
Lipid-Mediated Cell Surface Engineering
Jaina M. Patel, Vincent F. Vartabedian and Periasamy Selvaraj, Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA
Cell membranes provide not only a physical barrier between the extracellular and intracellular space, but they also contain many proteins, which serve as mediators of inside-out and outside-in signals essential for cell survival and functions. Therefore, these cell surfaces can be engineered to manipulate cellular functions. Lipid-mediated protein transfer allows for decoration of the cell surface by exogenous incorporation of proteins that are modified with hydrophobic tails into lipid bilayers. Protein transfer allows for controllable expression of functional protein on the periphery of cells in an easy, time-efficient manner, and can be performed in either a direct one-step incorporation method or an indirect two-step incorporation method. This technology has led to breakthroughs in designing tumor vaccines, targeted-drug delivery, enhancing function of killer immune cells, and engineering antigen-presenting cells.
Keywords
Glycosyl phosphatidylinositol (GPI) anchor; cholesterol anchors; palmitoylated protein A/G and tethered ligands; cancer vaccines
6.1 Introduction
The term “cell” was first coined in 1665, in Robert Hooke’s Micrographia [1]. Since then, breakthroughs in the field of cellular biology have given us not only a tremendous understanding of the cell and its components but also the ability to modify it through various means. In particular, our understanding of the cell membrane structure, its role in cellular function, molecules expressed on it, and their function have enabled us to engineer the cell surface for desired purposes.
In 1972, Singer and Nicholson [2] proposed the fluid mosaic model for cell membranes. The fluid mosaic model represents the cellular membrane as a fluid phospholipid bilayer decorated with proteins and oligosaccharides. This model also places an emphasis on the vast variety of structures that the phospholipid bilayer consists of, breaking the components down into three major categories: proteins, lipids, and oligosaccharides. Additionally, the lipids of the cell membrane exist as separate domains called lipid rafts. Lipid rafts are highly enriched with sphingolipids, complex glycosphingolipids, and cholesterol [3–6], and they play a vital role in cell membrane functions.
Cell membranes have been associated with a variety of cellular functions. Not only do they provide a barrier between interior cellular components and extracellular space, but cell membranes also are important for cell-to-cell communication and effector functions of the immune cell. Adhesion receptors found on the cell surface help to mediate these cellular communications as well as allow for targeted delivery of effector molecules. Additional cell surface receptors also bind to growth factors and cytokines allowing signals to be sent from outside the cell to inside further permitting communication between different cell types. These receptors selectively mediate communication between the extracellular and intracellular environment since not all molecules can pass easily through the cell membrane. These selective signals delivered by cell surface receptors determine cell survival, function, and in general, cell fate. Other cell surface expressed proteins, such as major histocompatibility complex class I (MHC I), also display internal peptide antigens on the cell surface facilitating immune surveillance by immune cells. This immune surveillance component of the cell membrane becomes important during infections, such as viral infections, as it is involved in detection and termination of sick and infected cells. Therefore, the cell membrane plays a vital role in determining the fate of the cell during disease state.
Consequently, cell membrane surfaces can be engineered to manipulate cell functions. Cell surface modifications serve as useful tools for a variety of applications such as targeting cells for drug delivery, activation, and amplification of immune cell responses, studying receptor functions, and development of cellular vaccines for tumor immunotherapy.
Genetic engineering techniques have been used to modify cell surfaces. These genetic engineering methods involve gene transfection in which the gene encoding the protein of interest is taken up within cells so that the cells translate and express the molecule of interest on the cell surface. Although this process is successful in leading to protein surface expression, many drawbacks exist: development of a culturable cell line or the availability of a large number of cells is needed; difficulty in transfecting many cell lines; the requirement of viral vectors; the amount of protein expressed by the cells is not controllable; and it is a time-consuming process. To counteract these drawbacks, surface engineering methods to exogenously place proteins on cell membranes have been developed. This phenomenon that decorates cell surfaces is known as “protein transfer” or “protein painting” [7].
One means of protein transfer, the focus of this chapter, involves attaching a lipid hydrophobic anchor to a protein and then exogenously adding the modified protein to the cells of interest. Such lipid-mediated surface engineering techniques allow for the anchored protein to be spontaneously incorporated into the cell membrane and into lipid rafts in particular [8], thus permitting the cell surface to be decorated by the protein in a controllable and time-efficient manner without any genetic modification.
Protein transfer to decorate cell surfaces can be divided into two main categories: the one-step and the two-step method. The one-step method consists of proteins that can naturally incorporate into lipid bilayers through hydrophobic chains (Figure 6.1), whereas the two-step protein transfer method requires incorporation of a scaffold onto the cell surface, which then in turn binds the protein of interest and thus allows for attachment to the cell surface (Figure 6.2). Thus, in the two-step protein transfer method, the protein of interest attaches indirectly to the cell surface. Of the one-step protein transfer methods, we will discuss cholesterol anchoring, palmitoylation modifications, glycosyl phosphatidylinositol (GPI)-anchoring, and use of hydrophobic sequences to anchor proteins on the cell membrane by protein transfer.
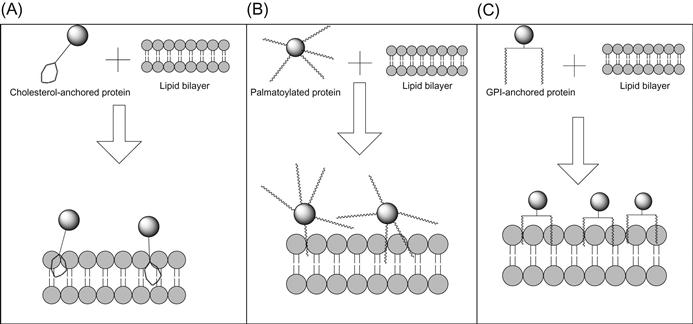
6.2 One-Step Protein Transfer
6.2.1 Protein Transfer Using Cholesterol-Modified Proteins
Cholesterol is an integral component of lipid rafts. With the depletion of cholesterol from cells, lipid raft domains disintegrate [6]. Cholesterol promotes lipid raft formation due to its ability to tightly pack with sphingolipids; however, other modified sterol structures that cannot tightly pack with sphingolipids disrupt lipid raft formations [9].
