
Advanced synthetic polymer biomaterials derived from organic sources
Abstract:
Synthetic polymers offer several advantages over their natural counterparts, including improved chemical resistance, tunability of their properties and mechanical durability. In this chapter, the properties and synthesis of several types of synthetic materials derived from organic polymers are reviewed. The use of these well-defined, highly ordered aggregates, as organised systems for bioactive molecule delivery as templates for the synthesis of metal nanoparticles and as micro-patterned scaffolds for directed attachment and growth of aligned cell monolayers, is discussed.
3.1 Introduction
Synthetic polymers are ubiquitous, having made their way into almost every facet of modern human existence, both as common materials and as functional elements in high-technology applications. In many cases, synthetic polymers have proved to be superior to natural polymers in terms of their composition, mechanical properties and degradation characteristics for a variety of medical implant and tissue engineering applications (Abbasi et al., 2001; Alperin et al., 2005; Ateh et al., 2006; Caracciolo et al., 2009b).
As with naturally occurring polymers, such as proteins, cellulose and natural rubbers, synthetic polymers are macromolecules comprised of a vast number of atoms joined together by covalent bonds. The nature of the repeating unit (monomer) and the specific bonds linking these units together determines the physical, chemical and biological properties of the resultant polymer. The spatial arrangement of the polymer (i.e. whether the polymer possesses a one-, two- or three-dimensional conformation) also significantly influences the properties and behaviour of the macromolecule. The degree of order within the polymer network (i.e. whether the polymer is amorphous or crystalline) is an equally important determinant of polymer behaviour. For example, linear, one-dimensional thermoplastic polymers possess strong intra-molecular covalent bonds and weak intermolecular van Der Waals bonds, which allow these macromolecules to be easily dissolved or remoulded via heating or chemical reaction. However, thermosetting polymers possess a three-dimensional conformation and an amorphous, highly cross-linked structure; these macromolecules are insoluble and cannot be remoulded without destroying the polymer backbone. Another good example of an amorphous polymer is synthetic elastomers, which are weakly cross-linked polymer networks comprised of long polymer chains that can alternately coil and uncoil, allowing them to regain their original shape after being stretched.
Since their inception in the 1930s and for over 50 years, the plastics market has been dominated by polymers produced from organic monomer sources (Gleria and De Jaeger, 2005). Organic polymers were the first plastics to be developed, due to their processability and reproducibility, valuable properties, and importantly, the relative abundance of low-cost precursors that were available as a result of buoyant oil markets (Yu et al., 2006).
In this chapter, several major classes of organic polymers will be reviewed in terms of their synthesis, fundamental and biological properties, and their utility in biomedical applications. The most recent advancements in the nanoscale applications of these macromolecules will be given greater attention than their more conventional medical uses. Given the abundance of original articles and high-quality review papers available in this area, only the most recent examples will be discussed. The structural and functional complexity of organic polymers is exemplified using the organic biopolymer poly(urethane).
3.2 Poly(ester)s and poly(ester) block copolymers
Aliphatic poly(ester)s are a widely studied family of biodegradable materials, which includes many poly(glycolic acid), poly(lactic acid), and poly(ε-caprolactone) homopolymers, and their block copolymers. Poly(glycolic acid) is a linear poly(ester), characterised by high crystallinity, high glass transition and melting temperatures, and low solubility in organic solvents. Poly(glycolic acid) is commonly synthesised via the ring opening polymerisation of glycolide. Their application as biodegradable and bioresorbable materials stems from their relatively high hydrophilicity and sensitivity to hydrolytic dissociation (Bhardwaj and Kundu, 2010).
As with poly(glycolic acid), poly(lactic acid) is synthesised via a ring opening polymerisation route from lactide, the cyclic diester of lactic acid. Depending on the stereochemistry of the lactide precursor, the resultant polymer may vary in its degree of crystallisation, from enantiopure semicrystalline (those synthesised from L- or D-lactides) to racemic amorphous structures (produced from D,L-lactides). These polymers possess different mechanical and thermal properties, depending on the synthesis technique used. Co-polymerisation of the relatively hydrophilic glycolide with the more hydrophobic lactide yields a range of polymers whose properties can be adjusted to suit a desired bio-application.
The polycondensation of hydroxyl acids, such as lactic acid, can also be used for polymer synthesis; however, several drawbacks need to be considered when using these polymers. The monomer contains water, which needs to be removed during the synthesis reaction in order to force the reaction equilibrium towards the formation of new ester bonds. As a result, it is a lengthy process, with water being produced during the condensation reaction. Furthermore, the products resulting from this polymerisation reaction are characterised by lower molecular weights. Despite these drawbacks, the technique shows promise, since lactic acid is a more cost-effective alternative starting material compared with the lactide monomer. Microwave radiation is being investigated for its potential to significantly enhance the reaction rate and extent of condensation, and to increase the molecular weight of the resultant macromolecules.
A rapid microwave-assisted synthesis of D,L-lactide from a racemic lactic acid monomer has also been reported. This process initially polycondenses lactic acid to form oligolactic acid, followed by a subsequent depolymerisation of the product to form lactones. In addition to the higher amount of lactone produced under these microwave conditions compared to that achieved using a conventional thermal reaction, the ratio of D,L-lactide to the total amount of L,L-, D,D- and meso-lactide was found to be significantly higher when using the microwave process and this increase became more pronounced over prolonged reaction times and increased temperature conditions. In addition, catalysts were found to further increase the degree of D,L-lactide production.
Several polymer systems have been identified that can provide unique mechanical properties. For example, a poly(L-lactic acid) soft matrix embedded with hydroxyapatite hard particles has been produced that demonstrates a mechanical behaviour dependence on the size and the volume fraction of its particulate inclusions (Balac et al., 2001). In other examples, poly(L-,actide) compounded with chitosan demonstrated shape memory behaviour, which was attributed to the viscoelastic characteristics of the synthetic polymer (Meng et al., 2009). Introduction of the chitosan was found to enhance the biodegradability and biocompatibility, while not significantly affecting the glass or melting transition temperature of the polymer. However, the shape recovery ratio was found to decrease with increasing chitosan concentration due to phase separation.
