
Cell Membrane Biology and Juxtacrine Signal Conversion
Mark L. Tykocinski, Department of Pathology, Anatomy and Cell Biology, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA, USA
Our contemporary view of the cell membrane pictures a highly dynamic structure endowed with a rich functional repertoire. This chapter describes the evolution of our understanding of membrane structure and function, and introduces the notion of ‘cell membrane emergence’ as a robust conceptual framework for bringing systems perspectives into this field. The notion of multi-tiered emergent states is of particular interest with respect to membrane proteins, in terms of their surface distribution, dynamic structure, and inbound and outbound signaling potential. Focusing on membrane protein-driven, intercellular ‘juxtacrine’ signaling, a scientific journey is showcased in which a succession of novel fusion protein paints and signal converter proteins have been developed for cell surface engineering and, in particular, juxtacrine signal conversion and rewiring of cellular networks.
Keywords
cell membrane biology; cell membrane emergence; juxtacrine communication; juxtacrine signal conversion; protein paints; signal converter proteins; auto-signaling; auto-apoptosis; auto-apoptosis; cell network modulation; cell surface protein interactome; autoimmune therapy; immunoregulation; cancer therapy
1.1 Introduction
The cell membrane, bulwark to the exterior and gateway to the interior, has emerged as a most remarkable biological structure. Complex in composition, organization, and biogenesis, it now reveals itself to be a dynamic, plastic, and functionally rich entity. As an ever more sophisticated experimental toolkit is deployed to probe the cell membrane’s complexities, our view of it is being enriched by the capacity to tease things apart—from single cell down to single molecule analytics—coupled to a growing ability to start to piece things back together again through system perspectives.
We are still a long way off from a “grand unification” that brings together the various perspectives and narratives of the biochemists, the biophysicists, the cellular physiologists, and the systems and computational biologists into a comprehensive, structurally, and functionally integrated view of the cell membrane. Cell surface proteomics, lipidomics, and glycomics are still in their infancy, and sophisticated tools to dissect and visualize static and dynamic macromolecular interplay are only now coming into play. Yet, though the cell membrane’s intricacies are yet to be unraveled, it has uncannily proven itself to be amenable to artificial manipulation. Quite remarkably, reductionism seems to work time and again, as simplistic interventions and modulatory inputs lead to predictable and utilitarian outputs. Somehow membrane engineering works despite the experimentalist’s indulgence in tunnel vision and conscious ignorance of the wider membrane biology panorama in its spatiotemporal complexity. Further, beyond the engineering of cell membranes, there are early successes in artificially mimicking them and recapitulating their functions in diverse and creative ways.
This chapter offers some historical highlights and perspectives on membrane biology, as a prelude to showcasing one early cell membrane engineering story line, directed toward the use of paintable and signal converter proteins to modulate cell membrane protein repertoires and their juxtacrine and autocrine signaling properties. Thinking about cell membranes is framed within a membrane emergence paradigm, with membrane engineering cast as a way to perturb and craft emergent cell membrane states for defined ends.
1.2 Cell Membrane Biology—Early Milestones
The history of cell membrane biology features a progression from simple lipid bilayer to complex macromolecular mixture, and from there to the current paradigm of a physiochemical entity that is intricate in substructure and dynamically molded by intrinsic and extrinsic factors. In the process, the cell membrane’s primitive “boundary” role, minimally tasked to envelop the cell’s constituents, has given way over time to a far richer constellation of functions, both outward and inward looking.
Our understanding of membrane biology has unfolded through several experimental narratives that have played out in parallel over the past century. Each narrative encompasses a series of historical milestones. These narratives are highly interwoven and relate to membrane composition and structure, membrane model systems, membrane protein structure and function, membrane biogenesis, and emergent properties of membranes.
A seminal insight into membrane composition and structure came in 1925, when Gorter and Grendel [1] proposed a lipid bilayer structure for cell membranes. This was built upon an interesting historical sequence of studies on the interaction of oil films with water, starting with Benjamin Franklin and furthered by Raleigh and then Langmuir [2]. The Gorter–Grendel lipid bilayer model was bolstered by a simple experimental observation: when lipids are extracted from erythrocytes, the area they cover at an air–water interface is twice as large as the original surface area of the cells. It became clear that the membrane bilayer results from the aggregation behavior of amphipathic phospholipids, in accordance with the hydrophobic effect, with the polar ends of the lipid molecules oriented toward the aqueous phase and the hydrophobic hydrocarbon regions organizing to prevent contact with the aqueous phase. The product is a planar biomolecular film separating two aqueous compartments. Proteins entered the picture in 1935 when Danielli and Davison offered a membrane model incorporating globular proteins [3]. Their image of a trilamellar sandwich, with globular proteins coating both surfaces of a lipid bilayer, was prompted by their attempt to accommodate the surface hydrophilicity of globular proteins. Their model was reinforced by the discovery back then of protein beta-sheet structure, which seemed to preclude protein penetration of the lipid bilayer. It was not until 1972 that the Danielli–Davison model was finally supplanted. In an alternative fluid mosaic model of cell membranes, Singer and Nicolson explicitly postulated two distinct classes of membrane proteins, peripheral and integral. Peripheral membrane proteins were defined as those removable with salt treatment or pH changes, whereas integral membrane proteins require detergents for removal, and importantly, can even span the entire membrane [4]. Various experimental findings had laid the groundwork for the Singer–Nicolson model, including the first high-resolution electron micrograph of a biological membrane by Palade [5], subsequently framed by Robertson into a “unit membrane” paradigm [6,7]; the elegant demonstration by Frye and Edidin [8], via experimental cell–cell fusion and fluorescein-labeled protein tracking, that membrane proteins can diffuse laterally in cell membranes; and the confirmation, by Hladky and Haydon [9], of the unit channel structure for Gramicidin A peptide.
Experiments with artificial membranes catalyzed this evolving understanding of membrane structure. A seminal milestone was the 1962 report by Mueller and Rudin [10] of the first artificial lipid bilayer. Such membrane modeling was taken to the functional level in 1969, when Huang [11] demonstrated that unilamellar lipid vesicles reconstituted with membrane proteins could be used to study ion flux. Since single membrane vesicles can be readily produced in large quantities, this technical achievement revolutionized transport studies, allowing insights into the kinetics of many channels, pumps, and transporters without having to know their structure. Next came the monolayer-derived planar bilayers of Montal and Mueller in 1972, virtually solvent-free synthetic membranes which allowed for the study of intrinsic properties of ion channels [12]. This technical advance set the stage for studying how lipid composition and distribution within cell membranes affects the activities of membrane proteins embedded in bilayers. In 1979, Schindler [13] described vesicle-supported planar bilayers, as synthetic membranes that are completely solvent free. The armamentarium of model membrane systems has continued to grow, including categories such as liposomes, sonicated vesicles, large unilamellar vesicles, black lipid membranes, lipid monolayers, high-density lipoprotein particles (reviewed in Ref. [14]), and dendrimersomes [15].
