
Cell Surface Enzymatic Engineering-Based Approaches to Improve Cellular Therapies
Ayman F. Abuelela, Kosuke Sakashita and Jasmeen S. Merzaban, Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal, Kingdom of Saudi Arabia
The cell surface represents the interface between the cell and its environment. As such, the cell surface controls cell–cell interactions and functions such as adhesion and migration, and will transfer external cues to regulate processes such as survival, death, and differentiation. Redefining the cell surface by temporarily (or permanently) modifying the molecular landscape of the plasma membrane affects the way in which the cell interacts with its environment and influences the information that is relayed into the cell along downstream signaling pathways. This chapter outlines the role of key enzymes, the glycosyltransferases, in posttranslationally modifying proteins and lipids to fine-tune cells, ability to migrate. These enzymes are critical in controlling the formation of a platform structure, sialyl Lewis x (sLex), on circulating cells that plays a central role in the recognition and recruitment by selectin counter receptors on endothelial cells that line blood vessels of tissues throughout the body. By developing methods to manipulate the activity of these enzymes and hence the cell surface structures that result, treatments can be envisioned that direct the migration of therapeutic cells to specific locations throughout the body and also to inhibit metastasis of detrimental cells such as circulating tumor cells.
Keywords
Glycosyltransferase; fucosyltransferase; sialyltransferase; core 2 N-acetylglucosaminyltransferase; migration; metastasis; stem cells; circulating tumor cell; leukocyte; adhesion
9.1 Introduction
Cell migration is central to many physiological and pathological functions in vivo from immunity to metastasis of cancer cells, to the homing of therapeutic stem cells to a target tissue, and tissue repair. In some cases, it would be desirable to promote migration of particular cells whereas in others inhibition of migration would be preferable. An understanding of the molecules involved in this migration paradigm will permit better development of technologies for therapies.
This chapter will start by introducing the principle of migration of cells to tissues/organs and how this process is controlled by a specific sugar decoration, mainly sialyl Lewis x/a (sLex/a), on cell surface proteins/lipids. This key epitope is created through the activity of specific glycosyltransferases (GTs). When these enzymes are stimulated to become expressed and/or active in a particular cell or are introduced ex vivo, this will result in migration of that cell to specific locations within the body where counter receptors to the epitope, the selectins, are expressed.
The chapter will focus on how controlling the expression and/or activity of these GTs makes it possible to (i) improve the migration of therapeutic cells (i.e., stem cells) to desired locations within the body in order to optimize therapeutic outcomes of disease, and also (ii) inhibit the migration of harmful cells (i.e., circulating tumor cells, CTCs) in order to control metastasis or inflammation. In this chapter, the role of other enzymes in the modification of cell surface architecture related to refining cell function(s) to benefit therapy will also be considered.
9.1.1 Multistep Paradigm of Cellular Migration
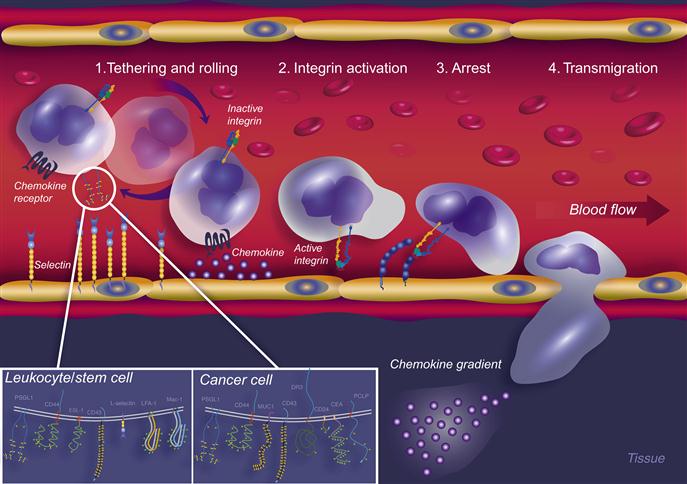
Nature has developed an extremely efficient mechanism for the delivery of circulating cells to specific sites in the body in immune processes such as inflammation and injury. An interesting analogy exemplifying cell movement within the blood stream is offered by Ehrhardt et al. [1], which states “Comparable to a human being plunged into a roaring river, a leukocyte (cell in flow) is exposed to high shears within the mainstream of the blood.” In whichever scenario, arrest and exit from the flow represent a particularly complex task for human or cell. The emigration of cells out of flow (such as in the blood circulation) is a sophisticated process controlled by various adhesion molecules. These comprise, in particular, selectins (E, P, and L), chemokines, and integrins, which function in a coordinated multistep cascade (Figure 9.1) [2–4]. The first essential event in cellular recruitment involves shear-resistant adhesion of flowing cells onto the endothelial surface, this being most efficiently mediated by the selectins and their respective ligands [5,6]. These interactions initially tether the cell in flow to the vessel wall and, in the context of vascular shear flow, cause the tethered cell to roll along the endothelium lining the inside of the vessel. Selectin-dependent tethering and rolling (Step 1) thus decreases cellular velocity below that of the prevailing hemodynamic stream and brings the cell into close physical proximity with the vessel wall. This process facilitates engagement of specific cell-borne chemokine receptors to pertinent chemokines present in the perivascular areas [7–9], thereby triggering activation through inside-out signal transduction events [10–13] leading to increased adhesiveness of integrin family members (Step 2). Adhesive engagements between the activated cell integrins and their cognate endothelial cell counter receptors then lead to arrest and firm adhesion of the cell to the vessel wall (Step 3) and, ultimately, the process of transmigration (Step 4) [14] to reach the extravascular space. This process was first described in the context of leukocyte recruitment to inflammatory sites and has subsequently been applied to describe other biological migration activities such as the movement of circulating hematopoietic stem progenitor cells (HSPCs) to the bone marrow during bone marrow transplants, and in the metastasis of the CTC. The migration of the cell out of circulation and into the target tissue or organ depends on each of these distinct classes of molecules acting sequentially, each step being a prerequisite for the next. In fact, it has been shown in numerous biological scenarios that the selectins, as the most effective mediators [15] of Step 1 interactions, are essential for recruitment of cells altogether and may be the gatekeepers of the multistep paradigm [16]. In fact, it is suggested that the selectins may even be able to substitute for the absence of chemokine receptor signaling events, in the case that chemokines are not present [17,18].