Many efforts have been made to combine the favourable properties of polylactide polymers with the biocompatibility, biodegradability and antimicrobial characteristics of chitosan via a variety of chemical and physical methods. One such example is the covalent immobilisation of chitosan on poly(lactide) membranes via UV cross-linking initiated with 4-azidobenzoic acid (Zhu et al., 2002). The hydroxyl and amino functionalities introduced by the presence of the chitosan were found to facilitate the subsequent chemical immobilisation of a variety of functional molecules, such as heparin, to modulate protein and cell adhesion. Chitosan has also been used to stabilise poly(lactide) scaffolds with a view to overcoming the synthetic polymer’s inherent limitations, such as its hydrophobicity and tendency to undergo rapid biodegradation into acidic products (Li et al., 2004). The incorporation of chitosan into the structure resulted in the attainment of suitable hydrophilicity and scaffold porosity conducive to cell attachment and growth. Films of a poly(lactic acid)-chitosan blend prepared by solution mixing and film casting were found to exhibit enhanced hydrophobicity, and in so doing increased the water vapour barrier of the antimicrobial chitosan material (Suyatma et al., 2004). However, the mechanical and thermal stability of chitosan was found to deteriorate upon poly(lactic acid) blending (Sébastien et al., 2006).
Another notable poly(ester) biomaterial, poly(ε-caprolactone), has been characterised as possessing sound biocompatibility, high hydrophobicity and a semicrystalline nature (Aghdam et al., 2010; Gea et al., 2010; Xie et al., 2007). In addition to its permeability to hydrophilic and hydrophobic drugs, poly(ε-caprolactone) has been found to be more hydrolytically stable compared to both poly(lactic acid) and poly(glycolic acid), and as such this biomaterial is being actively investigated for its use in the fabrication of solid and injectable implants, scaffolds for tissue engineering, and micro- and nano-particles for targeted delivery of biomolecules and in controlled drug release applications. The mild processing conditions associated with the relatively low glass transition and melting temperatures facilitate drug incorporation without jeopardising its chemical stability and potency. Another advantage of poly(ε-caprolactone) is that it is significantly cheaper compared to other poly(ester) precursors.
As with other poly(ester)s, the material properties of poly(ε-caprolactone)s and their polymerisation output can be significantly influenced via the introduction of a variety of initiators, such as benzoic, chlorinated acetic, maleic, succinic and adipic acids; catalysts, including lanthanide halides, zinc powder and lipase; and by adjusting the synthesis conditions (Tan et al., 2009; Wiesbrock et al., 2004) (Fig. 3.1). Hydrogen phosphonate initiators facilitate ring opening insertion polymerisation reactions, where the phosphate first reacts with carbonyl moiety of caprolactone to cleave the ester bond. The P-alkoxide of the resultant coordination intermediate is subsequently broken to produce an acyl-alkoxide bond. Alternatively, it has been suggested that trace amounts of water can also initiate the ring opening polymerisation of caprolactone, followed by transesterification between the oligo(ε-caprolactone) and the hydrogen phosphonate to generate an intermediate to catalyse the polymerisation (Tan et al., 2009). The microwave assisted synthesis was found to enhance both the reaction rate and the synthetic efficiency, compared to that obtained for other poly(ester)s, with the increase being attributed to the rapid temperature rise associated with the high microwave absorption by the ε-caprolactone. The initial rapid increase in temperature was followed by a plateau, which was attributed to the lower ε-component of the polymer compared to that of the monomer, similar to what has been reported for lactic acid and low molecular weight poly(lactic acid). Notably, temperatures in excess of 230 °C may lead to chain degradation becoming dominant over chain propagation processes (Persenaire et al., 2001).
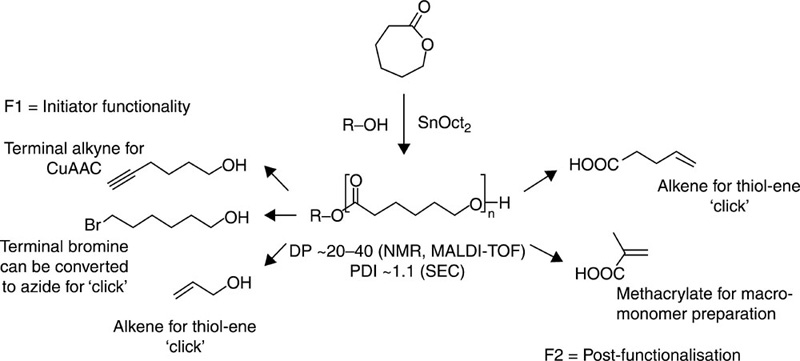
Poly(ε-caprolactone)s and their derivatives can be used for a wide variety of biopharmaceutical applications, such as formation of particles, monolithic implants and scaffolds. Biodegradable elastomers were prepared via the UV initiated cross-linking of trimethylene carbonate prepolymers with amounts of trimethylene carbonate and ε-caprolactone (Chapanian et al., 2009). The elastomers containing ε-caprolactone were found to be susceptible to lipase degradation, with both materials undergoing cholesterol esterase degradation and surface erosion. Trimethylene carbonate polymers were found to generate a better tissue response during biocompatibility studies. Biodegradable poly(ε-caprolactone) matrices reinforced with melt compounded bacterial cellulose pellicles showed significantly enhanced mechanical properties (Gea et al., 2010).
The relative hydrophobic properties of poly(ester)s places certain limitations on their use in biomaterials applications that require in vivo and in vitro biodegradability. In order to address this drawback, poly(ester)s have been copolymerised with polymers that contain highly hydrophilic functional units, such as poly(ethylene glycol) and poly(ethylene oxide). Furthermore, by conjugating strongly hydrophilic moieties onto hydrophobic chains, self-assembled polymeric micelles, vesicles and hydrogels can be fabricated, with the molecular weight of the resulting block co-polymer also having a significant effect on the configuration of the amphiphilic material.
3.3 Poly(2-oxazoline)s
Poly(2-oxazoline)s are chemically versatile biocompatible polymers (Agrawal et al., 2012). They are derived from both pristine and substituted 2-oxazoline via a ‘living’ cationic ring-opening polymerisation reaction. Their use as biomaterials is attracting more interest due to their processability and their ability to attain polymers of controlled molecular weight with low polydispersity (Kronek et al., 2012; Zhang et al., 2012a,b). These materials can be further modified to adopt the configurations of vesicles and vectors, micelles, hydrogels and scaffolds suitable for biomolecule and drug transport, gene transfection, controlled release and tissue engineering applications (Luxenhofer et al., 2012). Polymers possessing various material properties can be achieved by controlling the block arrangements and architectures, and processing conditions, such as properties of the solvent (Guillerm et al., 2012) (Fig. 3.2). Polymers derived from enantiopure and racemic precursors also differ in terms of their chemical functionality and physical conformation. Polymerisation of hydrophilic and hydrophobic oxazolines in a sequential fashion results in the production of an amphiphilic material that exhibits a tendency to self-assemble into polymeric micelles. These micelles show great promise for drug delivery, owing to the versatility of the functionalities available for conjugation that can be achieved by varying the polymerisation initiator. Fully amorphous branched 2-(3-ethylheptyl)-2-oxazoline homopolymers with a glass transition temperature below 0 °C can potentially be used as thermoplastic injectable materials.