Another narrative that has in parallel shaped the membrane biology story revolves around membrane protein structure. Glycophorin was the first integral membrane protein to be sequenced, back in 1975 [16]. That same year, Henderson and Unwin [17] reported the first electron microscopy-derived structure for a membrane protein, bacteriorhodopsin. Its remarkable structure, with seven transmembrane α-helices, supported the notion that α-helices are the secondary structure of choice for transmembrane proteins. The first X-ray high-resolution structure of a membrane protein, by Diesenhofer, Huber and Michel in 1985, reinforced the image of an α-helical transmembrane segment [18]. The β-barrel model for bacterial porin and the β-helix structure of Gramicidin A, which came later, came to be seen as exceptions. Yet another landmark was the high-resolution structure of a bacterial ion channel, solved in 1999 by MacKinnon, which linked protein backbone structure, as opposed to the expected amino acid residues, to ion selectivity [19].
Then there are the functional narratives, diverse and multifaceted. Back in 1855, Naegeli and Cramer ascribed to plant cell membranes an essential role in osmosis. In the last decade of the nineteenth century, Ernest Overton [20] framed the theory that “lipoid” membranes enclosing animal and plant cells control their osmotic properties and permeation of molecules, and posited fundamental membrane processes such as ion transport. A multitude of signaling insights and milestones have ensued, along with breakthroughs touching on diverse membrane functions that extend well beyond signaling.
1.3 Membrane Microdomains
The notion that there may be spatial separation of signaling reactions within membranes was first proposed for the cAMP system in 1981 [21]. Early on, the Singer–Nicolson fluid mosaic model stood in the way of the idea that cells might spatially and functionally compartmentalize signaling machinery, with skepticism over whether laterally diffusible molecules could be segregated effectively. However, this compartmentalization concept, instantiated in the paradigm of membrane microdomains, has by now entered the mainstream, taking the fluid mosaic model in a different direction. While proteins move laterally within membranes, membrane surfaces are in fact nonhomogeneous, instead displaying heterogeneity in substructure, molecular distribution, and inner and outer connections. Thus the “fluid mosaic” can be recast as a “mosaic of microdomains” [22].
Membrane microdomains have been defined as functional regions, of micron/submicron dimensions, that compartmentalize proteins, lipids, and signaling components into multimolecular assemblies [23,24]. Over the years, microdomains have been categorized in different ways, with the delineation of lipid rafts, tetraspanin webs (TEMs), and caveolae (reviewed in Ref. [25]). Lipid rafts, with an estimated size of 12–200 nm, showcase bilayer asymmetry with outer leaflets composed of primarily cholesterol that binds to glycosphingolipids and inner leaflets composed of saturated phospholipids [26,27]. TEMs are a distinct class of microdomains in which tetraspanins, each with a signature four transmembrane domain structure, serve as organizers of multimolecular complexes, organized through tetraspanin–tetraspanin interactions and selective associations with various other membrane proteins [28–30]. Caveolae are cholesterol-rich membrane invaginations comprising caveolin and cavin proteins, which themselves have significant functions. These various types of microdomains are not only dynamic in space and time, but they can also exchange molecular constituents amongst themselves [31–33].
Along with converging evidence for the actual physical existence of these evanescent structures [23,34,35], there have been early attempts to mathematically model them [36], keying in on relative rates of production, diffusion, and destruction of microdomain-localized signaling components. Such models attempt to factor in space and time, for instance, considering two defining spatiotemporal characteristics—the point at which the concentration of the activated signaling component is maximal and the concentration gradient down to the level in the surrounding milieu, with the gradient slope defining the boundaries of a given microdomain. With both parameters in constant flux, views of membrane microdomains are momentary snapshots, precluding simplistic steady-state assumptions. Factors underlying the formation and dissipation of these microdomains are complex and interdependent, and include cell shape, topology of the reaction network, diffusion coefficients, and intrinsic properties of the components involved.
The architecture of membrane microdomains, along with their mode of assembly, is starting to come into focus. This has been propelled by significant advances in the experimental tools available for tracking them, including the advent of super-resolution microscopy techniques, such as near-field scanning optical microscopy (NSOM), stimulated emission depletion (STED), structured illumination microscopy (SIM), total internal reflection fluorescence microscopy (TIRFM), Pointillism microscopy methods, and photoactivation localization microscopy coupled to pair correlation analysis, which are capable of nanoscale mapping of the cell surface landscape [37,38] (reviewed in Ref. [25]). These have been complemented by advances in single molecule imaging, such as fluorescence recovery after photobleaching-fluorescence lifetime imaging microscopy (FRET-FLIM) and multicolor single particle tracking (SPT), which provide insights into protein dynamics at the molecular level [39].
Formation of specialized membrane islands depends on an intricate array of protein–protein, lipid–protein, and lipid–lipid interactions. Beyond intramembranous molecular interplay, recent evidence points to a role for the cytoskeleton in creating membrane microdomain boundaries, with the intriguing implication of one particular integral membrane protein, CD317/tetherin, as a critical microdomain organizer. Unique structural features of CD317/tetherin, including its dual transmembrane and glycosyl-phosphatidylinositol (GPI) anchors and abilities to oligomerize and form disulfide-bonded parallel coiled coils, endow it with the potential to link integral membrane protein “pickets” to an actin cytoskeletal “fence,” at once delimiting lipid rafts and enabling their linkage into larger microdomains [22]. This provides an elegant protein structural underpinning to the actin cytoskeleton-based “picket-fence model” of microdomain organization, that proposes a hierarchical, three-tiered, mesoscale (larger than a nanometer, smaller than a micron)-domain architecture [40].
Much remains to be learned about the rules governing which surface proteins enter and exit the various kinds of microdomains. Typical lipid raft constituents, anchored to the outer leaflet, are GPI-anchored proteins [41]. Some proteins constitutively reside in rafts, whereas others are only transiently associated with them. Processes such as protein oligomerization can influence transit time, and coalescence of rafts impacts the dynamics. Special structural domains or motifs have been implicated in microdomain localization [42].
What emerges is a new image of the cell membrane, as a mosaic of structurally and functionally differentiated islands, with special abilities to connect to the cell interior and extracellular entities, all the while dissolving in and out. Out of this dynamism, discrete functionalities somehow emerge. Microdomains have been implicated in roles that go well beyond receptor signal acquisition. Outbound signals are empowered through microdomains as well. One of the more elegant microdomain stories has indeed come together around the “immunological synapse,” envisioning microdomain-based signaling elements on both the antigen-presenting cell (APC) signal sender and the T-cell signal receiver sides (reviewed in Ref. [25]). In addition to signaling, microdomains have been implicated in a variety of other aspects of cell dynamics and function such as membrane trafficking and regulated exocytosis. Membrane microdomains have also been implicated in pathogenic processes, the most straightforward being pathogen entry/egress and toxin entry. Human deficiencies in tetraspanin and other microdomain-associated proteins have emphasized the profound implications of perturbations in microdomain composition and biology (reviewed in Ref. [25]).
