
Noncovalent Functionalization of Cell Surface
Debjit Dutta, Srujan K. Marepally and Praveen Kumar Vemula, Institute for Stem Cell Biology and Regenerative Medicine, National Centre for Biological Sciences-Campus, UAS-GKVK, Bangalore, India
The cell membrane is a highly heterogenous and dynamic environment that dictates many biological functions like cell–cell and cell–niche communication, cell adhesion, migration, and differentiation. Thus, cell surface is a potential target for introducing different functional groups and molecular and therapeutic probes for diverse biomedical applications. In this chapter, we have discussed different noncovalent approaches such as membrane fusion, electrostatic and hydrophobic interactions using cationic and polymeric amphiphiles, and use of metabolic engineering and bioorthogonal click chemistry for the modification of cell surface. The advantages of noncovalent methods over covalent and genetic methods of modification and their applications in cell-based therapy, bioimaging, and tissue engineering have also been described. Future directions in terms of developing dynamic approaches to modify cell surfaces have been discussed in brief.
Keywords
Noncovalent methods; self-assembly; cell surface; electrostatic interactions; hydrophobic interactions; bioorthogonal chemistry; amphiphiles; cell therapy
5.1 Introduction
Surface of the cell plays a pivotal role in defining the cell fate. A wide range of functional biomolecules that anchored on the cell surface perform different roles. A cell communicates with other cells, environment, pathogens, and nutrients through its surface. In addition, cell migration is highly dependent on the cell membrane. Thus, cell fate could be modulated through engineering cell surface (Figure 5.1). Cell surface is a fertile ground to manipulate cell phenotypes and biological fates. While these manipulations are key tools to answer some of the basic biological questions [1], they could be instrumental in developing novel diagnostic and therapeutic systems [2] through engineering of cell surface. Applications of genetic tools for manipulating proteins and probing cellular functions have revolutionized biology and medicine. These tools were proven to be successful, but their applications are restricted due to their inability to introduce novel functionality into biomolecules or interrogate the distinct chemical entities that control cellular responses. To address these limitations, multiple research teams have developed a set of chemoselective reactions, known as bioorthogonal reactions, with minimal cross-reactivity and toxicity in biological systems.
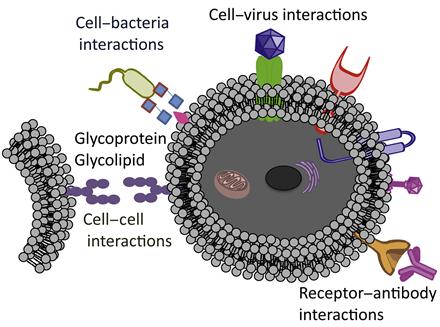
A number of synergistic approaches combining chemistry, material science, and biology have been applied to engineer and manipulate living cells. Desirable chemical functionalities and materials can be introduced onto the cell surface by covalent and noncovalent methodologies, as well as through specific biological recognition events, including antibody–antigen and ligand–receptor interactions. These strategies have been used in a wide range of applications that include designing the biomaterial scaffolds [3,4] to control cell fate, labeling of cells with molecular and nanoparticle probes to visualize cellular processes and molecular pathways [5–7], delivering diverse species into cells [8,9], and patterning cells for drug discovery [10,11]. To date, cell surface engineering has been primarily achieved through the use of molecular biology and genetic engineering [12]. However, perturbations of inherent cellular physiology with genetic engineering may significantly interfere with cellular functions. Thus, there is a need to develop tools that will provide simple alternatives to genetic and biosynthetic pathways.
Nongenetic cell surface modifications have been generally achieved using the following methods: (i) covalent conjugation of desired molecules to surface functional groups like amines [11,13–20], (ii) incorporation of amphiphilic polymers into the lipid bilayer membrane of cells by harnessing hydrophobic interactions [21], (iii) electrostatic interactions between cationic polymers and a negatively charged surface [15], and (iv) metabolic engineering of ketone- or azide-modified carbohydrates to display desired functional groups on the cell surface, followed by bioorthogonal click reactions to tether molecule of choice [22–24]. Schematic representation of some examples of engineering of cell surface is shown in Figures 5.2 and 5.3.
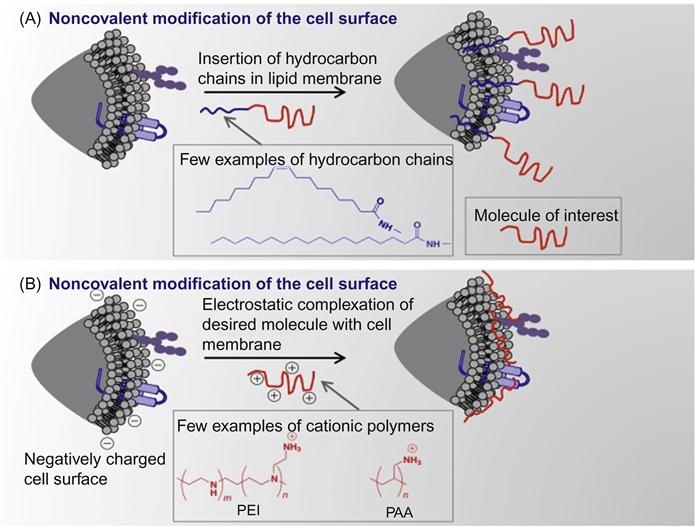
This chapter focuses on nanotechnology approaches that could be applied for the application of cell surface engineering and their limitations.