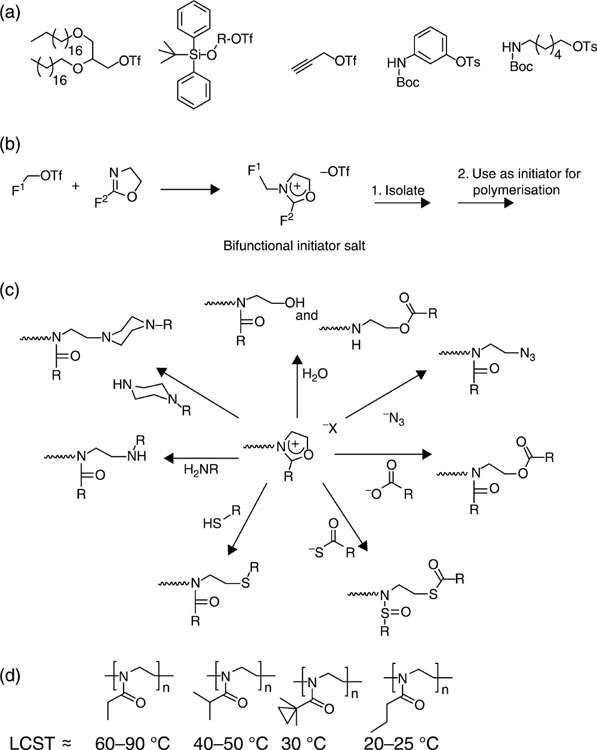

Hydrophilic poly(oxazoline)s have also been suggested as suitable poly(ethylene glycol) substitutes for the functionalisation of such nanocarriers of liposomes and active molecules to enhance their in vivo performance, particularly with regard to circulation time (Sedlacek et al., 2012). The use of poly(oxazoline)s rather than poly(ethylene glycol)s in such applications is advantageous, as it exhibits greater water solubility, chain flexibility, cytocompatibility, processability and low polydispersity. Furthermore, a wide variety of functionalities can be achieved from polymer derivatives, enhancing the subsequent complexation and conjugation potential. Drug conjugation products can also be achieved, such as high molecular weight trypsin and low molecular weight pyrimidine analogues, cytosine arabinose and modified 2-(3-ethylheptyl)-2-oxazoline.
3.4 Poly(alkyl carbonate)s
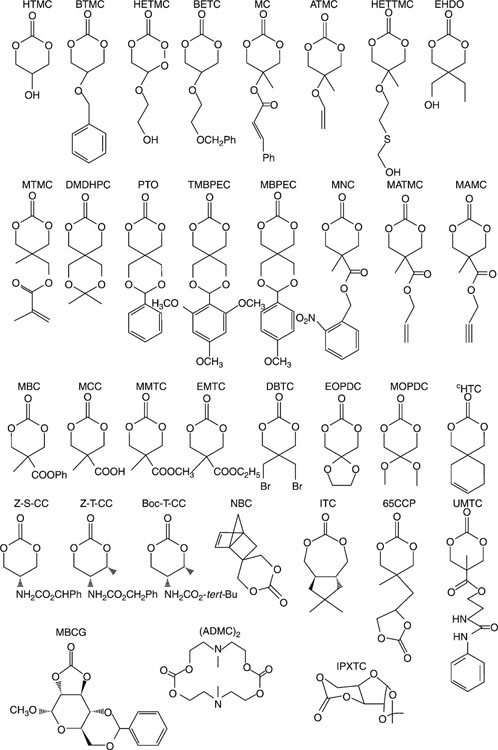
Poly(alkyl carbonate)s are characterised by sound biocompatibility and controllable material properties (Zhu et al., 1991). In vivo, these materials undergo slow biodegradation due to their highly hydrophobic nature, with the products of degradation being non-toxic, unlike the acidic by-products of degrading poly-(ester)s (Anderson, 2001; Sosnik et al., 2011; Watanabe et al., 2008). Poly(alkyl carbonate)s are commonly synthesised by the metal catalysed ring opening polymerisation of trimethylenecarbonate with other six-membered substituted ring derivatives (Feng et al., 2012; He et al., 2003; Zhang et al., 2009) (Fig. 3.3). The susceptibility of poly(alkyl carbonate)s to enzymatic degradation can be enhanced by modifying the chemical nature of the polymeric chain, yielding hydrophilic derivatives that possess terminal poly(ethylene glycol) monomethylether blocks (Feng et al., 2012; Yu et al., 2012). For example, poly(alkyl carbonate)s possessing a large number of amine functionalities have been designed for non-viral gene transfer applications (Seow and Yang, 2009).
3.5 Poly(ether)s
Poly(ethylene glycol) is a highly biocompatible poly(ether), which is soluble in aqueous solutions and organic solvents, which contributes to its biocompatibility and processability, respectively. The low toxicity and non-immunogenicity of poly(ethylene glycol), coupled with the ability for low molecular weight poly(ethylene glycol)s to obtain renal clearance render the biopolymer a popular choice for the surface modification of biomaterials, particles and micelles for active molecule transport, and for chemical and physical hydrogels. Poly(ethylene glycol) is not hydrolytically dissociated in vivo; however, its hydrophilic functionalities confer an enhanced water affinity and biodegradability to the polymer. Poly(ethylene glycol)s are fabricated via the polycondensation of ethylene glycol in the presence of acidic or basic catalysts, producing a lower molecular weight product. Ethylene oxide is employed as a precursor for the polymerisation of high molecular weight poly(ethylene glycol)s, and is a high efficiency process that allows the generation of heavier polymer molecules.