 ) and N-glycans are shown as green hexagons (
) and N-glycans are shown as green hexagons ( ).
).9.1.1.1 The Selectins
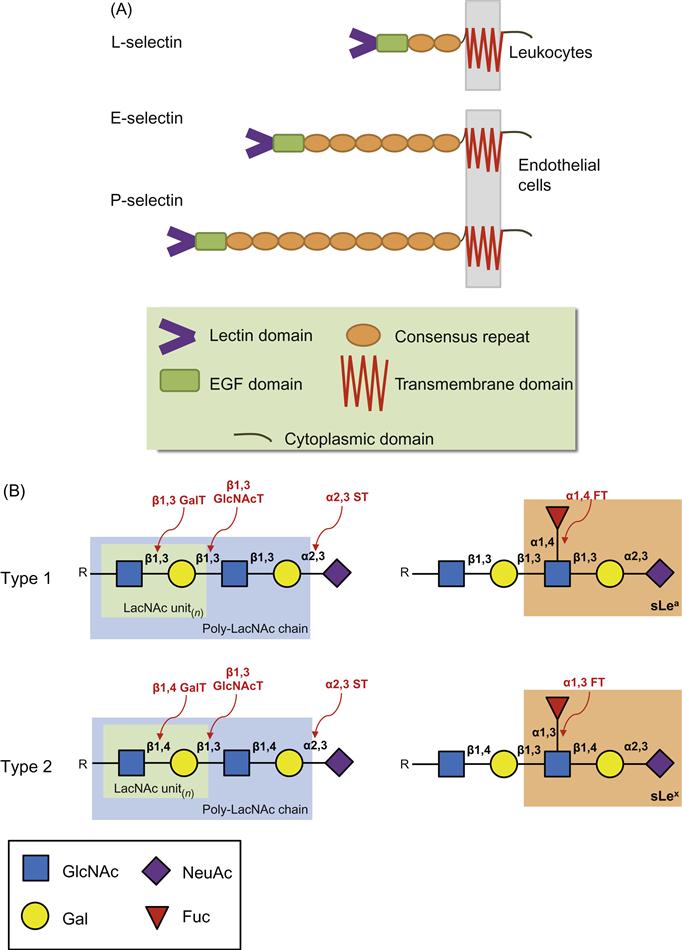
The selectins are a family of three C-type (Ca2+-dependent) lectins expressed exclusively by bone marrow-derived cells and endothelial cells: L-selectin (expressed on Leukocytes and HSPCs), E-selectin (expressed on Endothelial cells), and P-selectin (expressed on Platelets and endothelial cells). Due to their expression on the endothelium, E-selectin and P-selectin are often referred to as the “vascular selectins” whereas L-selectin is termed the “leukocyte selectin.” These molecules are type I transmembrane glycoproteins sharing a similar topology which comprises an amino-terminal lectin-like domain, an epidermal growth factor (EGF)-like domain, a variable number of consensus repeats (CRs; also known as complement regulatory proteins or “sushi” domains), a single membrane spanning domain, and a carboxy-terminal cytoplasmic domain (Figure 9.2A) [4,6]. The lectin domain primarily mediates ligand binding with minimal contribution provided by the EGF-like domain [19–22]. The number (and hence length) of sushi domains facilitates the recognition between selectins and their ligands as it allows a selectin to extend beyond the negatively charged cloud of the glycocalyx. The selectins bind to specialized carbohydrate determinants (Figure 9.2B), comprising sialofucosylations containing an α(2,3)-linked sialic acid substitution on galactose, and an α(1,3)-linked fucose modification on N-acetylglucosamine, prototypically displayed as the terminal tetrasaccharide sialyl Lewis x (sLex; or also to its isomer sLea) [16,23]. These determinants, also known as “CD15s,” may be displayed on either a protein scaffold (i.e., a glycoprotein) or a lipid scaffold (i.e., a glycolipid), and are recognized by mAbs such as CSLEX-1, KM93, and HECA-452 [24,25]. Although additional structural modifications, principally involving sulfation, increase the binding affinity of P-selectin and L-selectin to sLex, no known modifications are needed for optimal binding of E-selectin [26,27]. These characteristics of engaging rapidly and with high tensile strength to their ligands help to make the selectins the most important initiators of adhesion under flow. A brief introduction to each of the selectins is provided below.
