
Micro/Nano-Engineering of Cells for Delivery of Therapeutics
Oren Levy, Edward Han, Jessica Ngai, Priya Anandakumaran, Zhixiang Tong, Kelvin S. Ng and Jeffrey M. Karp, Division of Biomedical Engineering, Department of Medicine, Center for Regenerative Therapeutics, Brigham and Women’s Hospital, Boston, MA; Harvard Medical School, Harvard University, Boston, MA; Harvard Stem Cell Institute, Cambridge, MA; Harvard-MIT Division of Health Sciences and Technology, Cambridge, MA, USA
Cell-based therapies hold a great promise for treatment of a wide array of tragic diseases. Specifically, the use of cellular vehicle platforms emerges as a powerful strategy for targeted delivery of therapeutics to sites of disease. In this chapter, we first review the success and current challenges of different cell therapies, focusing on pancreatic islet transplantation as well as multiple types of stem cell therapies. We then shift our discussion to cellular delivery systems. Efficient cell targeting to desired sites is a crucial aspect towards effective use of such platforms. Accordingly, we thoroughly discuss a variety of cell engineering strategies to improve cell targeting to sites of interest. We then discuss key aspects of cellular delivery, including the types and characteristics of different nanosystems, candidate therapeutic agents and cell types suitable for this approach. Finally, we conclude with a case study of Trojan Horse cell-based cancer therapy.
Keywords
Stem Cells; Cell Therapy; Cell-based delivery platforms; Cell engineering
In this chapter, we discuss micro/nano-engineering strategies to enable efficient targeting of transplanted cells to diseased/damaged tissues that should improve the efficacy of cell therapy and permit the use of cells as drug delivery vehicles. To introduce the tremendous clinical potential and the challenges of cell therapy, we begin by describing a selection of current cell therapies and the importance of cell targeting. We will then discuss bioengineering approaches to modify the cell surface to improve cell targeting to desired sites. This will be followed by in-depth discussion of targeted cell-based drug delivery and its clinical potential.
12.1 Cell Therapy—Success and Current Challenges
12.1.1 Hematopoietic Stem Cell Transplantation
Over 10,000 bone marrow transplants are performed every year in the United States to treat patients with severe blood disorders [1]. Hematopoietic stem cells (HSCs), capable of giving rise to the entire hematopoietic system [2], are the key therapeutic component of the transplanted bone marrow units and are therefore widely used to treat many blood malignancies, including myeloma, leukemia, and lymphoma, as well as immune deficiencies and other hematological disorders [3–6]. In most cases, the patient’s own diseased hematopoietic system is first ablated and then reconstituted with healthy cells using HSC transplantation (HSCT). Following systemic transplantation, the first key cellular process that affects HSCT efficacy is the homing of HSCs to the bone marrow, where an optimal niche is provided for hematopoiesis [7]. Akin to leukocytes, HSCs roll on, adhere to, and migrate across the vascular endothelium at specific tissues, guided by chemotactic gradients [8,9]. Lack of homing capacity has been shown to drastically reduce HSCT efficacy [10]. After homing, engraftment is required to retain and maintain HSCs in a conductive microenvironment for long-term survival and proper function (e.g., self-renewal, differentiation). Enhancing homing and engraftment will likely improve efficacy and reduce the dosage of HSC infusion, allowing more patients to be treated with the same donor pool. This is important since HSCs are difficult to expand in vitro without inducing undesirable differentiation and cell death [11,12].
Because HSCT is typically allogeneic, a main challenge in HSCT is significant immune response following transplantation. Not only may the host immune response compromise the engraftment and survival of transplanted HSCs [13], patients are also at risk for graft-versus-host disease (GvHD), which can lead to death [14]. The risk of an immune response can be lessened via prophylactic immune suppression [15]. Haplotype matching based on the human leukocyte antigen (HLA) further ameliorates the risk, although this limits the number of potential donors [6]. Nowadays, HSCs can be efficiently harvested from multiple tissue sources, including cord blood, substantially increasing the number of patients who can benefit from HSCT [16,17]. Importantly, genetic modification enables the potential use of autologous HSCs: for some diseases, patients’ cells can be corrected ex vivo and returned to the same patient, providing a promising platform when matched donors are unavailable [18].
However, significant challenges still remain for HSCT. In addition to GvHD and poor engraftment of transplanted cells, disease relapse often occurs after allogeneic (unmatched), haploidentical (matched), or autologous transplantations [19]. Although continuous efforts are made to engineer cells to improve therapy, successful implementation into the clinic is challenging due to safety issues and preservation of engineered properties post-transplantation [20]. Genetic modification entails potential risks such as abnormal splicing, inactivation of tumor suppressor genes, or accidental oncogene activation, that may result in development of leukemia [21]. Nongenetic modifications may circumvent some risks while conferring better control over cell fate post-transplantation, as we will discuss in detail later.
12.1.2 Pancreatic Islet Transplantation
Another cell therapy in growing demand is pancreatic islet transplantation for treatment of type 1 diabetes (T1D) [22]. In T1D, the pancreas does not produce sufficient levels of insulin, mostly due to autoimmune destruction of beta cells, resulting in impaired glucose metabolism [23]. Patients with T1D need to constantly monitor their blood glucose and receive insulin injections to reduce hyperglycemia when necessary. Poor control of blood glucose may lead to hypoglycemia, which can be fatal, or chronic hyperglycemia, which may result in complications such as heart disease, kidney disease, and nerve damage [24,25]. Successful islet transplantation would restore autonomous, precise control of blood glucose, freeing patients from constant monitoring, and adjustment of blood glucose. Allogeneic islets are required for T1D [25], since the patient’s own islets are typically dysfunctional. Unfortunately, islets are composed of terminally differentiated cells and cannot be expanded ex vivo, while donor morbidity is clinically unjustifiable. Elaborate procedures for quickly obtaining fresh islets from cadavers are therefore required, especially given the fact that islets from at least two deceased donors are required to treat a single patient [26].