Looking beyond microdomain physiology and pathophysiology, one can begin to ponder theoretical opportunities to harness microdomain biology in the engineering of cell membranes for therapeutic purposes. Possibilities include purposeful perturbation of microdomain dynamics and selective alteration of microdomain-based protein and lipid repertoires. The therapeutic efficacy of currently available antibodies that are known to impact protein transit to microdomains hints at the future promise of microdomain targeting (reviewed in Ref. [25]).
1.4 Cell Membrane Emergence
It has been suggested that membrane microdomains, as locally restricted signaling reaction systems, constitute an emergent property of cells [36]. This notion can be generalized, beyond microdomains per se, to encompass the full array of membrane properties. Indeed, emergence offers a robust way to conceptually frame the panoply of structural and functional dimensions of cell membranes, as they play out in their spatiotemporal complexity. To capture this perspective, one could speak of cell membrane emergence. The phenomenon of emergence has been defined more generally as the way complex systems and patterns arise out of a multiplicity of relatively simple interactions. Emergence speaks to how novel and coherent structures, patterns, and properties arise during a process of self-organization in complex systems, with the emergent state being greater than the sum of its parts. Building upon this global concept, a cell membrane emergence paradigm would posit that membrane properties and states are malleable, in constant flux, and arise, sometimes in unpredictable ways, out of a dynamic interplay of a collection of intrinsic and extrinsic components. Further, the properties of cell membranes might reflect real-time “tuning” around quasi-steady emergent states, which can be shifted in incremental or quantum fashion by extrinsic perturbations, natural or artificial. Higher order complexity emerges, featuring spatial heterogeneity and flux over varying timescales.
At any moment in time, the physicochemical and functional properties of a cell membrane are dictated by two categories of drivers: (i) composition and structure, i.e., the component lipids and proteins and their organizational relationships, and (ii) influences extrinsic to the membrane proper, both intracellular (such as cytoskeleton and soluble cytoplasmic factors) and extracellular (such as matrix, signals arriving from a distance, and adjacent cells, viewed individually or as part of functionally integrated cellular networks). The process shaping a membrane’s emergent properties is inherently iterative, as membrane changes differentially affect the very drivers dictating its current state, for example, via alterations in cell–cell interactions that modify adjoining cells or in membrane trafficking phenomena that modify membrane flows to and from the cell interior.
Cell membrane emergence can be approached from two distinct angles—drivers of emergent membrane properties and the emergent membrane properties in-and-of themselves. Membrane proteins offer a tractable handle for latching onto membrane emergence phenomena. The emergent structural and functional properties of the membrane protein repertoire are dictated by multiple factors: (i) membrane protein composition (fashioned by endogenous protein production and membrane trafficking), (ii) membrane lipid composition, (iii) membrane protein dynamics (changes in protein structure, in turn affecting cytoplasmic, transmembrane, and extracellular portions of the respective proteins), (iv) membrane protein distribution (higher order oligomers, both homo and hetero, multiprotein complexes, and membrane microdomains), and (v) protein and nonprotein molecular factors, both intramembranous and extrinsic to the membrane (intra- and extracellular, e.g., extracellular matrix and juxtacrine, cell-based signals) that impact protein functionality within a given spatiotemporal context.
A promising future path for membrane biology can be intuited from two contemporary lines of investigation that each showcase just how elegant the intersection can be between membrane protein structural dynamics and cell membrane biology, with the two informing and playing off of each other. These two examples illustrate routes whereby cell membrane emergence manifests itself.
One line of investigation focuses on membrane fusion, and in particular, the intracellular machinery for fusing membranous organelles in the context of precisely timed release of neurotransmitters for synaptic transmission. A fundamental insight, emanating from the studies of Sudhof and Rothman, is that a thermodynamically spontaneous process of protein folding drives perturbation of the membrane bilayer and membrane fusion (reviewed in Ref. [43]). The beauty of this evolving story stems from its molecular detail, with SNARE proteins zipping up, and SM proteins organizing trans-SNARE complexes (i.e., SNAREpins) spatially and temporally. Beyond the informative conformational changes of these pivotal proteins, we are also learning about additional proteins that can actually control this process. Intriguingly, “grapple” proteins function contextually, and by their nature, can inhibit the process, activate it, or both depending upon the conditions [44]. This one mechanism has, in-and-of-itself, sweeping implications, as the orderly execution of membrane fusion goes well beyond neurocrine communication, impacting the exquisite compartmental organization of all cells, mechanisms of hormone release, and various other membrane fusion-based phenomena.
The interplay between protein dynamics and cell membrane biology has been showcased in another realm of investigation, this one centered around G protein-coupled receptors (GPCRs) (reviewed in Ref. [45]). GPCRs are highly dynamic and exist in a multitude of functionally distinct conformations. Insights into these conformations, and how ligands affect their energetics and rates of interconversion, have provided key clues into how GPCRs actually work and make it clear that their functional versatility is attributable, in no small part, to their structural plasticity. Thus, GPCRs exist as ensembles of discrete conformations with energetics that can be influenced by a range of factors (lipids, cytosolic signaling and regulatory proteins, ligands, pH, ions, oligomerization, and possibly transmembrane voltage gradients). Whereas in the membrane fusion setting, protein structural dynamics generate force, in the case of GPCRs, they drive conformational changes at the cytoplasmic ends of transmembrane segments, that in turn provide interaction interfaces for cytosolic proteins, including heterotrimeric G proteins, GPCR kinases and arrestins, as well as localization to specific signaling compartments.
The experimental enabler for connecting protein dynamics to emergent cellular architectures and functions has been the growing array of biophysical methods that can be deployed to study protein structure and dynamics. These include crystallography, nuclear magnetic resonance (NMR) spectroscopy, fluorescence spectroscopy, electron microscopy, and electron paramagnetic resonance (EPR) spectroscopy. Though ideally, protein kinetics should be studied in native cell membranes, methodological challenges remain, resulting in reliance upon simplified membrane model systems. Ultimately, a more complete picture of protein dynamics and function will demand an ability to monitor conformational changes in single receptor molecules as they function in a native cell membrane, with receptor dynamic measurements at timescales of milliseconds to seconds [45].