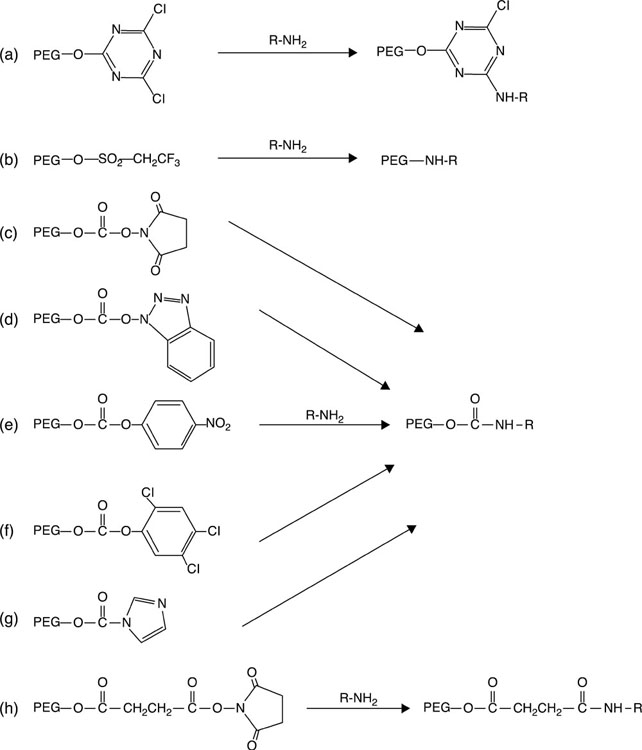
The distinctive solubility characteristic of poly(ethylene glycol), namely solubility in both aqueous and organic solvents, renders it suitable for end-group derivatisation and chemical conjugation to a variety of biological molecules, such as polypeptides, polysaccharides, polynucleotides and small organic molecules under mild physiological conditions (Roberts et al., 2012). First reported by Davies and Abuchowski in the 1970s for albumin and catalase modification, pegylation refers to the covalent attachment of poly(ethylene glycol) to molecules of interest (Banerjee et al., 2012). The process typically requires activation of the poly(ethylene glycol), where a derivative of the poly(ethylene glycol) with a functional group at one or both termini is prepared. The choice of the functional group is dependent on the type of available reactive group on the biological molecule to which poly(ethylene glycol) is to be conjugated (Fig. 3.4). For example, reactive amino acids, such as lysine, cysteine, histidine, arginine, aspartic acid, glutamic acid, serine, threonine, tyrosine, N-terminal amino group and the C-terminal carboxylic acid, are chosen for pegylation of proteins, whereas for pegylation of glycoproteins, reactive formyl moieties can be attained via oxidation of vicinal hydroxyl groups with periodate (Roberts et al., 2012).
3.6 Polypeptides
Polypeptides are biomaterials composed of repeating amino acid units linked by a peptide bond. Polypeptides can conform to different three-dimensional architectures, depending on their chemical composition (Fig. 3.5). Such versatility, coupled with their inherent biocompatibility and biological activity, make polypeptides ideally suited for drug and gene transfer applications and in the development of tissue scaffolds (González-Aramundiz et al., 2012; Grove and Regan, 2012; Bracalello et al., 2011; Tian et al., 2012). Polypeptides are formed via sequential reactions of protected amino acids. Strong inter- and intra-molecular hydrogen bonding between peptidic sequences results in a tendency to strongly aggregate, leading to incomplete acylation/deprotection reactions. This, in turn, retards the progress of the polymerisation reaction.
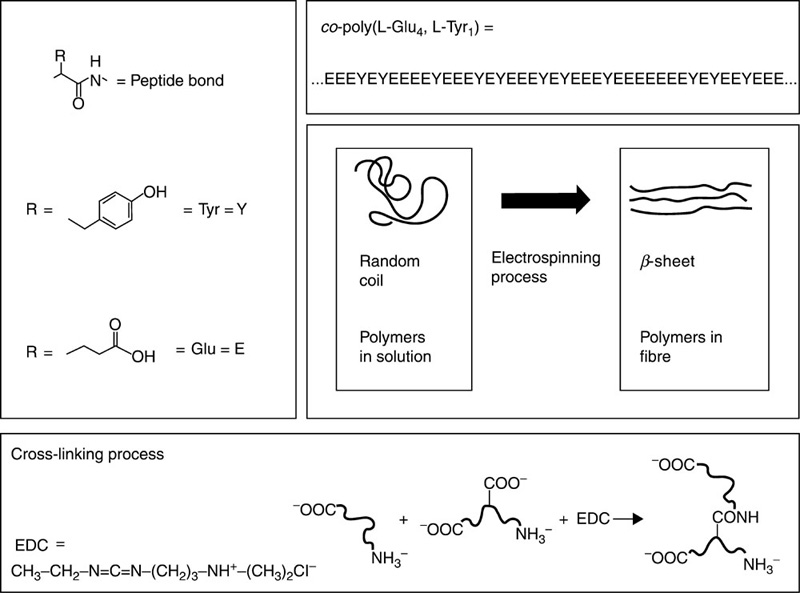
The natural biological functions of polypeptides have led to self-assembled elastin oligopeptides being considered for use as non-thrombogenic coatings and matrices for tissue engineering (Eldijk et al., 2012; Kim and Chaikof, 2010; Kyle et al., 2009; Li et al., 2010; Ulijn and Woolfson, 2010). Poly(aspartic acid) and poly(glutamic acid) are synthetic homopolypeptides being employed for drug and gene delivery applications, due to their biodegradable nature and pH sensitivity. These polymers and their benzyl derivatives have been further blended with poly(ethylene glycol), poly(ethylene oxide) and other hydrophilic polymer units to develop amphiphilic block polymers for the fabrication of micelles and vesicles. Benzyl derivatives of peptides, such as β-benzyl-L-aspartate or β-benzyl-L-glutamate N-carboxyanhydride are formed via a ring opening polymerisation reaction performed under different initiator conditions, with NaOH being employed for the deprotection of the benzyl moieties. The blending of poly(aspartic acid) with poly(lactic acid) has been reported to increase the hydrolytic dissociation rate of the latter polymer due to the presence of aspartic acid units. It has been found that the modification of surfaces with poly(aspartic acid) results in an enhanced cell adhesion and function of osteoblasts grown on polymeric scaffolds. Co-polymerisation of polypeptides with DNA complexes has resulted in the formation of materials with sound haemo- and cyto-compatibility properties.
3.7 Poly(anhydride)s
Poly(anhydride)s are a class of biodegradable polymers in which monomer units are linked via an anhydride bond, (RC(O))2O. Their use in the fabrication of drug delivery and biomedical devices has been attributed to their biocompatibility and their ability to degrade in vivo to form non-toxic diacid products that can be safely removed from the host system via normal metabolic activity (Carrillo-Conde et al., 2012; Lasne-Deschamps et al., 2012; Ojer et al., 2012; Rebouças et al., 2012). The rate of degradation of the polymer can be adjusted to take place over periods ranging from several days to years, a feature that makes poly(anhydride)s attractive for targeted drug delivery and controlled release applications. Poly(anhydride)s are generally synthesised via the melt polycondensation of acetylated dicarboxylic acid prepolymers, although the polycondensation of sebacic and 1,6-bis-(paraphenoxy) hexane diacids under microwave irradiation conditions has also been attempted in order to simplify and increase the speed of synthesis. Figure 3.6 shows the chemistry involved in the thiolene polymerisation of poly(anhydride)s (Lou and Shipp, 2012). High molecular weight polymers with a relatively high yield can be obtained under conventional polycondensation conditions.

