
Pushing the Bacterial Envelope
Strategies for Re-Engineering Bacterial Surfaces with Heterologous Proteins and Sugars
Samir Gautam1 and David A. Spiegel2, 3, 1Department of Cell Biology, Yale School of Medicine, New Haven, CT, USA, 2Department of Pharmacology, Yale School of Medicine, New Haven, CT, USA, 3Department of Chemistry, Yale University, New Haven, CT, USA
Over the past three decades, a powerful array of techniques has been developed for expressing heterologous proteins and saccharides on the surface of bacteria. Surface-engineered bacteria, in turn, have proven useful in a variety of settings, including high-throughput screening, biofuel production, and vaccinology. In this chapter, we provide a comprehensive review of methods for displaying polypeptides and sugars on the bacterial cell surface, and discuss the many innovative applications these methods have found to date. While already an important biotechnological tool, we believe bacterial surface display may be further improved through integration with emerging methodology in other fields, such as protein engineering and synthetic chemistry. Ultimately, we envision bacterial display becoming a multidisciplinary platform with the potential to transform basic and applied research in bacteriology, biotechnology, and biomedicine.
Keywords
Surface engineering; bacterial envelope; glycoengineering; heterologous expression systems; vaccine design; probiotics; cancer therapy; biocatalysis; high-throughput screening
4.1 Bacterial Surface Display
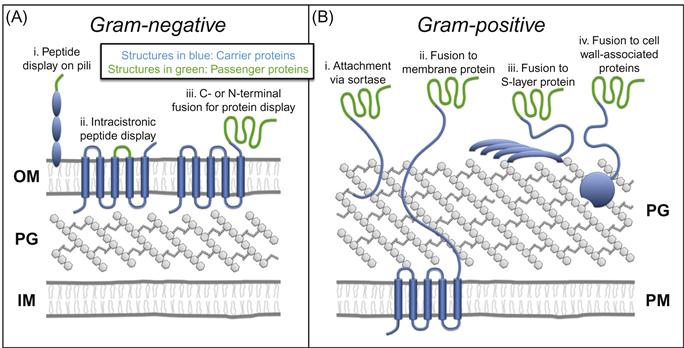
The bacterial envelope is a multilayered organelle that surrounds the cell, providing morphological structure and protection from environmental stressors. In gram-negative organisms, the envelope comprises an inner membrane (IM), an outer membrane (OM), and a periplasmic space in between that contains the peptidoglycan cell wall—a highly cross-linked mesh-like macromolecule that confers mechanical strength to the cell (Figure 4.1A). Gram-positive bacteria, in contrast, lack an OM and have a thicker, surface-exposed cell wall (Figure 4.1B). These structures have been studied intensely for over 60 years by bacterial cell biologists and antibiotic developers, among others, due to their critical roles in bacterial growth and survival [1,2].
As our understanding of the bacterial envelope has grown, however, so has its utility; we now possess a powerful set of techniques for expressing proteins on the cell surface that has transformed the envelope from a target of basic and pharmaceutical research into a versatile platform for biotechnological innovation. Since the first examples in 1986 [3,4], protein display in bacteria has matured into a robust, streamlined methodology with an expansive range of applications, including environmental remediation [5], biofuel production [6], biocatalysis [6,7], biosensing [8], protein library screening [9], cancer therapy [10], and vaccinology [7,11–14].
More recently, bacterial surface engineering has extended beyond the polypeptide paradigm to include sugars as well. This technology, referred to as “glycoengineering,” [15–17] involves heterologous expression of genes that mediate saccharide biosynthesis, enabling the display of a variety of nonnative proteoglycans, glycolipids, and polysaccharides on the surface of recombinant bacteria. Glycoengineered organisms, in turn, have been utilized as whole cell vaccines [18–21], anti-infective probiotics [22], and “living factories” for glycoconjugate synthesis [16].
In this chapter, we describe the repertoire of techniques that have been established for displaying heterologous proteins and sugars on the surface of bacteria. We then discuss how re-engineered bacteria have been exploited in basic and applied biology thus far. Throughout, we attempt to highlight recently developed technologies across disciplines that may prove useful in the context of bacterial surface display, improving existing applications and perhaps enabling new ones. Finally, we address an important caveat: despite the tantalizing clinical and biotechnological prospects for surface-engineered bacteria, their implementation outside the laboratory has been greatly hindered due to concerns over the environmental dangers of genetically modified organisms (GMOs). We therefore include a discussion of these theoretical risks and present a set of recombinant techniques for circumventing them (see Box 4.1).
4.2 Strategies for Re-Engineering Bacterial Surfaces with Heterologous Proteins
The majority of bacterial protein display methods employ a common strategy in which a heterologous protein of interest (passenger) is genetically fused to an endogenous surface protein (carrier). The structure of the cell envelope, in turn, determines where these extracellular passenger–carrier chimeras reside; in gram-negative organisms they associate with the OM (Figure 4.1A), while in gram-positive bacteria chimeras are usually attached to the peptidoglycan cell wall (Figure 4.1B) [30].
4.2.1 Gram-Negative Organisms
A number of recombinant techniques have been developed for expressing passenger–carrier fusion proteins on the surface of gram-negative species (Figure 4.1A). For peptides and small proteins up to approximately 60 amino acids [31], exogenous sequences may be inserted into (i) proteins that comprise surface appendages such as pili and flagella, (ii) extracellular loops of outer membrane proteins such as LamB and OmpA, or (iii) lipoproteins and virulence factors anchored to the surface [13,32]. However, for the display of larger proteins such as enzymes, passengers must be fused to the N- or C-terminus of carriers rather than intracistronically (Figure 4.1C,iii) [13].