 ) of the lactosamine with sialic acid (NeuAc,
) of the lactosamine with sialic acid (NeuAc,  )—ST—and the terminal N-acetylglucosamine (GlcNAc,
)—ST—and the terminal N-acetylglucosamine (GlcNAc,  ) of the lactosamine with fucose (Fuc,
) of the lactosamine with fucose (Fuc,  )—FT—are often correlated with cells that migrate or home to tissues where selectins are expressed.
)—FT—are often correlated with cells that migrate or home to tissues where selectins are expressed.L-selectin (CD62L) is expressed constitutively by all myeloid cells, virtually all B cells and naïve T cells, a subpopulation of memory T cells, NK cells, and both early and mature hematopoietic cells in the bone marrow. Unlike the other selectins, however, L-selectin is not expressed on endothelial cells [28]. Functionally, L-selectin is critical for the homing of naïve lymphocytes to high endothelial venules (HEVs) of secondary lymphoid organs [3,27]. L-selectin-dependent lymphocyte rolling on HEV requires localization of L-selectin to the tips of the microvilli characteristic of the surfaces of lymphocytes and other leukocytes, allowing for optimal leukocyte–endothelial interactions. Mapping of L-selectin domains by mAbs has revealed that the NH2 terminal nine amino acids are critical for ligand binding. The EGF-like and the two short sushi domains maintain the spatial conformation and are also important for ligand binding [27]. HEV-borne L-selectin ligands are collectively referred to as peripheral node addressins (PNAds) and include the glycoproteins GlyCAM-1 (glycosylation-dependent cell adhesion molecule-1), CD34, MAdCAM-1 (mucosal vascular addressin cell adhesion molecule-1), podocalyxin, Sgp200, endomucin, and endoglycan. All of these PNAds have mucin-like domains that carry O-glycans [27]. Although the primary role of L-selectin is in promoting lymphocyte homing, its expression by circulating neutrophils may facilitate the secondary tethering of these cells to an already rolling neutrophil at an inflammatory site (secondary tethering) [29,30]; this, however, may be a rare event of which the physiological significance has been questioned [31,32]. If L-selectin is to recognize them, these ligands must be modified by various GTs to give specific sialylated, fucosylated, and sulfated oligosaccharides.
P-selectin (CD62P) is a glycoprotein that is constitutively expressed within secretory granules of endothelial cells (Weibel–Palade bodies) and in α-granules of platelets. Upon activation by inflammatory mediators (histamine, thrombin, LPS), secretory vesicles fuse rapidly with the plasma membrane allowing expression of P-selectin on the surface of the platelet or endothelial cells within minutes [6]. Thus, P-selectin is often involved in early leukocyte recruitment during an inflammatory response. P-selectin is also constitutively expressed at low levels on the endothelium of thymus [33], lung and choroid plexus microvessels [34], bone marrow microvasculature [35,36], in postcapillary venules in skin [37], and on peritoneal macrophages [38]. The transcription of P-selectin can be induced by cytokines such as interleukin (IL)-4 and IL-13 [39,40]. Once expressed on the surface of endothelial cells, P-selectin is rapidly internalized by endocytosis [41]. In addition to mediating the homing of immune cells to inflammatory sites, P-selectin-mediated interactions are also involved in controlling T-cell progenitor (circulating thymic progenitor, CTP) migration from the bone marrow to the site of T-cell development, the thymus [33].
It is important to note that differences appear to exist between the responses of human compared with mouse endothelium to inflammatory cytokines. Whereas IL-1 and tumor necrosis factor-alpha (TNF-α) are potent inducers of P-selectin expression in mouse endothelium, these cytokines only induce the expression of E-selectin (and not P-selectin) in human endothelium [42]. Additionally, experiments in transgenic mice comparing human and mouse P-selectin genes have demonstrated that following injection of TNF-α, the expression of human P-selectin mRNA decreases whereas mouse P-selectin mRNA increases [43]. This suggests that E-selectin may be the more dominant selectin in human inflammation and disease and brings into question the extrapolation to human disease of the emphasis on P-selectin ligand interactions related to mouse models.