Like HSCT, allogeneic islet transplantation faces immune rejection and donor shortage. But unlike HSCs, islets are large multicellular aggregates containing multiple cell types and have to be administered locally instead of systemically (i.e., via the bloodstream) [27]. This greatly affects the repertoire of strategies for cell engineering. For example, genetic modification becomes more difficult since transfection efficiency will vary according to the location of the cell within the islet, whereas tissue engineered islet with defined compositional and structural features becomes instrumental for controlling cell fate and function following local transplantation [28]. However, while homing may be less relevant, engraftment and survival remain crucial for the success of islet transplantation: islets have to be vascularized quickly post-transplantation to maintain sufficient viability for long-term function [29].
Although the host immune response may be reduced by genetically downregulating foreign antigens [30–32], encapsulation inside biocompatible scaffolds such as alginate or porous pouches can sufficiently shield islets from both cellular and humoral components of the host immune system and thereby offer stronger and longer term protection [33–35]. Native islets deliver insulin directly into the hepatic portal vein; hence the gold standard has been intraportal implantation [36], which requires invasive surgery. Scaffold technologies may potentially enable minimally invasive [37] islet transplantation, such as subcutaneous implantation, given that a variety of problems will be addressed, such as capability of islets to detect changes in blood glucose, delivery of relevant amounts of insulin into the bloodstream, scaffold degradation over time, foreign body response against the scaffold, and nutrient transport to encapsulated islets [38]. Supplementing scaffold technologies with genetic strategies, such as accelerating vascularization by ectopically expressing pro-angiogenic factors [39], or promoting islet viability and functionality by ectopically expressing hormones [40] may be necessary to achieve sufficient efficacy of islet transplantation in the future [26]. An emerging paradigm is to derive pancreatic cells from precursors in vitro before transplantation [41,42]. Cells may then be administered systemically as single cells instead of aggregates, allowing them to efficiently home to and engraft at sites (e.g., hepatic or pancreatic capillary beds) where optimal islet physiology may be achieved.
12.1.3 Mesenchymal Stem Cell Therapy
Mesenchymal stromal cells, otherwise known as mesenchymal stem cells (MSCs), have emerged during the past two decades as promising candidates for cell therapy [43–45]. Unlike pancreatic islets, MSCs are readily isolated from bone marrow and other adult tissues without significant donor morbidity. Unlike HSCs, MSCs can be easily expanded ex vivo to obtain a sufficient quantity for cell transplantation [46]. Moreover, their immune-evasive and potent immunomodulatory properties following systemic infusion permit allogeneic transplantation and have prompted their use in over 380 clinical trials for treating multiple diseases, including lupus nephritis, GvHD, multiple sclerosis, and cardiovascular diseases [47]. While results from pre-clinical animal studies have been encouraging and hundreds of millions of allogeneic MSCs have been safely administered, clinical trials have produced only mixed results for efficacy endpoints and MSC translational potential has not yet been fully realized [48–51]. This could be attributed to the innate heterogeneity of MSC population, significant variability in their donor/tissue sources, manufacturing processes, as well as their diverse characterization methods [52]. One of the major challenges for MSC therapy is poor homing to diseased or damaged tissues, potentially due to minimal expression of key homing ligands on MSC surface [49,53,54]. Since the MSC secretome serves a crucial role in their therapeutic impact, increasing the number of transplanted MSCs at disease sites at the same total dosage will likely significantly improve the efficacy of MSC therapy [55].
12.1.4 Embryonic Stem Cells and Induced Pluripotent Stem Cells
Embryonic stem cells (ESCs) are perhaps the most promising yet most controversial cell type being investigated for therapy. Unlike adult stem cells, ESCs have an unlimited capacity for self-renewal, can differentiate into all three germ layers (ectoderm, endoderm, and mesoderm), and thus can generate almost any cell type [56]. However, adverse effects, such as formation of teratomas, and the many ethical issues that plague the use of ESCs, significantly hinder their translation into the clinic [57]. Numerous studies demonstrated the therapeutic potential of ESCs. For example, Min et al. [58] were able to significantly improve left ventricular function in post-infarcted rats after ESC transplantation. Brustle et al. [59] demonstrated that ESCs differentiate into oligodendrocyte and astrocyte precursors and induce axon myelination in a rat model of dysmyelinating Pelizaeus–Merzbacher disease. Besides cardiac and neural applications, ESC studies have spanned virtually every discipline of the biomedical field [60]. Better understanding of the mechanisms governing ESC differentiation and transformation may advance the development of safe and effective ESC therapies in the future.
Induced pluripotent stem cells (IPSCs) bypass ethical issues of ESCs and are also starting to be tested in clinical trials [61]. IPSCs are generated by reprogramming somatic cells to regain pluripotency [61,62], first demonstrated when Takahashi and Yamanaka [63] successfully reprogrammed adult fibroblasts into an embryonic-like state. Theoretically, this would enable a virtually limitless quantity of highly desirable ESC-like cells, while avoiding any ethical issues. One of the main questions is whether IPSCs are truly as pluripotent as ESCs [62]. Different groups have shown significant differences between IPSCs and ESCs. For example, the differentiation potential of IPSCs appears intrinsically lower than that of ESCs [64]. In addition, IPSCs seem to have an increased propensity to differentiate back into the cells they were formed from [65]. Despite these differences, IPSCs have been used in ample studies spanning a multitude of biomedical applications. For example, Wernig et al. [66] were able to demonstrate that neurons derived from IPSCs were able to functionally integrate into the brains of rats with Parkinson’s disease. Much of the potential of IPSCs remains untapped, however, making this field an exciting source of potential therapies.
Among the challenges discussed above, poor cell targeting to disease sites following transplantation seems to be a common drawback shared by all the aforementioned cell therapies. We will now discuss micro/nano-engineering approaches to control cell homing/targeting. These approaches mainly involve the cell surface, the key interface where the cell interacts with its environment during the homing process.
12.2 Cell Surface Engineering to Improve Cell Targeting
One of the major challenges in cell therapy is poor control over cell homing to disease sites following transplantation—to maximize therapeutic impact, delivering a sufficient amount of functional cells to target organs is crucial. Improving cell homing would also reduce the number of transplanted cells needed to achieve clinical improvement. This has enormous significance since cell expansion ex vivo has both biological and financial costs [48], and not every cell type can be expanded. For MSCs, which exert their immunomodulatory effects primarily via their secretome [55,67], increasing their bioavailability at the disease site is likely to significantly improve their therapeutic impact [68].