3.8 Poly(urethane)s
Poly(urethane)s are highly versatile biomaterials, which remain popular in many applications for their customisable chemistry, processability and bio- and haemo-compatibility (Xie et al., 2009). Their segmented block make-up allows these polymers to be formulated with a vast variety of physical and mechanical properties, stability and degradation rates. In addition, their compatibility with surrounding tissues can be conferred by the appropriate selection of poly(urethane) composition, blending and processing conditions (Kloss et al., 2009). Their popularity as biomaterials has not always been high; poly(urethane)s were recognised for their tissue and haemocompatibility in the 1970s and 1980s, and as a result they became the material of choice for long-term implantation applications, including cardiovascular devices and breast implants. Their stability fell under scrutiny in the late 1980s (Santerre et al., 2005). The relative sensitivity of poly(urethane)s to in vivo biodegradation led to their use in the fabrication of novel bioresorbable materials. An improved understanding of the mechanisms by which poly(urethane) biodegraded, obtained during this period, contributed to the development of more stable poly(urethane)s with finely tunable material properties. As a result, poly(urethane)s are now used in a broad range of medical applications, from biostable, inert devices and encapsulation coatings, such as those used in catheters, vascular grafts and artificial organs, to fully biodegradable materials for soft tissue engineering and controlled delivery of drugs and bioactive molecules, such as insulin-like growth factor-1 and hepatocyte growth factor (Attia et al., 2011; Nelson et al., 2011).
Poly(urethane)s are comprised of hard and soft segments, the former being composed of aromatic or aliphatic diisocyanates and chain extenders (diols), and the latter being composed of flexible polyols. The poly(urethane) R1–NH–CO–O–R2 is formed when the isocyanate functional group R1–N = C = O reacts with the hydroxyl group of a polyol R2–OH. The hard segments are stiff and immobile, acting as cross-links between the mobile softer segments. Aromatic isocyanates that are frequently employed include diphenylmethane diisocyanate and toluene diisocyanate, with hexamethylene diisocyanate and isophorone diisocyanate representing examples of aliphatic isocyanates. Diisocyanates can also be used to synthesise polyisocyanates possessing three or more –N = C = O functional groups, or reacted with polyols to form a prepolymer for the subsequent fabrication of poly(urethane).
Ethylene glycol, 1,4-butanediol, 1,6-hexanediol, cyclohexane dimethanol and hydroquinone bis(2-hydroxyethyl) ether are some notable chain extenders. Two key classes of polyols are commonly used: poly(ether)s, such as poly(propylene oxide), poly(tetramethylene oxide) and poly(tetrahydrofuran), and poly(ester)s, such as poly(caprolactone) and poly(carbonate) (Hashimoto et al., 2010). These polyols are commercially produced either via the ring-opening polymerisation of cyclic monomers or by the polycondensation of bifunctional monomers. In a quest to broaden the potential biomedical applications of poly(urethane)s, novel polyols are now being developed. For instance, polyols incorporating castor oil derivative have been proposed (Somani et al., 2006; Szelest-Lewandowska et al., 2003).
The choice of the chain extender and the chemistry of the hard segment, particularly with regard to the number of functional isocyanate groups and their reactivity, influences the mechanical properties and overall stability of the final poly(urethane) (Chan-Chan et al., 2010). For instance, aromatic diisocyanates-based poly(urethane)s discolour when exposed to visible light due to the presence of chromophores, whereas the more stable aliphatic diisocyanates-based materials do not discolour. An introduction of thiourea-containing diisocyanate and imide structures in the synthesis of poly(ethylene glycol)-based poly(urethane) has been shown to improve their thermal stability, chemical resistance and processability of the resultant thermoplastic (Kausar et al., 2011). The concentration of the hard segment within the polymer was shown to affect the extent of poly(urethane) crystallinity, hence affecting the extent of phase separation, and as such the mechanical and biodegradation properties of the polymer (Wang et al., 2003). Segmented poly(urethane) block copolymers of 4,4′-methylenediphenyl diisocyanate and 1,4-butanediol, and oligomeric ethoxypropyl poly (dimethyl siloxane) were found to consist of three micro-phases, namely an ethoxypropyl poly(dimethyl siloxane) matrix phase, hard domains and a mixed phase. The latter contained ethoxypropyl end group segments and dissolved short hard segments. The degree of phase separation was found to increase significantly as a function of hard segment content (Choi et al., 2010).
The type of chain extender has been found to profoundly influence the properties of the final polymer (Fig. 3.7). For example, bifunctional chain extenders produce thermoplastic materials, whilst polyfunctional extenders result in the production of thermosetting materials. Furthermore, the chain extenders influence the tensile strength, elongation and tear resistance, as well as flexural, heat and chemical resistance properties of the poly(urethane) materials (Kultys et al., 2009). The elasticity and strength of the biodegradable scaffolds used for cardiovascular tissue engineering were found to be higher in the case of poly(ester urethane)urea materials compared to poly(ether ester urethane)ureas, with the latter being more amenable to smooth muscle cell adhesion and proliferation (Guan et al., 2005).
Degradable poly(urethane) elastomers coated with gelatin, laminin or collagen IV and seeded with embryonic stem cell-derived cardiomyocytes hold great promise as bio-engineered cardiac grafts for the repair of damaged heart tissue in infarction patients (Alperin et al., 2005). Poly(urethane)s produced using glycine-leucine dipeptide sequence-based chain extenders were characterised by improved hard segment packing and hydrogen bonding, and as a consequence, they possessed a significantly higher initial modulus, yield stress and ultimate stress compared to poly(urethane)s containing a phenylalanine-based chain extender (Parrag and Woodhouse, 2010).
Similarly, urea-diol or aromatic amino-acid derivative chain extenders were used to enhance the hard segment association via bidentate hydrogen bonding or π-stacking interactions in bioresorbable aliphatic poly(ester urethane urea)s synthesised from poly(ε-caprolactone) diol and 1,6-hexamethylene diisocyanate or L-lysine methyl ester diisocyanate (Caracciolo et al., 2008, 2009a). This series of studies found that the symmetry of hard segment and hard segment cohesion modulated the extent of phase separation of soft and hard domains, having an important effect on the observed thermal and mechanical behaviour of the segmented poly(urethane)s, with materials based on 1,6-hexamethylene diisocyanate being stronger than those synthesised from L-lysine methyl ester diisocyanate. Poly(ester urethane urea)s produced using an amino acid derivative chain extender were more susceptible to degradation, due to the presence of hydrolysable ester bonds; however, the structure and exposure time were also found to influence the rate of polymer degradation.