6.2.1.1 Exogenously Added Cholesterol Incorporates into Membranes
Cholesterol-tethering of biomolecules has been used to paint the surface of cells. Exchange between cholesterol derivatives found in lipid suspensions with those in erythrocyte ghost membranes have been detected upon incubation for 24 h [10,11]. In order to further investigate the “spur” red cell phenomenon in which red blood cells from patients with spur cell anemia uptake cholesterol found in the serum, Cooper et al. [12] attempted to artificially reproduce the environment. They found that when phospholipids were sonicated with cholesterol, the cholesterol-to-phospholipid (C/P) molar ratio affected uptake of cholesterol in red cells that were incubated with the mixture. When the C/P ratio was less than 1.0, cholesterol was depleted from the red cells and when the C/P ratio was equal to 1.0, no change in cholesterol content in red cells was seen. However, when the C/P ratio was greater than 1.0, cholesterol was deposited into normal human red cell membranes and the spur cell phenomenon was detected [10–12]. As the C/P ratio was increased, increased incorporation of cholesterol in red cells was also detected in a dose-dependent manner over 24 h [12]. These results show that cholesterol can be exogenously added to incorporate into cell membranes.
6.2.1.2 Cholesterol-Tethering of Biomolecules to Membrane Surfaces
Due to the ease with which exogenously added cholesterol can incorporate into cell membranes, a method of membrane painting to modify the cell surface with biomolecules was established (Figure 6.1A). This method involves the use of cholesterol derivatives, such as N-(3β)-cholesterylglycine, that are tethered to biomolecules [13]. The addition of amino groups on cholesterol derivatives that decrease the cholesterol–phospholipid affinity in the donor membranes leads to enhanced incorporation of the cholesterol derivative in the accepting cell membrane vesicles [14]. Conjugation of a biomolecule, such as biotin or epitopes for anti-HA or anti-FLAG antibodies, to the cholesterol derivatives creates an amphiphilic complex. The amphiphilic nature allows for insertion of the cholesterol derivative of the biomolecule into lipid rafts, whereas the polar segment of the biomolecule remains exposed on the cell surface. Upon incubation of the cholesterol-tethered molecules for 1 h at 37°C in RPMI medium with 10% fetal bovine serum (FBS), stable dose-dependent incorporation is detected within lipid rafts of cellular membranes [13].
6.2.1.3 Application of Cholesterol-Tethering to Create Synthetic Receptors
Modification of cell surfaces by cholesterol-tethering of biomolecules allows for delivery of drugs or antitumor agents into target cells. This delivery system occurs when a cholesterol-tethered receptor bound to its ligand is internalized. Beforehand, a cholesterol derivative is tethered to the biomolecule receptor, which is then allowed to incorporate into lipid rafts of cells for 1 h. When the surrogate receptor (SR)-incorporated cells are incubated with ligand, nearly 100% of the cells bind to the ligand in a matter of 10 min. Upon receptor–ligand binding, internalization of the receptor–ligand complex was initiated, and as the concentration of SR added to the cells increased, the amount of internalization also increased upon ligand addition in a dose-dependent manner [13]. Furthermore, Hussey et al. [15] showed that internalization occurred partially within 10 min of incubation and completely within 4 h of incubation through a clathrin-mediated endocytosis pathway.
The length of the tethering linker between the cholesterol derivative and the biomolecule is imperative [16]. A longer linker allows for full insertion of the cholesterol derivative into the membrane, whereas the shorter linker prevents this. An 11-atom linker resulted in membrane insertion of a cholesterol derivative bound to biotin that was capable of internalizing its ligand, streptavidin, within 10 min [16].
Using this approach of creating synthetic cell surface receptors, Martin and Peterson [13] showed that tethering an HA epitope to a cholesterol derivative allowed for surface expression of the epitope on Jurkat cells. An anti-HA antibody complexed with a protein A-β-galactosidase conjugate bound the surface expressed HA epitope resulting in engulfment of the protein A-β-galactosidase complex. Furthermore, when the substrate to β-galactosidase was added to these Jurkat cells, fluorescence was detected intracellularly within 75% of the cells. These experiments suggest that not only can cholesterol-derived SRs be used to modify the cell surface, but also the added protein remains functionally active even when endocytosed within the cell. Similar binding and internalization studies were conducted with cholesterol-derived SRs bound to either FLAG epitopes or streptavidin ligands, such as biotin [13,16].
Cholesterol-derived SRs can be added to any cells that contain lipid rafts. Many multidrug-resistant tumors have been shown to produce increased levels of lipid raft components [16]. Therefore, the addition of cholesterol-derived SRs to these multidrug-resistant cancer cells via intratumoral injections should readily lead to the uptake and expression of the receptor on the tumor cell surfaces that contain increased lipid raft components. In this manner, many drugs or toxins can be preferentially targeted to these cancer cells as opposed to neighboring healthy cells that have fewer lipid raft components in comparison.
6.2.1.4 Advantages/Disadvantages of Cholesterol-Tethering
Cholesterol-tethering of biomolecules onto cell membrane surfaces provides for a simple method to express proteins on cell surfaces due to the ease with which cholesterol and cholesterol derivatives incorporate into the cell membranes. Since lipid rafts are commonly found on cells, albeit at different levels, incorporation of cholesterol-tethered biomolecules could occur on almost any cell surface. Through the production of SRs, this method can also lead to the engulfment of high molecular weight proteins, such as enzymes like A-β-galactosidase, toxins, or drugs, that remain functionally active within the cells, allowing for cell targeting via this mechanism [13,16].
However, the production of these cholesterol-tethered biomolecules is not an easy task; it involves many chemical additions and the linkage between the biomolecule and cholesterol derivative is vital for incorporation and ligand binding. Moreover, there is a possibility that lipids present in circulation may bind to these administered conjugates and block incorporation onto the defined target cells.
6.2.2 Surface Engineering with Palmitoylated Proteins
Palmitoylation is a process that involves the lipid modification of proteins in which a hydrophobic fatty acid palmitate group is added covalently to the protein of choice [17]. In the human body, palmitoyl groups are commonly found attached covalently to neurologic proteins and also play key roles in signal transduction of G protein and G protein receptors [18]. The unique reversibility of palmitoylation allows this process to play a role in not only the activation and deactivation of proteins [17,18], but also in protein localization to membranes and in interactions with other proteins [17]. In addition to its importance among biologic systems, palmitoylation is also gaining relevance as a protein-anchoring technique used for protein transfer, engineering, and vaccine creation.
In the body, palmitoylation often occurs in the cytoplasm after another fatty acid, such as a myristoyl group, has already been added [17]. However, procedures to modify proteins with palmitate in vitro also have been established. It has been shown that auto-palmitoylation of G proteins can be performed stoichiometrically in vitro via incubation with palmitoyl-CoA [19]. An additional preparation method involves using the reaction between the N-hydroxysuccinimide ester of palmitic acid with the protein of interest, forming a palmitate-derivatized protein [20]. Another commonly used method involves palmitoylation with palmitoyl chloride [21]. Enhancing the hydrophobicity of proteins by the addition of fatty acid palmitate groups allows for insertion of the exogenously added protein onto cell membranes (Figure 6.1B). The ability to create a more hydrophobic protein easily by palmitoylation in vitro makes palmitoylation an incredibly appealing technique for modifying cell surfaces by protein transfer.
