
Construction and Computation with Nucleic Acids on the Cell Surface
Alexey Y. Koyfman1 and Norbert O. Reich2, 1Department of Biochemistry and Molecular Biology, and Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, TX, USA, 2Biomolecular Science and Engineering Program, and Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA, USA
In this chapter, the authors summarize recent advances in using cells and their surfaces for engineering and computation applications. Many of these advances have a clear potential for translational applications, including imaging. Nucleic-acid based methods are scalable, modular, and rely on readily available materials. Examples and potential applications to drug targeting challenges are discussed. Finally, cell recognition, and the use of nucleic acids to mimic membrane components (e.g. channels and pores) are also described.
Keywords
Cell identification and targeting; DNA origami; DNA computation; DNA channels and pores
Cell surfaces provide a fertile ground for the construction of architectures with diverse applications. This chapter explores the capabilities of nucleic acids to perform computations on cell surface, attach nanostructures to cells, bridge cells, activate innate cellular responses, and build transmembrane channels and pores. We outline the multiple reasons researchers are interested in identifying the cell type, modifying a surface of a cell, and attaching an endogenous structure to the cell of choice. We also explain the basis for the emergent interest and new methods to rationally modify cell surfaces.
DNA is the preferred biological material to store and transfer genetic information. RNA can fold into intricate three-dimensional (3D) structures utilizing a number of naturally occurring structural motifs [1] and is used for enzymatic catalysis, and gene expression and regulation. Both nucleic acids show promise in bionanotechnology due to their defined length, unique base pair recognition capabilities, and possible base and backbone modifications.
In order to create sophisticated nanomachines, a complex asymmetrical structure has to be constructed. Since each position of the DNA sequence is addressable, DNA is a material that allows creating structures with broken symmetry—placing chemical groups at specific locations. In principle, any functional group can be placed anywhere in the DNA structure with high spatial precision. Chemistry is in a powerful position to enhance the capabilities of nucleic acid nanotechnology by adding tailored functionality through the selective modification of nucleic acid’s bases and backbone with chemical tags.
Small molecules such as cholesterol [2], peptides [3], enzymes [4], antibodies [5,6], quantum dots [7,8], inorganic materials [9,10], and carbon nanotubes [11] have been positioned at precise locations within DNA nanostructures.
DNA origami is a complex asymmetrical structure where every nucleotide is unique and addressable (Figure 8.1A and B). DNA origami, named after the Japanese art of paper folding, is composed of a multiple kilobase single-stranded DNA scaffold, often obtained from the M13 phage genome, which is folded into a designed shape with hundreds of short oligonucleotide staple strands (Figure 8.1A) [15]. The staples are short, single-stranded oligonucleotides that are complementary to sequences in multiple locations of the single-stranded M13 phage DNA scaffold and can therefore bridge distant sites. Therefore, it is possible to staple and fold the system into any 3D shape. DNA origami structures form in a one-pot thermal annealing reaction when scaffold and staple strands are mixed. DNA origami has been assembled into a variety of 3D shapes (Figure 8.1B) [16–18] and can be used as a nanoscale template to precisely position diverse components [19–21].
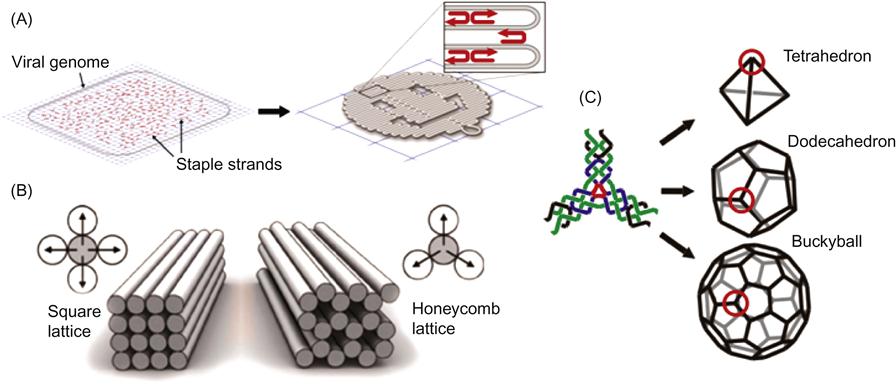
An asymmetric object like DNA origami composed of hundreds of unique oligonucleotides is expensive and not always desired. To minimize the number of DNA strands and simplify the design, symmetry has to be introduced (Figure 8.1C) [14,22–24]. Icosahedral viruses have C60 symmetry and structurally cannot be as programmable as DNA origami [25]. Tetrahedral, dodecahedral, and icosohedral DNA architectures were also developed with DNA [14] (Figure 8.1C). The advantage of this approach is that the same molecule can be expressed multiple times on the surface of symmetrical objects and can be used for certain applications when you do not want to position a specific molecule in one particular location. Also, only three oligonucleotides are needed to construct an intricate structure like a DNA icosohedron (Figure 8.1C) [14].