The field of membrane biology is now replete with hot topics, many of which could be framed within a cell membrane emergence paradigm. As a sampling:
• Lipid bilayer substructure and the process of membrane protein insertion [46,47]
• Bidirectional functional interplay between intrinsic lipids and membrane proteins, including transient lipid–protein complexes and inducible co-clustering [48–52]
• Membrane thickness, shape, curvature, pressure, tension, and fluidity as determinants of membrane function, including implications for fusion and fission of the lipid bilayer and positioning of membrane proteins [53–57]
• Role of cytoskeletal and other cytoplasmic and intramembranous proteins in molding membrane substructure and specialization [58–62]
• Supramolecular membrane protein cluster formation [63]
• Coordination between transmembrane and GPI-anchored proteins [64]
• Single molecule membrane biology [65]
• Quantitative cell surface proteomics, both global and membrane substructure specific [66–68]
• Membrane alterations as drivers and reporters of pathogenesis.
Things will get more complicated before they get simpler. Lipidomics has started to hint at the vast complexity of cellular lipids, and the same holds for glycomics and the surface glycoprotein and glycolipid repertoires. Furthermore, variability in membrane composition and structure among different cell types, along with associated microdomain architectures, have hardly been explored.
Back in 1962, Kauzman and Tanford theorized about the effect of hydrophobicity on protein folding and stability, which was a first step to frame thinking around the emergence of membrane properties. Clearly, things have come a long away since then. The stage is now set for designing membrane engineering strategies with complex, emergent membrane states as a conceptual framework. One can think of surface engineering driven emergence as an overlay on existing, physiologically driven emergent membrane states. From a functional perspective, two pivotal emergent membrane states are the potentials for receptor (inward signal flow) and juxtacrine (outward signal flow) signaling. Attention will now be turned to the latter, in detailing a line of investigation where the early dividends have been flexible cell surface engineering tools and novel therapeutics, and with the ultimate goal of modulating cellular networks now coming into sight.
1.5 Juxtacrine Signaling and Rewiring Cellular Networks
Juxtacrine signaling is a form of intercellular communication that depends on direct interaction of adjoining cells [69–72]. This cell contact-dependent mode of signaling, typically mediated by paired interactive proteins on apposed cells, is at the heart of diverse biological processes, in particular those requiring tight regulation and spatial control of stimulation of one cell by another (reviewed in Ref. [73]). The simplest juxtacrine links involve just a pair of interacting cells, for example, an APC interfacing with a T cell of the immune system. The triggering of T-cell receptors by APC-anchored major histocompatibility complex (MHC) antigenic peptide complexes represents a classic two-party juxtacrine signal encounter. Yet, even in this seemingly straightforward situation, greater complexity quickly arises as third-party cells enter the picture, along with an array of paracrine (soluble factor mediated) and more distant endocrine immunomodulatory signals. Thus, juxtacrine intercellular signaling is best thought of in a more encompassing way, as operating within cellular networks of varying complexity that frequently evolve in multistep fashion over varying timescales. Morphogenesis and developmental patterning exemplify how juxtacrine signals can cascade and effect phenotypic changes across a field of cells, with juxtacrine Delta-to-Notch serving as the classical paradigm [74–78].
Over the years, our laboratory has focused on strategies for engineering cell surfaces, with the express aim of modulating juxtacrine signaling potential. The ultimate goal is to alter, and even rewire, cellular networks by altering juxtacrine cues among the component cells. Interestingly, the concept of emergence also has relevance to such cellular network engineering, in a sense creating what amounts to a hierarchical series of emergent states that can continuously evolve. That is, cell surface engineering elicits emergent juxtacrine signaling arrays; the receiver cells manifest emergent phenotypes that influence other cells in the network; and the very network itself displays its own emergent properties. The “cellular choir” acquires its new voice. The emergence within engineered cells is a function of not only the particular molecules in play but also the particulars of the engineering method itself.
Emergent heterogeneity arising via juxtacrine signaling can be mathematically modeled [79]. It is not just a matter of how already networked cells are communicating among themselves but also how they embrace other cells outside of the immediate network. Advances in cytomics, systems analytics, and mathematical modeling should begin to enable the decoding of new cellular configurations and functionalities within such emergent cellular networks.
Much of juxtacrine signal engineering to date has revolved around tailoring the cell’s membrane protein repertoire, by either adding, deleting, or otherwise modifying selected proteins with defined intercellular (trans) signaling properties. This membrane protein-focused engineering can be accomplished by a variety of means [80–84]. Genetic approaches may deal with individual genes, gene groupings, or synthetic gene circuits [80] (reviewed in Ref. [69]). Inherent limitations of gene transfer for certain applications have prompted the addition of nongenetic strategies to the cell surface engineer’s toolbox, primarily ones that are protein transfer based. Mirroring synthetic cells, synthetic scaffold-based platforms, featuring juxtacrine signaling molecules detached from their endogenous cell backgrounds and immobilized on various matrices, are being developed as juxtacrine signaling surrogates [69,85].
Of note, juxtacrine signals are not restricted to surface glycoproteins. Platelet-activating factor (PAF), a biologically active phospholipid expressed on the surface of endothelial cells at inflammatory sites, mediates juxtacrine stimulation of leukocytes [86]. Another variant is molecular intersection between juxtacrine and paracrine signaling, with the membrane-anchored ligand virtually the same as, and often a precursor of, the secreted one [71]. The juxtacrine signaling molecule can even be an interloper, as an extraneous soluble factor that is immobilized via cell surface proteoglycan [87,88].
The design and deployment of synthetic cells with altered juxtacrine signaling potential is not without its complexities. Changes in juxtacrine signal arrays are being layered onto a backdrop of other regulatory inputs, encompassing bystander cells, extracellular matrix, and various paracrine, endocrine, and even autocrine signals. Additional complexity emanates from the interactive membrane proteins themselves, for instance, from their frequent capacity to signal bidirectionally. Another intriguing phenomenon that modulates juxtacrine signaling is “cis inhibition,” wherein ligand and receptor are also neighbors on the same cell, and one interferes with the juxtacrine trans activating potential of the other. This serves to modulate signaling dynamics in significant ways [89,90]. There are also peculiar phenomena that influence the signal array, such as the internalization of membrane-associated juxtacrine signals through transcytosis by an adjacent cell [91,92].
1.6 Protein Painting, Artificial Veto Cell Engineering
Our laboratory’s path toward cell surface engineering and juxtacrine signal modulation started back in the 1980s with the development of what we termed protein painting strategies. The application was immune cell engineering, and more specifically, the engineering of APC. At the time, we were building on a discovery made several years earlier in the field of immunoregulation—that APC, typically immune activating, can also function as immune-inhibiting “veto” cells. This was a twist on one of cellular immunology’s central dogmas, namely, that APC, such as dendritic cells, drive T-cell activation. This is mediated by surface-associated MHC·peptide antigen complexes and costimulators which provide APC-anchored first and second T-cell-activating juxtacrine signals, respectively. Thus, while the focus had traditionally been on the APC role as a T-cell activator, the early immunology literature was hinting at another APC functional dimension, casting it as a potential T-cell inhibitor. Thus emerged the concept of an immunoregulatory APC Trojan horse, a cell inhibiting a second cell that recognizes it [93,94].