5.2 Methods of Cell Surface Engineering—Applications and Recent Developments
5.2.1 Targeting Ability of Surface Engineered Cells
Cell therapy involving systemic infusion and targeted delivery of progenitor/mesenchymal stem cells (MSCs) and other cell types to damage and diseased tissues hold tremendous promise for the treatment of a number of diseases such as cardiomyopathy, cancer, and fibrosis [25]. MSCs are capable of differentiating into multiple lineages and produce extracellular matrix (bone, cartilage, fat) and can also secrete immunomodulatory cytokines to reduce inflammation. Although they are currently being tested in more than 100 clinical trials for treatment of multiple diseases, including graft versus host disease, myocardial infarction, multiple sclerosis, and skeletal tissue repair, the major challenge in MSC therapy is the delivery of MSCs efficiently to targeted location. Systemically administered therapeutic MSCs have poor expression of homing ligands or lose them during the culture expansion and only less than 1% of culture-expanded MSCs can actually home to the target tissue [26]. Therefore, efforts have been made to introduce cell homing ligands onto cell membranes. Cell homing ligands (on the homing cell) and receptors (on the endothelium) allow homing cells to tether, roll, adhere, and then transmigrate on endothelium as part of the cell homing cascade. A great effort has been made to achieve targeted delivery of cells, and a wide range of methods such as genetic and enzymatic engineering [27–29] treatment with cytokines [30] and chemical approaches have been developed [31,32].
5.2.2 Noncovalent Immobilization of Agents on the Surface of Cells
Although a covalent approach offered a method to modify the cell surface for longer duration, often, covalent functionalization might interfere with the function of native proteins or other molecules that are present on the cell surface. Thus, to overcome these potential limitations, a robust noncovalent approach to transiently engineer the cell surface has been developed [33]. In this method, unilamellar vesicles composed of biotinylated lipid were fused with the cell membrane to anchor biotin on the cell membrane of MSCs (Figure 5.4). Intercalation of the lipid vesicles with lipid bilayer of the cell membrane is a robust phenomenon; it offers numerous opportunities to immobilize any desired molecules on the surface of the cell membrane. The biotinylated surface of the cell was further hybridized with multivalent streptavidin and followed by biotinylated SLeX (Figure 5.4). As cell membrane undergoes a flip–flop process, lipids in the membrane are rearranged and recycled. Thus, modification on the cell surface through noncovalent liposomal hybridization offers a transient cell surface medication and avoids nonspecific modification of cell surface molecules. In addition, transient immobilization of adhesion ligands offered a great advantage through allowing for initial adhesion and cytokine activation, not impeding subsequent extravasation.
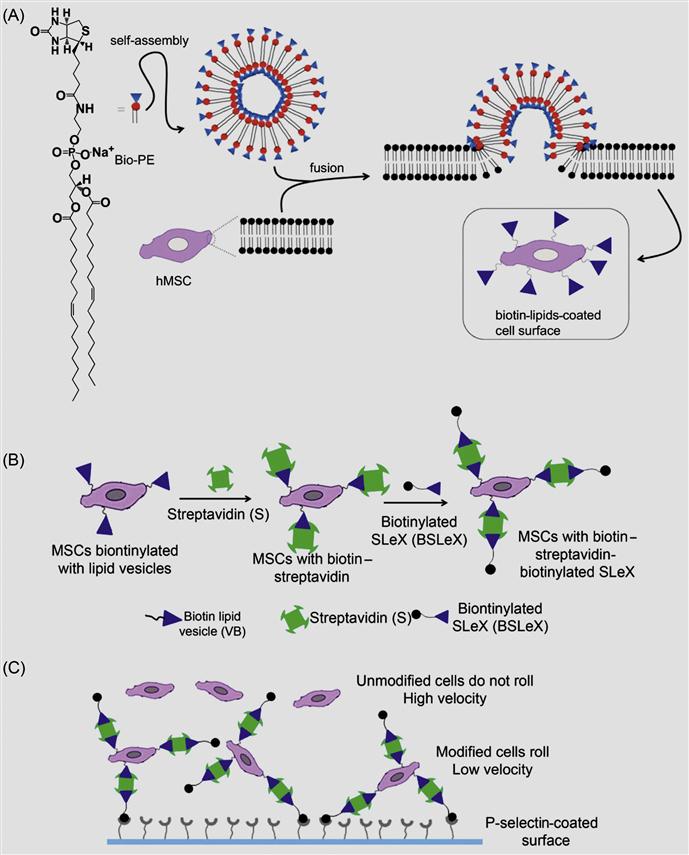
Furthermore, the rolling response of the SLeX-modified MSCs with P-selectin-coated substrates has been examined using a flow chamber assay. MSCs modified with SLeX showed considerably lower velocities on immobilized P-selectin substrates compared to phosphate buffer saline (PBS) treated cells (Figure 5.5). SLeX-modified MSCs exhibited ~eightfold less velocity compared to unmodified MSCs at a shear stress of 0.5 dyn/cm2 which suggested the effect of noncovalently modified surface with SLeX.