E-selectin (CD62E) is a glycosylated glycoprotein that recognizes diverse groups of glycoconjugates on hematopoietic and cancer cells. Unlike P-selectin, E-selectin is not presynthesized in endothelial cells but, rather, is transcriptionally regulated by mediators including TNF-α, IL-1β, IFN-γ, and LPS. However, exceptions to this rule exist including the observations that vascular beds within the skin [37,44] and bone marrow [36,45] constitutively express E-selectin. Expression can occur as early as 2 h after stimulation and decline within 24 h [46]. Despite this delayed expression (as compared to P-selectin), E-selectin overlaps with P-selectin temporarily enhancing leukocyte recruitment during an inflammatory response and functionally leading to a dramatic decrease in the rolling velocity (slow rolling), and enhancing the probability of adhesion [47] and the subsequent steps of the cascade (including integrin activation) [48]. Even following the downregulation of P-selectin, E-selectin continues to enhance leukocyte–endothelial interactions leading to slower rolling velocities and improved adhesion [49]. When the stimulus wanes, E-selectin becomes internalized and degraded in lysosomes [50]. Physiologically, E-selectin-mediated interactions are involved in controlling leukocyte recruitment to injury and inflammatory sites [51], steady-state, tissue-specific homing in cutaneous tropism of skin-homing T cells [52,53], HSPC entry into bone marrow [45,52,53], as well as metastasis of CTCs.
9.1.2 E-Selectin Ligands in Therapy and Disease
Emphasis on E-selectin-mediated interactions and the regulation of migration of human cells in flow is crucial to understanding many physiological phenomena in human disease and therapy since: (i) E-selectin responds differently from P-selectin to stimuli such as inflammatory cytokines, (ii) E-selectin contributes dramatically to decreasing the velocity of the cells in flow for an extended time, and (iii) E-selectin is constitutively expressed in some vascular beds such as the bone marrow, this being important both for an understanding of how to enhance migration of blood stem cells to bone marrow and also for how to inhibit the metastasis of CTCs to this site.
9.1.2.1 E-Selectin Binds Specific Selectin Ligands on HSPCs to Guide them to the Bone Marrow
The vascular endothelial cells within the bone marrow constitutively express E-selectin and vascular cell adhesion molecule-1 (VCAM-1)[36]. Numerous studies in mice, including intravital microscopy studies, have demonstrated the importance of E-selectin, VCAM-1, and stromal cell-derived factor 1 (SDF-1) in mediating the entry of cells in flow (such as HSPCs and immune cells found in the blood) to the bone marrow [54–56]. These findings have established that homing to bone marrow involves a multistep cascade initiated by E-selectin binding to its ligands on the HSPCs to mediate tethering and rolling, followed by SDF-1 binding to CXCR4 (on HSPC) leading to activation of VLA-4 (on HSPC), resulting in firm adhesion mediated by VLA-4 binding to VCAM-1 and subsequent transmigration. The Step 1 effectors of this cascade, E-selectin and its ligands, play a fundamental role in the recruitment of circulating cells to the bone marrow as demonstrated by many studies and reviewed by Sackstein [16].
Studies in both humans and mice indicate that E-selectin is constitutively expressed on marrow endothelial cells [36,54,57], and intravital studies have revealed that HSPC migration to marrow occurs at specialized microvascular beds expressing E-selectin [54]. These and several other independent lines of evidence [45,58–62] have highlighted a central role for E-selectin-dependent interactions in HSPC recruitment to marrow. E-selectin-dependent binding of HSPC to marrow microvessels, either concurrent with transmigration or subsequently within “vascular niches,” could affect hematopoietic activity and with differential effects depending on the target glycoprotein ligand(s). Importantly, it is known that ligation of P-selectin glycoprotein ligand 1 (PSGL-1) can suppress hematopoiesis in mouse [63] and human [64] HSPCs. This fact, together with evidence that non-PSGL-1 and E-selectin ligands can also trigger apoptosis and growth inhibition of HSPCs from human and mouse [64], highlights a broader role for E-selectin receptor/ligand interactions in hematopoiesis. Indeed, recent work by Winkler et al. demonstrated that E-selectin is a crucial component of the vascular HSC niche that actively induces hematopoietic stem cell (HSC) proliferation and if E-selectin is removed from the equation, HSC quiescence is maintained [65]. This suggests that in the absence of a specific E-selectin-mediated proliferative signal, greater numbers of HSCs remain dormant.
Control of this process may be through the expression upon “metabolic activation” [66] of a specific E-selectin ligand with an ability to bind E-selectin and induce the observed proliferation and commitment.
9.1.2.2 E-Selectin/E-Selectin Ligand Interactions Involved in CTC Metastasis, Regulation, and Maintenance
Metastasis is the culmination of an elaborate cascade of events in which a cancer cell breaks away from the primary tumor, enters blood vessels, and travels through the body until it finally extravasates through the vessels of a distant organ to establish a secondary colony. In order to successfully establish a metastatic clone at a distant site, any CTCs must survive the targeted abuse present in the blood stream that may lead to induction of apoptosis or necrosis as well as avoid elimination by immune cells. CTCs that possess these enhanced survival capabilities (and have overcome this “microevolutionary” process) will generate metastatic colonies in distant organs and even “self-seed” [67] the primary site with more hostile tumor cells. Research focused on elucidating the molecular mediators used by CTCs to adhere to the endothelial cells lining vessels at a target site, i.e., bone marrow, may lead to therapies that can be used to dissuade metastasis.