8.1 Computation of Cell Identity
Currently it is possible to accurately identify an unknown cell type using nucleic acid computations. An easily identifiable DNA fluorescent nanorobot targets only a specific receptor or multiple receptors on the cell. Alternatively, it is possible to target a specific cell type known to be in the cellular mixture by attaching a directed nanostructure.
A cell membrane contains many cell surface proteins also known as surface antigens that are unique to each cell type. Over 200 types of cell are histologically identifiable in humans. Furthermore, various cell types express multiple cell surface markers, also known as “clusters of differentiation” (CDs) [26]. These surface antigens can be used to identify both the cell type and the functional state of the cell. Healthy and diseased cells can be also classified by inventories of their cell surface markers [27,28]. Most identified cell surface markers are not exclusively expressed on the target population of diseased cells. A marker overexpressed on the surface of cancer cells is often also expressed at a lower concentration on the surface of some normal cells. The distribution of any given protein type on a cell surface is not uniform. Cells will have very common “housekeeping” antigens and sometimes very unusual antigens [29]. Cells can be triggered to perform a certain function or behave in a certain manner upon binding by antibodies or drugs. Some drugs target disease-causing cells by binding to a specific receptor. Unfortunately, some disease-causing cells do not have unique markers and therefore drugs also bind to non-targeted healthy cells and cause “off-target” toxicities [30]. This is sometimes the case for cancer therapy utilizing antibody–drug conjugates (ADCs). For example in leukemia, both healthy and diseased subpopulations of white blood cells display surface markers that are indistinguishable by the current single-receptor antibody therapy, leading to serious complications [31] In comparison, a more practical and less risk-prone approach would simultaneously assess multiple surface receptors to pinpoint specific disease cells and enhance diagnostic accuracy [32,33].
The ability to specifically label a particular and narrow cell type subpopulation within a much larger population of related cells for the purpose of cellular elimination [34], isolation and analysis [35], and imaging [30] has many potential benefits. This challenge could be readily addressed in a direct manner if targeted subpopulations could have some unique cell surface marker against which antibodies could be raised [30].
8.1.1 Accurate Identification of the Cell Type
Accurate identification of the cell type depends on having sufficient information about what uniquely defines that cell. Although a single, cell-specific antigen is sometimes known, it is frequently necessary to evaluate multiple surface antigens to pinpoint a specific cell. Since a cell contains a number of different cell surface antigens, a molecular device that is able to bind multiple receptors will be able to pinpoint a specific cell more accurately. Four to five unique antigens would suffice to specifically identify a particular cell within a multicellular population [36].
Sergei Rudchenko, Milan Stojanovic, and coworkers at Columbia University reported in Nature Nanotechnology [36] that they have harnessed the power of DNA to identify a cell by performing computations directly on the cell surface. Blood cells were chosen for the experimental design since these cells are well understood [37]. Blood cells are often characterized using flow cytometry according to their different levels of expression of the CDs. Lineages and stages of differentiation in blood cells are defined by the presence or absence of multiple cell surface markers. The team of scientists designed molecular robots that can identify multiple cell surface antigens, thereby effectively labeling more specific subpopulations of cells. Each probe molecule is composed of an antibody and a synthetic DNA duplex. While the targeting antibody plays an identification role, the DNA executes the computation (Figure 8.2). Cells can be labeled in 15 min using this technique. The researchers also showed that the targeted DNA strand could be programmed to fluoresce in the cellular solution. These molecular robots can essentially label a more specific subsets of cell populations therefore allowing for more targeted therapy. Researchers could either label cells that they want to target or avoid the labeled cells.