12.2.1 Affinity-Based Cell Targeting
Different bioengineering approaches have been exploited to modify the cell surface to enhance cell targeting to desired sites. A generalized concept is to trigger receptor–ligand interactions between the surface of transplanted cells and the cells at the target site. Cells can be engineered to overexpress biological or artificial receptors or antibodies to specifically target cells expressing the corresponding ligands or antigens [69] (Figure 12.1).
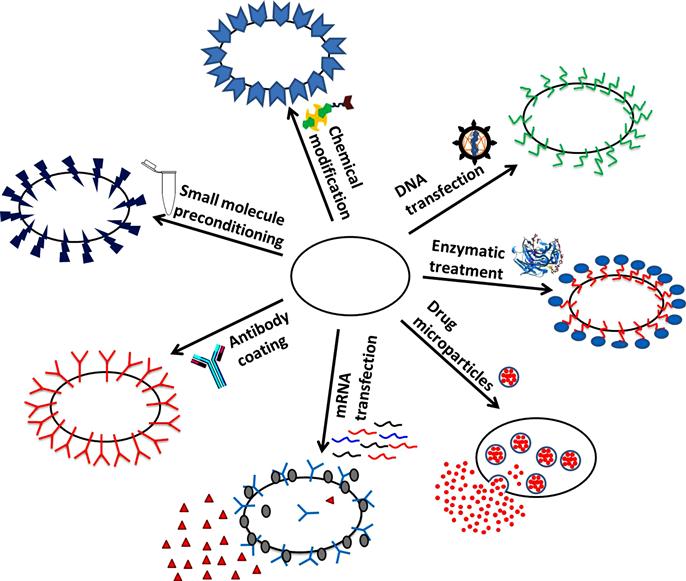
Whole and fragmented antibodies have been incorporated into cell surfaces to enhance cell localization to target sites. For instance, coating MSCs with whole antibodies against MAdCAM increased their homing to inflamed bowel and improved therapeutic efficacy on inflammatory bowel disease [70]. Likewise, in adoptive immunotherapy, autologous T cells may be modified with antibodies ex vivo and redirected to target specific cells, such as cancer cells. Whole antibodies have been engineered to create chimeric T-cell receptors (TCRs), which signal the T cell to launch a killing response upon binding to malignant cells [71,72].
Fragmented antibodies, such as single chain variable fragments (scFv) antibodies which do not contain the Fc domain but retain the high specificity and affinity of whole antibodies, have also been used to modify the cell surface [73]. Omitting the Fc domain eliminates potential binding of Fc receptors onto the cell surface and facilitates vector design for genetic engineering, but removes the signaling ability intrinsic to the Fc region. Fragmented antibodies have been engineered to create bispecific adaptors (diabodies) that link the specificity of two antibodies into a single agent and act as a bridge between two cell types. Cells have been genetically modified to secrete diabodies which bind tumor cells at one end and bind/activate T cells on the other, directing T-cell cytotoxic activity specifically toward tumors and significantly inhibiting tumor growth [74–76]. Fragmented antibodies, instead of whole antibodies, are preferred for this approach since they penetrate tissues better due to their reduced size [77].
These two approaches represent how both membrane-anchored and secreted factors may enhance cell targeting via the cell surface. The same concept extends to receptors beyond antibodies. We will next discuss how such factors may be imparted to cells that natively underexpress them.
12.2.2 Bioengineering Approaches to Accomplish Affinity-Based Cell Targeting
12.2.2.1 Genetic Modification
Genetic modification has been employed to introduce new genes or to knock out specific genes to confer added functionalities to cells. The stability and specificity of genetic modifications have made it an attractive technique to achieve enhanced cell homing through cell surface modifications. The use of viral vectors such as adenoviruses and lentiviruses, as well as nonviral reagents such as lipid and polymeric micro/nanoparticles, has been widely explored to deliver DNA/RNA to cells to overexpress specific receptors to achieve improved homing to target sites.
12.2.2.1.1 Viral Vectors
Viral vectors have been utilized as gene delivery vehicles to overexpress receptors on cell surfaces due to their high transfection efficiencies. Yu et al. [78] found that transducing MSCs with lentiviral vectors encoding the gene for the chemokine receptor CXCR4 resulted in enhanced engraftment and neuroprotection in rats subjected to middle cerebral artery occlusion. This was facilitated via interactions between CXCR4 and, its ligand, SDF-1, which is expressed along the ischemic boundary of the brain, and was shown to promote the migration of CXCR4-MSCs toward the ischemic zone [78]. In another study, increased homing of MSCs to the brain in a malignant glioma model was induced by transducing cells with retroviruses encoding EGFR [79]. Ectopic expression of EGFR on the surface of MSCs resulted in increased targeting toward its ligands EGF, HB-EGF, and TGF-alpha, all of which are present on glioma cells [79]. Retrovirally induced CXCR4 overexpression was also demonstrated to improve MSC homing to infarcted myocardium, also resulting in improved cardiac function following cell treatment [80]. In another study, retroviral delivery to increase CCR1 expression on MSC surface prior to transplantation increased cell viability, migration, and engraftment to infarcted myocardium [81]. Furthermore, viral-induced expression of integrin alpha 4 on MSC surface was shown to target MSCs to bone in immunocompetent mice, where MSCs exhibited differentiation into osteoblasts and osteocytes in the growth plate of recipient mouse limb bones [82]. Other studies have used genetically modified affinity-based targeting to increase delivery of systemically administered MSCs to tumor sites. For example, MSCs have been genetically modified with adenoviral vectors to express artificial receptors that can bind to the tumor cell marker erbB2 [69]. These erbB2-receptor-modified MSCs were retained in high concentrations in the lungs of erbB2-expressing mice, and they were targeted toward ovarian tumors in ovarian xenograft tumor models [69]. In another study, monocytes were adenovirally transduced to express a chimeric CD64 receptor to target CEA-expressing tumor cells, resulting in significantly reduced in vivo tumor growth rates in xenograft studies [83].