Our seminal first step was to demonstrate that veto cells can be generated artificially through cell surface engineering. For expediency, we first invoked gene transfer as the means to neo-express surface proteins that can send inhibitory or pro-apoptotic juxtacrine signals to target T cells. Our group coined the term coinhibitor to characterize such surface-associated, trans-signaling inhibitory proteins, drawing a parallel between them and their well-established counterparts, the costimulators [95]. As defined, costimulators and coinhibitors alike mediate APC-to-T cell contact-dependent signaling, and operate alongside and coordinately with MHC·peptide antigen complexes to modulate antigen-specific T cells. In one case, there is T-cell activation, in the other inhibition. Having established that artificial veto cells can indeed be produced [82,95], we next sought to replicate this genetic feat via protein transfer. This led us to a series of technical advances in the protein transfer field, as we designed new classes of protein paints for cell surface engineering.
The first protein we invoked for juxtacrine coinhibition was the lymphoid surface molecule CD8. As it turned out, this classical cis-acting cytotoxic T-cell costimulator receptor could be co-opted to serve as a coinhibitor ligand, simply by artificially enforcing its expression on APC [96,97], thereby drawing upon its capacity to send inhibitory reverse signals through T-cell resident MHC class I, its cognate counter receptor [98]. This unusual choice of CD8 for coinhibition was prompted by our earlier demonstration, applying antisense RNA technology to T cells for the first time, that deletion of CD8 from alloantigen-presenting cloned CD8+ T cells abrogates their veto function [99]. Our description of CD8 as coinhibitor, along with our formulation of the artificial veto cell paradigm [82,96,99–101], was amply confirmed by other investigators [102–104], albeit occasionally recast under new names [105–108].
From CD8, we turned to a more classic inhibitory surface protein, this one with the capacity to trigger T-cell apoptosis. The Fas ligand (FasL, CD95L, CD178): Fas (CD95, TNFRSF6) juxtacrine signaling axis plays a key role in shaping T- and B-cell repertoires and maintaining peripheral tolerance [109]. Naturally expressed FasL expresses veto-like function [110]. Chen et al. [111] and others proceeded to show that enforced FasL expression could induce alloantigen-specific T-cell tolerance and clonal deletion in vivo [105,110,112–115].
In principle, protein transfer offers a number of advantages over gene transfer, including finer control of surface protein levels, simplification of surface protein co-expression, shorter half-lives of exogenous protein on modified cells, and ultimate translatability into therapeutic applications in the clinic. We therefore shifted from gene to protein transfer for APC surface engineering. The concept is simply to exogenously coat cell surfaces by painting proteins on them. Over the years, the palette of fusion protein paints has grown, evolving from ex vivo to in vivo paints.
The first fusion protein paints were GPI-modified derivatives of transmembrane proteins, produced via chimeric gene expression. GPI proteins are “paintable” by virtue of their amphiphilicity, since they (i) remain soluble as pseudo-micelles after depletion of solubilizing detergents, and can thus be added back to cells without lysing them; and (ii) spontaneously incorporate into cell membranes. Tykocinski et al. [116] and Caras et al. [117] originally identified the GPI modification signal sequence within decay-accelerating factor (DAF; CD55), a natural GPI-anchored protein. The next step was to convey GPI anchors to proteins of interest by appending the DAF signal sequence to their carboxyl termini. The juxtacrine trans-signaling functionality of artificial GPI proteins, once appended to cell surfaces, was amply validated [118–123], creating the means to generate protein paints at will.
GPI protein paints, however, were hard to scale up, given the need to purify them from complex membrane lysates of transfectants. We therefore developed an alternative class of protein paints that can be produced as soluble recombinant proteins and are thus more scalable. These consisted of two-component protein complexes in which protein·Fcγ1 derivatives are conjugated to chemically palmitated protein A, with both components produced efficiently as soluble recombinant proteins.
A series of immunobiological studies validated the functionality of both GPI protein paints and protein·Fcγ1:palmitated-protein A conjugate paints. Painting of peptide antigen-loaded MHC class I·GPI complexes sensitized cellular targets to cytotoxic T-cell-mediated destruction, with the ability to titrate optimal antigenic density [119]. Moving from first to second APC-to-T juxtacrine cell signal, costimulator·GPI derivatives could be used to engineer tumor cell surfaces and thereby enhance their immunogenicity [120,124,125]. Significantly, the ability to finely titrate costimulator·Fcγ1:palmitated-protein A conjugates onto APC surfaces via protein painting allowed us to uncover costimulator receptor activation thresholds [126]. Moreover, it enabled the generation of effective anti-cancer immunity by painting tumor cells in situ with a combination of four costimulator·Fcγ1:palmitated-protein A conjugates [127]. Coinhibitors, such as FasL, could also be painted, which allowed for studies of how responding T cells quantitatively integrate opposing (costimulator and coinhibitor) juxtacrine signals concurrently delivered by APC [111].
1.7 Trans Signal Conversion
This early ex vivo cell surface engineering using membrane-incorporable protein paints beckoned the development of next-generation fusion protein paints that could be injected systemically and would home to their cellular targets in vivo. We referred to these generically as signal converter proteins, or SCP, since, once arriving at the target cell surface, they convert juxtacrine signals. We proceeded to develop a series of distinct types of multifunction signal converters, featuring a spectrum of signal-converting functionalities. Each new SCP seems to offer the unexpected, whether by its unusual potency, multiplicity of cellular targets, or mechanisms of action not anticipated from the known functions of the chimeric protein’s components. Thus, SCP function can be framed in terms of emergence, beginning with the emergent states of the SCP-painted cell membrane and extending to the cellular targets of the converted juxtacrine signal and the cell network in which they are together embedded. Coupling a diverse and inherently multifunctional SCP repertoire to multicellular networks, one gets a tapestry of emergent cell states, matrices of cellular interaction, and ultimately, therapeutic outputs.
The following sections showcase a series of signal converter paradigms, which together highlight the robustness of this cell surface engineering modality. As the library of SCP has continued to grow, the original simple paradigm—conversion of juxtacrine signals between two interacting cells—has given way to more intricate ones. Converted juxtacrine signals can be redirected to third-party cells, and even to receptors on the very same cell, creating auto-signaling loops at the cell surface. A given SCP can in fact operate in more than one mode, perhaps converting signals between two cells in one context and redirecting them in another. In some instances, this functional multidimensionality allows for markedly different therapeutic applications for the same SCP.