Autodisplay is one such system commonly used for the display of large proteins. The strategy involves fusion of passenger proteins to autotransporters, a class of virulence factors that includes IgA1 protease from Neisseria gonorrhoeae, MisL from Salmonella enterica serovar Typhimurium, and AIDA-I (adhesin involved in diffuse adherence) from Escherichia coli [7]. During normal expression, autotransporters insert into the OM, forming a porin-like β-barrel structure with their C-terminal domain. The N-terminal domain, connected to the β-barrel via a long, flexible linker is subsequently translocated through the pore to access the extracellular milieu. Therefore, autotransporters may be ‘hijacked’ to allow surface display by replacing the native N-terminal domain with a heterologous passenger [33]. A notable advantage of this approach is the lateral mobility of carriers within the OM, which enables multimerization of passenger proteins, as has been demonstrated with surface-displayed streptavidin [34], nitrilase from Klebsiella pneumoniae [35], and sorbitol dehydrogenase [36].
The other major system for expressing large proteins at the bacterial surface involves fusion to ice nucleation protein (INP), an abundant lipoprotein present in the OM of many plant pathogens [13,37]. A unique property of INP is its mechanism of surface expression; rather than threading through the IM in an unfolded state like the majority of bacterial proteins, INP is translocated fully folded. Thus, INP-mediated surface display can accommodate passenger proteins that associate with bulky cofactors, such as heme- and diflavin-containing cytochrome P450 enzymes [13,38].
Since their introduction in the 1990s [39,40], autodisplay and INP-based expression systems have developed into highly optimized, well-characterized biotechnological platforms. Passenger–carrier chimeras expressed using these methods (i) efficiently traverse the IM and periplasm to reach the cell surface in great abundance (more than 100,000 molecules per cell in the case of autodisplay [41]), (ii) reside stably at the surface without substantial detachment and loss of the recombinant protein, (iii) resist degradation by proteases within the periplasmic space, and (iv) do not disrupt the integrity of the cell envelope or induce growth defects [42]. Given these properties, along with the well-defined genetic techniques required for autodisplay- and INP-based expression, surface display in gram-negative bacteria can be considered a straightforward and robust class of techniques.
4.2.2 Gram-Positive Organisms
Whereas surface display in gram-negative bacteria is achieved through fusion to carrier proteins expressed on the OM, a different strategy must be undertaken in gram-positive organisms, which lack this outer vestment (Figure 4.1B). The most common method involves attaching proteins directly to the surface-exposed peptidoglycan cell wall, using a class of enzymes called sortases (Figure 4.1B,i) [43,44]. These membrane-associated enzymes recognize specific pentapeptide sorting motifs in secreted proteins, cleave within the motif, and covalently attach the N-terminal portion to the cell wall [45]. Thus, fusion of a passenger protein to the N-terminus of a sortase substrate protein enables efficient expression on the gram-positive cell surface. Sortase-mediated display of heterologous proteins has proven especially robust both because of the covalent nature of attachment, and because of the rigid peptidoglycan substrate, which ensures stability of surface expression [11].
Alternatives to sortase-based surface display have been explored as well. For example, passengers fused to transmembrane proteins that extend from the plasma membrane through the cell wall and into the extracellular space are effectively displayed at the cell surface (Figure 4.1B,ii) [46]. Surface expression can also be achieved using chimeras between passengers and proteins that comprise the bacterial cell surface layer (S-layer), a self-assembling crystalline lattice that coats several species of bacteria (Figure 4.1B,iii) [47–52]. Notably, the absence of an OM on gram-positive organisms removes a significant obstacle to protein translocation, facilitating the display of large proteins when using sortase-mediated, transmembrane fusion, and S-layer fusion protein methodologies [11].
A final, somewhat unique approach involves fusion to autolysins—cell wall degradative enzymes that bind non-covalently to components of the gram-positive cell wall (Figure 4.1B,iv) [53–57]. The key advantage of this methodology is that autolysin-passenger chimeras may be expressed in a model organism (e.g., E. coli), purified, and adsorbed to the cell wall of a wild-type gram-positive strain. Thus, using this strategy, gram-positive bacteria can be re-engineered with heterologous surface proteins, but remain genetically unchanged, thereby circumventing the potential environmental risks of GMOs (see Box 4.1) [55].
4.2.3 Spores
Certain bacterial species, such as those within the Clostridium and Bacillus genera, display a unique behavior known as sporulation, in which bacteria transform from metabolically active (vegetative) cells into dormant structures (spores) in response to nutrient depleted conditions [12]. During this process, bacterial DNA becomes cocooned within a multilayered wall of peptidoglycan and concentric protein shells, rendering the DNA impervious to toxic chemicals, radiation, and extremes of pH, temperature, and dehydration [12]. Spores are thus able to survive in inhospitable environments for extended periods—in one report, 25 million years [12,58]. Despite the spore’s extraordinary inertness, its surface proteins remain functional (in part due to their attachment to a stable matrix, see Section 4.3.4), ready to sense nutrient repletion, and germinate into a fully active state [59]. Numerous methods have been established for displaying heterologous proteins on these remarkable structures, all involving fusion to carrier proteins expressed at the spore surface [12]. These carriers include CotB, CotC, and CotG in Bacillus subtilis; [60]. BclA and BclB in Bacillus anthracis [61]; and Cry1Ac in Bacillus thuringiensis [62]. Spores re-engineered by these means have proven useful as biosensors [8,63–69], biocatalysts [70], and vaccines [71,72].
While the applications of spores are discussed in more detail below, it is worth describing here the unique advantages spores offer over conventional bacteria in three biotechnological settings. First, similar to vegetative cell systems, spores are well suited to biosensing—the detection and reporting of analytes in solution (see Section 4.3.5.1)—because of their sensitivity, low cost, ease of use, and small size. The exceptional stability of spores, however, allows them to be used outside of the laboratory in harsh natural environments, where conventional cellular systems cannot survive [70]. Second, spores are unique in that proteins need not traverse a membrane in order to be expressed on the cell surface; instead, proteins are attached to the outermost layer of the spore, which is assembled within the cytoplasm of the mother cell during sporulation [73]. This property allows expression of multimers such as streptavidin [74], proteins with problematic hydrophobic domains that preclude membrane translocation [60], enzymes requiring cofactors [60], and very large proteins [31]. Finally, the spore’s ability to germinate confers an advantage in the context of vaccine delivery, as it remains stable during storage at room temperature, but then transforms once delivered to the nutrient-rich environments of the tissue into vegetative cells with potent immunostimulatory properties (see Section 4.3.1) [75,76].