Recently, human IL-2, a cytokine commonly used in anticancer therapy, was palmitoylated and protein transferred to incorporate onto cytotoxic T cells (CTLs) [22]. These protein-transferred CTLs were then injected into mice along with EG7-OVA lymphoma cells. Mice that received protein-transferred CTLs had decreased tumor growth when compared to mice that received unmodified CTLs. Furthermore, intratumoral injection of palmitoylated-IL-2 in mice with melanoma led to increased overall survival, decreased metastasis to the lungs and increased levels of splenic and tumor infiltrating IFN-γ producing T cells compared to mice that received unmodified IL-2 intratumorally [22]. Therefore, the use of palmitoylated cytokines, such as IL-2, has the clinical potential to be used as an antitumor therapy.
6.2.2.1 Palmitolyation to Create Surrogate Cell Surface Receptors
Originally, artificial receptors were incorporated onto B cells by palmitoylating antibodies. Peacock et al. [20] showed that antibodies can be palmitoylated and subsequently inserted into a cell membrane. These membrane-bound antibodies can act as receptors, and upon binding to target antigen can induce an immune response by delivering signals to the decorated cells. In the study, protein 315, a secreted antibody against dinitrophenyl (DNP) lacking the Fc region, was palmitoylated before being incubated with murine splenocytes at 37°C. The palmitoylated antibody spontaneously incorporated onto splenocyte cell membranes in cell media, specifically via the palmitate groups since nonpalmitoylated proteins did not incorporate. Incorporation of the palmitoylated antibody on cell membranes was concentration and time dependent, and the incorporated palmitoylated antibody was still able to bind the DNP antigen and induce a polyclonal antibody response specific to DNP [20]. However, the degree of palmitoylation was also important for functional antigen binding. Increasing palmitoylation led to decreased ability of the incorporated antibody to bind antigen [20]. Furthermore, through fluorescence photobleaching recovery (FPR), palmitoylated-antibody receptors that incorporated exogenously onto B cells were found to have similar membrane properties as surface immunoglobulin (Ig) expressed naturally on B cells [23]. Therefore, exogenously decorating B cells with palmitoylated antibodies could provide insight into how surface antibodies are able to exhibit an antigen-dependent immune response.
This antibody-anchoring technology was further developed to create “SRs” on the cell surface. The establishment of SRs allows for the creation of “custom-designed killer cells” in which palmitoylated antibodies incorporated onto cell membranes provide specificity for target cells [24]. Either palmitoylated F(ab′)2 against mouse Ig or intact anti-TNP antibody was incorporated onto the membrane of nylon wool nonadherent spleen cells from mice. The antimouse Ig decorated cells were then incubated with syngeneic B cells stimulated with LPS, while the spleen cells decorated with anti-TNP antibody were incubated with TNP-modified EL4 T-lymphoma cells. Both sets of decorated cells demonstrated the ability to lyse their targets, while their undecorated counterparts did not. Furthermore, neither set of decorated cells exhibited the ability to lyse cells not displaying the antigen specific for their SR, demonstrating the specificity of the SR. Of particular interest, this SR-mediated cellular cytotoxicity (SRMCC) occurred even when the SR did not have an Fc region, suggesting that SRMCC is independent of Fc receptor interactions [25]. Although SRMCC was not detected when NK cells were depleted by anti-asialo GM1, SRMCC could induce lysis of NK-resistant cells. This suggests that NK cells, while not the sole effector cells, play a key role in SRMCC [24]. Thus, incorporation of palmitoylated SRs onto cells provides a means to engineer killer cell surfaces to express receptors that can specifically target cells expressing particular ligands.
6.2.3 Surface Engineering with GPI-Anchored Proteins
Proteins are expressed on the membrane of a cell through multiple mechanisms. Aside from commonly found transmembrane anchored proteins, lipid anchored proteins also exist. Lipid anchoring of proteins via glycophosphatidyl inositol (GPI) is predicted to be found in 460 proteins [26]. The GPI-anchor which anchors proteins to the outer leaflet of the cell membrane does not span the entire lipid bilayer as transmembrane proteins do [3]. This anchor consists of two or three hydrophobic fatty acid chains that insert into the outer leaflet of the lipid bilayer exposing the anchored proteins to the outside environment. The core structure of the GPI-anchor is conserved in eukaryotes and consists of a phosphoethanolamine group, followed by a glycan region, followed by a phospholipid tail. The phosphoethanolamine group is covalently attached to the C-terminal end of the GPI-anchored protein (GPI-AP) by an amide bond. The conserved GPI backbone structure is “EtN-P-Man-Man-(EtN-P)Man-GlcN-PI,” where EtN is ethanolamine, P is phosphate, Man is mannose, GlcN is glucosamine, and PI is phosphatidylinositol. The glycan region can vary between species and proteins via side chain modifications; however, the core backbone is conserved [27,28]. Inositol attaches the glycan region to the phospholipid tail, which consists of two long fatty acid chains that are either saturated or unsaturated, and vary in length. This phospholipid tail is responsible for inserting into the lipid bilayer and anchoring the GPI-AP to the cell surface. In addition to serving as a membrane anchor, other functions of the GPI-anchor include signal transduction, membrane trafficking, and targeting proteins to lipid rafts [27].
The discovery and purification of the enzyme phosphatidylinositol phospholipase C (PI-PLC) from the bacteria, Bacillus cereus, in 1976 [29] further led to the discovery of the GPI-anchor as a means for membrane-anchoring proteins. PI-PLC treatment led to the release of many proteins from tissues, thus suggesting that these proteins are anchored via a phosphatidylinositol group. Moreover, phase separation with Triton X-114 (TX-114) can be used to characterize GPI-APs [30]. At 0°C, the nonionic detergent TX-114 forms a homogenous mixture with GPI-APs and an aqueous solution; however at 37°C, TX-114 separates away from the aqueous phase. GPI-APs are found in the TX-114 phase, whereas other transmembrane and soluble proteins are found in the aqueous phase; therefore, TX-114-mediated phase separation also verifies the presence of a GPI-anchor [30]. Upon further characterization, the structure of the GPI-anchor found in many GPI-APs became evident. However, some GPI-APs are resistant to PI-PLC and phospholipase D cleavage. These GPI-APs contain an extra fatty acid chain (palmitate) attached to the inositol ring, thus they have a total of three fatty acid chains anchoring the protein to the cell membrane [31].