Endothelial cells expressing E-selectin, such as the bone marrow or activated inflamed vessels, are able to capture CTCs displaying E-selectin ligands on their surface, and initiate the adhesion/metastasis cascade that leads to invasion of the tumor cell (Figure 9.1). These ligands have also been implicated in cancer stem cell regulation and maintenance relating to epithelial-to-mesenchymal transition (EMT) and other events necessary for metastatic growth. E-selectin-dependent interactions have been established in promoting metastasis of many cancers including breast, pancreatic, colon, and prostate [68–72]. sLex/a epitopes are significantly upregulated on cancer cells compared with normal cells and this may play a role in promoting CTC adhesion during metastasis [68,72,73]. This increased expression of selectin ligands is due, in part, to altered regulation of the GTs responsible for creating the sLex/a epitope [74,75]. Thus, it is necessary to identify not only the glycoproteins and glycolipids presenting sialo-fucosylated glycans but also the GTs responsible in the upregulated expression of selectin ligands in order to target their downregulation. The role of E-selectin ligands and relevant GTs will be discussed in the following section.
9.1.3 Role of GTs in Controlling Cell Migration
As illustrated in Figure 9.1, the migration of cells to a specified site is dependent on the interaction of a number of ligands and receptor pairs expressed both on the surface of the migrating cell and on the surface of the endothelium, these working together in a coordinated multistep fashion to orchestrate the delivery of cells to a specific site. Although many molecules are involved in organizing the steps involved in this cascade of events, the selectin binding to its ligand(s) literally “gets the ball (cell) rolling” and on its way to enter a tissue/organ. This initial interaction is the prerequisite for the remaining steps in the cascade and is dependent on the careful glycosylation of selectin ligands. The glycan structure most common for the recognition of ligands by selectins is the sLex (or its stereoisomer sLea) addition to the N-/O-glycan on the protein (Figure 9.2). GTs expressed in the ER and Golgi act in an assembly line fashion by adding one glycan at a time to a growing carbohydrate chain whereby the product of one GT reaction becomes the substrate for the following GT. All GTs are type II transmembrane-Golgi/ER localized enzymes with a short NH2 terminal tail in the cytosol followed by a transmembrane domain, which is linked to the catalytic domain in the Golgi/ER lumen by a short stem region. Generally, each GT uses only one of several sugar nucleotide substrates (including UDP-Galactose (Gal), UDP-Glucose (Glc), UDP-N-acetylgalactosamine (GalNAc), UDP-N-acetylglucosamine (GlcNAc), GDP-fucose (Fuc), GDP-mannose (Man), UDP-xylose (Xyl), or CMP-sialic acid (SA)). Each GT also uses one and sometimes more glycan acceptor substrates and, as they are stereospecific, these enzymes are only capable of forming one specific glycosidic bond (i.e., either an α or β anomer) [76]. Glycan synthesis is the consequence of a series of ordered, GT-dependent events that are characterized by glycan polymer elongation or branching. In general, it is the suite of GTs expressed by a cell that will dictate the glycan structures made. However, the expression of specific GTs does not permit prediction of the glycan structures that will be synthesized. Many factors can affect the activity and ability of the GTs to create a given structure such as the availability of sugar nucleotides, the relative location of the GT in the Golgi stacks in relation to the substrates that it can recognize, and even the possibility of modifications of the glycan product by naturally expressed glycosidases [5,77]. In the context of the glycan structures that confer selectin counter receptor activity on a cell, the GT suite will therefore participate in the control of selectin-dependent leukocyte recruitment. Although all three selectins recognize the sLex epitope, each selectin requires particular fine structural details of this epitope or of the peptide backbone to confer optimal binding affinities.
In order for selectin ligands to be formed, GTs responsible for creating the sLea/x structures must be endogenously expressed and active. The native display of these glycan structures on selectin ligands requires the expression and activity of various GTs and sulfotransferases, including the action of α1,3- or α1,4-fucosyltransferases, α2,3-sialyltransferases, β1,4-galactosyltransferases, and β1,6-N-acetyl-glucosaminyltransferases on either a protein scaffold (i.e., N/O-glycans on glycoproteins) or a lipid scaffold (i.e., glycolipids). Specifically, this specialized carbohydrate motif comprises sialofucosylations containing an α(2,3)-linked sialic acid substitution on galactose, and an α(1,4)-linked (for sLea) or α(1,3)-linked (for sLex) fucose modification on N-acetylglucosamine, of either a type 1 (for sLea) or type 2 (for sLex) lactosamine unit, creating a terminal tetrasaccharide sLea or sLex structure, respectively (Figure 9.2). Although E-selectin binds both sLea and sLex, it binds sLex with a higher avidity [78] and, with the exception of human metastatic cancer cells, most human hematopoietic cells and “normal” stem cells prototypically express only type 2 lactosamine units [79,80]. Understanding the expression and regulation of GTs in the creation of E-selectin ligands will guide efforts to manipulate the migration of human cells in circulation. Indeed, knowledge of which GTs are required to create E-selectin ligands has allowed researchers to manipulate cells in order to create ligands on cells ex vivo with the intention of helping direct the migration of therapeutic cells (such as HSPCs) to specific tissues (such as the bone marrow) that express the corresponding selectins [59,79]. Such work will be discussed in the following section.