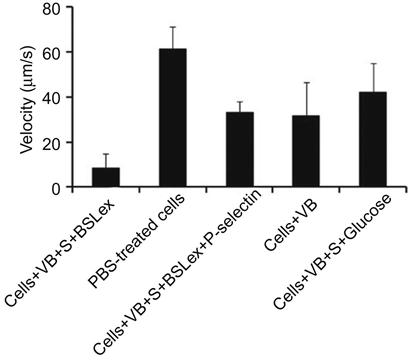
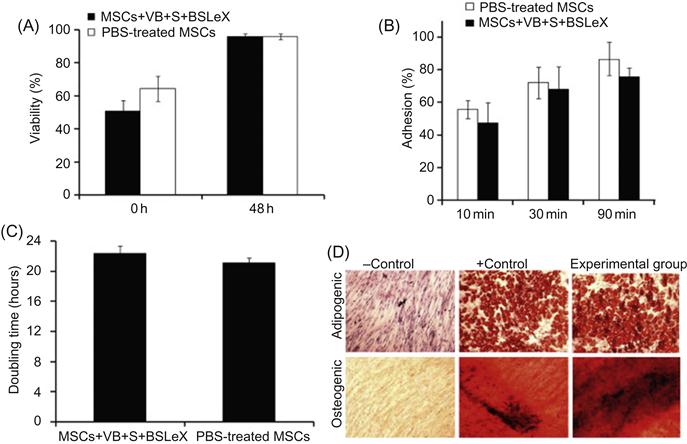
Inherent properties of stem cells are sensitive to minor modifications of cell machinery and microenvironment. Thus, it is important to investigate the effect of cell surface modification through liposomal fusion on the cell fate. Therefore, Sarkar et al. [33] have thoroughly characterized the phenotype of MSCs after modification through liposomal fusion (Figure 5.6). Viability and proliferative rates of the modified MSCs were comparable to unmodified MSCs. In addition, the adhesion kinetics of modified MSCs and untreated MSCs are similar on tissue culture polystyrene plates, which indicated that inherent properties of the cell surface did not compromise due to noncovalent liposomal fusion. Another important characteristic of MSCs is the ability to differentiate into multiple lineages that did not compromise modification of the cell surface with biotinylated lipids and subsequent complexation of streptavidin and biotinylated SLeX. Modified MSCs were efficiently differentiated into osteogenic and adipogenic lineages as shown by alkaline phosphatase activity and Oil Red O staining, respectively (Figure 5.6). Those results clearly suggested that modifying the cells with rolling ligands through a noncovalent lipid vesicle approach did not affect the cell phenotype and thus cells may maintain their normal functional characteristics that will be important for cell therapy. This is a compelling example for the broad scope of cell surface modification through noncovalent approaches.
5.2.3 Noncovalent Cell Surface Modification Followed by Covalent Chemistry to Enhance the Cell Interactions
As described in previous sections, where cell surface molecules were covalently reacted with biotinylated-N-hydroxyl-succinimidyl (NHS) followed by immobilization of a molecule of interest through noncovalent complexation, often it may lead to nonspecific functionalization of cell surface molecules. To overcome this limitation, Dutta et al. [34] have developed an alternative method where low-molecular-weight amphiphile with a desired functional group was noncovalently fused with the cell membrane; subsequently, they have covalently conjugated those immobilized functional groups selectively with complementary reactive groups (Figure 5.7). In this approach, they have eliminated the nonspecific functionalization of biomolecules that are present on the cell surface.
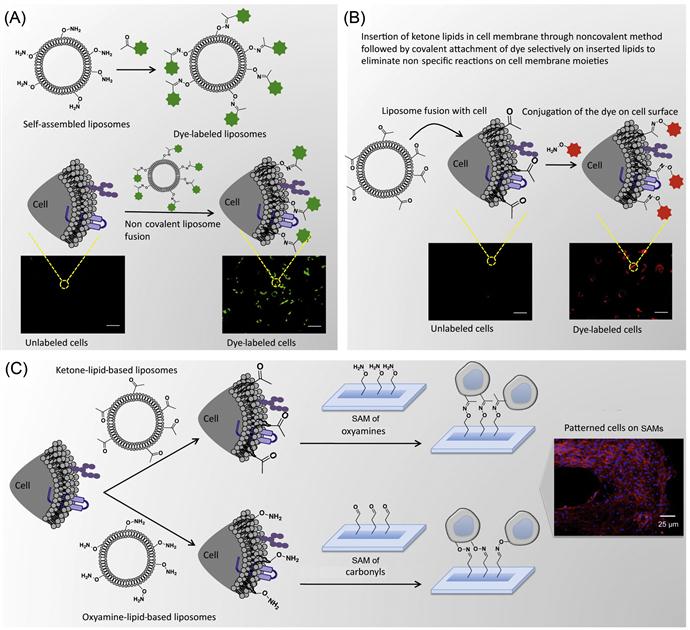
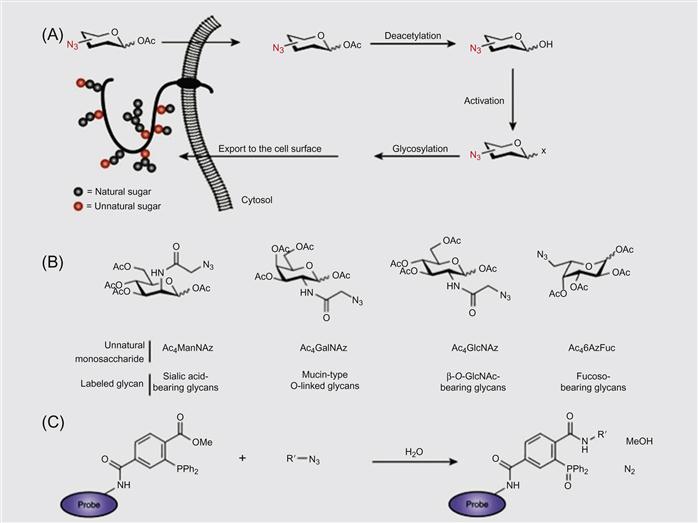
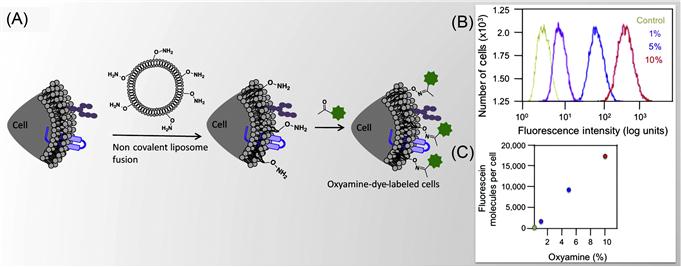
Dutta et al. [34] have presented a method to tether chemoselective ketone and oxyamine groups from mouse fibroblast surfaces by liposome fusion toward the goal of rewiring the cell surface. The modified cells were patterned on solid surfaces using bioorthogonal oxime bond formation and without the use of any cell-adhesive ligand (Figure 5.7) and could be quantified using flow cytometry (Figure 5.8). The ketone and oxyamine groups act as synthetic cell surface receptors and can immobilize a wide range of ligands and probes for subsequent use in cell targeting and tracking. Thus, rewiring cell adhesion was achieved using chemoselective oxime bond formation and without using any cell-adhesive ligands such as Arg-Gly-Asp (RGD) and fibronectin. Through avoiding the use of any additional biomolecules in this strategy, one can eliminate the potential long-term stability and degradation limitation in complex cell culture media or cell targeting in vivo. This methodology also bypasses any metabolic or genetic approach to modify the cell surface, which holds the risk of perturbing any cellular processes by altering cellular physiology.