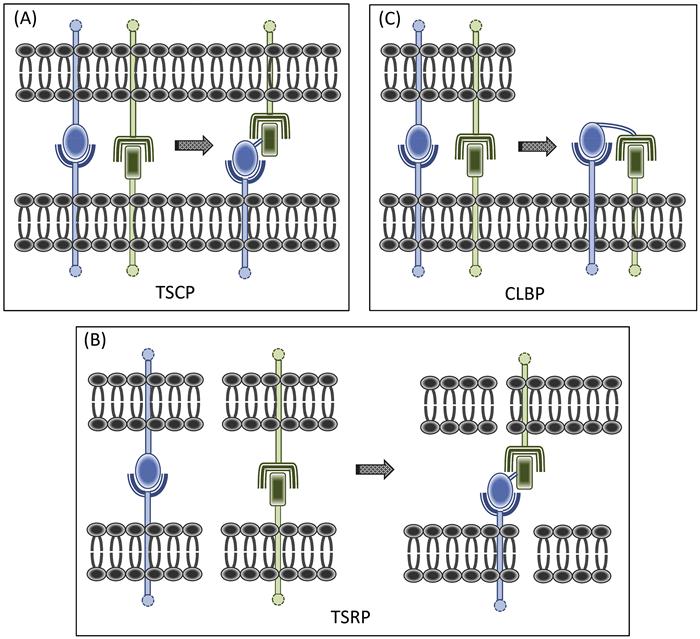
Trans signal converter proteins (TSCP) were developed first (Figure 1.1A). The concept was to combine within a single fusion protein homing and effector elements, together geared toward converting a juxtacrine signal between two cells. Judicious selection of the homing element can elicit yet additional functions that reinforce the effector element. Our paradigmatic artificial veto cell-generating TSCP, CTLA-4·FasL [128], illustrated this point well. The CTLA-4 (CD152) component serves a dual purpose, targeting the protein to APC surfaces bearing its counter receptors [B7-1 (CD80) and B7-2 (CD86)], and in the process, passively blocking their costimulator function. The FasL (CD95L) component sends a pro-apoptotic trans signal to its cognate Fas receptor (CD95), upregulated on activated T cells. Thus, this TSCP, with a costimulator receptor (CoSR)·coinhibitor (CoI) configuration, exchanges an active inhibitory (apoptosis-inducing) signal for a T-cell-activating costimulator one which is being passively blocked.
CTLA-4·FasL’s functional potential may go even further. First, given that increased B7 triggering of the CD28 costimulator receptor on T cells interferes with Fas-mediated apoptosis [129], CTLA-4·FasL, in mimicking the B7 costimulator blockade of CTLA-4·Ig [114,130], can potentiate its own pro-apoptotic, FasL-mediated activity. Second, by anchoring FasL to cell surfaces, CTLA-4·FasL leverages the higher efficacy of surface-associated FasL [131,132]. Third, as a two-faced, Janus-like entity, CTLA-4·FasL can be viewed from the opposite perspective, as anchoring CTLA-4 to activated T-cell surfaces. This creates the potential for two interesting CTLA-4-driven phenomena: reverse juxtacrine signaling, back through B7 ligand [133] and trans-endocytosis of B7 ligands away from APC surfaces [134,135].
CTLA-4·FasL exhibits striking potency in vitro, when compared to CTLA-4·Ig or soluble FasL, alone or in combination [128,136–138]. Interestingly, CTLA-4·FasL not only deletes activated T cells but also induces anergic proliferative hyporesponsiveness in them [136]. CTLA-4·FasL-mediated inhibition of allogeneic responses was documented in the in vivo context as well, with evidence for antigen-induced cell death of alloreactive cells [137]. The demonstration of CTLA-4·FasL in vivo efficacy has also been extended to animal models of autoimmune and alloimmune pathogenesis, including autoimmune diabetes [139] and cardiac and corneal transplantation [140,141].
An important mechanistic insight into CTLA-4·FasL function was the recognition of its ability to prevent the upregulation of anti-apoptotic c-FLIP which typically accompanies T-cell activation [142], serving to coordinately potentiate the fusion protein’s own FasL pro-apoptotic signal [138]. Interestingly, this upregulation of an anti-apoptotic cytoplasmic protein, along with early post-activation apoptosis, does not occur when the T cells are simply treated with the two fusion protein components, alone or in combination. Thus, this functional activity represents an emergent property that is dependent on the chimeric structure of the perturbing agent and illustrates yet another nexus between SCP and the concept of emergence.
For all the SCP described herein, chimerization provides functional dividends that cannot be recapitulated by simply delivering the fused components as a combination of separate molecular entities. New mechanisms and functions are emergent within fusion proteins. The growing list of chimerization dividends for SCP include abilities to: (i) elicit new, sometimes unexpected, functionalities (as exemplified by CTLA-4·FasL effect on c-FLIP); (ii) co-localize targeted molecules on cell membranes, sometimes generating macromolecular assemblies associated with enhanced function; (iii) deliver ligands to pre-selected cell types and organ sites in vivo, thereby concentrating therapeutic agent at the right location (as exemplified by Fn14 (fibroblast growth factor-inducible 14 kDa protein)·TRAIL (TNF-related apoptosis-inducing ligand) targeting TRAIL to inflamed TWEAK+ (TNF-related weak inducer of apoptosis) endothelium, vide infra); (iv) tether ligands to membranes, thereby eliciting higher order ligand function (as exemplified by the various FasL-containing SCP that yield membrane-anchored FasL); and (v) enhance functional avidity by enforcing multimerization (discussed later in this chapter).
1.8 Redirecting Juxtacrine Signals
TSRP were a logical next step, as we created a new class of signal converters with the capacity to redirect converted signals to third-party cells, outside of the original cellular dyad (Figure 1.1B). In contrast to the situation with TSCP, where the cell being modulated bears receptors for both the blocked and converted signals, for TSRP, these two receptors reside on different cells.
We developed Fn14·TRAIL as a paradigmatic TSRP, with inhibition of inflammation in mind. This fusion protein bridges two well-studied TNF superfamily signaling axes, TWEAK-to-Fn14 (CD266) and TRAIL (CD253)-to-TRAIL-R (TRAIL-R1, CD261; TRAIL-R2, CD262; TRAIL-R3, CD263; TRAIL-R4, CD264), which are linked to inflammation and immunoregulation, respectively [143,144]. TWEAK, a type II transmembrane protein, is expressed on a range of immune and nonimmune cell types [145] and is upregulated on activated monocyte/macrophages. TWEAK’s counter receptor, Fn14 (fibroblast growth factor-inducible 14 kDa protein), is also widely expressed on many cell types, with the notable exceptions of T and B cells, and is overexpressed in tissue injury. Membrane-anchored TWEAK, functioning as a juxtacrine signal [146], contributes to autoimmune inflammatory disorders not only by promoting pro-inflammatory cytokine production but also through its pro-angiogenic effect and ability to increase the permeability of the neurovascular unit in the context of autoimmune encephalomyelitis [147,148]. Indeed, antibody-mediated blocking of this juxtacrine signaling axis ameliorates autoimmune encephalomyelitis manifestations in rodent models [149]. TRAIL (CD253) too has been linked to autoimmunity, with the documented capacity of this pro-apoptotic type II juxtacrine signaling protein to inhibit disease manifestations in autoimmune animal models [112,150–154].