Overall, the materials were also characterised by sound anti-thrombogenicity and satisfactory cytocompatibility, with a demonstrated potential for use in the fabrication of nonporous films and microfibre/nanofibre and highly-porous scaffolds effectively mimicking the protein fibres of native extracellular matrix (Caracciolo et al., 2009b, 2011). L-lysine diisocyanates are synthesised by phosgenation of amine-terminated lysine esters, which have a lower vapour pressure compared to other aliphatic diisocyanates, such as hexamethylene diisocyanate. As such, poly(urethane)s based on lysine derivatives can be used in applications where inhalation toxicity, handling and processing are important considerations (Cossu et al., 2011).
Innovative chain extenders, such as 4,4′-isopropylidinedi-(2,6-diiodo-phenol) and N,N′-bis(3-hydroxypropxyl)-2,3,5,6-tetraiodoterephthalamide, can be employed to combat the radiolucent property of poly(urethane)s, the latter being a limiting factor in medical applications in minimally invasive operations and where the noninvasive evaluation of the implanted material or device is required (Kiran et al., 2009; Qu et al., 2011). In addition to high radiopacity, sound thermal stability, favourable mechanical properties and versatility in their chemical synthesis, the advantage of using iodine-containing diols over commercial radiopaque additives, such as barium sulphate, zirconium dioxide or bismuth halides, lies in the non-cytotoxicity of the resultant biomaterial.
The nature of the soft segment is an important consideration, particularly with regard to its chemical composition, molecular weight, proportion of primary hydroxyl groups, functionality and viscosity. Linear, difunctional poly(ethylene glycol) segments, for example, result in soft, flexible and elastic poly(urethane)s, whereas high functionality initiators yield poly(urethane)s of higher rigidity. Poly(ether)urethanes synthesised from higher molecular weight poly(ethylene glycol), hence containing the highest soft segment content, also demonstrated comparatively high hydrophilicity, low tensile strength and high elongation at break compared to polymers derived from low molecular weight poly(ethylene glycol) (Sarkar et al., 2010, 2011). Similarly, the weight composition of the polyol component influenced the sorption behaviour of castor oil derivative/poly(ethylene glycol) and toluene diisocyanate-based poly(urethane), with the diffusion coefficient decreasing with an increase in chain length of the soft segment (Somani et al., 2006). Poly(urethane)s with longer poly(caprolactone) soft segments and a molecular mass of 2000 were also demonstrated to be semi-crystalline, whereas polymers with shorter polyol chain lengths were found to be unable to undergo crystallisation (Wang et al., 2003). Introduction of arabinitol-based diols into the structure reduced the crystallinity, which was found to influence the degradation pattern of dithiodiethanol and 1,6-hexa-methylene diisocyanate-based polymers under physiological conditions, with the ratio of reactive units also playing a significant role (De Paz et al., 2010). Aqueous solutions of poly(ether urethane), consisting of poly(ethylene-butylene), poly(ethylene glycol) and poly(propylene glycol) segments, exhibited thermogelling behaviour at critical gelation concentrations and non-toxicity of the copolymer towards mouse fibroblast cells (Nam Nguyen et al., 2011).
Generally, poly(urethane)s are prepared via the polyaddition of diols and diisocyanates, whose high reactivity has limited the introduction of functional groups onto poly(urethane) backbones. Poly(ether) polyols are often chosen for their variability in terms of molecular weight, viscosity, functionality and composition, with poly(tetrahydrofuran) being particularly popular as a result of its low temperature flexibility, low content of extractable substances, microbial resistance and hydrolytic stability (Basko et al., 2011). However, tetrahydrofuran in its monomer form has no reactive pendant group in its structure, with the resulting poly(tetrahydrofuran) being unsuitable for modification once it is incorporated in the poly(urethane) backbone. Hence, the introduction of an elevated and controllable amount of functional groups that do not interfere with the poly(urethane) chemistry is highly desirable and can be achieved via side-chain functionalisation (Yang et al., 2011). As such, functionalised poly(tetrahydrofuran)s have been obtained via the cationic ring-opening copolymerisation of tetrahydrofuran and functionalised cyclic ether, namely glycidyl propargyl ether (Basko et al., 2011). Once synthesised into linear poly(urethanes), the resulting alkyne side groups present along the main chain were used as latent modification sites for further functionalisation by a consecutive reaction with azides, leading to a vast array of possible functionalised biomaterials.
In another study, poly(hydroxyurethane)s, fabricated by the polyaddition of bifunctional carbonates with diamines, have been used to synthesise poly(urethane)s bearing hydroxyl groups in their side chains. The reactivity of the hydroxyl groups also allowed for further esterification and silylation of poly(hydroxyurethane)s which, when reacted with functional isocyanates, could produce poly(urethane)s with superior mechanical properties, biocompatibility and controlled solubility. Poly(urethane)s bearing urethane groups in the side chains were prepared by the addition of isocyanates to the hydroxyl groups in poly(hydroxyurethane), prepared by the polyaddition of a bifunctional cyclic carbonate with 1,12-diaminododecane (Ochiai et al., 2007b). Addition of 2-methacryloyloxyethyl isocyanate resulted in polyurethane-bearing methacrylate groups through urethane linkages, with both polymers being thermally cross-linkable (Ochiai et al., 2007a).
Furthermore, urethanisation of the aforementioned poly(hydroxyurethane) with 3-(triethoxysilyl)propyl isocyanate resulted in a poly (urethane) with a triethoxysilyl group, which has potential as a precursor for the synthesis of a poly(hydroxyurethane)-silica hybrid. The incorporation of polar oxyethylene groups into the side chains of the poly(vinyl ether) soft segments was found to produce poly(urethane)s with a highly hydrophilic nature and thermo-responsive functions (Hashimoto et al., 2010). Biodegradable poly(urethane)s, bearing a varied content of hydrophilic segments and reactive pendant amino groups, were prepared by adjusting the molar ration of poly(ethylene glycol) ester of NH2-protected-(aspartic acid) to poly(ε-caprolactone) diols (Xie et al., 2007).