4.2.4 Outer Membrane Vesicles and Bacterial Ghosts
Outer membrane vesicles (OMVs) and bacterial ghosts (BGs) are acellular derivatives of the gram-negative envelope that contain the parent cell’s surface-expressed proteins [77–79]. OMVs consist of outer membrane-derived bilamellar vesicles, 50–250 nm in diameter, which are continuously elaborated by growing gram-negative bacteria [77]. Efficient surface display on OMVs has been achieved by fusing passenger proteins to the E. coli toxin ClyA, which becomes concentrated on these structures during vesiculation [80]. OMVs have particular utility as vaccine delivery vehicles because they are stable during prolonged storage [81], and strongly immunogenic (by virtue of the many immunostimulatory components of the OM these vesicles retain), but do not present the same environmental safety hazards as intact cells because they are unable to propagate (see Box 4.1) [82–84].
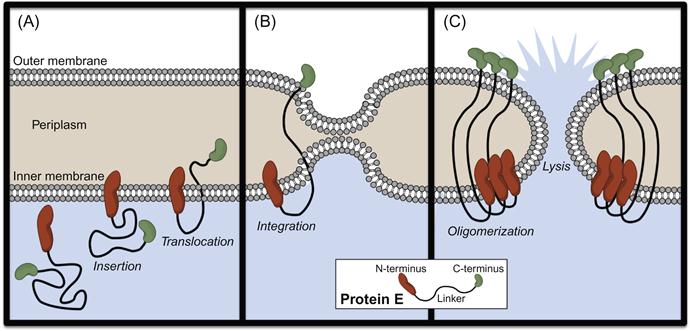
BGs, in contrast, are complete gram-negative bacterial envelopes produced through heterologous expression of the bacteriophage-derived lysis gene E (Figure 4.2). Upon expression, Protein E inserts into the IM via its N-terminus, while the C-terminal domain translocates across the IM (Figure 4.2A) and integrates into the OM (Figure 4.2B), inducing fusion of the two membranes. Protein E oligomerization leads to the formation of a “lysis tunnel” (Figure 4.2C), through which the cytoplasm is released extracellularly. However, due to the controlled nature of the lytic event and the effective seal between IM and OM, cell envelope morphology and periplasmic content are maintained [78]. Most commonly, anchoring of foreign proteins on BGs is achieved through fusion of passengers with the outer membrane protein, OmpA. Finally, like OMVs, BGs are excellent vehicles for vaccine delivery because of their potent immunostimulatory properties and inability to replicate [85,86].
4.3 Applications of Bacteria Expressing Heterologous Surface Proteins
Bacterial surface display has lent itself to a host of biotechnological and biomedical applications. These can be organized into four categories: (i) display of antigens in vaccine design, (ii) expression of antitumor proteins for cancer therapy, (iii) immobilization of enzymes for biocatalysis, and (iv) a set of related applications that involve binding between surface proteins and molecules in solution (biosensing, biosorption, and library screening). These topics will be discussed here only in brief; please see focused reviews for greater detail [6,9,11–13].
4.3.1 Vaccines Against Infectious Disease
In general, vaccines have two components: a pathogen-associated antigen that serves as the target for adaptive immune responses and an adjuvant—a material that potentiates and tunes immune responses to the co-delivered antigen [87]. Uptake of these components by antigen presenting cells (APCs) such as dendritic cells represents the crucial initiating step in the generation of immune responses that protect against future infection.
Surface-engineered bacterial vaccines are constructed by expressing a pathogen-associated antigen on an inactivated bacterium, live-attenuated pathogen, or food grade (nonpathogenic) bacterial species. The bacterial vector itself is able to serve as an adjuvant due to the numerous immunostimulatory pathogen-associated molecular patterns (PAMPs) present within the cytoplasm and cell envelope. These include bacterial DNA, peptidoglycan, gram-positive lipoproteins, gram-negative lipopolysaccharide (LPS), and a set of PAMPs associated with viable cells (vita-PAMPs) which includes mRNA and cyclic dinucleotides [88].
Localization of antigen at the bacterial surface per se appears to be important for the immunogenicity of these recombinant vaccine vectors. In almost all direct comparisons between surface, cytoplasmic, and secreted expression systems, surface-engineered vaccines have proven most efficacious (even when normalized to magnitude of expression) [89–100]. One reason for the improved immunogenicity of surface-anchored antigen is that it is efficiently co-delivered with adjuvant (bacterial PAMPs) to APCs, which is essential for generating effective immune responses [101]. Furthermore, surface expression produces a multivalent display of antigen on the cell surface, which is known to promote cross-linking of B-cell receptors and stimulation of humoral immune responses [102].
Bacterial surface display has been especially useful in the development of mucosal vaccines with several advancing as far as clinical trials [7,11–14]. Table 4.1 provides a summary of the bacterial vectors used in mucosal vaccinology, and a catalog of the pathogens these vaccines have targeted.