T cells which have been genetically modified with viral vectors to express chimeric TCRs that recognize tumor markers have also been evaluated. For instance, peripheral blood-derived human T cells were transduced with retroviruses encoding chimeric TCR specific to CD30 and were found to specifically target CD30+ lymphoma cells. Following engraftment with CD30+ lymphoma cells, the TCR-modified T cells secreted high levels of IFN-γ to induce killing of specific tumor cells [84]. Furthermore, human lymphocytes which have been genetically engineered to express tumor-specific chimeric receptors have been translated to phase I clinical trials [85] due to their high potency against tumors in animal models, which has resulted in partial or complete remission of cancer [72,86]. Despite their high transfection efficiency, viral vectors are challenged by several safety issues such as immunogenicity and potential oncogene activation, resulting in the search for and development of nonviral vectors to genetically engineer cells [87].
12.2.2.1.2 Nonviral Vectors
A primary challenge in nonviral transfection is overcoming the electrostatic repulsion between negatively charged nucleic acids and the negatively charged cell membrane to facilitate uptake of DNA/RNA by cells. Common transfection methods include reagent-based methods such as cationic lipids or polymers, and device-based methods such as electroporation. Despite the lower transfection efficiency of nonviral vectors in comparison to viral vectors, the reduced immune response and toxicity of nonviral vectors have made them attractive gene delivery vehicles [88–94].
Kim et al. demonstrated that MSCs transfected with plasmids encoding the gene for CXCR1 through electroporation resulted in superior migration of MSCs toward gliomas. The increase in migration was attributed to the binding interaction between CXCR1 and IL-8, which is released by glioma cells [95]. MSCs have also been genetically modified through electroporation to express scFv antibodies on their surface to target EGFRvIII, which is highly expressed in many glioblastoma (GB) tumors. The modified MSCs were found to bind to U87-EGFRvIII cells at high levels in vitro, and they experienced a sevenfold increase in retention in human U87-EGFRvIII expressing tumors in vivo [96].
Finally, peripheral blood human T cells, which were transfected via electroporation with chimeric TCRs specific to CD20, were found to preferentially migrate towards CD20+ lymphoma cells. Following coculture with CD20+ lymphoma cells, the TCR-modified T cells exhibited cytotoxic characteristics through an increased secretion of IFN-γ [97].
Recently, we have demonstrated the use of lipofectamine to deliver in vitro transcribed mRNA to control MSC phenotype following transplantation [98]. Unlike delivery of DNA, which could result in stable transfection but risks integration of foreign DNA into the genome, mRNA transfection elicits a transient, yet functionally significant, expression of genes of interest. Via transfection with pseudouridine-modified mRNA (to increase translation efficiency), we have expressed sialyl Lewis X (SLeX) and PSGL-1 on MSC surface, resulting in improved homing of systemically transplanted MSCs to healthy and irradiated bone marrow as well as to distant sites of local inflammation [98]. The transient effect of mRNA transfection may not be desired for all clinical applications. However, this may be partially addressed by administering multiple infusions to patients and further research is needed to fully elucidate the translatability of this approach into the clinic.
Genetic engineering has the potential to stably and specifically modify cell surfaces to achieve increased cell targeting to different disease sites. However, this approach raises potential safety risks, mostly due to potential integration of transfected DNA into the genome of the host cell and the use of viruses for delivery. An alternative approach, which is safer and may result in more transient cell surface alteration, is chemical or enzymatic modification of the cell surface prior to transplantation.
12.2.2.2 Chemical Modification
Another technique to modify the cell surface is to chemically conjugate molecules or nanoparticles (NPs) onto existing functional groups on cell surface proteins, polysaccharides, or lipids. It is also possible to metabolically or genetically introduce new, reactive functional groups onto the surface of cells to act as anchors for other molecules to remodel the cell surface [99]. However, chemical conjugation is a simpler and more transient method to modify the cell surface.
12.2.2.2.1 Surface Functional Groups
Functional groups such as thiols and amines are naturally present on the cell surface and are readily available for covalent conjugation. Other functional groups such as aldehydes and ketones have been used as anchors for surface-modifying molecules; however, they must first be exposed through chemical or enzymatic treatment of cell surface carbohydrates. Primary amine groups have been utilized as sites for covalent conjugation through biotinylation, in which the amine groups are reacted with amine-reactive biotin, such as N-hydroxy-succinimide biotin derivatives. The cell surface can then be functionalized with other biotinylated molecules or NPs through a streptavidin bridge [100,101]. We previously found that covalently coupling SLeX to the surface of MSCs through a streptavidin–biotin bridge resulted in a robust cell rolling on P-selectin-coated substrates in vitro, as well as an increased homing efficiency to inflamed tissue in vivo, without compromising the intrinsic properties of MSCs [102,103].
12.2.2.2.2 Lipid Insertion
Another approach involving the chemical conjugation of molecules to the cell surface takes advantage of the hydrophobic portion of the cell membrane to immobilize antibodies onto the cell surface. This two-step method consists of first coating cells with palmitate-conjugated proteins which are stabilized onto the cell membrane through the insertion of the hydrophobic palmitate moiety into the phospholipid bilayer. The palmitated proteins, which have high binding affinity for the Fc region of antibodies, allow cell surfaces to be functionalized with a variety of antibodies. These antibodies then act as artificial receptors for soluble or cell surface antigens [104]. Kim et al. [104] showed that cells coated with palmitate-conjugated protein A followed by rabbit anti-mouse IgG demonstrated enhanced adhesiveness to surface-bound IgG-positive mouse B cells in vitro. Others investigated MSCs coated with palmitated protein G (PPG) followed by antibodies against ICAM-1 and found an increased binding of anti-ICAM-1 MSCs to activated endothelial cells in flow chamber studies [105]. Similarly, Lo et al. [106] recently attached PSGL-1 (via PPG) to MSC surface, resulting in MSC tethering and rolling on stimulated endotheilal cell monolayers. Finally, Dennis et al. [107] focused on enhancing the engraftment of pre-chondrocytes to cartilage defects by coating the cell surface with lipidated protein G and antibodies which are specific to cartilage matrix antigens. Painted cells which were injected into rabbit cartilage explants with a partial thickness defect preferentially bound to the exposed cartilage material within the defect.
Another example of cell painting to improve cell homing involves binding antibodies utilizing the interaction between glycosylphosphatidylinositol (GPI) and cell surface proteins [108]. Sheep erythrocytes surface loaded with GPI-anchored scFv antibodies against CD-40 were found to specifically target and form rosettes with CD20-positive cancer cells [109]. Finally, Kean et al. [110] improved cell homing of MSCs to sites of myocardial infarction (MI) by coating MSCs with MI-specific peptides which have palmitic acid tails to facilitate integration into the cell membrane. Peptide-coated MSCs which were injected into mice with MI were localized at the heart in higher numbers than native MSCs [110].