Functionalisation of poly(urethane)s via the incorporation of low molecular weight diols into the hard segment of the polymer using click chemistry has also been attempted (Fournier and Du Prez, 2008). Recently, click chemistry has been attracting attention in organic and polymer chemistry for post-polymerisation modification reactions and step-growth polymerisations (Fournier et al., 2007; Gauthier et al., 2009; Kolb et al., 2001). For example, linear poly(urethane)s with alkyne groups along the backbone synthesised by reacting two different alkyne-functionalised diols with a diisocyanate compound, were subjected to Huisgen 1,3-dipolar cyclo-addition using a variety of azide compounds (Fournier and Du Prez, 2008). As a result, poly(urethane)s with varying degrees of functionalisation were obtained. The resultant functional groups could be used to selectively bind a variety of proteins.
Phenyl azide groups introduced into biodegradable poly(urethane)s containing free side hydroxyl groups via 4-azidobenzoic acid conjugation, were capable of binding mouse IgG under ultraviolet irradiation in 3 minutes (Yang et al., 2011). Importantly, the bound mouse IgG retained its biological activity and could further bind the labelled anti(mouse IgG), with potential applications in immunofluorescence assay and related fields. More knowledge regarding the molecular processes involved in protein adsorption and binding, however, is required for the development of an effective means to control interfacial interactions (Yaseen et al., 2008). For instance, human serum albumin adsorption was found to be little affected by differences between the surface chemistry of the commercial poly(urethane)s and a novel poly(carbonate-urea)urethane matrix containing silsesquioxanes, the latter poly(urethane) being significantly rougher due to the presence of the silsesquioxane hard segments. However, fibrinogen adsorption was much greater on the poly(urethane) surfaces, indicating a strong surface effect.
The ability to withstand or undergo controlled hydrolytic dissociation is an important consideration for in vivo poly (urethane) applications (Braun et al., 2011). Thus, hydrophobicity and ability to resist hydrolysis for extended periods of time is imparted on those poly(urethane)s intended for long-term implantation, whereas those materials designed to perform as biologically reactive scaffolds are synthesised to be more hydrophilic, with their aptitude to absorb biological fluids and undergo hydrolytic and oxidative degradation modulated via hydrophilic/hydrophobic balance. This balance can be achieved with varying ratios of the hydrophilic-to-hydrophobic segment, where hydrophilic segment can be based on poly(ethylene oxide-propylene oxide-ethylene oxide) diols and the hydrophobic segment based on poly(ε-caprolactone) diol (Gorna and Gogolewski, 2002). Tetrahydrofuran ether-based poly(urethane)s demonstrated excellent hydrolysis and microbial resistance compared to poly(ester)-based materials, due to the highly vulnerable ester linkages of the latter, yet they were found to be more susceptible to oxidative degradation and heat ageing (Cozzens et al., 2010; da Silva et al., 2010). Poly(carbonate urethane)s have been demonstrated to be more resistant to oxidative degradation compared to the aforementioned poly(urethane) types (Christenson et al., 2004). Interestingly, their hydrolytic degradation can be effectively modulated using shielding carbonate chemistry, to different degrees by using hard segment chemistry.
The in vitro degradation studies of amphiphilic alternative block poly(urethane) copolymers based on poly(3-hydroxybutyrate-co-4-hydroxybutyrate) diol and poly(ethylene glycol)-diisocyanate demonstrated how the chain extender and soft segment composition and length influenced the stability of the materials in different media, with responses ranging from surface erosion to diffusion bulk collapsing (Pan et al., 2009). Intelligent mixing of soft segments with conventional hard units can result in the production of poly(urethane)s possessing increased elastomeric properties without having to compromise their oxidative and hydrolytic stability or biocompatibility of the poly(urethane)s. Incorporation of modest amounts of poly(tetra-methylene oxide) chains into poly(isobutylene)-based poly(urethane) significantly increased both the tensile strength and elongation of the material, the resultant polymer possessing mechanical properties approaching that of conventional thermoplastic polymers. The resultant materials also exhibited excellent biocompatibility and oxidative/hydrolytic stabilities far superior to the most oxidatively stable commercial poly(urethane)s (Jewrajka et al., 2009).
The rate of biodegradation of poly(urethane)s is crucial to their in vivo performance, as they must retain their physical properties under ambient conditions during the intended integration period. Integration of biomaterials into the surrounding tissue is frequently accompanied by a local inflammatory response, whereby circulating monocytes are recruited by constitutive or inflammatory signals in the tissues (Polati et al., 2009). Resultant tissue-activated monocyte-derived macrophages can attach to the surface of the indwelling biomaterial for the duration of the implantation, their activity being fundamental to wound healing (McBane et al., 2011). Both macrophages and neutrophils are actively involved in material biodegradation via a co-mediated mechanism involving the release of oxidative and hydrolytic agents such as hydrogen ions and hydrogen peroxide, and cholesterol esterase, carboxyl esterase and serine proteases, respectively (Zhao et al., 1991). Characterisation of the degradation mechanism of polymeric scaffolds and drug delivery systems is therefore critical in evaluating their clinical relevancy, with minimal transient inflammatory response and controlled degradation to soluble non-cytotoxic breakdown products that can be removed by physiological processes being fundamental performance criteria (Hafeman et al., 2011). In vitro studies on several relatively oxidation-resistant poly(carbonate urethane)s demonstrated that pre-lreatment with H2O2 modulated degradation by esterases (McBane et al., 2007). It was found that chemically altering the material surface affected cell function and hence the macrophage-mediated degradation, with material composition being a contributing factor.
It is important to note that in vitro degradation is, at best, an approximation of in vivo conditions, as it is difficult to account for all the complexities of the in vivo environment. For example, poly(ester urethane) scaffolds prepared from lysine-derived polyisocyanates have been demonstrated to degrade faster under in vivo compared to in vitro conditions, indicating that cell-mediated degradation is taking place (Hafeman et al., 2008; Zhang et al., 2000). A study on scaffolds synthesised from lysine triisocyanate or a trimer of hexamethylene diisocyanate demonstrated that in buffer, the hydrolytic dissociation of ester bonds to yield α-hydroxy acids was the dominant mechanism, with esterolytic media only modestly increasing the degradation rate. However, in oxidative media, hexamethylene diisocyanate scaffolds showed a modest increase in degradation rate, whereas lysine triisocyanate scaffolds degraded six times faster, approximating their in vivo rate in rat excisional wounds. The propensity of lysine triisocyanate scaffolds to oxidative degradation was further corroborated via detection of myeloperoxidase expressed by mouse macrophages at the material surface (Hafeman et al., 2011).