6.2.3.1 Expression of Proteins on the Cell Surface by Protein Transfer of GPI-APs
Purified GPI-APs contain the unique property of being able to reincorporate into cell membranes and still retain their biological activity. In 1984, Medof et al. [32] showed that the purified GPI-AP, DAF, was able to reincorporate into erythrocyte membranes by simply incubating purified DAF with erythrocytes. This reconstitution of DAF prevented complement protein C4b2a activity on the erythrocytes in a DAF dose-dependent manner. Furthermore, in the acquired disorder paroxysmal nocturnal hemoglobinuria (PNH), the absence of DAF in erythrocytes from PNH patients leads the erythrocytes to be susceptible to complement-induced lysis. However, this lysis is not detected when exogenous DAF is reconstituted back into erythrocyte membranes [33]. Additionally, in PNH patients, another complement regulatory protein, homologous restriction factor (HRF), later known as CD59, is also absent from erythrocyte membranes. Incubation of purified radiolabeled HRF with erythrocytes led to insertion of HRF into erythrocyte membranes and the inserted HRF remained biologically functional in preventing complement lysis by C5b-9 in vitro [34–38]. Oligodendrocytes that also lacked CD59 became resistant to complement-induced lysis after exogenously incorporating purified CD59 into their membranes as well [39]. Another complement regulatory protein which behaved similarly to DAF and CD59 is C8-binding protein (C8bp). In a comparable manner, C8bp, which is also not found in erythrocytes obtained from PNH patients, can be inserted into erythrocyte membranes exogenously after purification of the protein and again inhibit complement-induced lysis of erythrocytes [40].
Additionally, adhesion molecules such as lymphocyte function-associated antigen 3 (LFA-3) are also found to be deficient on erythrocytes obtained from PNH patients [41]. LFA-3 is expressed as a GPI-anchored form in erythrocytes, whereas nucleated cells express both GPI-anchored forms and polypeptide anchored forms [42]. Consequently, partial deficiency of LFA-3 was observed in nucleated cells from PNH patients. The interaction between LFA-3 on erythrocytes and its receptor CD2 on T cells induces formation of T cell rosettes [43]; however, erythrocytes from PNH patients did not form rosettes upon incubation with CD2 expressing Jurkat cells. Interestingly, when exogenous purified GPI-anchored LFA-3 was added back to erythrocytes from PNH patients in vitro, rosettes were formed, suggesting that protein transfer of exogenous LFA-3 leads to stable as well as functional LFA-3 expression on the cell surface [41]. Also, exogenous incorporation of purified CD16B (Fcγ receptor III), another naturally occurring GPI-AP, onto both a human T cell leukemia and B lymphoblastoid cell lines in vitro demonstrated that protein transferred CD16B on nucleated cells can still retain its function and mediate endocytosis of its ligand [44]. Another GPI-AP, Thy-1, was able to exogenously incorporate onto murine Thy-1-negative cells in vitro. After this protein transfer, staining of the exogenously incorporated Thy-1 was similar to staining patterns of endogenously expressed Thy-1 and the lateral mobility of both endogenously and exogenously inserted Thy-1 was similar as determined by fluorescence recovery after photobleaching (FRAP) [45]. Furthermore, even nonmammalian GPI-APs, such as the membrane form of variant surface glycoprotein (mfVSG) expressed on the parasite Trypanosoma brucei, can also be purified and exogenously incorporated onto cell surfaces with similar lateral mobility as endogenously expressed mfVSG [46]. However, PI-PLC treatment of purified GPI-APs decreases the insertion levels of these proteins onto cell membranes suggesting that protein transfer is due to the presence of the GPI-anchor [36,41,45]. All of the above examples show that purified GPI-APs can be incorporated via the GPI-anchor into cell membranes by simply exogenously adding the purified GPI-APs and that after incorporation onto cell membranes, the biological function and lateral mobility of the protein remains similar to that of the constitutively expressed protein counterpart.
6.2.3.2 Characteristics of GPI-AP Protein Transfer
Protein transfer of GPI-APs onto cell membranes (Figure 6.1C) occurs in a concentration-dependent manner. As higher concentrations of GPI-APs are added to the membranes, increased incorporation occurs [32,44,47]. However, a saturation point is expected to occur in which all GPI-AP sites on the membrane will be occupied and no more GPI-APs can incorporate by protein transfer without disrupting the integrity of the membranes. GPI-anchor-mediated protein transfer also results in surface expression on 100% of the cells incubated, as shown by flow cytometry [48]. Furthermore, GPI-anchor-mediated protein transfer is time dependent. As the time for protein transfer is increased, increased expression of GPI-APs on membrane surfaces occurs. Incorporation was detected in as little as 20 min postincubation of GPI-APs with membranes and incorporation levels increased with incubation time for up to at least 4 h [32,44,47]. Medof et al. [32] noticed that incorporation of radiolabeled DAF into erythrocyte membranes occurred best at 37°C followed by 20°C with no detectable DAF incorporation occurring at 0°C. Poloso et al. [47] detected a similar phenomenon in which protein transfer occurs best on cell membrane vesicles at 37°C followed by 25°C followed by 4°C. These observations suggest the necessity of membrane mobility for the incorporation of GPI-APs. Plasma membranes are most fluid at 37°C compared to lower temperatures; therefore, the dynamics between GPI-AP micelles and the lipid bilayer occurs more readily at 37°C [49].
Optimum incorporation also occurs without the presence of certain serum proteins. Incorporation of GPI-APs is inhibited by the presence of human serum lipoproteins, such as HDL and LDL, and fatty-acid-binding proteins, such as bovine serum albumin (BSA) and orosomucoid [32,41,44]. High concentrations of ovalbumin on the other hand do not affect incorporation of GPI-APs onto cell membranes [32,44]. Once incorporated onto the cell surface by protein transfer, GPI-APs remain stably integrated into the cell membrane of erythrocytes and cannot be removed by washing even with high salt concentrations [32]. After protein transfer, GPI-APs can however be removed by solubilization with detergents, such as NP-40. However, incorporation onto live or irradiated nucleated cells leads to loss of incorporated proteins under culture conditions [48].
Protein transfer leads to the incorporation of exogenously added GPI-APs into lipid rafts of cell membranes [8,50]. After Triton X-100 lysis of cells that have been protein transferred with GPI-APs, the GPI-APs are detected in detergent insoluble microdomains as are other lipid raft specific proteins [50]. Furthermore, enhanced incorporation of green fluorescent protein (GFP)-labeled GPI-anchors into lipid rafts was also associated with the presence of lipid raft-associated molecules (cholesterol, sphingomyelin, and dipalmitoyl-phosphatidylethanolamine) further proving that protein-transferred GPI-APs associate with lipid raft structures [8]. Upon initial incorporation of GPI-APs onto cell membranes by protein transfer, the GPI-APs are widely distributed throughout the cell membrane as observed by confocal microscopy [51]. However, hours after incorporation, the GPI-APs localize into lipid raft microdomains as detected by confocal microscopy or by observing association of GPI-APs with lipid raft domains after lysis with Triton X-100 [50,51].