9.1.3.1 GTs Involved in Creation of E-Selectin Ligands
For the most part, the enzymes responsible for forming E-selectin ligands are similar to those responsible for both L-selectin and P-selectin ligand formation in that they require sLex structures. However, unlike L- and P-selectin ligands, the sLex cap can occur on a number of different glycans from N-glycans to glycolipids. In addition, analysis of FXnull mice (mice with a null mutation of the FX locus that encodes an enzyme within the de novo pathway for GDP-fucose synthesis) by Lowe’s group suggests that E-selectin ligands are much more sensitive than P-selectin ligands to loss of fucose, this further highlighting the importance of fucosylation to the formation of functional E-selectin ligands [81].
9.1.3.2 FTs and E-Selectin Ligand Formation
α1,3-fucosylation is imperative for E-selectin ligand activity and a number of human fucosyltransferases (FTs) have been identified that are able to modify terminal lactosamines. These include FT-III, FT-IV, FT-V, FT-VI, FT-VII, and FT-IX [82,83]. FT-III to FT-VI are all able to modify either sialylated or unsialylated type 2 lactosamines resulting in sLex or Lex (Figure 9.2) structures, respectively, while FT-III (and FT-V) is also able to modify type 1 lactosamines resulting in sLea or Lea.
Analyses of mice with targeted mutations in FT genes reveal that only two of these enzymes are required to form functional selectin counter receptor activity in leukocytes. These studies all support the observation that FT-VII and FT-IV cooperate in the synthesis of fucosylated glycans required for all leukocyte selectin ligand activity [84–86]. Specifically FT-VIInull mice have leukocytosis and their blood leukocytes have reduced E- and P-selectin ligand activity that effectively compromises the recruitment of leukocytes to inflammatory sites in in vivo mouse models of inflammation, such as the thioglycollate-induced peritonitis model [85] and the contact hypersensitivity model (CHS) [84,86]. However, residual recruitment is still evident (particularly with myeloid cells) and is due to FT-IV activity. CHS response studies performed in mice deficient in FT-IV, FT-VII, or both enzymes revealed that, although lymphocyte (Th1 and Tc1) recruitment was absent in FT-VIInull animals, both enzymes needed to be knocked out to inhibit the recruitment of myeloid cells. A subtle decrease in the myeloid cell CHS response was seen in FT-IVnull mice whereas lymphocyte recruitment was unaffected. Overall, these observations support the hypothesis that FT-VII dependent fucosylation of selectin ligands accounts for most of the selectin-dependent adhesion in leukocytes especially within the lymphocyte fraction. The dependence of selectin ligand formation on FT-VII is also paralleled in studies with human T lymphocytes [87–89]. Indeed, studies suggest that FT-IV acts preferentially on glycolipids whereas FT-VII acts on glycoproteins [90] and, interestingly, gangliosides are able to mediate tethering and rolling [91–93] on endothelium. However, glycoproteins are better at facilitating firm adhesion under physiological flow conditions [93] which could also be related to signaling through the glycoprotein selectin ligands to activate integrins directly [17]. Recent studies focused on assessing knockdowns of FT-IV and -VII in human cells suggest that an additional α1,3-fucosyltransferase, FT-IX, is important in decorating E-selectin ligands on human leukocytes but not mouse [94,95]. This is significant since the selectin ligands, particularly those that bind E-selectin, vary between different leukocyte cell populations and among species [96].
9.1.3.3 ST3GalTs and E-Selectin Ligands
α2,3-sialic acid linkages to terminal galactose residues are crucial in the formation of sLex epitopes. The contribution of sialylation to the formation of selectin ligands is well documented as sialidase treatment of leukocytes under static conditions in vitro completely abolishes binding of P-selectin and E-selectin to selectin ligands [97]. This linkage is catalyzed by the activity of the sialyltransferases (STs) ST3GalT-I, -II, -III, -IV, -V, and -VI. Only ST3GalT-III, -IV, and -VI have been shown to sialylate type 2 lactosamines and ST3GalT-IV and ST3GalT-VI are implicated in E-selectin (and P-selectin) ligand formation [97–100]. In vivo inflammatory studies of ST3GalT-IVnull mice reveal a mild but significant defect in selectin ligand function, including a mild reduction in E-selectin-dependent rolling, an increase in E-selectin-dependent rolling velocity, a decrease in L-selectin-dependent rolling during inflammation (but no effect on L-selectin-dependent rolling on HEVs [101]), and no contribution to P-selectin ligands [97,102]. Since sialylation is so crucial to selectin binding but yet the phenotype of the ST3GalT-IVnull is so mild, it is suggested that other STs, particularly ST3GalT-VI, contribute to selectin ligand formation. With the generation of the ST3GalT-VInull and ST3GalT-IV/ST3GalT-VI double knockout mouse, the role of these STs appears to be critical for the generation of functional selectin ligands, especially in the case of the formation of P-selectin ligands (ST3GalT-VI is involved in E-selectin-mediated capturing but not in E-selectin-dependent rolling) [100]. Implications for the involvement of additional STs involved in selectin ligand synthesis and function are still warranted, especially for E-selectin and L-selectin.