Table 4.1
Examples of Surface-Engineered Bacteria in Mucosal Vaccinology
| Bacterial Vector | Pathogenic Target |
| Salmonella spp. [103,104] | Dengue virus [105], Hepatitis B virus [106], Japanese encephalitis virus [107], Rotavirus [108], Sendai Virus [109], Transmissible gastroenteritis coronavirus [100,110,111], Bacillus anthracis [112,113], Enterotoxigenic E. coli [100,114,115], Helicobacter pylori [116], Listeria monocytogenes [117], Streptococcus pneumoniae [118,119], Yersinia enterocolitica [120], Yersinia pestis [121,122], Plasmodium falciparum [123–125], Porphyromonas gingivalis [126], Trichinella spiralis [123,127] |
| Bordetella pertussis [128] | Enterovirus 71 [129], Influenza virus [130], Haemophilus influenzae [131], Neisseria meningitidis [132], Schistosoma mansoni [133] |
| Escherichia coli | Borrelia burgdorferi [134], Enterohemorrhagic E. coli [134,135], Enterotoxigenic E. coli [136,137], L. monocytogenes [134], Salmonella spp. [138], Y. enterocolitica [120], P. falciparum [135] |
| Shigella spp. | Enterotoxigenic E. coli [139–143] |
| Vibrio cholerae [144] | Attaching and effacing E. coli [145], Enterotoxigenic E. coli [146] |
| Lactic acid bacteria [147–150]: Lactobacillus spp., Leuconostoc spp., Pediococcus spp., and Streptococcus spp. | Human immunodeficiency virus [151,152], Human papillomavirus [153–158], Influenza virus [159], Rotavirus [160–163], SARS Coronavirus [164], B. burgdorferi [165], Clostridium tetani [166], Enterotoxigenic E. coli [167–170], H. pylori [171], N. meningitidis [172], Proteus mirabilis [173], Salmonella enteritidis [174–176], Streptococcus agalactiae [177], Streptococcus mutans [178], S. pneumoniae [179–184], Streptococcus pyogenes [185], Yersinia pseudotuberculosis [186], Giardia lamblia [187], P. falciparum [188–190] |
| Staphylococcus spp. [191] | Respiratory syncytial virus [192–194], Salmonella spp. [138] |
| B. subtilis spores [12] | B. anthracis [195,196], Clostridium perfringens [197], C. tetani [198,199], Enterotoxigenic E. coli [199], Clonorchis sinensis [200,201], Schistosoma japonicum [202] |
| OMVs [82] | S. pneumoniae [203] |
| BGs [85,86] | Foot-and-mouth disease virus [204], Hepatitis B virus [205], Human immunodeficiency virus [206], B. anthracis [112], Chlamydia trachomatis [207,208], N. meningitidis [209–211], P. falciparum [99] |
Lactic acid bacteria include Lactobacillus, Leuconostoc, Pediococcus, and Streptococcus spp.
Several arguments have been proposed for why surface-engineered bacteria have proven such effective mucosal immunogens. The first derives simply from the size of the bacterial vector, which at >500 nm in diameter is considered “particulate.” Delivery of antigen by such particulate vehicles is known to greatly improve antigen presentation by APCs, leading to stronger elicitation of immune responses at the mucosa than the same antigen in soluble form [88]. Second, the intrinsic adjuvanticity of bacteria provides a potent immunostimulatory signal to overcome the bias toward tolerance that exists in mucosal immune tissue [212]. Third, the transient colonization and/or mild infection induced by live bacterial vaccines leads to a sustained, local generation of antigen at the mucosa that greatly improves delivery to APCs [213]. Finally, bacterial vaccines are uniquely efficient at gaining access to APCs within mucosal immune tissue, by virtue of surface properties that prevent trapping within the mucus layer [214], and surface adhesion proteins and carbohydrates that promote attachment to mucosal epithelium and translocation into immune inductive sites [215,216]. With respect to bacterial surface display, it is worth noting that surface expression of targeting proteins, such as antibodies that bind to proteins on epithelial cells, has been shown to further increase adhesion and translocation of bacterial vaccine vectors into immune tissue [193,217].
4.3.2 Anticancer Therapeutics
The use of live bacteria in cancer therapy has fascinated researchers since the early nineteenth century, when tumor regression was observed in a set of cancer patients after they contracted gas gangrene (an aggressive soft tissue infection caused by C. perfringens) [10,218–220]. The anticancer effect of bacteria has since been traced to a number of intrinsic properties of these organisms, such as their ability to home to tumor tissue with up to 1000-fold selectivity over healthy tissues [221]. In the case of C. perfringens, this ability arises from the fact that spores selectively germinate within the core of solid tumors, which is hypoxic due to poor vascularization [222]. Facultative anaerobes like Salmonella species and L. monocytogenes, meanwhile, accumulate in cancer tissue due to entrapment in the tortuous tumor vasculature [223], chemotaxis toward nutrients released from dying cancer cells [224], and preferential growth in the tumor’s unique metabolic milieu [224], where they are also protected from attack by white blood cells due to local immunosuppression [225,226]. Upon accumulation in the tumor, bacteria promote cell death both by competing for nutrients and stimulating normally suppressed intratumoral immune cells to attack cancer cells [225].
With the advent of genomics and recombinant technology, antitumor bacteria—which had largely remained curiosities in the field of cancer treatment—have now been transformed into finely tuned therapeutic devices through surface expression of proteins with antitumoral properties. For instance, the invasin adhesion protein from Y. pseudotuberculosis, which mediates entry of the bacterium into nonphagocytic mammalian cells, has been expressed on the surface of nonpathogenic E. coli-based therapeutic strains to promote invasion into cancer cells [227]. This strategy was utilized to deliver “trans-kingdom” RNAi in a mouse model of colon cancer to silence expression of the epithelial oncoprotein, β-catenin [228]. In another example, S. enterica serovar Typhimurium was engineered to express the apoptosis-promoting protein, Fas ligand, on its cell surface. The modified bacterium was shown to inhibit growth of primary breast and colon cancers as well as pulmonary metastases in mice [229]. Finally, bacteria have been engineered to express cytotoxic prokaryotic proteins such as cytolysin A, a surface-localized pore-forming toxin, producing strains that significantly inhibit tumor growth and metastasis in murine models of colon and lung cancer [230,231].