12.2.2.3 Enzymatic Modification
It is also possible to directly modify existing cell surface molecules through enzymatic transformations. This method was employed to improve HSC homing and engraftment by ex vivo fucosylation of cord blood in order to achieve an increase in HSC tethering to activated endothelial cells expressing P-selectin and E-selectin [111]. This concept was further applied to human MSCs, where enzymatically modifying native CD44 glycoforms into hematopoietic cell E-selectin/L-selectin ligand resulted in targeting of modified MSCs to bone marrow in a murine model [112,113].
12.2.2.4 Physical Targeting
Physical targeting strategies utilize appropriate devices or procedures to enhance cell targeting to a specific site and may involve cell surface modifications. Although the concept of using devices or procedures to enhance cell targeting appears simple, a number of issues associated with gaining physical and direct access to organs severely complicate this method. While guiding devices exist, such as catheters, which can directly introduce cells into the brain [114,115], heart [116], and into wounds [117] with positive therapeutic outcomes, other physical methods exist like loading cells with magnetic compounds to redirect cell localization via application of a magnetic field to the target site. The initial purpose of using magnetic compounds was to monitor cell migration in vivo with magnetic resonance imaging; however, it has since evolved to be used to redirect cell localization both in vitro and in vivo. Nakashima et al. [118] showed that magnetically labeled NK cells targeted human osteosarcoma cells under a magnetic field in vitro. Other groups managed to magnetically target MSCs in animal models. For example, iron oxide-labeled MSCs were shown to specifically concentrate in the liver when external magnets were placed over it [119]. The same group showed that bone marrow stromal cells injected into the cerebrospinal fluid were able to aggregate on the surface of the spinal cord nearest to the magnetic field [119]. Another study focused on delivering magnetic NP-loaded endothelial cells to the steel surfaces of intra-arterial stents in a rat carotid artery stenting model [120]. Loading cells with nano/microparticles can be used beyond cell localization, especially for drug delivery applications.
12.3 Cell-Based Drug Delivery
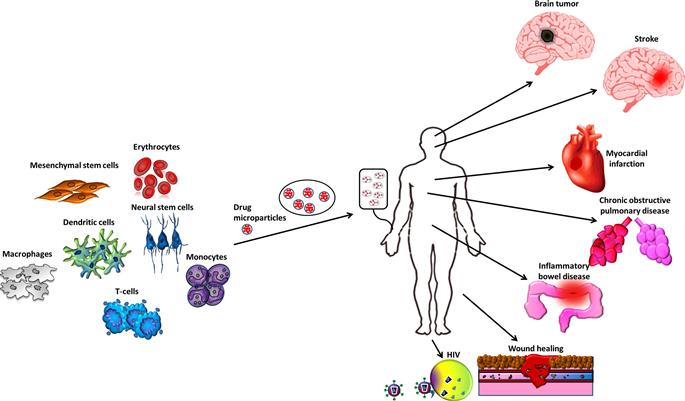
The ability of cells to target specific tissues presents new opportunities for drug delivery. Harnessing cells as vehicles for drug delivery potentially overcomes challenges faced by current treatments. Local administration of therapeutics is undesirable when the disease is systemic or requires invasive access to target sites. Meanwhile, challenges of systemic administration include host toxicity, uncontrolled drug levels at desired sites, premature inactivation of the drug [121], and fast clearance of the agent from the blood, consequently requiring frequent repeat dosing. The usage of drug carriers is critical in the field of drug delivery and has been researched intensively [121]. Encapsulation of therapeutic agents in micro/nanoparticles increases safety by reducing toxicity and increases efficacy by protecting the drug from early degradation, permitting sustained therapeutic effects of systemically administered drugs. However, targeted delivery, which maximizes efficacy by releasing drugs only at disease sites, remains challenging. Particles passively target tissues where the vasculature is leaky (e.g., liver, kidney, and tumor). Although immobilizing ligands on the particle surface can actively target particles to specific tissues and counter passive targeting, excessive active targeting limits the depth of tissue penetration, and may also compromise the drug release kinetics [122]. In contrast, cells have homing and migratory properties; they can target and penetrate specific tissues even without modification. Therefore, cells with known or controllable tissue targets following transplantation emerge as a promising platform for targeted drug delivery. Like a Trojan horse, a cellular carrier protects the drug, infiltrates into the target tissue, and locally releases therapeutic cargo. Sustained drug release may be achieved by loading cells with drug-containing particles [123]. Not only does this arm the cell with additional therapeutic capabilities, its fate and phenotype may also be controlled by the released drug, like in the case of MSCs [55,98]. Herein, we will discuss several nanosystems, cell types, delivery methods, and disease models that utilize this strategy (Figure 12.2).
12.3.1 Nanosystem Types and Key Characteristics
The largest group of particulate carriers is NPs which encompass nanospheres, nanocapsules, micelles, dendrimers, nanocrystals, and nanogolds. NPs are chosen for their small size, high surface-to-volume ratio, and surface functionality. The most commonly used NP materials are polymers and lipids, which typically encapsulate hydrophobic drugs. Polymer-based NPs are made from synthetic polymers such as poly(lactic acid) (PLA) and poly(lactide-co-glycolide) (PLGA) or natural polymers such as gelatin, alginate, collagen, and chitosan [124–126]. PLA and PLGA NPs are especially attractive since their degradation time can be tuned from days to months based on copolymer configuration and molecular weight [127]. However, without special treatments to render their surfaces hydrophilic, they are unstable in the aqueous biological environment and are unsuitable for systemic delivery. Lipid-based NPs typically have an amphiphilic coating to stabilize their hydrophobic cores in an aqueous environment. There are two common types of lipid-based NPs: solid lipid NPs [128] which consist of a solid lipid core and lipid nanocapsules [129] which have an oily (liquid) center [130]. Although lipid-based NPs may be more biologically stable, tuning their biodegradation in vivo is challenging. Other lipid-based NPs include micelles and liposomes, with liposomes being particularly attractive for their ability to encapsulate hydrophilic molecules [131].