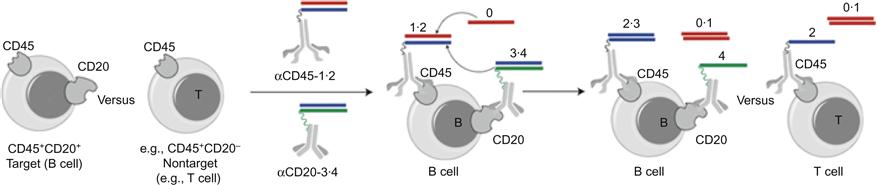
The computation YESCD45YESCD20 would only label B cells and avoid T cells (Figure 8.2). YESCD45YESCD3 would only label T cells, and the computation YESCD3YESCD20, is a negative control, since no cells display these two markers at the same time. No labeling was observed in YESCD3YESCD20 two-step computation which indicated that the T cells did not exchange DNA oligonucleotides with B cells through diffusion or through direct physical contact between B and T cells. Rudchenko et al. present a simple method, where a number of straightforward components are assembled on the cell surface to compute the cell type.
8.1.2 Logic-Based Cancer Targeting and Therapy
A related DNA-based approach [38] combines the structure-switching properties of DNA aptamers with toehold-mediated strand displacement reactions. DNA Nano-Claw quantitates multiple cancer cell surface markers, produces a diagnostic signal, and autonomously induces therapeutic operations. The targeted therapeutic effect is triggered by these structure-switchable aptamer conjugates to induce photodynamic therapy (PDT) [39,40].
Both DNA-based methods are easy to reproduce and reliable because they use standard antibodies and commercially available synthetic DNA. Both systems are also modular and scalable in principle. There is a wide range of possible clinical applications if molecular robots work in mice studies and human clinical trials. For example, cancer patients could benefit from more targeted chemotherapeutics. Also, drugs for autoimmune diseases could be more specifically tailored to impact disease-causing autoimmune cells and not the immune cells needed to fight infection.
With advancement of DNA computations, molecular probes may be used to find viable bloodstream targets. However, in order to proceed to the routine in vivo use of molecular DNA computational robots, a few major barriers have to be addressed. DNA robots have to be distributed well within the body. Molecules with mass below 500 Da and relatively low polarity are traditionally used in medicine [41]. Unfortunately, nucleic acids do not diffuse rapidly due to a mass well above small molecules. Also DNA by itself does not cross membranes well. For other in vivo targets alternative delivery methods will be required.
8.2 Targeted Transport of a Payload to a Cell Surface and Activation of Innate Cellular Response
DNA nanostructures capable of sensing cell surface antigens were used to transport molecular payloads to cells and activate intracellular transformations.
The ability to organize materials into defined shapes is a core goal of bionanotechnology. Cell surface engineering [42] seeks to localize nanoscale materials onto cellular membranes. The defined organization of nanoscale objects on the cell surface has a number of biomedical applications. Some examples include the organization of cells into predictable architectures on surfaces [43,44] and the delivery of diverse molecules to cells [45]. Cells have been assembled into microtissues using DNA-mediated interactions [46].
8.2.1 Cellular DNA Patches Targeted to Cancer Cells
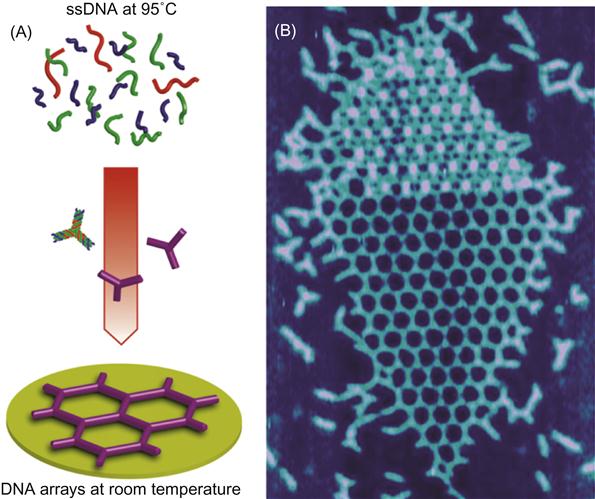
Self-assembled DNA arrays were assembled from three single-stranded DNA oligonucleotide strands into hexagonal units repeated every 30 nm (Figure 8.3A). Hexagonal DNA arrays assemble when the three strands cool down from boiling water temperature to room temperature. The size of the DNA patch is controllable with concentration of divalent magnesium ions from compact multilayers (Figure 8.3B) and into extended networks spanning several microns [23].
Self-assembled DNA arrays can be directed to the surface of cells through biotin–streptavidin (STV) interactions and specific antibody–cell surface interactions [24] (Figures 8.4 and 8.5). The versatile cargo-carrying ability of arrays is explored for directing cell–surface interactions, cell–cell bridging, and positioning multiple cells onto a DNA fabric (Figure 8.4).