A number of other non-surface related genetic modifications have been shown to potentiate the anticancer effects of bacterial vectors as well, many of which could conceivably be combined with surface display to create more effective, multimodal therapeutics. For instance, Salmonella spp. have been designed to secrete pro-inflammatory cytokines such as IL-2 that promote adaptive immune responses [232], as well as cytotoxic agents such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which directly induce tumor lysis [233]. A second effective modification is expression of genetic circuits that selectively activate gene expression in response to cues in the tumor microenvironment [234]. Such circuits have been used to couple cytotoxin production to arrival at tumors, which reduces the off-target toxicity of these tissue-damaging proteins. Third, through expression of prodrug converting enzymes, tumor-localized bacteria can be used to activate chemotherapeutic prodrugs in situ, increasing drug concentrations in the poorly accessible tumor core, and reducing collateral damage to healthy tissue. The efficacy of this approach has been confirmed not only in murine models but also in patients as well, as phase I trials have shown that an attenuated Salmonella strain, VNP20009, expressing the E. coli cytosine deaminase gene—which converts the prodrug 5-fluorocytosine into the active chemotherapeutic 5-fluorouracil (5-FU)—homed to tumors and generated significantly elevated intratumoral 5-FU levels with respect to serum [235].
Of course, intravenous administration of live bacteria is not without risk of side effects. For instance, preliminary tolerability studies with VNP20009 showed dose-dependent elicitation of cytokines, such as IL-1β, IL-6, and TNF-α, which were associated with hypotension, fever, thrombocytopenia, anemia, diarrhea, nausea, and vomiting [236]. However, through detoxification of pro-inflammatory bacterial components such as LPS (see Section 4.3.1), attenuating mutations to virulence factors and genes in central metabolic pathways (see Box 4.1), titration of tolerable doses, and careful clinical monitoring post-administration, the immunotoxicity of live bacteria may be minimized, allowing these potentially valuable therapeutics to be tested further in clinical settings.
4.3.3 Vaccines Against Cancer
In addition to serving as devices designed to directly kill cancer cells, genetically engineered bacteria have also proven a highly effective platform for development of anticancer vaccines [237–241]. In actuality, these two approaches overlap in many ways; for example, vaccine vectors such as L. monocytogenes colonize tumors and induce direct cytotoxicity, while conversely, oncotherapeutic bacteria lead not only to tumor damage but also to tumor-specific adaptive responses due to release of cancer cell antigens during bacterially induced lysis [240,242]. Nevertheless, in cancer vaccinology, elicitation of immune responses is the primary objective, the ultimate goal being immune-mediated clearance of both primary tumors and metastases as well. In this context, the immunostimulatory properties of the live bacterial vector are leveraged to break the inherent tolerance to cancer-associated antigens that exists because of their similarity to self proteins and their residence in an immunosuppressive tumor milieu [243]. Notably, translation of bacteria-based anticancer vaccines to the clinic has recently begun, with phase I trials demonstrating the safety of a promising listerial vaccine against cervical carcinoma [244].
Bacterial surface display has been used in the design of anticancer vaccines extensively as a means of antigen expression. For instance, T-cell epitopes from NY-ESO-1, a commonly expressed tumor antigen, were inserted into the fimbrial proteins of S. enterica serovar Typhimurium to create a vaccine capable of generating robust antigen-specific T-cell responses in mice [245]. Similarly, the E7 oncoprotein (which is involved with the development of cervical cancer) and the 37 kDa oncofetoprotein (which is found in many human cancers) were surface expressed on lactic acid bacteria to generate effective mucosal vaccines against human papillomavirus-induced tumors [246–249]. Further, it was shown in a mouse model of cervical cancer that the immunogenicity of live E7-expressing bacterial vaccines was greatly potentiated when the vector simultaneously secreted the pro-inflammatory cytokine, IL-12 [155]. This important finding illustrates the potential of combining multiple recombinant modifications in the same therapeutic. It also suggests that vaccine-generated cytokine secretion is a strategy that could, in principle, boost the immunogenicity of any live vaccine.
In addition to antigen expression, bacterial surface display has been used to improve targeting of bacterial vectors to tumors, in an analogous manner to the targeting of vaccines to mucosal immune tissue (described in Section 4.3.1). On an attenuated Salmonella vaccine strain, for example, surface display of single chain antibody fragments specific for carcinoembryonic antigen—a glycoprotein overexpressed on many epithelial-derived cancers—was found to significantly improve intratumoral accumulation of the recombinant bacteria in vivo [250].
4.3.4 Biocatalysis
The use of microbially expressed enzymes for (bio)catalysis of chemical reactions has a long history that includes the ancient practices of bread and cheese making, beer and wine fermentation, and, more recently, antibiotic production. The value of enzymes in chemical synthesis lies in their potential to catalyze reactions with remarkable rapidity in a highly chemo-, regio-, and enantioselective manner under mild conditions, while minimizing the toxic waste generation, protection/deprotection steps, and energy expenditure involved in conventional synthetic chemistry [251,252]. Furthermore, through directed evolution and other protein engineering techniques (described in Section 4.3.5.3), modified enzymes may be designed to (i) remain stable at elevated temperatures in solution with organic solvents, (ii) accept nonnative substrates, and (iii) catalyze novel reactions [251]. These properties have led to a rapidly growing interest in the use of biocatalysts in industrial production of detergents, food, cosmetics, textiles, fine chemicals, biofuels, and pharmaceuticals [253–255]. In fact, over 500 commercial products are produced using enzymes, generating an industrial enzyme market valued at US$5.1 billion as of 2009 [255].
Immobilization of enzymes on the bacterial cell surface offers key advantages over the use of free enzyme in biocatalysis. First, attachment of enzymes to a solid matrix, such as the bacterial envelope, vastly increases their stability [7,256]; in one example, a surface-expressed organophosphorus hydrolase retained 100% catalytic efficiency for an astounding 45 days in culture [257]. Second, the production of enzyme on the cell surface obviates the need for protein purification, which can be laborious and costly. Third, once genetic engineering of a biocatalyst strain is complete, there is access to a nearly infinite supply of enzyme, requiring only culture in inexpensive growth medium—again greatly reducing the price of production. Enzyme display on the cell surface also has advantages over cytoplasmic expression, in that it allows use of substrates and generation of products that are impermeable to lipid membranes.