9.1.3.4 C2GnTs and Functional E-Selectin Ligands
C2GnT (core 2 β(1,6)-N-acetylglucosaminyltransferase) isoenzymes (C2GnT-I, -II, -III) create the core 2 O-glycan branch [103] by adding GlcNAc to the Galβ1,3GalNAc core 1 structure expressed on serine or threonine residues. Capping of the core 2 branch with a sLex moiety creates the P-selectin counter receptor. Analyses of C2GnT-Inull mice, however, show that it is the isoenzyme essential for PSGL-1 modifications on leukocytes [104–106]. Despite the essential role of C2GnT-I in P-selectin ligand formation, C2GnT-Inull mice possess a relatively mild phenotype, showing only a partial reduction in selectin ligands and essentially no effect on lymphocyte homing [104], reflecting the possible redundancy of the C2GnT isoenzymes. It has been suggested that C2GnT-II and/or C2GnT-III contribute to selectin ligand formation and cell trafficking [107,108]. C2GnT-I and C2GnT-III are expressed primarily by lymphocytes while C2GnT-II is associated with goblet cell mucin production in the intestinal epithelium. A single knockout of any of these enzymes and double knockout of C2GnT-I and C2GnT-III had no effect on the trafficking of lymphocytes in an inflammatory mouse model of the large intestine. However, mice deficient in C2GnT-I and C2GnT-II or all 3 C2GnT enzymes were not able to overcome a helminth infection, suggesting that C2GnT-dependent modifications on lymphocytes may play a role in migration of lymphocytes (to the large intestine) [108].
In the context of E-selectin ligands, a number of studies indicate that cells deficient in C2GnT-I are able to bind to E-selectin, although at a slightly reduced level compared with wild-type cells [104,107,109]. These studies reveal that 75–80% of E-selectin binding is maintained in the absence of C2GnT-I, suggesting that either the isoenzymes are compensating for the loss of C2GnT-I or that other E-selectin ligands (besides PSGL-1) are dominant mediators of this interaction.
9.2 Use of Enzymes to Modify Cell Surface Carbohydrates on Proteins and Lipids to Enhance Migration to Tissues
Direct injection of therapeutic cells into target tissue sites is only practical for a limited number of applications including pancreatic beta islet transplants through the portal vein into the liver or intracoronary infusions of myoblasts into damaged heart muscles. For the vast majority of therapeutic cellular-based therapies that are targeting systemic multifocal sites of injured or inflamed tissues or organs, intravenous transfer is the routine method of administrating cells. Therefore, it is crucial that the efficient homing of donor cells to the site of intended action be ensured. Since only a subset of intravenously injected cells may engraft in the intended location due to the absence of the key homing molecules on infused cells, the development of methods for improving therapeutic cell trafficking is a high priority. One relatively simple cell engineering method uses the GT enzymatic machinery responsible for creating elaborate glycan structures on cell surface proteins to program the in vivo trafficking of systemically delivered cells. Through the introduction of GTs important for creating the key carbohydrate structure, sialyl Lewis x/a (sLex and sLea), on the cell surface, infused cells can then be attracted by the cognate selectin counter receptor expressed on the endothelium and direct its subsequent migration. These GTs can be introduced genetically using expression vectors containing the GT gene of interest, induced through cytokine signaling mechanisms, or using recombinant active GTs to add on glycans ex vivo.
9.2.1 Genetic Approaches for Modification of Cell Surface Glycoconjugates
It is possible to introduce into cells genes that are required to achieve the formation of the glycoconjugates on the surface of cells that render selectin ligand binding.
Among the 13 family members recognized in the human genome, eight FTs (FT-III, FT-IV, FT-V, FT-VI, FT-VII, FT-IX, FT-X, and FT-XI) are reported to possess the alpha-1,3-fucosyltransferase activity which potentially could confer formation of the sLex structure with distinctive substrate specificities [110,111]. In particular, it has been shown that FT-III, FT-IV, FT-V, FT-VI, and FT-VII are involved in the formation of the selectin ligand and by inhibiting the expression of these FTs in various cell types including leukocytes [95], prostate cancer cells [71], and gastric cancer cells [112], selectin binding is inhibited. Consequently, FTs can be considered as good candidate genes to introduce to therapeutic cells and enhance sLex formation. The mode of introduction of FTs into cells could be either transient or stable. Since the formation of the sLex moiety is a prerequisite only in the initial step of the transition of cells to a tissue, transient expression of FT could adequately satisfy this requirement. The benefit of the transient expression strategy compared with stable expression through viral vector-mediated gene integration is that the former approach avoids genetic disturbance of the genome and so minimizes the likelihood of spontaneous oncogenesis.
Successful physical approaches for the delivery of plasmid vectors into cells for transient expression have been reported using lipofection [113,114] or electroporation [115,116], in both cases with good transfection efficiency, high viability and recovery of transfected cells, and without any significant effect on capability for differentiation or proliferation.