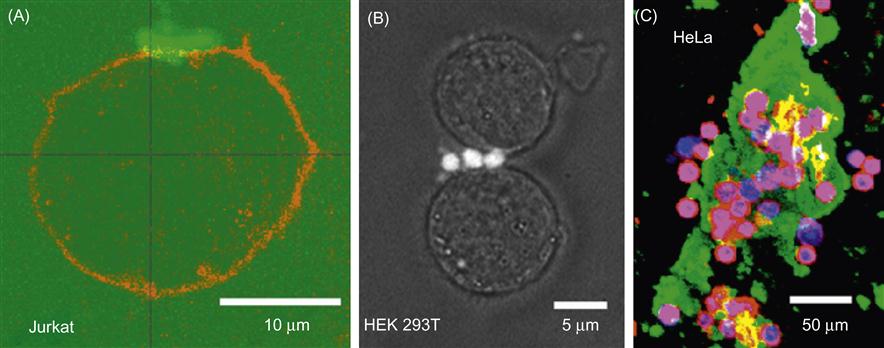
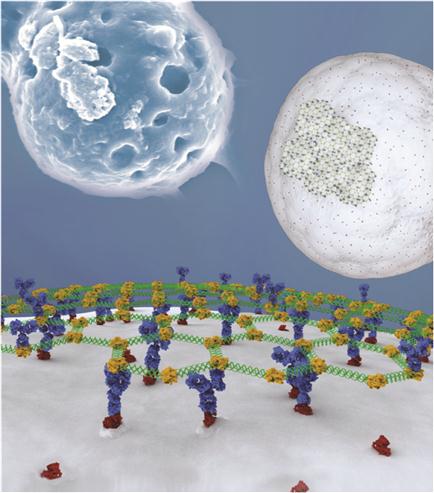
We present two strategies to attach hexagonal DNA arrays to human cells. First, biotin-modified arrays were bound to biotinylated cancer cells using STV as a bridging component. Our second strategy for directing DNA arrays to cancer cells builds on the previous strategy but uses interactions between native cell-specific surface proteins and their antibodies. This approach provides a basis for directing the assembly of distinct cell types through the use of various cell-specific surface markers. Epidermal growth factor (EGF) receptors (EGFRs) are overexpressed on the surface of many human cancer cells, at levels of up to 106 per cell [47,48]. We used two EGFR-expressing cell lines (HEK 293T and HeLa) and one nonexpressing control cell line (Jurkat) for attachment of DNA patches through EGFR antibody. The biotin-modified arrays attached to antibodies were bound to native EGFRs using STV and antibodies as bridging components (Figure 8.5).
Micron-sized DNA–biotin–STV–Ab arrays bind to cancer cells and envelop multiple cells (Figures 8.4 and 8.5). Thus, interactions mediated by DNA assemblies as well as DNA hybridization [46] may be used to direct tissue engineering. Large numbers of antibodies would likely be required for efficient bridging of cells. Arrays optimized to uniquely bind multiple surfaces could form the framework for organizing cells on surfaces and inside 3D matrices. By incorporating gold and magnetic iron oxide nanoparticles in nucleic acid nanostructures [49], it is possible to assemble 3D tissue cultures based on magnetic levitation of cells [50]. The geometry of the cell mass can be further manipulated by spatially controlling the magnetic field. A large number of repeating units in a hexagonal DNA array provide an opportunity for loading many copies of identical or different molecules for cellular delivery or interconnection of cells. The generic biotin–STV modification allows the anchoring of a variety of biomolecules like peptides or aptamers to arrays, which might serve to identify or induce the fate of stem cells [51].
Further, since DNA networks can be systematically varied in size [15,17,23], it is possible to identify various cells by both the size and color of the fluorescent DNA patches. This could potentially be used for cancer cell screening. Having many fluorophores positioned very closely to each other on a DNA scaffold enhances sensitivity to lower intensity light used for array detection. It is possible to uniquely color fluorescent multifunctional DNA patches, identifying the location of tethered small molecules, nanoparticles, or other cargo.
DNA nanoarrays provide a number of strategies to specifically label cell surfaces with functionalized micron-sized patches, to deliver materials to cell surfaces, and to engineer cell/cell networks. The porous and periodic nature of the DNA material could be useful for tissue engineering. Functionalized nanoarrays could be used for screening cancer cells, the control of stem cell fate, and the controlled activation of immune response.