A wide array of enzymes have been expressed on the surface of bacteria, including oxidoreductases (e.g., cytochrome P450 enzymes [38,258]) and various hydrolases (e.g., glycosyltransferases [259], phosphatases [260], lipases [261], other esterases [262]) [13,33]. Organisms re-engineered with such enzymes have been used to degrade organic pollutants, generate fuels from renewable energy sources (e.g., plant biomass), and manufacture chemical compounds for pharmaceutical and industrial purposes [6].
4.3.5 Interactions Between Surface Proteins and Solutes
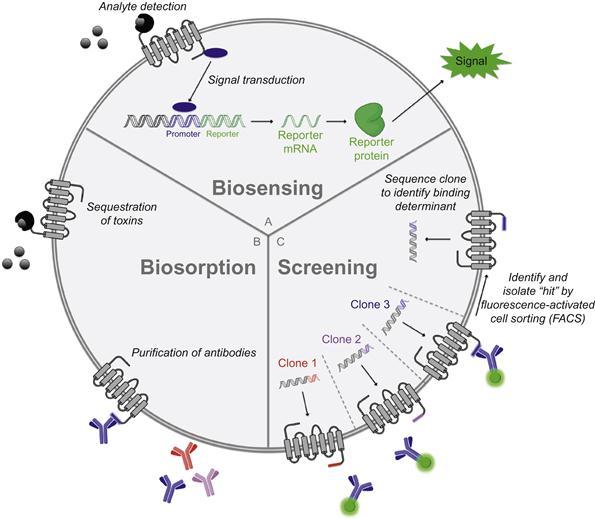
In this section, we discuss applications of bacterial surface display that are based on binding between a surface-bound protein and a molecule in solution, which may be a protein, a small molecule, or an element. A myriad of inventive uses for this binding interaction have been established, falling under the headings of biosensing, biosorption, and screening (Figure 4.3).
4.3.5.1 Biosensing: Binding for Purposes of Detection
Over the last two decades, researchers have endeavored to genetically engineered bacteria that serve as devices for detecting and quantifying molecules in solution. To this end, an entire class of “two-component biosensors” (Figure 4.3A) has been devised. These bacteria express (i) a genetic sensor that binds to an analyte and transduces the binding event into a dose-dependent change in activity of a promoter and (ii) a reporter gene driven by said promoter that produces a measurable signal (e.g., luciferase, green fluorescent protein, or β-galactosidase) [263]. In one notable example, E. coli was converted into a biosensor for arsenite-containing compounds through transformation with a plasmid containing the ars operon, which mediates bacterial resistance to such toxins. This regulatory circuit works as follows: in the absence of arsenite, the protein arsR represses the ars promoter, but upon binding the metalloid, it unbinds the promoter to permit expression of several genes that detoxify and extrude arsenite-containing compounds. Thus, by expressing a promoterless bacterial luciferase (luxAB) gene [264] downstream of the ars promoter in E. coli, a simple biological sensor for arsenite could be constructed. Remarkably, due to the sensitivity of arsR for its substrate and the signal amplification inherent in such operons, this re-engineered bacterial strain was capable of detecting arsenite at subattomolar concentrations—1000 times below the detection limits of conventional techniques [263,265]. Similarly designed biosensors have been used to measure concentrations of (i) various heavy metals and organic toxins in the setting of environmental monitoring [266], (ii) nutrients in the study of soil microbe metabolism [267], and (iii) analytes with potential clinical relevance such as quorum-sensing molecules [268] and antimicrobial drugs [12,269].
To date, two-component biosensors have been constructed only with intracellular sensing mechanisms. However, surface-engineered bacteria have been used as the basis for a novel ELISA-like quantitative immunoassay. In this approach, single chain variable fragments (scFvs) of antibody proteins are expressed on the surface of bacteria, analogous to the immobilization of antibodies on ELISA plates. Samples are incubated with these recombinant bacteria, and antibody-bound analyte is then separated from unbound analyte via centrifugation. Quantification of bound analyte is performed using horseradish peroxidase–antibody conjugates or other detection mechanisms. Along these lines, Chen et al. [270] have constructed scFv-expressing bacteria capable of measuring concentrations of the cardiac glycoside drug, digoxin, in the nanomolar range. The success of this approach indicates the enticing possibility that such diagnostic devices may complement or even replace traditional immunoassays like ELISA due to their cost efficiency, ability to self-regenerate, and relatively high sensitivity.
4.3.5.2 Biosorption: Binding for Purposes of Purification
Bacteria expressing peptide or protein ligands for soluble binding partners have been used as effective chromatographic media for purification of a range of molecules (Figure 4.3B). For example, biosorption of heavy metal toxins to metal-binding peptides on the surface of E. coli and Staphylococcus species has been used for environmental remediation of polluted water [5]. Meanwhile, expression of peptide epitopes has enabled isolation of single antibody clones from polyclonal serum samples (produced in immunized rabbits)—an approach that may serve as an alternative to hybridoma development for accessing monoclonal antibodies [271].
The scope of this technology, in fact, is likely to broaden further in light of a recent landmark study in protein engineering, which showed that novel proteins capable of binding to ligands of interest can be rationally designed in silico [272]. The authors of this study described a general computational method for designing small molecule-binding sites and applied it to create a protein capable of binding to the steroid digoxigenin. Through standard directed evolution methodology (described in Section 4.3.5.3), the group was able to obtain an optimized binder with picomolar affinity, and exquisite selectivity for digoxigenin over the related steroids digitoxigenin, progesterone, and β-estradiol. With this ability to program ligand specificity into synthetic proteins, it will, in theory, be possible to design biosensors and biosorbents for virtually any small molecule—a truly revolutionary advance.