Similar to the erythrocyte-incorporated proteins described earlier, these protein-transferred GPI-APs expressed on nucleated cells and cell membranes retain their functional biological activity in comparison to endogenously expressed forms of the proteins [44,47,48,52]. Not only do exogenously incorporated GPI-APs retain the functional extracellular activity of the GPI-AP, but they can also retain functional intracellular activity [50,51]. Exogenously incorporated GPI-APs onto cells led to association of the GPI-AP with active intracellular kinases [50,51]. Upon antibody crosslinking of exogenously incorporated GPI-B7-1, DAF, or CD59, phosphorylation induced by active kinase activity was detected [50,51] as well as an induced transient Ca2+ flux signal [51].
It has also been observed that cells genetically transfected to express the GPI-anchored form of the cytokine, GM-CSF, not only expressed functional surface-bound GM-CSF that was susceptible to PI-PLC cleavage, but GM-CSF was also detected in the supernatant in a soluble form. This soluble GM-CSF was released by either shedding or proteolytic cleavage and was not released by secretion [53]. Similarly, IL-2 was detected in the supernatant of cultured cell lines transfected to express GPI-IL-2; however GPI-IL-12 was not secreted when expressed on CHO cells [54,55]. It is not clear as to how only IL-2 was released; however, proteins differ in protease cleavage sites and also may differ in shedding capabilities. Therefore, this dual ability of cytokines to not only be expressed on cell surfaces by a GPI-anchor but also to be shed into the extracellular space could lead to a cytokine gradient allowing for the infiltration and activation of immune cells.
More importantly, after protein transfer of GPI-APs onto the lipid bilayers, the newly incorporated GPI-APs remain functionally active. Although it has been stated that naturally occurring GPI-APs can be reincorporated exogenously by protein transfer and retain their biological functional activity, even recombinant GPI-APs, such as GPI-B7-1 and GPI-IL-12, after protein transfer retain their biological functional activity [47,48,52,56,57]. Protein-transferred GPI-B7-1 and GPI-IL-12 incorporated on tumor cells and tumor membrane vesicles (TMVs) retained their ability to induce T cell proliferation.
6.2.3.3 Advantages and Limitations of GPI-AP Protein Transfer
Modification of membranes and cell surfaces provides a wide range of application possibilities. Protein transfer allows for membrane modification in an easy, quick, and efficient manner compared to the traditional genetic engineering approaches. (1) With protein transfer, purified GPI-APs incorporate onto cell membranes in a matter of minutes with maximal incorporation occurring around 2–4 h [47], whereas genetic engineering approaches take much longer from the order of days to months to establish cultured transfected cell lines. (2) Genetic engineering makes modifying some cell lines extremely difficult especially when primary cell lines, such as bone marrow progenitors and primary cultures, are hard to transfect. Also, protein transfer can be performed on small cell numbers and does not require cell division to occur. (3) Using protein transfer, the amount of incorporation can be finely controlled by adjusting the amount of exogenously added GPI-APs. (4) Finally, multiple GPI-APs can be incorporated onto the same cell membranes by protein transfer, whereas double and triple transfections by genetic engineering are more difficult to attain [58].
However, membrane anchors are known to affect the functionality of external domains of some proteins [59]. It has been noted that PI-PLC treatment of some GPI-APs renders some antibodies nonresponsive to the protein even if the antibody binds the GPI-AP in the presence of the GPI-anchor [60–62]. Therefore, the addition of a GPI-anchor can affect the structural conformation and function of proteins. Also, if important antibody epitopes are found at the C-terminal end of a protein, the addition of the GPI-anchor at the C-terminus may alter the antigenicity of the protein. Similarly, the GPI-anchor attaches the protein close to the membrane with less than 10–14 Å space between the protein and the membrane surface as determined by fluorescence resonance energy transfer (FRET) [62,63]. This close proximity to the membrane surface may alter accessibility of important antibody-binding sites. Therefore, it is imperative that the functionality of each GPI-AP be tested before use. A bottleneck of the protein transfer method using GPI-APs is the purification of GPI-APs. Purification using immunoaffinity chromatography is the best defined manner for obtaining pure fractions of GPI-APs; however, optimization of the process for each individual GPI-AP is time-consuming. Despite these disadvantages, when optimal purification of GPI-APs that retain their biological functionality is attainable, protein-transferred GPI-APs provide an efficient method to controllably modify membrane surfaces.
6.2.3.4 Protein Transfer of Antigen-Presenting Cells with GPI-Anchored MHC Complexes
During viral infections, CTL responses are important to combat infected cells. For a CTL to become activated, the T cell has to be engaged with MHC I that expresses the viral peptide; however, many viruses downmodulate the expression of MHC molecules on the cell surface to escape from the immune system. Therefore, Huang et al. [64] constituted a method to control the amount of MHC–peptide complexes expressed on a cell by protein transfer. Their model system utilized a HLA-A2.1 MHC complexed with a hepatitis B virus (HBV) peptide that induces a dominant T cell response. They constructed a GPI-anchored form of HLA-A2.1 by attaching a GPI-anchored signal sequence to the heavy chain and contransfected Schneider S2 Drosophila melanogaster cells with the beta 2 microglobulin gene. Upon purification of this HLA-A2:GPI, they were able to show that the MHC complex incorporated into HLA-A-negative and HLA-B-reduced cells in a time, temperature, and concentration-dependent manner. They were able to detect incorporation by flow cytometry within 1 min of incubation with cells; however, maximal incorporation occurred after 1 h incubation at 37°C. Additionally, incorporation was inhibited by the presence of 10% FBS, but if FBS was added after protein transfer, then no affect on cell surface expression of the MHC complex was noticed. Peptide was loaded onto the GPI-MHC molecules at 4°C overnight before protein transfer leading to stable expression of the MHC–peptide complex and MHC–peptide specific cell lysis by CTLs after protein transfer. These results showed that protein transfer with GPI-anchored MHC complexes could be used to express desired quantities of specific MHC complexes on cell surfaces. Thus, antigen-presenting cells (APCs) can also be decorated with desired MHC–peptide complexes allowing further studies of APC and T cell interactions to elucidate specific peptide-mediated T cell activation and tolerance.