Enzyme catalysis can be used as a tool for the controlled disassembly of biodegradable poly(urethane)s for drug delivery and soft tissue engineering (Woo et al., 2000). Recently, poly (urethane) s used to construct biodegradable scaffolds with remodellable features were synthesised by incorporating a glycine-leucine dipeptide sequence, the latter being the selective site of cleavage of several matrix metallo-proteinases, hence enabling enzyme-triggered degradation (Parrag and Woodhouse, 2010). Pancreatic lipase has also been demonstrated to initiate urethane-bond hydrolysis in 1,6-hexamethylene diisocyanate based poly-(urethane)s, with 1,4-di-S-benzyl-D,L-dithiothreitol and triethylene glycol soft components (Ferris et al., 2010). The study of L-tyrosine based poly(urethane)s with varied soft segments showed that poly(ethylene glycol) based polymers underwent soft segment oxidative degradation, whereas those based on poly(caprolactone) underwent hard segment degradation (Sarkar and Lopina, 2007). The presence of the amino acid based chain extender afforded the polymer an enhanced extent of enzymatic degradation when exposed to the proteolytic enzyme α-chymotrypsin.
Another study investigated the effect of different hard segment structures and esterase activity in sensitising of poly(carbonate urethane)s towards enzyme-catalysed hydrolysis by cholesterol esterase (Tang et al., 2003). Irrespective of the hard segment chemistry, the enzyme dose response was found to decrease with increase in polymer hard segment content. However, the dependence on enzyme concentration varied vastly between poly(urethane)s of different hard segment chemistry, with 4,4-methylene biscyclohexyl diisocyanate-based poly(urethane)s displaying the most dramatic dependence compared to those polymers synthesised from 1,6-hexane diisocyanate or 4,4′-methylene bisphenyl diisocyanate. Despite being the most stable polymer of those tested, at the low enzyme concentration of 80 units per mL, the 4,4-methylene biscyclohexyl diisocyanate-based biopolymer underwent a catastrophic breakdown when the cholesterol esterase concentration was increased to 400 units per mL. Although the latter concentration is considerably higher than that encountered in vivo, the accelerated degradation experiment was shown to be a useful tool in the investigation of long-lerm polymer biological stability. When such a concentration of cholesterol esterase was used to incubate commercial poly(ether urethane) and poly(carbonate urethane) over a 36-day period, only a small loss in surface soft segment content was found, indicating the surface confinement of enzymatic hydrolysis (Christenson et al., 2006). The study suggested oxidation and not enzymatic hydrolysis to be the principal mechanism to which in vivo degradation of poly(ether urethane) and poly(carbonate urethane) should be attributed, as demonstrated previously using 20% H2O2/0.1 M CoCl2 treatment and antioxidant agents (Christenson et al., 2004). Overall, both studies demonstrated poly(carbonate urethane) to be more stable than poly(ether urethane).
In addition to chemical reactivity, the processing conditions and the resulting physical configuration of poly(urethane)s are known to significantly influence their material properties, and as such their extent of biodegradation and the way in which they interact with ambient environments. Slowly degrading poly(ester urethane)s made into porous (mesh) and nonporous (film) scaffolds using electrospinning and solvent casting, respectively, were characterised by varied morphology, pore size and filament diameter. Although both configurations were found to be cytocompatible, the mesh scaffold was more favourably disposed towards cell attachment and viability (Henry et al., 2007). The mechanical properties and degradation behaviour of a random configuration of electrospun poly(carbonate urethane) scaffolds intended for the growth of a tissue-engineered annulus fibrosus disc component were compared to those of aligned nanofibres. Although the tensile strength and initial modulus of the aligned scaffolds were found to be significantly higher than those of random fibre scaffolds prior to degradation, these parameters notably reduced upon wetting. Similar changes were not observed for the random scaffold configuration (Yeganegi et al., 2010). In a separate study, pore morphology, orientation and porosity were found to influence scaffold degradation in an aqueous buffer (Guan et al., 2005). Scaffolds with open and interconnected pores were fabricated from poly(ester urethane)urea and poly(ether ester urethane)urea by thermally induced phase separation using dimethyl sulfoxide as a solvent.
Although frequently used to enhance the polymerisation process or impart desired properties onto poly(urethane)s, the solvents used in polymerisation processes need to be chosen carefully since many of them are toxic. Such additives may not be fully eliminated from the final product, and therefore may undermine the resultant polymer’s biocompatibility and clinical applicability. For example, ionic liquids used in the microwave-assisted synthesis and processing of poly(urethane)s often have higher boiling points than more toxic, but more volatile solvents, and therefore may not be effectively removed. Microwave-assisted polymer fabrication has been reported to result in a higher degree of polymerisation compared to that of conventional methods, with the non-polar decalin leading to a higher extent of polymerisation than obtained using polar solvents such as N,N-dimethylacetamide or chlorobenzene, probably due to the thermal degradation of the growing polymer chain. Furthermore, microwave processing has been suggested as the technology capable of overcoming solubilisation limitation of poly(urethane)s stemming from the large amount of strong interchain hydrogen bonds generated.
3.9 Conclusion
Synthetic versatility and tunability of the properties of organic polymeric biomaterials ensure their leading position in a variety of biomedical applications that require stimuli-responsiveness, site targeting and spatio-temporal delivery of biological entities and cells. The flexible chemistry and relative ease with which functional units can be appended onto a polymer backbone allows for precise control over their porosity, microsphere size and cross-linking behaviour, as well as their in vitro and in vivo compatibility and biodegradation kinetics. Indeed, relatively minor alterations to an organic polymer via addition, subtraction, self-reaction or cross-reaction with other macromolecule and non-polymer units produces biomaterials that contain characteristics that allow the conception of novel applications, together with the advancement of existing pharmaceutical and medical products.
In addition to their use as solid implants, injectable organic polymeric materials hold considerable promise for bone tissue regeneration applications that have distinct advantages compared to the traditional highly invasive orthopaedic, craniofacial and periodontal procedures. Injectable microspheres and gels, such as those based on poly(L-lactide-co-glycolide), poly(propylene fumarate), poly(ethylene glycol) and poly(L-lactide-co-glycolide)–poly(ethylene glycol) copolymer can be used to completely fill irregularly shaped bone defects and set in situ, providing a suitable platform for cell attachment, differentiation, proliferation and extracellular matrix formation. Although these are promising applications, the currently available injectable and in situ settable materials still require substantial improvement with regard to their clinical handling properties before they can be used in patients. Their reactivity in vivo also requires further investigation. As researchers learn more about these and other exciting materials that are derived from organic polymers, they will be able to overcome current technical barriers and facilitate translation of these promising biomaterials into clinical settings.