which constructs containing vascular endothelial cells have been implanted is small diameter blood vessels. Implanted artificial blood vessels fail to be endothelialized by the host for more than a few millimetres. In the case of small diameter blood vessels, lack of endothelialization leads to blood clotting and rapid failure. In human trials of such constructs, the entire device is of autologous origin (L’Heureux et al., 1993, 2006). In the canine experiment, mentioned above, although the smooth muscle cells were allogeneic, the endothelial cells had to be autologous. In that case, endothelial cells were isolated from liposuction fat derived from the prospective host (Williams et al., 1994). Although such cells might not be identical to small vessel aortic endothelial cells, they were found, after conditioning to the fluid shear to be expected after implantation, to perform satisfactorily.
Another method for vascularizing an organ in an autologous manner is implantation of the construct in a host location rich in blood supply and inducing vascular development in the implant by using growth factors, followed by transfer to its ultimate site. To do this, the organ would probably have to start as a primordial structure, which could survive as vascularization was established, and then grow within the host. While this approach will provide an autologous vascular bed, it is likely to involve multiple connections to the host. If an organ is to be transplanted, it is desirable to limit the number of inlets and outlets to minimize microvascular surgery during transplantation. Vascularization of tissue engineered constructs represents one of the greatest challenges to the development of the field and considerable effort is being expended to find a solution (Novosel et al., 2011).
Methods are available now to obtain endothelial cells comparatively easily, but strategies will have to be devised to establish a vascular network rapidly. One possible method is inosculation (Black et al., 1998), in which vessels preformed by allogeneic cells are populated from the host. The allogeneic endothelial cells would have to be destroyed before implantation, leaving only basement membrane channels. Another approach is growing the organ within its ultimate host, using the host circulatory system to supply nutrition, in much the same way as in some early experiments, where organs (among them an ear) were grown in the mouse (Cao et al., 1997). A third is to develop machines that would grow the vascular system within a preformed tissue-engineered implant. However, our current understanding of the mechanisms involved in the development of blood vessels, particularly small arterioles and venules, is inadequate to allow us to do this as yet.
8.6 Conclusions and future trends
Despite experience with organ transplantation, implantation of tissue engineered products containing live cells has thus far shown no clinical evidence for immune rejection. Limited testing for specific mechanisms of rejection have shown no evidence of either humoral or cell-mediated reaction to the implants. The major reason for the lack of immunogenicity lies in the lack of antigen-presenting cells in the implants, and inclusion of such cells has been found experimentally to confer rejectability (Rouabhia, 1996). These results are consistent with those of Erdag and Morgan (2004), who compared the immunological response of grafts of intact skin and tissue engineered skin constructs. In a transplanted organ, endothelial cells are a major source of antigen-presenting cells and acute rejection may be seen as an attack on the vascular system that rapidly extends to other cells. It has also been found that fibroblasts in a scaffold-based three-dimensional culture show a selective response to γ-interferon, which, although it induces molecules associated with antigen presentation in many cell types under suitable culture conditions, does not do so in this case.
While the low immunogenicity of many cell types applies to applications such as treatment of non-healing wounds, where the long-term survival of the cells of the implant is not important, a long-term, low grade immunogenic response may be important if the implant is expected to provide a permanent function. In long-term implants, it is likely that the cells of the graft will slowly be replaced. However, in this case, there is much interest in autologous products and in developing means to circumvent an immunological reaction. In some cases, this may achieved through a suppressive effect of the cells, such as mesenchymal stem cells, themselves (Klyushnenkova et al., 2005), through their secretion of prostaglandin (Cui et al., 2007) or through indoleamine dioxygenase (Jalili et al., 2010; Bahar et al., 2012). Another approach is to design immunological evasion systems based on those used by other biological system such as reducing the expression of MHC1 and immunostimulatory molecules on the cell surface using the Kaposi’s sarcoma herpes virus immunomodulatory gene MIR2 (Thakur et al., 2011) or an intracellularly expressed antibody against MHC class 1 (Doebis et al., 2006). Other strategies have been to repel T-cells using SDF-1 (Papeta et al., 2007) and to kill them by transfecting the implant cells with fas ligand (Xie et al., 2008) or using the immunosuppressive effects of CTLA4 (Dai et al., 2006).
8.7 Sources of further information and advice
Much of the literature in this field is primary, the remainder being generated within companies. A series of early articles indicating that skin substitutes were not immunogenic include Hefton et al. (1983); Hull et al. (1983); Sher et al. (1983); Thivolet et al. (1986); Palmer et al. (1991); Otto et al. (1995). The demonstration that adding antigen-presenting cells to constructs confers rejectability is given in Rouabhia, (1996) and Briscoe et al. (1999). Methods for obtaining, comparatively conveniently, autologous endothelial cells is described by Williams et al. (1995).
8.8 Acknowledgements
PCR amplification of DNA to determine the persistence of implanted fibroblasts was performed by Kathryn MacKenzie. Determination of lymphocyte activation by γ-interferon production was performed by Dr L. Oliver at the Mayo Clinic. Figures 8.3 and 8.4 were previously published by the Journal of Investigative Dermatology. Immunoblots to determine humoral responses to Dermagraft were performed by E. Pinney. RNA for expression array analysis was prepared by Dr K. Liu.