6.2.3.5 Protein Transfer of GPI-APs in Tumor Immunotherapy
Another application of protein transfer using GPI-APs is in tumor immunotherapy. Cancer cells have the ability to continuously evade the immune system; therefore, finding a way to activate the immune system against these cells is key to combating tumors. One method in which tumor cells evade the immune system is through the downregulation of costimulatory molecules, such as B7-1, on tumor cell surfaces. This downregulation renders tumor-specific T cells anergic and nonresponsive to the tumor. Therefore, tumor cells have been transfected to express B7-1 in order to improve the immunogenicity of cell-based therapeutic vaccines. However, in order to transfect tumor cells with immune stimulatory molecules, the establishment of primary tumor cells lines from patients’ tumor tissue is required. However, tumor cells derived from patients are extremely difficult to culture; therefore, McHugh et al. [52] proposed the use of protein transfer to express GPI-anchored B7-1 on tumor cells or on TMVs. The TMVs that are derived from the homogenization of tumor cells still contain tumor-associated antigens along with a lipid bilayer, thus they can be modified with GPI-APs by protein transfer. It was shown that TMVs derived from EG7 tumors, a murine T cell lymphoma line, could be protein transferred to express GPI-B7-1 on the surface. They found that these protein-transfer-modified TMVs could protect against challenge with EG7 tumor cells. Furthermore, B7-1-modified TMVs led to T cell proliferation and cytolytic T cell activity in vaccinated mice ex vivo against the parental tumor [52].
Additionally, protein transfer was also shown to be conducted on TMVs derived from surgically removed human tumor tissue. These TMVs were stably incorporated with GPI-B7-1 by protein transfer in a concentration-, time-, and temperature-dependent manner. These incorporated TMVs were functionally active in stimulating T cell proliferation [47].
Many cytokine therapies, such as the administration of the cytokine IL-2, have been used as a tumor immunotherapy in order to activate immune cells against the tumor [65]. Although cytokine administration has worked in activating the immune system, systemic toxicity also results which impedes the success of the treatment. Therefore, human TMVs modified by protein transfer to express GPI-cytokines were used as a method of delivery of not only the cytokine in a less toxic manner, but also tumor-associated antigens [47,57]. Using protein transfer, purified GPI-anchored IL-12 was incorporated onto human and murine TMVs from mammary carcinomas, renal cell carcinomas, melanoma, and B cell lymphoma cultured cell lines, and the resulting TMVs expressed functionally active IL-12 that could induce T cell proliferation and IFN-γ secretion. IL-12 also worked well in combination with CD80 or CD40 to enhance T cell proliferation and IFN-γ secretion [57].
Taken together, protein transfer of costimulatory molecules and cytokines that have been converted into GPI-anchored forms has led to a promising approach for inducing antitumor immunity. Because GPI-anchored cytokines are attached to the membrane surface, a slow-release depot of the cytokine is ensured circumventing the systemic toxicity associated with administration of soluble cytokines.
6.2.4 Protein Transfer Using Proteins with Hydrophobic Domains
Another type of one-step protein transfer involves the incorporation of hydrophobic amino acid sequences into the lipid bilayer of cell membranes. Using this protein transfer method, the coding region of the superantigen toxic shock syndrome toxin-1 (TSST1) was attached to the hydrophobic sequence of the transmembrane portion of the proto-oncogene c-erb-B-2, forming a TSST1-TM molecule [66]. This TSST1-TM also had a histidine-tag attached to the N-terminal end for purification using a nickel-agarose column. Flow cytometry analysis showed that purified TSST1-TM incorporated onto multiple tumor cell line membranes after incubation for 4 h at 37°C in cell medium that contained 2.5% fetal calf serum. Furthermore, modified tumor cells were able to induce in vitro human peripheral blood lymphocyte proliferation and mice vaccinated with TSST1-TM-modified mitomycin C-treated tumor cells had decreased tumor growth after challenge with parental tumor cells [66]. Therefore, TSST1-TM incorporated onto tumor cells by protein transfer remained functional and exhibited antitumor responses.
As opposed to using GPI-anchor attachments to incorporate proteins onto cell membranes, this method that attaches hydrophobic sequences to proteins allows for the expression of modified prokaryotic proteins. Attachment of GPI-anchors to proteins requires a eukaryotic processing system, whereas this method does not [67]. Therefore, prokaryotic proteins, such as TSST1, can be modified and purified using a prokaryotic system without affecting the protein conformation and processing of the original molecule. This allows for functional production and incorporation of the modified prokaryotic protein.
6.3 Two-Step Protein Transfer
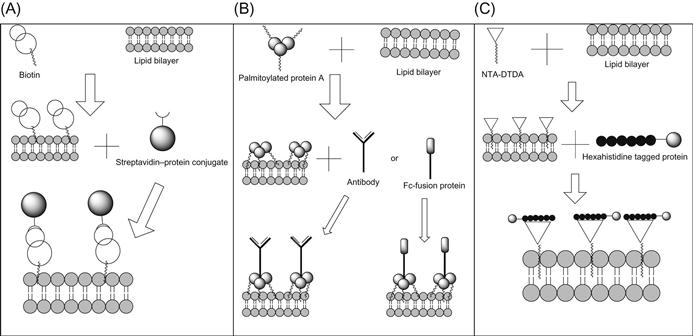
Two-step protein transfer can be implemented by using many different techniques, such as biotin–streptavidin, palmitoylated protein A (pal-protein A) or through the use of metal chelators (Figure 6.2).
6.3.1 Biotin–Streptavidin-Mediated Two-Step Protein Transfer
The biotin–streptavidin system has been implemented in many different ways in detection, purification, immunoassays, nucleic acid hybridization assays, DNA sequencing, etc. [68]. Biotin, or vitamin H, is found as a cofactor in many carboxylation reactions. It binds tightly to its ligand, avidin or streptavidin, with a high affinity constant [68–70]. Avidin was isolated from egg whites, whereas streptavidin was found in the bacteria Streptomyces [71]; however, they both form tetramers [68,69]. Since streptavidin does not bind specifically to many molecules other than biotin, and since streptavidin and biotin are highly resistant to dissociation, streptavidin is widely used to bind biotinylated proteins [68]. Using this biotin–streptavidin system, the protein transfer technology ProtEx was developed.
ProtEx is a technology designed to exogenously decorate the surface of cells through the use of the biotin–streptavidin system [70,72,73]. In this method (Figure 6.2A), cells are first biotinylated with Sulfo-NHS-LC-Biotin, which results in the attachment of biotin onto the cell membrane. Then the protein of choice, which is conjugated with core streptavidin (SA), is added to the biotinylated cells, allowing for stable interaction between streptavidin and biotin to occur and thus resulting in cell surface expression of the protein of choice [70,72,73].
An advantage of using the ProtEx system is that cell surfaces can be engineered within 2 h to stably express the protein of choice without toxicity to the cell. Due to the ability of biotin to remain stably incorporated on the cell surface for weeks [70], the ProtEx technology also allows for stable cell surface expression of the protein of choice. The extracellular domain of CD80 attached to SA (CD80-SA) was found to be stably incorporated onto biotinylated cells with a half-life of over 10–12 days in vitro and in vivo [73], whereas the kinetics of the expression of the extracellular domain of Fas ligand bound to SA (SA-FasL) was cell dependent [70]. Furthermore, the ProtEx technology allows for the protein of choice to exist as tetramers due to the nature of streptavidin which may play a beneficial role in some receptor–ligand interactions. However, for ProtEx to work, the addition of streptavidin to the protein of choice must not affect the functionality of the protein or the ability of streptavidin to bind to biotin. This restriction may limit the variety of proteins that may be used with the ProtEx technology.