5.2.4 Programmed Cell–Substrate and Cell–Cell Assembly
Cellular communication in a three-dimensional (3D) environment often requires physical contact between neighboring cells that is mediated by the surrounding extracellular matrix [35]. Efforts to mimic the 3D cellular environment and assembly have been mostly limited to using artificial solid scaffolds as platforms to seed cells and manipulating them by external forces and tools [36–39]. Recently, chemists and material scientists have begun to develop minimal invasive ways to engineer cell surfaces by biorecognition moieties that act as synthetic cell surface receptors to control cell–cell and cell–substrate interactions. Several approaches, including the use of dielectric forces [40–42], laser-guided writing [43,44], surface manipulation [45], and a number of lithographic printing techniques [11,46,47], have been integrated with 3D scaffold designs to produce multitype cellular arrays or 3D cell clusters or spheroids.
A DNA-based approach has been used for the programmable assembly of 3D cellular structure, which may find applications in synthesizing artificial tissues [38]. Recently, Gartner and Bertozzi [38] have demonstrated the assembly of DNA-modified cells into microtissues in a well-defined manner (i.e., structures were controlled by varying the DNA site density on the cell surface, cell concentration, and DNA sequence) as demonstrated in Figures 5.9 and 5.10. Briefly, by adjusting the stoichiometry (i.e., 1:50) of two Jurkat cell populations that were modified with complementary DNA strands, single cell clusters were produced where the limiting cell type was surrounded by cells added in excess. DNA strands were introduced to the cell surface by Staudinger ligation between phosphine-modified single-stranded DNA (ssDNA) and azide-modified cell surfaces where the azide functionality was introduced by metabolic engineering of peracetylated N-α-azidoacetylmannosamine (Ac4ManNAz) and subsequent display of the corresponding sialic acid in the cell membrane. Interestingly, using fluorescently labeled DNA, they showed that DNA strands are clustered at the cell–cell interface, confirming their specific role in cell–cell assembly. Significantly, such assembled cell clusters could be isolated and purified using flow cytometry and subsequently used as the core for further iterative cell assembly (i.e., adding another layer of DNA-modified cells).
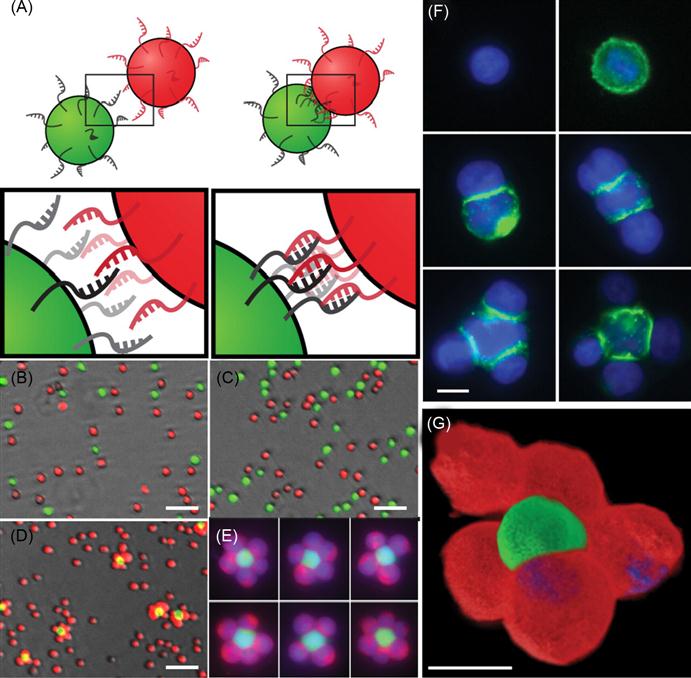
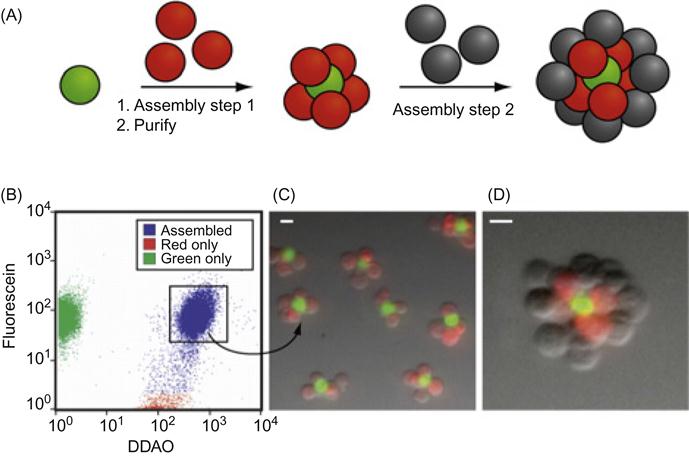
More importantly, they have demonstrated that cells within the assembled microtissues communicate via a paracrine mechanism, which is a prerequisite for the development of therapeutically functional tissues. Specifically, in a proof-of-concept model, a CHO cell line engineered to express growth factor interleukin-3 (IL-3) was used as a first building block and the second was an untransformed hematopoietic progenitor cell line (FL5.12) whose survival and replication depend on the presence of IL-3. Cell composites were assembled using DNA hybridization at the interface and then embedded within a 3D agarose matrix. After 16 h, the cell structures underwent accelerated growth due to the presence of IL-3. By contrast, when CHO cells lacking the gene that codes IL-3 were used in the microtissue, FL5.12 cells showed no growth and instead underwent a morphological change corresponding to the absence of IL-3-induced apoptosis. Additional advantages of using DNA assembly in such systems include their reversibility and versatility with respect to engineering multiple orthogonal cell–cell interactions. In addition to ssDNA, self-assembled cargo-carrying DNA arrays can also be attached to the surface of cells. Reiche and colleagues attached hexagonal DNA arrays on cells using streptavidin and antibody as bridges [48]. In their study, it was observed that one or two DNA array patches often attached to one cell and that single DNA arrays often to bridged cells. Occasionally, micron-sized DNA arrays seemed capable of binding to and enveloping multiple cells into larger cellular structures. These larger structures may have the potential to form the foundation for the construction of tissues or organs for transplantation in tissue engineering.