Nucleic acids represent an important class of therapeutic hydrophilic molecules. Safety concerns over viral vectors have shifted research focus toward nonviral vectors [132]. Cationic liposomes and cationic polymer-based vectors interact with negatively charged molecules such as DNA while maintaining an overall positive charge on the surface, enabling interaction with the negatively charged cell surface and subsequent endocytosis, thus preventing DNA from systemic enzymatic degradation. Cargo is released into the cytoplasm via several mechanisms of endosomal escape such as membrane disruption/reassembly [133]. Similarly, cationized polysaccharides such as spermine-dextran [134] and spermine-pullulan [135] can be recognized by cell surface receptors to promote internalization, with biodegradability serving as another advantage. Although recent in vivo results have shown toxicity and low transfection efficiency, charged polysaccharides still demonstrated remarkable potential for use in tissue regeneration and cancer target therapy [91,136,137].
Another promising nanosystem is carbon nanotubes (CNTs) [138,139], which can immobilize and intracellularly deliver small molecules, nucleic acids, and proteins as long as the cargo can adsorb onto the CNT surface, typically by pi–pi stacking interactions [140,141]. Unfortunately, studies have also associated CNTs with cell toxicity [142], reducing its potential for clinical translation.
Protein adsorption on the surface of particles was originally viewed as undesirable as it promotes clearance by phagocytes. Efforts have since shifted toward using cellular binding and internalization properties to their advantage, such as to target specific cell types or to develop cellular drug carriers [143]. Internalization efficiency of particles by cells depends on cell type, particle size, surface chemistry, surface topography, shape, and mechanical properties [89]. Additional ligands can be conjugated to the particle surface to promote internalization via specific endocytic pathways [144].
12.3.2 Candidate Therapeutic Agents and Cell Types for Cell-Based Delivery
The types of therapeutic agents to be encapsulated in particles or cells depend not only on the disease of interest but also their stability during encapsulation and release kinetics after encapsulation. Encapsulation in cells warrants extra attention because the released drug may affect the cell and vice versa. For example, cell-based delivery of cytotoxic chemotherapeutics requires careful tuning of release kinetics, since excessive burst release may kill the cellular carrier before it reaches the target tissue [145]. An appealing strategy is to deliver prodrugs—drugs that are administered as inactive molecules, but are converted into their active form after specific chemical or enzymatic activity intracellularly or extracellularly. For instance, one can use the resident enzymes present in cells to convert prodrug into diffusible drug that can be released into the circulation [146]. Many therapeutic cargos, including nucleoside/nucleotide analogs [147], glucocorticoid analogs [148–151], enzymes [152–154], toxins [155,156], mRNA [98], peptides [157–167], and antisense peptide nucleic acids [168–171], are available as prodrugs.
An ideal cellular carrier must not only withstand its therapeutic cargo but also home to target tissues for efficient drug delivery. The most obvious, yet valuable, approach is to harness the innate homing properties of specific cells. Examples include leukocytes and stem cells that are known to home to tumors and sites of inflammation [172–175]. However, the innate homing properties may be suboptimal; hence cell surface engineering can be applied to maximize cell delivery to specific sites, as discussed in the previous section. The next section reviews a selection of cell types and their tropism. Discussion of cancer applications will be deferred to the following section, where we provide a case study on how multiple cell studies have been used as Trojan horses to target tumors.
12.3.2.1 Erythrocytes
One of the deficiencies of using pure drugs or drug delivery nanosystems is their short life spans—they are quickly cleared by immune cells, liver, and kidneys. Frequent injections of the drug or nanosystem are required to resupply the effective dosage. Known for their long circulating life spans (up to 120 days), erythrocytes or red blood cells (RBCs) have been harnessed for sustained drug delivery [176]. Drugs or drug-loaded particles may be conjugated as “backpacks” onto RBC surfaces where immune-evasive molecules reside [177] or encapsulated within RBC membranes [178]. Both strategies “camouflage” the drugs from clearance pathways. RBCs also have other advantages such as abundance (~5.4 million cells/mm3 blood), size and shape uniformity [176], as well as long life span in circulation [146].
Particularly, RBC modification can be used in blood disorders. Fibrinolysis, the process of breaking down blood clots, is used to treat conditions due to hypercoagulation such as stroke and brain ischemia. Coupling fibrinolytic agents such as tissue-type plasminogen activator (tPA), streptokinase, and urokinase onto RBCs have been shown to sustain fibrinolytic activity without affecting RBC circulation [179–181]. Furthermore, when an antibody against collagen was attached on the RBC membrane with streptokinase, RBCs were able to attach to immobilized collagen and lyse the overlaying fibrin clots [182]. This approach may potentially be used to target sites where integrity of the vascular endothelium is compromised, since coagulation can be triggered by exposure of basement membrane proteins (e.g., collagen) to blood.
Dexamethasone is a potent glucocorticoid drug that has anti-inflammatory and immunosuppressant properties. Unfortunately, dexamethasone treatment has severe adverse effects, including redistribution of fat, muscle wasting, acne, bruising, thinning of skin, osteoporosis, exacerbation of diabetes mellitus, suppression of growth in children, and cataract. In light of these systemic effects, Pierige et al. developed a dexamethasone prodrug that utilizes enzymes in RBCs for its activation. The prodrug is encapsulated into autologous RBCs. When applied as a therapy, it is dephosphorylated by RBC enzymes and releases active dexamethasone. Owing to the slow, yet sustained, release of effective doses of dexamethasone, patients fighting chronic obstructive pulmonary disease [149], cystic fibrosis [150], and bowel inflammatory disease [183–186] were able to see benefits from treatments without adverse effects [147].
Although utilizing RBCs as drug carriers may achieve sustained systemic delivery, circulation times of “camouflaged” drugs were not as long as expected, typically lasting less than a week in stark contrast with the 120-day life span of unmodified RBCs [178]. Biomolecules on the RBC membranes that are responsible for prolonged circulation may be irreversibly damaged during RBC modification. In particular, RBCs lack tissue penetration properties and are unsuitable for delivering drugs deep into solid tissues. Consequently, targeted cell-based drug delivery strategies favor immune and stem cells, mostly for their innate tropism toward inflamed, hypoxic, and cancerous tissues [68].