By connecting the TWEAK:Fn14 and TRAIL:TRAIL-R signaling axes via chimeric Fn14·TRAIL, we were introducing a protein that could at once bind to surface TWEAK at inflammatory sites (via its Fn14 component) and trigger inhibitory, pro-apoptotic TRAIL-R receptors on third-party (Fn14-negative) activated T cells in these locations (via its TRAIL ligand component) [155]. Thus, as a juxtacrine signal converter, Fn14·TRAIL replaces a pro-inflammatory signal with one that knocks out bystander-activated T cells. There are other potential dividends of this fusion protein: (i) Fn14·TRAIL in effect tethers TRAIL to the cell surface, leveraging the fact that surface-associated TRAIL is more functional than its soluble counterpart [156]; and (ii) as a TWEAK blocker, Fn14·TRAIL likely interferes with both innate and adaptive immunity and TWEAK-driven pro-inflammatory activities.
This constellation of pleiotropic Fn14·TRAIL functions all point to autoimmunity as an ideal therapeutic application. This proved to be the case, as we demonstrated that Fn14·TRAIL prevents auto-antigen-induced autoimmune encephalomyelitis in mice, with an efficacy that is substantially greater than that for either of its component parts (as Fn14·Ig and soluble TRAIL derivatives) when used in isolation or in combination [155]. This seminal study further suggested that Fn14·TRAIL, through TWEAK blockade, can reverse the enhanced permeability of the blood–brain barrier in diseased animals, providing yet another explanation for the high therapeutic efficacy of this fusion protein in the encephalomyelitis model.
Fn14·TRAIL, as well as the other SCP described in this chapter, are type I·II fusions, that is, a type I membrane protein at the amino terminus is linked to a type II membrane protein at the carboxyl terminus. This type I·II structural configuration results in a fusion protein that retains the extra-cytoplasmic ends of the respective constituent proteins, without constraining them. The added advantage is that one can draw upon members of both the immunoglobulin (predominantly type I) and TNF (predominantly type II) superfamilies, which are broadly represented in the cell membrane protein repertoire.
Fn14·TRAIL also features the interesting kind of stoichiometries that can fortuitously emerge when one couples trimeric TNF superfamily members to nontrimeric type I membrane proteins, which favor higher order multimer formation. Our three-dimensional modeling suggested that trimerization, enforced by trimeric TWEAK on the otherwise monomeric Fn14 component, yields a neo-Fn14 trimer that can in turn stabilize the active, trimeric state of the fused TRAIL component [155]. Similarly, our CTLA-4·FasL modeling suggested that interplay of dimeric CTLA-4 with trimeric FasL allows for a hexameric configuration, representing at once a dimer of FasL trimers and a trimer of CTLA-4 dimers [128]. This is functionally significant, since hexamerization empowers FasL and perhaps other TNF family members [131,157].
1.9 Creating Auto-Signaling Loops
For TSCP and TSRP, the receptor for the converted signal resides on second- and third-party cells, respectively. However, the receptor for the converted signal can just as well reside on the same cell that bears the surface signaling ligand that is being converted. This idea led us to yet another SCP category, designed to redirect converted signals back to the same cell, thereby generating auto-signaling loops. By judiciously choosing the component parts of such CLBP (Figure 1.1C), one can take advantage of forward and reverse signaling features of the fusion protein’s surface molecular targets to amplify the converted signal. Thus, the CLBP is being configured to double as both signal converter and dual co-signaling agent.
Autocrine signaling has traditionally been framed in terms of secreted soluble factors modulating the very cell that produced them. This type of signaling calls for clustering of like cells in order to achieve higher local concentrations of the secreted signal, and engenders a community effect that encourages identical cells to respond coordinately as a group. This contrasts with auto-signaling mediated by surface-associated molecules, where the demand for cellular congregation is mitigated, as each cell carries its own triggers in immediate proximity to the cognate receptors. Thus, CLBP, by co-opting juxtacrine signals and converting them into autocrine ones, create the opportunity for community independence. The cell can become a lone ranger.
Our first paradigmatic CLBP, CD40·FasL, was geared toward engendering autoinhibition of activated effector T cells [158]. The CD40 component binds to CD40L (CD154) neo-expressed on activated T cells, while the FasL (CD95L, CD178) component triggers the pro-apoptotic Fas receptor on these cells. The FasL:Fas signaling axis was an attractive choice for this fusion protein mediated, auto-apoptosis induction strategy, since data had already pointed to physiologic auto-apoptotic “suicide” signaling [159–162] between the Fas and FasL that are concurrently expressed side by side on activated T cells during an immune response [159,163,164]. CD40·FasL artificially recapitulates this scenario, by converting the outbound immune-activating signal emanating from CD40L into a pro-apoptotic FasL signal redirected back to a neighboring Fas receptor on the same cell.
The logic for pairing these particular proteins was several fold. First, cell selectivity was achieved by choosing CLBP components whose counter receptors (CD40L and Fas) are neo-expressed, and acquire full functionality, following T-cell activation [165,166]. Second, the two chosen signaling pathways had a fortuitous connection, in that reverse signaling through CD40L sensitizes activated T cells to Fas-mediated apoptosis [167]. Thus, CD40·FasL can function as a dual-triggering agent, driving a CD40L reverse signal that potentiates the Fas-mediated pro-apoptotic signal. Third, CD40·FasL tethers FasL to cell surfaces, thereby invoking surface-associated FasL’s higher efficacy [131,132]. This collection of mechanistic features exemplifies the kind of elegant design that is possible in pairing SCP components, where multiple mechanisms can be leveraged to achieve functional synergies and signal reinforcement.
We experimentally validated CD40·FasL’s cis autoinhibitory “loop-back” mechanism and immuomodulatory activity [158]. Evidence for the autocrine mechanism came from both soft agar clone formation and cell dilution analysis, showing that CD40·FasL inhibition does not require intercellular contact. Of note, the idea that cis loop-back signaling can be artificially elicited using fusion proteins dove-tailed with our prior finding that painting of CoS·Fcγ1:palmitated-protein A conjugates can yield auto-activating T cells [168].
1.10 SCP Therapeutic Flexibility
While the paradigmatic TSCP, TSRP, and CLBP fusion proteins were designed as distinct SCP categories, it turns out that the dividing lines are in reality blurred, with considerable functional overlap. That is, a fusion protein developed for redirecting juxtacrine signals to third-party cells can in a different cellular context trigger auto-signaling. In turn, this has served to extend the therapeutic reach of individual SCP.