Engineered cell–cell interactions in a bottom-up tissue engineering approach can also be achieved by presenting bioorthogonal functional groups on the cell surface. Dutta et al. [49] have developed a simple liposome fusion method to display ketone or oxyamine functional groups from cell surfaces to generate 3D spheroid assemblies and multilayered tissue-like structures (Figures 5.11 and 5.12). Tissue-like networks were also formed on the surface with geometric control. Surfaces were patterned with cell-adhesive and nonadhesive regions to generate multilayered sheets and patterned tissue structures, respectively. Ketone- and oxyamine-tailored liposomes were cultured with separate fibroblast populations, resulting in membrane fusion and subsequent presentation of chemoselective sites for oxime conjugation from the surface. Culturing these groups on a solid support (~105 cells/ml) and in a layer-by-layer deposition manner gave rise to multilayered, tissue-like cell sheets, which were characterized by confocal microscopy. Cells cultured without the ketone and oxyamine groups did not form any multilayered structures. Spatial control on tissue formation was demonstrated using a micro-contact printing method where a variety of patterns and geometries were produced on a gold substrate. Employing SAMs and microfabrication techniques, hexadecanethiol (1 mM in ethanol) was printed on a gold surface. The surface was then backfilled with tetraethylene glycol (1 mM in ethanol, 16 h) to render the remaining regions inert to nonspecific protein absorption. Fibronectin, a cell-adhesive protein, was then added (10 mg/ml in calf bovine serum (CBS) 2 h), adhering only to the hydrophobic, patterned areas. When liposome fusion occurs to display complementary ketone and oxyamine groups from cell surfaces, multilayered 3D cell patterns were formed. The assembled tissue-like structures were generated in different shapes such as circular, bar, and squares. The strategy was also extended to form 3D cocultures of hMSCs and fibroblasts. The 3D cocultures were generated with two different cell types, in this instance, hMSCs and fibroblasts. Using this method, they retained individual cell phenotype properties that were demonstrated through differentiating hMSCs into adipocytes [49].
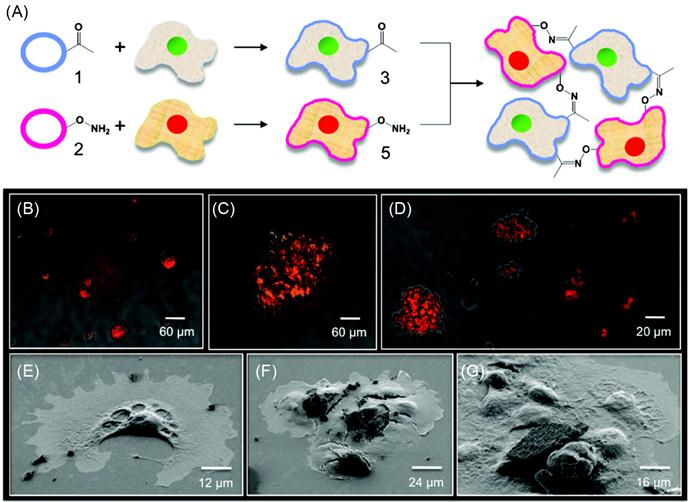
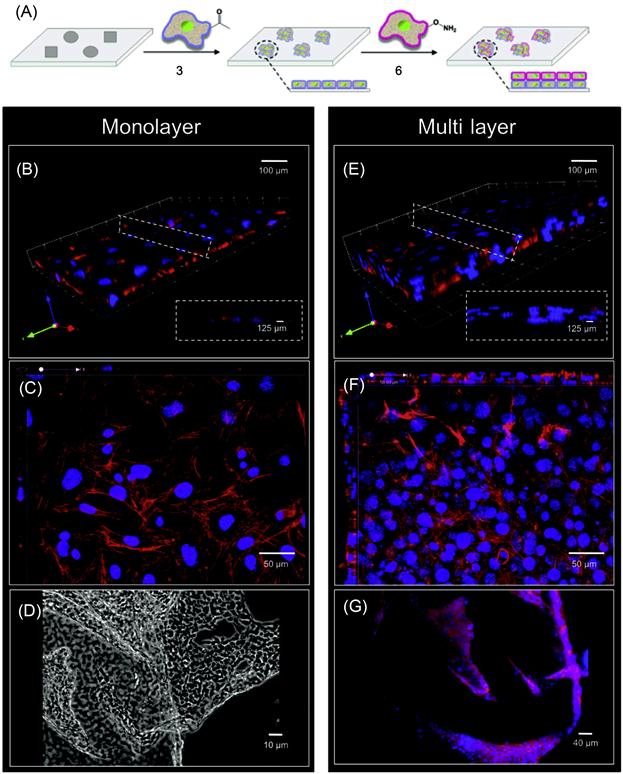
5.2.5 Anchoring Biomolecules on the Cell Surface Through Insertion of Hydrophobic Chain into the Cell Membrane
Although hydrophobic interactions are inherently weak in nature (in the order of 2–5 kBT in vacuum), they play a prominent role in many biological processes like lipid bilayer formation, protein folding, and protein–protein recognition [50]. In a seminal work by Dennis and coworkers [51] using a lipid bilayer system, MSCs were treated with palmitated protein G where the palmitate chain was incorporated into the lipid bilayer via hydrophobic interactions and protein G provides generic binding sites for antibodies (Figure 5.13). In a proof-of-concept work, intercellular cell adhesion molecule-1 (ICAM-1) was conjugated onto MSCs, which enabled the cells to bind to ICAM-1, a critical adhesion molecule expressed on activated endothelium. These chemical approaches to tailor cell surfaces with functional ligands are broad and generic and are not limited to only MSCs and the ligands mentioned herein.