12.3.2.2 Stem Cells
12.3.2.2.1 Neural Stem Cells
Neural stem cells (NSCs) exhibit tropism toward intracranial gliomas and medullablastomas in animal models [187–189]. However, NSCs are only found in the brain [190] which poses significant donor morbidity. The challenge of harvesting enough autologous NSCs for clinical applications is a downfall of using NSCs. Other cell types—fetal brain, adult allogeneic brain, and ESCs—are being investigated as potential alternatives for autologous NSCs [191].
12.3.2.2.2 Mesenchymal Stem Cells
MSCs exhibit tropism toward sites of injury [192] and tumors [174,175], and their homing efficiency can be enhanced by genetic, chemical, enzymatic, and other modifications as previously described. Unlike NSCs and other stem cells, MSCs can be feasibly obtained at sufficient numbers for clinical applications since they can be isolated from many adult tissues [193–202] at significant yields using minimally invasive methods and, more importantly, can be readily expanded ex vivo in large scales. Recently, our group has explored the potential of using MSC as cellular carriers via several bioengineering approaches. For instance, MSCs carrying intracellular dexamethasone-loaded PLGA microparticles could program the phenotypes of the MSC carrier as well as neighboring cells [103]. We have also demonstrated the use of mRNA modification on MSCs to suppress local inflammation via targeted delivery of the anti-inflammatory cytokine interleukin-10 following systemic MSC administration [98]. Moreover, MSCs can also be genetically modified to promote wound repair [203,204].
12.3.2.3 Leukocytes
Leukocytes, also known as white blood cells, possess natural homing properties toward tumors as well as other disease sites due to their major role in the immune response. Isolation of leukocytes is minimally invasive (via blood) and relatively straightforward. Consequently, multiple types of leukocytes have been implicated for Trojan horse delivery of therapeutics [205].
12.3.2.3.1 Monocytes
Monocytes are one of the first cells recruited by damaged, infected, or diseased tissues. After arriving at the site, monocytes differentiate into macrophages that phagocytose pathogens and recruit additional immune cells to the disease site [206]. At tumor sites, monocytes may become tumor-associated macrophages (TAMs), which constitute up to 70% of the tumor mass in breast cancer [206]. As drug carriers, monocytes may infiltrate into the hypoxic core of tumors and force surrounding TAMs to prevent proliferation [200,207–210], tumor neoangiogenesis [158–160,201], invasion [161,164,167,207], and metastasis [207,211–218] of malignant epithelial cells. An advantage of using monocytes or macrophages as transport vehicles is their innate phagocytic capability which provides high efficiency in particle loading. The use of macrophages as cellular carriers for NPs arose from the observation that systemically administered of NPs for imaging were ingested by endogenous macrophages that migrated and accumulated in and around tumors [219–221]. Another advantage of macrophages as cellular carriers is their ability to cross the blood brain barrier (BBB). For instance, macrophages preincubated with NPs containing a retroviral drug and administered in a murine model of HIV-1 encephalitis were observed to cross the BBB, increase local drug concentration for 14 days, and thereby suppress HIV-1 replication [222,223].
12.3.2.3.2 T Cells
T cells play a key role in adaptive immunity, for instance by mediating cellular responses against infected or cancerous cells [224]. T cells have potential as chaperones of surface-attached therapeutic cargo, supported by their previous use in clinical trials for adoptive T-cell therapy in cancer, ease of harvest, and well-established protocols for their genetic modifications and expansion [225]. They have also shown promising infiltration into tumor lesions by crossing the barriers imposed by endothelial and stromal tissues [226].
12.3.2.3.3 Dendritic Cells
Dendritic cells (DCs) are antigen-presenting cells, mostly found in the lymph nodes. After a local infection, DCs process and present antigens on their surfaces in order to activate naïve T cells and B cells [227]. Cell–cell interactions between T cells and DCs occur through direct contact [228] and paracrine signaling. There have only been a limited number of studies employing DCs as cellular vehicles. For example, DCs have been employed to systemically deliver oncolytic reovirus for killing melanoma cells and shown effective protection for the virus against the preexisting antiviral immunity [229]. Interestingly, the principle of “antigen presentation” on DCs was mimicked by immobilizing receptors and co-stimulatory ligands onto synthetic particles to activate adaptive immunity [230].
12.3.3 Case Study: “Trojan Horse” Cell Therapy for Cancers
Tumors are characterized by chaotic vasculature, high interstitial pressure, and dense extracellular matrices, presenting major obstacles for treatment via systemically administered drugs or drug-loaded particles [126]. Often, drugs and particles are unable to penetrate into the tumor and, as a result, are rapidly cleared. An example is GBs, which are extremely malignant brain tumors, known for their rapid growth and vascularization [231]. Current treatments revolve around surgery, radiation, and chemotherapy with DNA alkylating agents such as temozolomide (TMZ). However, there are several limitations with current treatments such as the poor effectiveness of TMZ for TMZ-sensitive GB patients as well as GB resistance against chemotherapy and radiotherapy. In addition, glioblastoma stem cells (GSCs) possess infiltrative, proliferative, and progressive characteristics, rendering them more resistant to hypoxic and acidotic tumor microenvironments [232]. Although GBs are highly vascularized, penetrating the BBB is yet another challenge toward effectively delivering therapeutic agents to the tumor. In fact, the BBB prevents uptake of large molecules and of over 98% of small-molecule drugs available [233]. Likewise, NP transport efficiency across the BBB is poor. To deliver antitumor agents across the BBB and deep into GBs, cellular carriers—namely monocytes and stem cells—are attractive candidates due to their ability to infiltrate into gliomas [234–238].
Known for their tropism toward intracranial tumors, NSCs have been used to transport toxic molecules [239–244], proliferation inhibitors [245], anti-angiogenic agents [246,247], and cytokines with toxic molecules [240]. Additionally, NSCs have been genetically modified to yield cytokines such as IL-18, IL-2 [248–250], IFN-γ [251], and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [252–254] for modifying increased immune response at gliomas. NSCs have also been engineered to secrete cytosine deaminase (CD) that activates the prodrug 5-fluorocytosine (5-FC), so that cytotoxic effects of the systemically administered drug are localized within the tumor [189]. Off-target homing of antitumor NSCs is potentially dangerous as antitumor agents can harm healthy cells. To reduce off-target actions, overexpression of a pH-sensitive viral fusogen on the NSC surface has been used to fuse NSCs specifically with tumor cells, since tissue pH is lower at tumor sites [255]. These engineered NSCs carried a suicide gene, allowing selective killing of any syncytium formed by the NSCs and residual, unfused NSCs.