4.3.5.3 Screening: Binding for Purposes of Identification
Similar to the more commonly used phage display technique [273], bacterial surface display enables the identification of proteins and peptides with desirable binding attributes by linking the identity of displayed proteins with their genetic sequence. To achieve this, large sets of heterogeneous bacterial clones are generated (by transforming bacteria with surface display vector libraries containing either cDNA or randomly mutated sequences), subjected to iterative rounds of screening and amplification, and sequenced to identify the surface protein(s) responsible for mediating binding interactions (Figure 4.3C). The use of bacteria rather than phage in such screens is advantageous because of the density of surface protein expression, which, at >100,000 per cell, dwarfs the few molecules displayed per phage [274], and greatly enhances the sensitivity of screens. Additional advantages are that bacteria, in contrast to phages, replicate autonomously and are of sufficient size to be detected by fluorescence-activated cell sorting (FACS) instruments, which allow quantitation of fluorescence of individual cells at rates upwards of 30,000 per second. This technology enables highly efficient identification of peptides and proteins with desired properties. For instance, through surface display of peptide libraries, the linear epitopes of antibodies have been rapidly determined through incubation of fluorescently labeled antibody with individual clones and FACS-mediated selection of antibody-bound bacteria (Figure 4.3C) [275,276]. Similar approaches have been taken to discover peptides with a range of targets including tumor cells [277,278], human signaling proteins [279,280], neural stem cells [281], viral proteins [282], etc. [9,283–287]. In addition, novel inhibitors for enzymes such as cathepsin G and trypsin have been culled from surface-displayed peptide libraries based on inhibitor enzyme binding affinities [288,289].
One particularly powerful application of combinatorial bacterial display is directed evolution, a process through which proteins are subjected to rounds of random mutagenesis and screening (often through FACS sorting) to identify “evolved” proteins with improved or unique function [290]. The use of bacteria is essential for directed evolution experiments because of their short generation time, amenability to genetic manipulation, and compatibility with sorting instruments.
Surface display-based directed evolution has found several applications thus far. In one example, antibodies with improved affinity were generated through anchored periplasmic expression (APEx) of antibody libraries in E. coli, permeabilization of the OM, incubation with fluorescent antigen, and FACS sorting of labeled cells [291–294]. Enzymes with novel activity have also been evolved using bacterial surface display [295–301]. Importantly, identification of hits in such screens differs from previously described methods, in that clones are not identified through interaction with fluorescent binding partners, but through attachment of fluorescent reaction products to the cell surface. For instance, in the development of an endopeptidase with novel substrate specificity, a library of bacterial clones displaying enzyme variants was incubated with a peptide substrate containing a BODIPY fluorophore on one side of the scissile bond and a quencher on the other. Clones expressing an enzyme capable of cleaving the bond would electrostatically capture an unquenched BODIPY-containing cleavage product on their membrane (by virtue of the product’s +3 overall charge), allowing for rapid detection and isolation by FACS [298].
4.4 Strategies for Re-Engineering Bacterial Surfaces with Heterologous Sugars
As described above, surface display of exogenous proteins is accomplished through fusion of passengers to extracellular carriers. Heterologous expression of glycans, meanwhile, is more challenging because their biosynthesis is not template directed. Thus, installation of nonnative sugars on the bacterial cell surface must be achieved indirectly, via recombinant expression of enzymes that mediate synthesis, attachment, and remodeling of oligosaccharides, or deletion of endogenous genes involved with these processes (see Box 4.2 for a detailed description of endogenous bacterial glycans).
Three basic strategies have been used in bacterial glycoengineering. In the first, the operon encoding the entire metabolic pathway responsible for synthesis of a given sugar is cloned and transferred to a carrier strain (Figure 4.4A). This approach has been used to express Pseudomonas aeruginosa capsular polysaccharide [18,307–309] and Shigella LPS [310–313] in a number of heterologous species. The second strategy involves site-specific N-linked glycosylation of target proteins (Figure 4.4A). This is accomplished through expression of (i) the genes required for biosynthesis of the nonnative sugar; (ii) PglB, an oligosaccharyltransferase cloned from Campylobacter jejuni that catalyzes attachment of glycans to an asparagine residue within the glycosylation sequon, D/EYNXS/T; and (iii) a modified surface protein containing the heptapeptide glycosylation sequon [15–17]. A third strategy entails glycan remodeling through addition and/or deletion of enzymes that modify cell surface oligosaccharides—most commonly LPS, as described in the following section (Figure 4.4A) [314,315].
4.5 Applications of Bacteria Expressing Heterologous Surface Sugars
4.5.1 Vaccines Against Infectious Disease
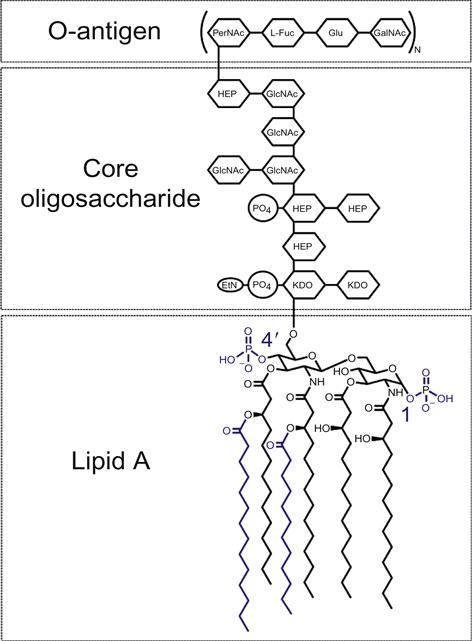
Glycoengineering methods have found a number of useful applications—the most successful, perhaps, being vaccine design. One effective vaccine platform involves the heterologous expression of carbohydrate antigens (such as glycoproteins [19], capsular polysaccharides [18,307–309,316–319], and LPS [311–313,320]) on the surface of attenuated vaccine strains. Analogously to the recombinant protein-expressing vaccines described above, the bacterial vector serves as a potent adjuvant stimulating adaptive immune responses against the pathogen-associated glycoantigen. The second major approach involves attenuation of pathogens through genetic modification of their LPS, allowing direct vaccination with avirulent pathogenic strains. Common molecular alterations to LPS, diagrammed in Figure 4.5, include addition of 4′-phosphate groups (through deletion of the phosphatase, LpxF) or of fatty acids to produce hexa-acetylated Lipid A (through expression of acyltransferases, LpxL, and LpxM) [314]. In contrast to the immunoevasive native glycoforms, re-engineered LPS variants potently activate toll-like receptor 4 (TLR4), leading to the elicitation of pro-inflammatory immune responses that mediate rapid clearance of the bacteria, and generation of adaptive immunity against the bacterial vector [315]. This method has been applied to construct attenuated vaccines for Shigella flexneri [324], Y. pestis [21,325,326], Salmonella spp. [327,328], Francisella tularensis [20,329], Leptospira interrogans [330], Neisseria meningitides [331], Moraxella catarrhalis [332], and K. pneumoniae [333].