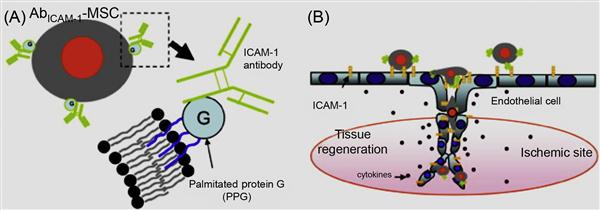
Cell surface hydrophobicity and cell surface charge have been recognized as measurable physicochemical variables for evaluating cellular adhesion to surfaces. Amphiphilic polymers, such as polyethylene glycol (PEG) conjugated phospholipids (PEG-lipid) and poly(vinyl alcohol) bearing hydrophobic alkyl side chains (PVA-alkyl), have been used in cell surface modification [52–56]. In these methods, the hydrophobic alkyl chains of amphiphilic polymers are spontaneously anchored into the lipid bilayer membranes through hydrophobic interactions (Figure 5.14). These spontaneous insertions can be monitored by a surface plasmon resonance (SPR) when a PEG-lipid solution is applied to supported lipid membranes that are formed on an SPR sensor. The spontaneous incorporation is greatly affected by the length of phospholipid alkyl chain length.
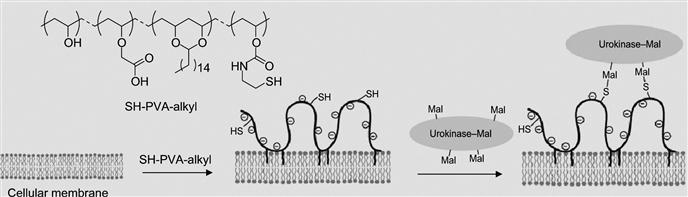
The other method is the formation of an intermediary polymer layer through hydrophobic interactions using amphiphilic polymers, such as PEG-lipid and PVA-alkyl, that bear a wide range of functional groups. The functional groups that are introduced on the cell surface through amphiphilic polymers can be used for the immobilization of bioactive substances (Figure 5.14). In one example, PVA-alkyl presenting SH groups were used for the immobilization of bioactive substances displaying maleimide groups. In a similar manner, PEG-lipids presenting maleimide at the end of the PEG chain was also used for the immobilization of bioactive substances tethering SH groups without cytotoxicity.
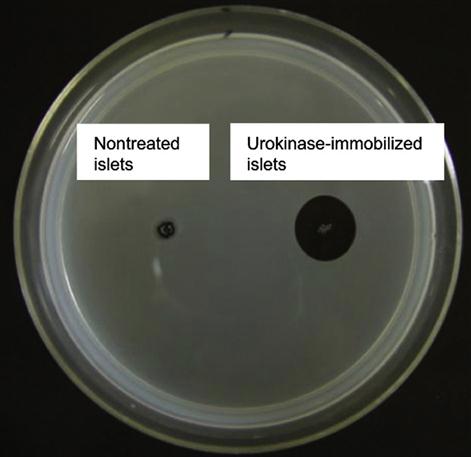
By utilizing this approach, Totani et al. have developed a promising system that is urokinase-immobilized islets for the potential treatment of diabetes. Although transplantation of islets of Langerhans is a promising method to treat insulin-dependent type I diabetes, innate immunological response activated by blood coagulation plays a key role in the loss of islets at the beginning. Thus, to overcome this limitation and to inhibit blood coagulation on the islet surface immobilization, a plasminogen activator, urokinase, was immobilized on the islet surface via a PVA derivative with alkyl chains and thiol groups (Figure 5.14). When the PVA derivative was added to an islet suspension, the alkyl side chains spontaneously anchored into the lipid bilayer membranes of islet cells. Subsequently, urokinase modified with maleimide groups was immobilized onto the islet surface by thiol/maleimide covalent bonding with the PVA layer. Urokinase-immobilized islets exhibited fibrinolytic properties, indicating that blood coagulation can be controlled on the islet surface. Furthermore, fibrinolysis activity of surface-immobilized urokinase was determined by the fibrin-plate-based assay (Figure 5.15). Urokinase activates plasminogen in the fibrin gel to form plasmin. Plasmin triggers a proteolytic cascade that results in fibrin gel degradation, which forms a transparent window in the gel plate. A large transparent window (2.5 cm2) was observed around the spotted urokinase-islets, which reflected the fibrinolytic activity of urokinase immobilized on the islets. This is an elegant example of versatility and efficiency of noncovalent immobilization of biomolecules on the cell surface.