8.2.2 DNA Crosslinked Receptors Trigger Robust Signaling Responses
Antigen-mediated crosslinking of IgE bound to its receptor, Fc![]() RI, on mast cells stimulates degranulation, phospholipid metabolism, and cytokine production leading to inflammation and allergies [52]. Spatial organization of the clustered IgE–Fc
RI, on mast cells stimulates degranulation, phospholipid metabolism, and cytokine production leading to inflammation and allergies [52]. Spatial organization of the clustered IgE–Fc![]() RI complexes affects the assembly of the transmembrane signaling complexes. DNA three-way junctions with tunable lengths were utilized to bring IgE–Fc
RI complexes affects the assembly of the transmembrane signaling complexes. DNA three-way junctions with tunable lengths were utilized to bring IgE–Fc![]() RI complexes and crosslink them together [53]. Sil et al. reported that the trivalent DNA ligands triggered robust signaling responses, stimulated phosphorylation of Fc
RI complexes and crosslink them together [53]. Sil et al. reported that the trivalent DNA ligands triggered robust signaling responses, stimulated phosphorylation of Fc![]() RI subunits, and lead to degranulation [53].
RI subunits, and lead to degranulation [53].
8.2.3 DNA Origami Barrels Activate Signaling Pathways in Target Cells
A recent paper described a method for creating tiny machines out of strands of protein and DNA [54]. Each DNA origami nanorobot measures 45 nm long×35 nm wide, roughly the size of a virus. Both virus and DNA origami barrel can be thought of as molecular delivery trucks, virus delivering nucleic acids, and DNA barrel transporting medications to specific cells in the body. The nanoscale DNA origami barrel releases Fab antibody fragments in the presence of target cells in vitro.
To characterize the system, the authors delivered fluorescently labeled Fab antibody fragments to cells. DNA origami barrel showed highly specific binding to the cells that displayed the correct combination of surface antigens in mixed cell populations and whole blood leukocytes.
The constructed DNA origami barrel consists of two domains that are covalently attached in the back by single-stranded scaffold hinges (Figure 8.6). The two domains stay locked when DNA strands in the front are base paired. DNA origami barrel opens when DNA aptamer-based lock binds the antigen keys (Figure 8.6). This lock system was inspired by aptamer beacons [55] and structure-switching aptamers [56], which typically undergo target-induced switching between an aptamer–complement duplex and an aptamer–target complex (Figure 8.6). Both aptamers have to recognize their targets in order for DNA origami barrel to open and expose the inner surfaces. The DNA robot could be programmed to activate in response to a single type of key by using the same aptamer sequence in both lock sites. Alternatively, different aptamer sequences could be encoded in the locks to recognize two inputs. Both locks needed to be opened simultaneously to activate the robot. The robot remained inactive when only one of the two locks was opened. Two different aptamers specifically binding two different protein antigens were designed. This lock mechanism is thus equivalent to an AND logic gate that requires the presence of two different protein targets to be activated. When both aptamers bind their targets, a conformational change opens up the barrel and releases the cargo.
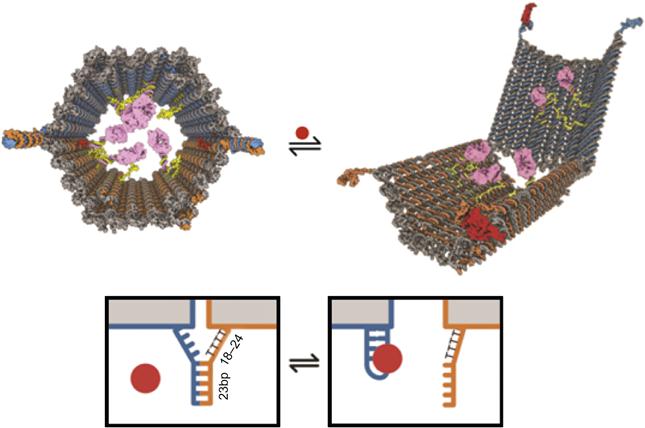
To test the generality and robustness of the aptamer-encoded logic gating, six different DNA barrels were designed using pairwise combinations of aptamer locks drawn from a set of three well-characterized aptamer sequences: 41t, against platelet-derived growth factor (PDGF) [57]; TE17 [58]; and sgc8c, against protein tyrosine kinase 7 [59].
Various leukemia cells opened DNA origami barrels only with the right aptamer locks. No DNA barrels were activated on Burkitt’s lymphoma cells (Ramos). An acute myeloid leukemia cell line (Kasumi-1) activated all robots. An aggressive lymphocytic NK-type leukemia (NKL) cell line [60] activated DNA barrels with two 41t locks, two TE17 locks, and with one 41t and one TE17 lock. Jurkat cell line, isolated from acute T cell leukemia and acute lymphoblastic leukemia (CCRF-CEM), activated DNA barrels with two TE17 locks, two sgc8c locks, as well as one TE17 lock and one sgc8c lock. Neuroblastoma cell line (SH-SY5Y) activated DNA barrels with two sgc8c locks.