Many applications of the ProtEx technology have been utilized in both autoimmune or transplantation settings, and in tumor settings. During immune homeostasis, Fas-FasL interactions play a strong role in maintaining a healthy immune environment that can fight invading organisms without being detrimental to the host. However, during many graft transplants, the immune system identifies the graft as a foreign agent and thus rejects the graft. In order to prevent graft rejection, Yolcu et al. [72] showed that by using biotin–streptavidin two-step protein transfer, FasL can be expressed on donor splenocytes and aid in preventing allogeneic pancreatic islet graft rejection. SA-FasL was added to biotinylated splenocytes and cotransplanted with islet cells into mice. Mice that received splenocytes decorated with SA-FasL had prolonged survival compared to mice that received unmodified splenocytes. The suggested mechanism behind this method was that interaction between alloreactive T cells and FasL-decorated splenocytes led to apoptosis of the Fas-expressing alloreactive T cells. Therefore, decoration of cells using SA-FasL by the biotin–streptavidin system can be used in preventing graft rejection. Bone marrow cells have also been decorated with SA-FasL in order to induce tolerance of grafts [74]. To expand the usage of SA-FasL, killer regulatory T cells were also developed using the biotin–streptavidin two-step protein transfer method. In this method, regulatory T cells decorated with SA-FasL, termed killer Tregs, were able to postpone autoimmune diseases such as diabetes [75], inflammatory colitis [76], and graft-versus-host disease [77].
On the other hand, in a tumor setting, the ProtEx technology has been used to decorate tumor cells with CD80-SA. Since the costimulation molecule, CD80 or B7-1, is often absent from many tumor cells, tumors can evade the immune system. Therefore, addition of the costimulation molecule on the tumor surface activates the immune system to recognize and attack tumor cells. Singh et al. [73] decorated human tumor cells with CD80-SA; these CD80-decorated tumor cells were then able to activate T cells to recognize and kill unmodified tumor cells in an antigen-specific manner. Furthermore, this two-step protein transfer method was used to induce protection against a B cell lymphoma line, A20, in mice. Mice were injected with A20 cells followed by irradiated A20 cells decorated with CD80-SA. These mice developed protection against tumor growth compared to control mice that received unmodified irradiated A20 cells [73]. Therefore, this ProtEx technology to decorate the cell surface through the use of the biotin–streptavidin system has proved to be an efficient method in preventing autoimmunity and graft rejection, as well as in preventing tumor growth.
6.3.2 Palmitolylated Protein A-Mediated Two-Step Protein Transfer
Although creating an SR on cells using palmitoylated antibodies as a one-step protein transfer method often is an effective approach, it is not desirable under some circumstances. Too much palmitate derivatization of antibodies, especially within the antigen-binding sites, can decrease affinity of the antibody for the specified target antigen, and the site and extent of palmitoylation can cause incorporation onto the cell membrane in a nonoptimal orientation for antigen binding. For these reasons, a need for creating another method for coating cells with specific receptors arose. In 1993, a solution was developed that involved the use of pal-protein A as an intermediate between the cell surface and the antibody [78], thus resulting in a two-step protein transfer method (Figure 6.2B). It was proposed that using unmodified antibodies bound to pal-protein A, which in turn was incorporated in the cell membrane, would yield a correctly oriented receptor. This is because protein A binds to the Fc region of the antibody [79,80] thus ensuring that the antigen-binding sites on the antibody are available for antigen binding. Furthermore, the antibody remains unmodified and thus retains the original affinity for antigen.
Using this two-step SR method, pal-protein A incorporated onto T cell hybridomas in a concentration-dependent manner, whereas non-pal-protein A did not [78]. Incorporated pal-protein A was able to bind rabbit IgG as detected by flow cytometry suggesting that palmitoylation of protein A did not inhibit incorporation or binding ability to IgG. To expand on this concept, T cell hybridoma cells decorated with pal-protein A followed by antimouse IgG were able to form conjugates with murine B lymphoma cells that express surface mouse IgG; these intercellular conjugates were proportional to the amount of pal-protein A/mouse IgG coating the cell; however, when nonspecific IgG was bound to pal-protein A-coated cells, decreased binding to target cells resulted [78].
Using pal-protein A for the formation of SRs is a useful method for quickly preparing cells coated with antibodies of different isotypes and specificities without having to palmitoylate each individual antibody. It also orients the antibody so that the antigen-binding sites are facing out and are able to interact with the antigen specifically. Therefore, using a two-step pal-protein A incorporation followed by antibody binding can lead to specific cell–cell interactions allowing for the creation of effector cells that can target a specific cell population.
Furthermore, this pal-protein A two-step protein transfer method can be expanded to bind Fc-fusion proteins. Using this method, the costimulation molecule, B7-1, was fused with the Fc region from the IgG1 antibody. This fusion protein can be used to “paint” the cell surfaces of cells that have been treated with pal-protein A. In this system, pal-protein A incorporated onto the cell surface in a concentration-dependent manner, whereas unmodified protein A could not [81]. Then the human B7-1·Fcγ1 fusion protein was incubated with protein A-modified cells so that the Fc region of the fusion protein bound to protein A on the cell surface. This consequently led to surface expression of the B7-1 fusion protein in a concentration-dependent manner [81]. Furthermore, B7-1 expressed on the cell surface remained functionally active and could lead to proliferation of PHA-activated or anti-CD3-activated T cells as well secretion of IFN-γ and IL-2 upon T cell stimulation [81]. Further analysis using this two-step protein transfer showed that preassembled B7-1·Fcγ1:pal-protein A could be incorporated onto tumor cells not only ex vivo, but also in vivo by intratumorally injecting the preassembled complex into mice that had L5178Y-R lymphomas [82]. These injected mice showed B7-1 expression on the lymphoma cells 1 h after administration as detected by flow cytometry. In another study, mice that received intratumoral adoptive transfer of ex vivo amplified tumor-specific T cells protein transferred with B7-1·Fcγ1:pal-protein A had increased tumor regression compared to mice that received control-modified T cells [83]. The antitumor efficacy of this vaccine was also seen in mice that had established 4T1 (a mammary carcinoma cell line) tumors, in which prolonged survival of mice that received protein-transferred T cells was observed over mice that received control-modified T cells [83]. Moreover, three other Fcγ1-fusion-costimulator molecules, 4-1BBL, CD48, and CD40L, along with B7-1 were also produced and individually incubated with pal-protein A. These preassembled complexes were then injected intratumorally together to form a “tetra-costimulator” system of protein transfer. Mice that received this tetra-costimulator complex showed 45% tumor regression of L5178Y-R lymphomas with evidence of induction of tumor-specific CTLs [82]. These studies show that this two-step protein transfer method involving pal-protein A and a fusion between costimulator molecules and the Fc region of IgG1 can be used as an effective method to induce local expression of proteins in vivo.