4.5.2 Anti-Infective Probiotic Therapy
Surface glycoengineering has also been used to develop a potentially revolutionary class of anti-infective probiotics: avirulent bacterial strains expressing oligosaccharide receptors for the bacterial exotoxins that cause cholera, shigellosis, and traveller’s diarrhea, which together claim hundreds of thousands of lives per year worldwide [22,334–338]. The rationale for this approach is that exotoxins are sequestered by receptor decoys on probiotic strains in the gut lumen rather than binding to their targets on colonic epithelial cells, which induces diarrheal disease. Due to the density and multivalency of receptor expression on the surface of probiotic strains, exotoxins are intercepted in vivo with extraordinary efficiency. In one study, 1 mg of recombinant bacteria was shown to neutralize over 150 μg of shiga toxin, a potent exotoxin released by Shigella spp. that leads to dysentery. In mice, twice daily administration of the therapeutic strain provided 100% protection against otherwise fatal shigellosis, even in killed form. Thus, in addition to being cost effective (given that large-scale production can be achieved through simple fermentation), this therapeutic approach is highly effective and safe. Furthermore, these re-engineered probiotics apply no selective pressure for evolution of resistant pathogenic strains because they specifically intercept disease-causing toxins rather than killing the pathogen itself. Recently, similar principles have guided the development of a probiotic expressing the glycan receptor for adhesion proteins on uropathogenic strains of E. coli. Prophylactic treatment with the recombinant strain abrogated binding of the pathogen to the urinary epithelium, preventing development of urinary tract infection in mice [339]. Unfortunately, despite the great promise such glycoengineered probiotics hold for the treatment of infectious disease, they have been slowed in their clinical development due to apprehension surrounding the release of GMOs (see Box 4.1).
4.5.3 Glycoprotein Production
The final application of glycoengineering relates to production of recombinant glycoproteins. The importance of this process is highlighted by the fact that approximately 70% of therapeutically relevant proteins have saccharide modifications, including catabolic enzymes used for treating inborn errors of metabolism and erythropoietin, which is used in the treatment of chronic kidney disease [340]. These carbohydrate moieties affect several pharmacological properties of glycoproteins, including in vivo activity, half-life, proteolytic stability, and tissue targeting [341–346]. Glycoproteins are also used as vaccines; in fact, some of the most commonly used vaccines—including those against H. influenzae, N. meningitidis, and S. pneumoniae—consist of capsular polysaccharide–protein conjugates. Conventional methods for producing such compounds involve isolation of polysaccharide capsule from pathogens, purification of recombinant carrier proteins, chemical coupling of polysaccharides to carrier proteins, and purification of glycoconjugates—a typically low yielding and expensive process [15]. Alternatively, researchers have pursued total synthesis of glycans, but despite having led to elegant chemical developments [347,348], these approaches have been hampered by the extreme chemical complexity of oligosaccharides, deriving from the stereoisomeric diversity of monosaccharide subunits, numerous possible glycosidic linkages, and frequent branching.
Heterologous expression of N-linked glycosylation systems in E. coli, meanwhile, allows for scalable, inexpensive production of easily purifiable, homogeneous glycoconjugates, with site-specific installation of glycans on target proteins. While still in need of improvements (such as identification of bacterial oligosaccharyltransferases that can attach glycans with non-reducing sugars to acceptor proteins), bacterial glycotagging technology has the potential to revolutionize the production of therapeutic and immunomodulatory glycoproteins in the future [16,17].
4.6 Conclusion
Owing to the self-regenerating nature of the bacterial vector, its immunogenic properties, strong genotype–phenotype linkage, and amenability to genetic manipulation, bacterial surface display has proven an extraordinarily versatile and economical biotechnological system. Applications of these strategies in vaccinology have led to agents in clinical trials, while catalytic “biofactories” have played a key role in industrial and pharmaceutical synthesis, and bacteria-based screening technologies have driven forward basic research. However, we believe the surface engineering paradigm may be further improved by combining heterologous expression of protein and saccharides with the many complementary technologies elucidated above including computational protein engineering. Even further, genetic surface display can benefit from integration with an exciting set of methods emerging in chemical biology that allow installation of synthetic compounds on the cell surface (see Box 4.3) [364,365]. Together, these powerful techniques would impart incredible versatility to the surface engineering platform, transforming the cell envelope into a veritable tabula rasa, able to accommodate all manner of protein, sugar, and synthetic chemical in virtually any combination. With these tools, for instance, one may imagine constructing an anticancer therapeutic strain that expresses (i) a tumor antigen to trigger adaptive immune responses; (ii) an oligosaccharide targeting moiety to promote uptake by APCs and efficient antigen presentation (e.g., the DC-SIGN ligand, Sialyl-LewisX) [366–368]; and (iii) a chemotherapeutic payload that is released upon arrival in the tumor microenvironment [225]. Thus, through interdisciplinary efforts involving synthetic chemists, bacteriologists, and biomedical engineers, we imagine the development of a new class of “synthetic organisms”—chemically and genetically re-engineered bacteria that will serve as novel multimodal therapeutics, biotechnological devices, and tools to advance fundamental research.
Acknowledgment
We deeply appreciate the contributions of Dr. Thihan Padukkavidana toward the completion of this manuscript.