DNA origami barrels were also designed to activate signaling pathways in target cells. DNA barrels were loaded with Fab antibody fragments known to bind human CD33 and human CDw328 and induce growth arrest in leukemic cells [61]. Upon recognition of the surface antigen PDGF on cells from a patient with aggressive lymphocytic NKL, the barrel structure opened, allowing the antibody payload to bind to cell surface receptors and stopped the growth of the targeted NKL cells.
DNA origami barrels were also loaded with a combination of antibodies against a T cell surface receptor and flagellin, the protein making up bacterial flagella. Flagellin is known to promote T cell activation [62]. DNA nanorobots collected flagellin from the solution environment of the cells and induced activation of T cells.
Much work remains to take these ideas toward successful drug delivery. For example, DNA origami barrels might be unstable in the bloodstream in the presence of nucleases and other enzymes. It might be necessary to chemically modify the DNA backbone to increase resistance to degradation. Also, aptamer-encoded locks may lose specificity and efficiency in protein-rich serum. In addition, delivery into cells is required for many intracellular targets. DNA strands and large DNA nanostructures have difficult penetrating biological membranes. In order to cross the membrane and increase cellular uptake, amphiphilic molecules or peptides could be displayed on the surface of DNA origami barrels. Another concern is that DNA origami is a complex structural molecule that requires hundreds of unique oligonucleotides to assemble. The cost to synthesize sufficient amounts of customized long-scaffold DNA has to decrease. On the other hand, it may be necessary to minimize the number of DNA strands and simplify the design to increase the production of the nanostructure as a medicine [14,22–24].
Currently, we treat cancer with chemical and nuclear weapons: chemotherapy and radiation. Maybe we need to switch to smarter, more targeted therapeutics. The aim of smart drug delivery systems is to administer smaller drug doses to patients while offering improved therapeutic efficiency and fewer side effects compared with conventional drug delivery methods. Chemotherapy might shut down cancer cells but also shuts down all sorts of vital processes in the human body that we want to keep going. The toxic chemotherapy molecules lead to hair loss, nausea, and other side effects. By contrast, nanorobots could deliver medication directly to cancerous tumors. Nanorobots this small can interact directly with the surface of individual cells. The amount of medicine would be drastically decreased, since it is being delivered directly to target cells.
8.3 DNA Channels and Pores Spanning Lipid Membrane
Various kinds of DNA origami channels and pores were constructed and inserted into the lipid membrane using chemically attached hydrophobic molecules. DNA origami transmembrane channel and pores conduct an electrical current proportional to the potential that is placed across the membrane. The channel is similar to what is observed in many naturally occurring protein pores and shows stochastic gating—conformational fluctuations between open and closed states. Attaching transmembrane nanostructures to cells will provide a new type of sensing device and help manipulate or augment cell functions.
8.3.1 Transmembrane DNA Channel
A transmembrane channel made entirely from DNA and DNA-bound cholesterol moieties was constructed [2]. The inspiration for the design of the DNA transmembrane channel came from the bacterial protein pore α-hemolysin [63,64]. Streptococcus bacteria excretes α-hemolysin and perforates a target cell in order to leach iron through the resulting channel for the bacterium to consume [65]. Similar to α-hemolysin structure, the DNA channel structure consists of a channel that spans the lipid bilayer and a barrel-shaped cap that binds to the surface of the membrane.
The channel consists of two parts: a stem that penetrates through a lipid membrane and a barrel-shaped cap that attaches through cholesterol molecules to the membrane. The channel comprises six DNA helices that form a hollow tube. The interior of this tube acts as a transmembrane channel with a diameter of 2 nm and a length of 42 nm. The stem of the channel runs through both stem and barrel (Figure 8.7A). Adhesion to the lipid bilayer is mediated by 26 cholesterol molecules that are attached to the surface of the barrel (Figure 8.7A).

Transmission electron microscopy (TEM) images of purified DNA channels with unilamellar lipid vesicles confirmed the membrane binding (Figure 8.7B). Synthetic DNA channels bind to lipid bilayer membranes in the desired orientation in which the cholesterol-modified face of the barrel forms a tight contact with the membrane and the stem appears to protrude into the lipid bilayer (Figure 8.7A and B).