A case example is CTLA-4·FasL. It was first designed to be an immunoregulatory TSCP for the treatment of autoimmunity and transplantation diseases. In this context, it introduces an APC-to-T cell coinhibition signal (FasL/CD95L) in place of a costimulation one (B7-1/CD80; B7-2/CD86). However, we demonstrated that this very same fusion protein can function as a CLBP in instances where its cognate counter receptors, CD80/86 and CD95L, are co-expressed on the same cells, e.g., on malignant B cells. Here, CTLA-4·FasL elicits auto-apoptosis [169]. A variation on this theme is seen with our other FasL-containing SCP, CD40·FasL. Developed originally for creating auto-apoptotic loops in activated autoimmune and alloimmune T cells, CD40·FasL was subsequently shown to have utility for eradicating transformed T cells expressing the cognate counter receptors, CD40L (CD154) and Fas (CD95). In this case, we showed that it draws mechanistically on the same CLBP mode of activity [169]. Of note, despite the focus on its auto-apoptotic activity, there is no reason why CD40·FasL can not in parallel bridge and drive Fas receptor-dependent apoptosis in neighboring cells.
Fn14·TRAIL is also multifaceted. Originally conceived of as a TSRP for interfering with a pro-inflammatory intercellular signal (TWEAK) and redirecting a converted inhibitory signal (TRAIL) toward autoimmune T cells, Fn14·TRAIL has since emerged as a cancer therapeutic. Neo-expression of this fusion protein’s counter receptors (TWEAK and TRAIL-R) on various solid tumor types, including those of breast, liver, pancreas, brain, and colon [170–174], has made it possible to reinvent Fn14·TRAIL as a CLBP to elicit tumor cell auto-apoptosis, as we have reported for hepatocellular carcinoma [175].
These examples illustrate how cancer cells, like activated T cells, can be prime targets for individual multifaceted SCP, and how therapeutic agents for autoimmunity can be reinvented as anticancer therapeutics. While we have keyed in on the auto-apoptotic mechanism in each instance, other supportive antitumor mechanisms come into play. For instance, as a TWEAK blocker, Fn14·TRAIL can interfere with various TWEAK functions linked to tumor progression, such as promotion of proliferation, invasion, angiogenesis, and inflammation. Fn14·TRAIL can also be cast as a next-generation soluble TRAIL derivative, more effective than the soluble TRAIL which has been under active development for cancer therapy [176]. The fusion protein’s higher efficacy stems from its cell-associating and multimerization properties. This cancer therapeutic paradigm—a fusion protein inducing tumor cell auto-apoptosis—is now being extended through the use of tumor cell-directed antibody TRAIL fusion proteins [177].
We introduced the idea of an “SCP function bar code” to capture the multiple functions of individual signal-converting fusion proteins [84]. The mechanistic profile of each SCP, coded in this fashion, evolves over time, as new functional capabilities surface. SCP functional richness stems in part from that of membrane proteins themselves, an example being Escher-like surface receptor–ligand functional duality. That is, many surface proteins, regardless of whether they were initially labeled as “receptors” or “ligands,” turn out to function as both, sending signals both inward and outward in signal-receiving and signal-sending modes, respectively. Molecular pairs associated with bidirectional signaling in the immune system include CD40L (CD154):CD40 (TNFRSF5), OX40L:OX40 (CD134), GITRL (TNFSF18):GITR (CD357; TNFRSF18), CD80/86:CTLA-4/CD28, and CD70:CD27 (TNFRSF7). The continuously growing list of “receiver” proteins that can send (forward signaling) and “sender” proteins that can receive (reverse signaling) seems to make bidirectional signaling a rule rather than an exception. The implication for SCP is that any given one can simultaneously function as an intercellular signal blocker, a reverse signaling ligand, and a competitive inhibitor of the receptor function.
1.11 The Cell Membrane Frontier
Introducing multifunctional SCP into multicellular networks in vivo creates a rich tableau of possibilities, some unexpected. Complexity in this setting emerges from a variety of factors, one of which is the promiscuous expression of many cognate counter receptors targeted by SCP. The dual binding capacity inhering in SCP contributes to target selectivity, but this is not absolute, with each SCP often able to touch more than one cellular network. This means that individual SCP can in principle impact more than one critical network node (for instance, via CLBP-mediated auto-apoptotic cell deletion) or intercellular link (for instance, via TSCP- or TSRP-mediated juxtacrine signal conversion) within one or more cellular networks. Further complexity arises from the constant flux in the in vivo cellular landscape, at the individual cell and cell population levels. Cell surface protein displays, along with the conformations and topologies of their component proteins, are dynamic, and SCP-elicited cell membrane emergence is layered onto this background flux.
Future SCP design will be guided by an ever-expanding knowledge base pertaining to cell surface protein repertoires, membrane protein dynamics, and functional cellular webs. Interesting leads will come from the growing compendium of single function antibody therapeutics [177]. A fully annotated cell surface protein interactome (CSPI), mapping surface protein interaction networks and elaborating on those of particular relevance to juxtacrine communication, would provide a useful framework for charting SCP possibilities. Such a database could readily build on ongoing initiatives to catalog the full human interactome and disease-associated protein–protein interactions [178,179], as well as more focused exercises, such as keying in on the independently folding modular domains of proteins and mapping their interactive potential in a “fragmentome” [180]. The very definition of protein–protein interaction will need to be revisited, as confounding concepts such as “protein stickiness” [181] and functional “promiscuity and evolvability” enabled by protein conformational dynamism [182] gain traction. The SCP designer will clearly have to navigate a large matrix of juxtacrine-associated protein pairs in selecting those that are most targetable. The SCP library of the future will not only more fully embrace this large set of protein pairings but will likely contain SCP classes that go well beyond those featured in this chapter.
Fundamental advances in membrane biology will translate into entirely new classes of membrane-targeted therapeutics. Membrane protein dynamics and membrane microdomain physiology will provide the substratum, going beyond cell surface protein expression profiling per se. In the case of SCP, one can envision targeting selected active conformations of membrane proteins or modulating the trafficking of selected proteins into and out of membrane microdomains. The concept of induced “spatial mutation,” purposefully changing the arrangement of membrane proteins on a cell surface, sets the stage for such therapeutic membrane modulation [183]. The tools for protein transfer will evolve in parallel. The possibilities include protein painting strategies that can introduce proteins into selected membrane subcompartments, or ones that can efficiently paint cells on the move within the blood circulation or other tissue compartments. The use of targeted liposomes for in vivo painting of circulating cells points to this type of next-generation cell surface engineering [184].
Ultimately, the cell membrane emergence paradigm may itself inform membrane-directed therapeutics design. Emergent membrane protein fingerprints, juxtacrine interactions, and cellular network states could eventually all become druggable. Deciphering emergent phenomena within the cell membrane could actually pave the way for simpler, smartly targeted interventions. By grappling with complexity, one can sometimes arrive at simplicity. There is now the prospect of bringing systems membrane biology together with therapeutics design, SCP among them, in compelling new ways.