6.3.3 Lipid-Metal Chelator-Mediated Two-Step Protein Transfer
Another approach for two-step protein transfer utilizes lipid-metal chelators (Figure 6.2C). In this system, nitrilotriacetic acid ditetradecylamine (NTA-DTDA) [84] or 3(nitrilotriacetic acid) ditetradecylamine (NTA3-DTDA) [85] are used. The hydrophobic chains of DTDA allow for insertion of the metal–chelator complex into cell membranes leaving the NTA head groups available for subsequent binding to hexahistidine-tagged proteins or peptides [84–87]. NTA3-DTDA allows for an anchor that is more stable than NTA-DTDA due to the presence of three NTA head groups [85]. Using this two-step protein transfer method, two different hexahistidine-tagged molecules, B7-1 and CD40, were able to simultaneously incorporate onto tumor cells [84], and hexahistidine-tagged peptides could be used to incorporate onto lipid membranes [88,89].
The lipid-metal chelator two-step protein transfer has many applications. This method was used to study low-affinity receptor–ligand interactions [90]. Due to the fluidity of the lipid bilayer, when NTA-DTDA is incorporated onto cell surfaces and engrafted with hexahistidine-tagged CD40, B7-1, or CD4, the ligands are able to move in order to better interact with the respective receptor multimerically, thus allowing for lower-affinity receptor–ligand interactions to be further studied. Moreover, this two-step protein transfer method can be used to directly decorate tumor cell surfaces with hexahistidine-tagged costimulatory molecules, B7-1 and CD40, to induce protection from tumor challenge [84] as well as enhance the immunity of liposome complexes by engrafting hexahistidine-tagged flagellin-derived peptides onto the liposome surfaces [86]. Another approach for using this two-step protein transfer is to target both liposomes and plasmid DNA-lipoplexes to specific cell types or tumor locations in vivo [85,87–89]. Liposomes were targeted to dendritic cells in vivo by engrafting hexahistidine-tagged antibody fragments against CD11c and DEC-205 (molecules expressed on DCs) onto the liposomes to specifically target either antigens or cytokines to DCs [85]. Anticancer drugs, such as doxorubicin, can also be loaded into liposomes that are then modified with NTA3-DTDA and a hexhistidine-tagged peptide to target the drug to NIH-3T3 cells [89]. In a similar manner, plasmid DNA-lipoplexes can also be targeted to specific cells [88] and liposomes can be targeted to the tumor site in vivo by using hexahistidine-tagged peptides specific for proteins expressed in the vasculature formed by tumors [87]. Therefore, the lipid-metal chelator two-step protein transfer allows for specific targeting of anticancer drugs and molecules to tumor cells.
6.4 Summary
Cell surface engineering by lipid-mediated protein transfer provides an efficient and quick manner to control the expression of surface proteins on cell membranes. Both one-step and two-step lipid-mediated protein transfer methods are quick and efficient ways to engineer the cell surface. While the modified protein of interest incorporates directly in the one-step method, indirect expression of the protein of interest results in the two-step protein transfer method. Both methods allow for controllable expression of functional proteins on the cell surface. The two-step protein transfer method is useful when hydrophobic modifications of the protein of interest render the protein dysfunctional. Therefore, the two-step protein transfer can be used to maintain functionality of membrane-incorporated proteins.
Modification of cell surfaces by expressing new proteins has many advantages over genetic engineering approaches. Genetic engineering approaches have been quite successful in expressing proteins on the cell surface; however, genetic engineering requires fast dividing cell lines or a large number of cells. It is difficult to transfect primary cells especially when low numbers of cells are available. Also, many primary cells, such as immune cells and primary tumor cells, are resistant to transfection. In genetic engineering, the amount of surface expression cannot be controlled and to achieve high surface protein expression of the transfected cells, rounds of selection or sorting of high expressing cells has to be performed. Thus genetic engineering of the surface is a very time-consuming method. It also becomes difficult to express high levels of multiple proteins on the cell surface by transfection.
On the other hand, protein transfer allows for controlled expression of protein on the surface of the cell as well as on cell membrane vesicles. Protein transfer can be performed quickly within a few hours and can lead to surface expression of functional protein. Since the lipid anchor attached to the proteins can incorporate on any lipid bilayer, protein transfer results in protein expression on 100% of the cells. Also, multiple functional proteins can be incorporated simultaneously on the cell surface by protein transfer. However, each protein that undergoes protein transfer needs to be modified with lipid tails so that it can insert exogenously into the lipid bilayer of cells and membrane vesicles. This modification should not interfere with the function of the protein; therefore, each modified protein needs to be tested for functionality before use. Furthermore, division of cells that are protein transferred will lead to the diffusion of the protein among the dividing cells. Therefore, it may be beneficial to use cell membrane vesicles or irradiated and mitomycin C-treated cells with protein transfer so that cell division does not diffuse protein surface expression.
Many applications for using protein transfer exist. One of the most widely used protein transfer applications involves enhancing antitumor immunity. Many applications have been made to enhance the immunogenicity of tumor cells by protein transferring immune-stimulating molecules, such as the costimulatory molecules B7-1 and B7-2 or cytokines such as IL-12, onto tumor cells or TMVs. This modification enhances antitumor immune responses and also transforms tumor cells into APCs. Protein transfer can also be used to target cells by creating artificial receptors on membrane surfaces. Through this manner, delivery of drugs or toxins can be targeted to specified cells that express incorporated receptors on their surface. On the other hand, these artificial receptors can also be used to target the incorporated cells or lipid vesicles toward other cell types in order to stimulate either killing or activation of the target cells or induce uptake by cells, such as APCs. Furthermore, protein transfer can be used to engineer APCs to activate T cells by incorporating exogenous costimulatory or activating proteins or by incorporating more adhesion molecules on the APC cell surface. However, conversely, protein transfer can also be used to induce killing of T-cells by exogenously modifying the APC cell surface with immune inhibitory molecules.
In summary, lipid-mediated cell surface engineering can be achieved by a variety of methods. These approaches to manipulating the cell surface have the potential to be used in studying cellular receptor functions, developing delivery vehicles for drugs and designing cellular vaccines.
Acknowledgments
This work was supported by NIH grants R01 CA138993 (to P. Selvaraj) and F31 CA165632 (to J. M. Patel). The authors thank Archana V. Boopathy for critical reading of the manuscript.