8.3.2 Charge-Neutralized Transmembrane DNA Pores
A DNA origami nanopore composed of six interconnected DNA duplexes was constructed [66]. The DNA-based nanopore structurally mimics the amphiphilic nature of protein pores in order to insert into the membrane. Membrane proteins feature an outer hydrophobic surface. DNA nanobarrels have to overcome the unfavorable energetic interaction between the hydrophobic environment of the membrane and the hydrophilic, negatively charged phosphate groups in the outer pore wall. To ensure insertion into the membrane, the pore was chemically modified with alkyl groups, which mask the negatively charged oligonucleotide backbone (Figure 8.7C). This modification allows the insertion of the DNA pore into the hydrophobic environment of the lipid membrane. The DNA barrel contains the 2.2-nm-long outer hydrophobic ring (Figure 8.7C, magenta), which matches the thickness of the lipid bilayer. A total of 72 alkyl-phosphorothioates groups (12 per each duplex) make up the hydrophobic belt. DNA origami nanopores, inserted into lipid bilayers, were tested using single channel current analysis for ionic current. After membrane insertion, DNA nanopores supported a steady transmembrane flow of ions through a single pore.
A total of 26 cholesterol molecules were placed in the strategic positions on the surface of the DNA barrel [2] (Figure 8.7A). A total of 72 alkyl-phosphorothioates groups make up the hydrophobic belt of the DNA origami pore [66] (Figure 8.7C). In order to simplify nanopore design and minimize chemical intervention, a smaller number of chemical tags with greater hydrophobicity were used for membrane anchoring [67]. Only two porphyrin-based hydrophobic tags achieve the otherwise energetically unfavorable anchoring of the negatively charged DNA nanopore into the hydrophobic core of lipid bilayers. Porphyrin has a large aromatic core and a van der Waals surface area approximately 12 times higher than ethane. The aromatic porphyrin tags can also be powerful visualization tags since they are chromophores with a fluorescence emission at 656 nm. Moreover, inserting porphyrins into lipid membranes leads to fluorescence shift and confirms membrane anchoring.
The engineered DNA transmembrane channels and pores demonstrated electrical conductivity using an integrated chip-based setup. The synthetic DNA channel was further engineered to display gating behavior similar to natural ion channels. The mutant was designed by incorporating a single-stranded heptanucleotide protruding inside the transmembrane tube shown in red in Figure 8.7. The transmembrane current gating behavior depended on fine structural details of the synthetic DNA channel. It is challenging to modify the geometry of natural biological pores made from protein. On the other hand, the geometry of synthetic DNA origami objects and their chemical properties can be tailored for custom sensing applications of desired materials [66–69]. The possibilities with this system are endless, because one can easily vary the sequence and the corresponding structure, analogous to site-directed mutagenesis in biology. The DNA transmembrane channel was used to study unzipping of DNA hairpins and unfolding of guanine quadruplexes [70,71]. Single-stranded DNA molecules fit through the designed 2-nm central pore of the DNA channel, whereas larger DNA complexes such as hairpins or quadruplexes did not. Hairpins and quadruplexes had to unzip or unfold to pass through the DNA channel and the kinetics of the unfolding was monitored. The channel can also distinguish differing DNA lengths. DNA origami nanopores were used for the detection of λ-DNA molecules [68]. DNA molecules can be used to create designer channels and pores with adaptable diameter, shape, and surface functionality. Thus, synthetic DNA channels can be custom made as sensing devices for desired analyte molecules.
Synthetic DNA channels spanning lipid membranes are a crucial first step toward harnessing ion flux for creating sophisticated nanodevices. The inspiration for DNA channels can be found in nature by the rich functional diversity of membrane machines such as rotary motors, ion pumps, and transport proteins. The transmembrane channel and nanopore designs can be utilized as artificial pores with tunable voltage-gating and ion-selective permeation properties. Possibly, a DNA machine can be engineered to actively transport matter across a membrane by utilizing DNA-based motors [72–74]. Synthetic DNA channels could be engineered to have antimicrobial properties and used for molecular detection applications, and DNA sequencing, and research into nanofluidics and controlled transmembrane transport. DNA nanostructures can be used for sophisticated drug delivery applications in medical therapies targeting specific cell types and inserting a transmembrane pore for drug delivery much like a virus would [75,76,82].
8.4 Advances in Microscopy
The biological processes occurring on the surface and inside the cell are complex and dynamic. In order to achieve a comprehensive understanding of the molecular mechanisms underlying these biological processes, both temporal and spatial resolution are required.