5.3 Electrostatic Interactions Mediated Cell Surface Modification
5.3.1 Engineering of Cell Surface by Harnessing Electrostatic Interactions
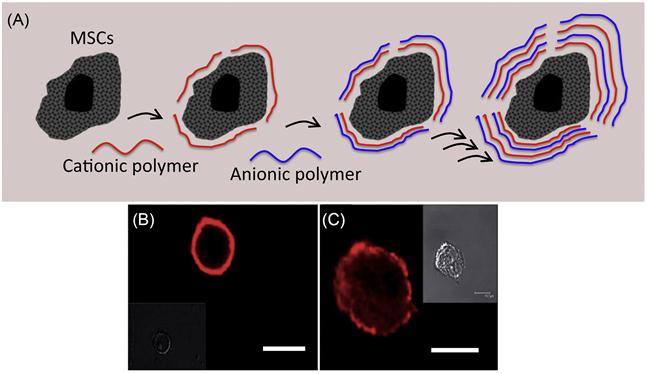
The cell membrane is intrinsically negatively charged due to a high composition of negatively charged lipids such as phosphatidylserine and phosphatidylinositol lipids. The possibility of constructing a thin polymer membrane on the surface of cells through electrostatic interactions between negatively charged cell surfaces and cationic polymers and then further modifying them using a layer-by-layer technique of anionic and cationic polymers [57–61] has been explored by several research groups. The ionic polymers that have been employed are poly(allylamine hydrochloride), poly(styrene sulfate), poly-L-lysine, and poly-(ethyleneimine). A layer-by-layer method is a simple and attractive technique to modify cell surfaces (Figure 5.16) [62]. The outermost layer of the polymer controls the surface properties. The thickness of the layer is also controllable by the number of polymer layers. However, most polycations, such as poly-L-lysine and poly-ethylene-imine, can be extremely cytotoxic and severely damage the treated cells. Although cationic polymers are efficient to interact with the cell membrane, often they rupture the cell membrane through electrostatic interactions. This leads to severe cell toxicity. To avoid cationic-polymer-associated cytotoxicity, attempts have been made to minimize electrostatic interactions between cationic polymer and cell surface through modulation of charge on the polymer. However, such weak electrostatic interactions often do not yield desirable effects due to reduction of residence time of polymer on the cell surface. Thus, it is extremely important to carefully design the polymers to achieve a balance between toxicity and efficiency.
5.3.2 Noncovalent Cell Surface Modification for Bioimaging Applications
Cell surface modification can facilitate the characterization of a number of cellular processes and signal transduction pathways in terms of identifying key proteins and signaling molecules [63]. One major application of engineering cell surfaces can be in modifying the cell membrane with molecular and detection probes for subsequent tracking during postinfusion in cell therapy [64]. Chemical reporters have been incorporated to the cell membrane using the metabolic pathways [14]. Orynbayeva et al. [65] engineered chromatic polydiacetylene(PDA) polymer patches onto the cell surfaces for imaging structural perturbations of the membrane bilayer (Figure 5.17). Lipid vesicles comprising of 1,2-dimyristoyl-sn-glycero-phosphoethanolamine and 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] were fused with the cell membrane, and PDA was formed in situ upon ultraviolet irradiation. The tethered nanopatches of PDA on the cell surface had an intense blue color. The structural perturbations of the membrane bilayer due to the conformational changes in the conjugated (ene-yne) polymer backbone led to a blue-to-red transition of the conjugated PDA. During this blue-to-red color transition, the initial nonfluorescent PDA also emitted fluorescence at 560 and 640 nm. The PDA nanopatches can therefore be used to visualize the cell bilayer perturbations induced by, for example, lidocaine, polymyxin-B, cholesterol, and oleic acid and to screen toxic pesticides and other environmentally toxic small molecules.
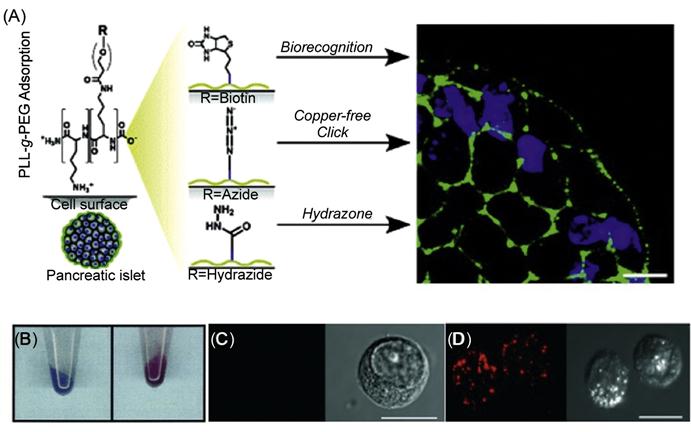
In another study, Wilson et al. [66] have used cationic graft copolymers to noncovalently engineer pancreatic islet surfaces using electrostatic interactions for subsequent labeling and visualization (Figure 5.17). Specifically, poly(L-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) copolymers where the polymer backbone was terminally functionalized with biotin, hydrazide, and azide moieties were used to reengineer the cell surfaces with streptavidin-, aldehyde-, and cyclooctyne-labeled probes.