Monocytes or macrophages can also cross the BBB, but unlike NSCs, monocytes are isolated with much less invasive procedures. Choi et al. envisioned loading monocytes with gold nanoshells such that they home to the tumor site, differentiate into TAMs, migrate and deliver the nanoshells into hypoxic regions of the tumor. Once in place, near-infrared illumination would be used to heat the nanoshells and destroy the TAMs and adjacent tumor cells [205]. Nanoshell-based photothermal ablation therapy has shown promising results toward reducing tumor in mice, with over 90% remission rate [256]. Combining these concepts, monocyte carriers of gold nanoshells were successfully used to ablate human breast carcinoma tumor spheroids [172,205,219]. In another study employing a murine glioma model, cyclodextrin-based NPs were internalized by TAMs. Due to their tumor-tropic properties, the TAMs migrated within and around the tumor, demonstrating their capability to distribute therapeutic agents across the tumor [257]. T cells have also been used for nanoshell-based photothermal ablation [258], demonstrating the general effectiveness of leukocytes as tumor-homing cellular carriers.
Another use of T cells as a cellular vehicle followed from observations that viral particles can “hitchhike” on the T-cell surface, be taken up by tumor cells in vivo, and be released at the tumor site [259]. Extending the success of this strategy, numerous groups have used tumor antigen-specific T cells to carry oncolytic viruses to tumor deposits [260–262]. The same strategy can be applied to deliver NPs for cancer diagnostics and therapeutics, where the use of viruses is undesirable. Such agents include small-molecule drugs, antibody–drug conjugates, aptamers, and magnetic imaging agents [263,264]. The Irvine group used thio-reactive maleimide groups to crosslink nanocarriers, surface proteins, or lipids to the T-cell surface for up to a week. Their in vivo results showed successful biodistribution of T-cell-coupled NPs in tumor models, while systemic injection without cellular carriers resulted in fast clearance of the particles in liver and spleen [265].
MSCs are yet another cell type that exhibits tumor-tropic properties. Systemically administered MSCs carrying intracellular PLA and lipid nanocapsules demonstrated successful delivery of nanocapsules to gliomas [175]. NPs have also been conjugated onto the MSC surface using biotin–streptavidin bridges without attenuating MSC tropism toward tumor spheroids [266]. In multiple studies, MSCs were genetically engineered using viral and nonviral vectors to secrete immunostimulators such as IL-2, IL-12, IFN-beta, IL-18, and IL-7 to suppress tumor growth in glioma and other cancer models [203,250,251,267–276]. MSCs are also being used to deliver oncolytic adenoviruses to tumor sites [277,278]. More complex systems combining gene modification and prodrugs such as the CD/5-FC system (also applied on NSCs) have been tested on MSCs [279,280]. Another system uses herpes simplex virus thymidine kinase (HSVtk), which activates the chemotherapeutic prodrug ganciclovir [281]. Retrovirally transfected MSCs carried HSVtk to the tumor site, such that systemically administered ganciclovir was preferentially activated in the tumor, resulting in tumor suppression in GB patients [281]. The same system appeared to be less efficacious when NSCs are used instead [241,282–284]. Yet another combination involves rabbit carboxylesterase and CPT-11, in which genetically modified adipose-derived MSCs were used to deliver carboxylesterase that enzymatically cleaves the CPT-11 prodrug to release the active drug SN-38 to combat brainstem gliomas [285]. Despite these advances, thorough investigation is required to fully elucidate the interaction between MSCs and tumor environment, especially when MSCs have been suggested to promote tumor growth [286], and clinical trials have not shown long-term engraftment of MSCs in tumors [287].
While most cellular drug delivery vehicles are still in early stages of research, one cell type has been successfully translated into FDA-approved therapy. DCs have become a part of FDA-approved therapeutic cancer vaccine (against prostate cancer), in which antigen-loaded autologous DCs are injected into patients [288]. In addition, another group has proposed a method to track cells that have been loaded with NPs. Noh et al. encapsulated near-infrared fluorophores and iron oxide NPs together with model antigens such that cells carrying the NPs could be tracked in real time. Injection of those DCs resulted in tumor suppression [289]. DCs also show tropism toward lymph nodes which enables the use of DCs to carry immunosuppressive drugs to lymphoid tissues in order to suppress T-cell proliferation [290].
Apart from solid tumors, blood cancers may also be treated with cellular carrier. In a pilot clinical trial, autologous RBCs were loaded with asparaginase so they could remove asparagine—a nonessential amino acid—from blood. Asparagine has been shown to increase lymphoblastic proliferation which is crucial for the progression of lymphoblastic leukemia [291].
12.4 Concluding Remarks
Cell therapy provides hope for treating many tragic diseases. Harnessing the innate therapeutic properties of certain cell types, such as MSCs, and using cells as vehicles for targeted delivery of therapeutics are promising strategies to further advance cell-based therapy. However, for cell therapy to fully realize its clinical potential, better control over cell fate following transplantation must be achieved. Bioengineering approaches, such as genetic, enzymatic, or chemical surface modifications, may enable specific delivery of systemically infused cells to desired organs, although this has yet to be tested in humans. Cells loaded with micro/nanoparticles containing specific drugs can then be directed to diseased organs, generating high local levels of therapeutics in the desired location, minimizing systemic toxicity, and significantly improving treatment efficacy. Tailoring drug release from cells has potential to provide a highly regulated and sustained therapeutic impact, and multiple cell infusions can further maximize the effect. Furthermore, mRNA engineering can be utilized to simultaneously but transiently control multiple cell properties, enabling improved cell homing and delivery of soluble biologics to distant diseased sites, while avoiding potential risks associated with DNA transfection. Targeted delivery of biologics is a major challenge that potentially could be overcome by use of cell-based approaches.
To circumnavigate current challenges in cell therapy, a concerted effort from clinicians, cell processing professionals, cell biologists, and bioprocess engineers is required [292]. Developing large quantities of cells that are immune evasive and that can be readily engineered to accurately deliver therapeutics including biologics to distant desired sites would have immense clinical potential.