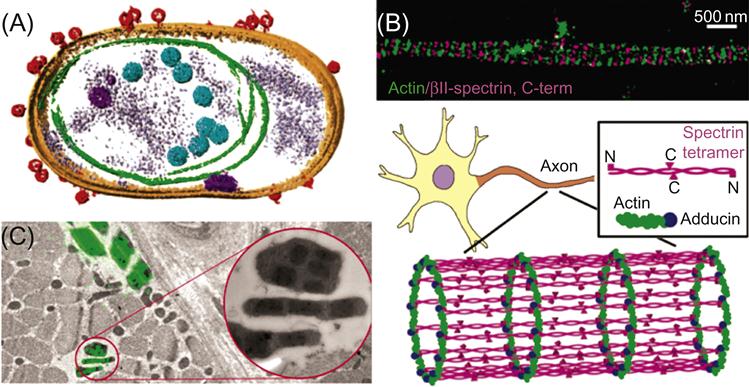
Cryo-electron tomography (cryoET) [77] provides the spatial resolution. Using cryoET, 3D volumes of near-native frozen hydrated structures are obtained at molecular resolution (Figure 8.8A). Recent cryoET advances vividly demonstrate inner structures of the flagella [79,80] and attachments of viruses to cells (Figure 8.8A) [75,77,81,82].
On the other hand, fluorescence light microscopy provides the temporal resolution. It provides time-resolved dynamics of fluorescent proteins of interest in living cells.
Recent advances in fluorescence light microscopy have further improved the spatial resolution by an order of magnitude [81–84]. Super-resolution fluorescence microscopy imaging, which applies switchable probes, as in PALM and STORM [85,86] has enabled a better structural understanding of the elaborate arrangement of cellular components (Figure 8.8B) [78].
Combining and integrating fluorescence light microscopy and electron microscopy (termed correlative microscopy) is an emerging powerful approach to visualize dynamic cellular processes, since it unites the strengths of the two complimentary imaging modalities (Figure 8.8C) [87,88]. On one hand, correlative microscopy provides temporal resolution and dynamic information available from optical microscopy. On the other hand, spatial resolution and molecular-level details are provided by cryoET (Figure 8.8C).
Advances in emerging techniques like cryoET, super-resolution, and correlative microscopy will help us pinpoint interactions of nucleic acid nanostructures with cell membrane and elucidate the intricate arrangement of structural components on the cell surface.
8.5 Conclusion
In this chapter, we summarize the most recent advances in the engineering and computing on the cell surface. In particular, we focus on translational research with the most promising potential applications and emerging imaging techniques.
In order to create more targeted therapeutics, it is necessary to clearly identify the target. Diseased cells often have a number of overexpressed cell markers. Unfortunately these markers are sometimes expressed in healthy cells. In order to correctly identify a diseased cell, more than one marker has to be located on the cell. Recently reported DNA-based computational methods for cellular identification are modular, scalable, and use standard antibodies and synthetic DNA. However, in order to proceed as a therapeutic drug, cell targeted DNA robots have to be able to find their target first. We need to switch to smarter, more targeted therapeutics in order to treat diseases with fewer side effects. The nanoscale machines can interact directly with the surfaces of individual cells of interest and deliver smaller drug doses compared with conventional medicine. In order to increase the production and decrease the cost, it may be beneficial to simplify the design of a nanostructure.
There are still a number of challenges before successful drug delivery is viable. We have to address major barriers like large molecular mass, stability in the blood, and crossing cellular membranes. Chemical stability of DNA nanostructures has to be improved in the presence of bloodstream nucleases and other digestive enzymes. It might be necessary to chemically modify the DNA backbone to increase resistance to degradation. In order to cross the membrane and increase cellular uptake, amphiphilic molecules or peptides could be displayed on the surface of DNA origami objects. For some targets fully or partially eliminating the negative charge of the DNA backbone [66,67] is an option. For other targets, alternative delivery and modification methods will be required.
A treasure trove of the natural membrane machines like rotary and ion pumps, and transport proteins provides the inspiration for DNA channels and pores. Synthetic DNA origami channels spanning lipid membranes can be engineered to harness ion flux with tunable voltage-gating and ion-selective properties. Artificial DNA pores can be designed to actively transport payload across the lipid bilayer. DNA nanostructures are about the size of a virus and can be used for sophisticated drug delivery applications. Currently it is possible to recognize a specific cell type, insert a transmembrane pore, and deliver the cargo. Synthetic DNA channels can also be custom made with adaptable diameter, shape, and surface functionality for detecting desired analyte molecules.