5.3.3 Noncovalent Cell Surface Engineering to Manipulate Cell Biological Fate
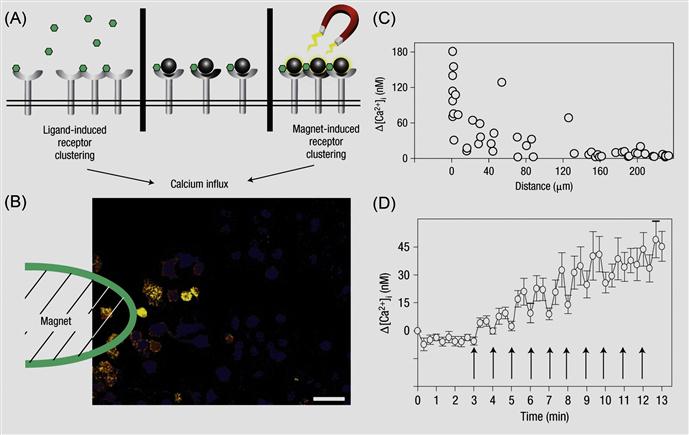
Externally applied chemical moieties and materials can be engineered to cell surfaces to control cell biology. The idea is to stimulate and reversibly control different cell signaling pathways by physical tools that are normally biochemically regulated. This can be achieved by linking different molecular probes to the cell surface. Often native cell surface is not amenable to external stimuli manipulation. In such instances, transiently engineering the cell surface by immobilizing an additional agent on the cell surface would enable to manipulate the cell surface and cell fate through an external stimulus. Ingber and coworkers [67] have demonstrated the use of magnetic nanoparticles on the cell membrane to regulate cellular signaling pathways (Figure 5.18). Specifically, mast cells express membrane high-affinity IgE receptors (FcεRI) that can bind to the Fc region of IgE molecules. Multivalent antigens like dinitrophenyl (DNP) that bind to IgE can then induce FcεRI receptor oligomerization on the cell membrane whereas monovalent antigens will fail to do so. The FcεRI receptor clustering quickly triggers an intracellular signaling response characterized by a rapid rise of cytosolic Ca2+, which in turn leads to the local inflammatory response of mast cells. Instead of using an antigen ligand to induce such signaling pathway, the authors immobilized DNP-attached magnetic nanoparticles on IgE-tethered mast cells [68]. The authors ensured that only one DNP molecule was attached on each particle so that receptor clustering did not occur. When an external magnetic field is applied, magnetic nanoparticles clustered on the cell membrane, which caused the oligomerization of FcεRI receptors, a process that could be visualized by the local Ca2+ concentration change. Such magnetic nanoparticles mediated cellular signaling is reversible, switching on and off the magnetic field can directly correspond to a local Ca2+ concentration oscillation. This is an elegant example for modulation of intracellular properties through controlling an altering cell surface by an external stimulus.
5.4 Advantages and Limitations of the Noncovalent Modification of the Cell Surface
5.4.1 Advantages of Noncovalent Modification of the Cell Surface
Cell surface engineering has primarily been a subject of molecular biology and genetic engineering. Although genetic manipulation is an efficient method to modulate the cell surface, potentially it might disrupt the inherent cell properties. Often it might lead to unforeseen and unwanted effects. Thus, selective engineering of cell surface without the need of genetic manipulation is a welcoming approach to modulate the cell surface features and properties. Typically, NHS-activated esters and cyanuric chloride derivatives are frequently used to form covalent bonds to the cell surface functional groups such as amines. In addition, maliemide derivatives can be used to react with thiol groups of membrane molecules. However, although covalent modification provides a robust functionalization approach, degree of functionalization needs to be highly optimized to avoid deactivation of membrane molecules such as proteins, lipids, and receptors. Thus, potential toxic effects may be exerted on membrane biomolecules [68]. Enzymatic treatment and metabolic introduction via sialic acid biosynthetic pathway (Bertozzi group) have been employed to add multiple functional groups such as biotin, azido, and ketone groups to living cell surfaces [14,15]. However, these technologies are limited to the introduction of specified small molecules to cells and may perturb cell physiology. Thus, noncovalent methods to transiently engineer the cell surface could overcome these limitations. Liposome fusion can serve as an important tool among many other techniques that have been previously described to modify and tailor cell surfaces with biologically relevant molecules, significantly contributing to tissue engineering fields in generating 3D tissue constructs and in the field of cell-based targeted therapy.
5.4.2 Limitations of Noncovalent Modification of the Cell Surface
Although noncovalent modification of cell surfaces does not perturb the cellular machinery of the cell, the noncovalent forces like hydrophobic and electrostatic interactions holding the molecular probes to the cell membrane are intrinsically weaker in nature. This can result in a comparatively unstable system leading to temporal sensitivity. While this approach is optimal for transient surface medication, engineering surface through noncovalent modifications might not be retained in the longer term. Thus, an extensive optimization needs to be done to increase the longevity of the engineered cell surfaces.
5.5 Conclusions and Future Perspectives
Noncovalent methods for cell surface engineering provide efficient and facile approaches toward modulating cellular functions and finding solutions to fundamental biological problems. It has potential uses in a wide range of fields of biomedicine, tissue engineering, regenerative medicine, wound healing, cell-based therapies, imaging, and diagnostics. In future, coupling multiple noncovalent forces in a single system that would enhance the longevity of the system could enable the development of more elegant and robust methods of engineering the cell surfaces with different molecular probes. Efforts should also be made in designing dynamic systems, where molecules of choice could be conjugated and released from the cell surface using external stimuli. The eventual design and manufacture of these systems would need further collaborations between biologists and engineers for such products to make it to the marketplace.
Acknowledgments
PKV thanks Department of Biotechnology, Government of India for Ramalingaswami Re-Entry Fellowship, and Institute for Stem Cell Biology and Regenerative Medicine (inStem), Bangalore for financial support.