One potentially intriguing approach to overcome the relatively lower efficiency of transient transfection is the use of a nonintegrating version of foamy virus-based vector [117], which has been shown to transduce stem cells rather effectively [118]. Although this vector system is still under development, it could permit a high efficiency by virtue of being a virus-based transduction while at the same time keeping the genome of the target cells intact.
Stable expression of exogenous genes can be achieved using genomic integration mediated by virus-based vectors. Various viral vector systems have been used for transduction of cells including lentivirus-, retrovirus-, adenovirus-, adeno-associated virus-, or baculovirus-based vectors to achieve long-term and stable expression of exogenous genes [119,120].
Although ex vivo enzymatic treatments are generally viewed as being more desirable in terms of simplicity and efficiency, it would seem reasonable to employ a viral vector approach when the introduction of transgenes or correction of a genomic mutation is required.
9.2.2 Cytokine and Growth Factor Induced Expression of GTs Within Cells
Several cytokines are implicated in the regulation of the key GTs involved in selectin ligand synthesis of leukocytes and other circulating cells, such as hematopoietic stem/progenitor cells, as they undergo differentiation and specialization.
In T cells, expression and regulation of GTs important in forming P- and E-selectin ligands require antigen-stimulated activation [121–124]. Following this finding, a number of studies focused on defining how key cytokines involved in the activation of T cells affect the expression of GTs that create sLex epitopes on E- and P-selectin ligands. In vitro activation studies using cytokines that induce the polarization of CD4+ and CD8+ T cells toward activated TH1 (TC1) or TH2 (TC2) subsets demonstrated that the GTs C2GnT-I, FT-VII and ST3GalT-IV and -VI are differentially regulated depending on cytokines used. TH1 cytokines, including IL-12, -2, -15, and TGF-β tend to enhance the expression of C2GnTs [107,125,126], FT-VII [127–131], and ST3GalTs [127,132] and induce binding to E-selectin and P-selectin, while TH2 cytokines, such as IL-4 and -5, repress their expression. Based on their potent impact in vitro, these cytokines were expected to modulate homing receptor formation under in vivo conditions. Although these cytokines appeared to regulate C2GnT-I in vitro, there was no effect on the selectin ligand formation and proliferative responses in the absence of these cytokines in vivo [133].
Other signals have been implicated in the regulation of selectin ligand formation, such as the vitamin A and D derivatives. A recent study illustrated that exposure of human T cells to physiological levels of retinoic acid (RA) and 1,25D(3) completely eliminated E-selectin ligand activity and subsequent skin-homing abilities, whereas the inactive/precursor forms of vitamins A and D had little inhibitory action [134]. Analysis of GTs involved in forming sLex structures revealed that FT-VII and ST3GalT-III were significantly reduced upon exposure to these vitamin derivatives. This finding is intriguing as RA is currently being used successfully as a differentiation therapy for the treatment of acute myeloid leukemia (AML) subtype 3. In addition to inducing the differentiation of the diseased leukemic cells, it may also influence the ability of the leukemic blasts/stem cells to migrate to the bone marrow where E-selectin is expressed by inhibiting the GTs involved in creating E-selectin ligands.
In hematopoietic stem/progenitor cells and myeloid cells, G-CSF (granulocyte-colony stimulating factor) significantly enhanced the expression of sLex on HSPCs and their subsequent ability to bind E-selectin and home in vivo. This was accompanied by an associated increase in the expression of the GTs, ST3GalT-IV, FT-IV, and FT-VII [135]. Studies on human CD34+ cord blood stem cells exposed to dimethyl-prostaglandin E2 (dm-PGE2) have demonstrated that this treatment increases HSPC numbers and enhances their homing [136–138]. However, to date, this enhanced homing has not been correlated with measurements of GT or sLex expression or selectin binding.
Immune blood cells, such as lymphocytes and macrophages, are known to secrete inflammatory factors that could potentially influence cancer cell metastatic progression. In particular, the proinflammatory cytokines, TNF-α, and IL-6 have been reported to be elevated in the blood serum of patients diagnosed with advanced stage breast cancer and this has been correlated with an increase in the number and size of metastatic sites [139]. These cytokines have also been shown to promote the growth and invasiveness of breast, colon, and prostate cancer cells [140–142]. In addition, cancer cell chemokine receptors have also been implicated in playing a role in identifying the destination of metastases [143]. It can be inferred that these cytokines lead to enhanced expression of GTs involved in the creation of sLex epitopes on ligands of these cancer cells, which results in enhanced adhesion to selectins [140]. This process will be discussed later.
9.2.3 Use of Recombinant GTs ex vivo
GTs expressed in the ER and Golgi act in an assembly line fashion by adding one glycan at a time to a growing carbohydrate chain decorating a particular glycoprotein. Depending on which GTs are expressed in a particular cell, a diverse set of structures can be made. In order for selectin ligands to be formed, GTs responsible for creating the sLex structure need to be endogenously expressed and active, as outlined in Figure 9.2. The knowledge of which GTs are required to create particular structures allows researchers to manipulate cells in order to create ligands on cells’ surface ex vivo with the intention of helping to direct the migration of therapeutic cells to specific tissues that express the corresponding selectins [59,79,144].
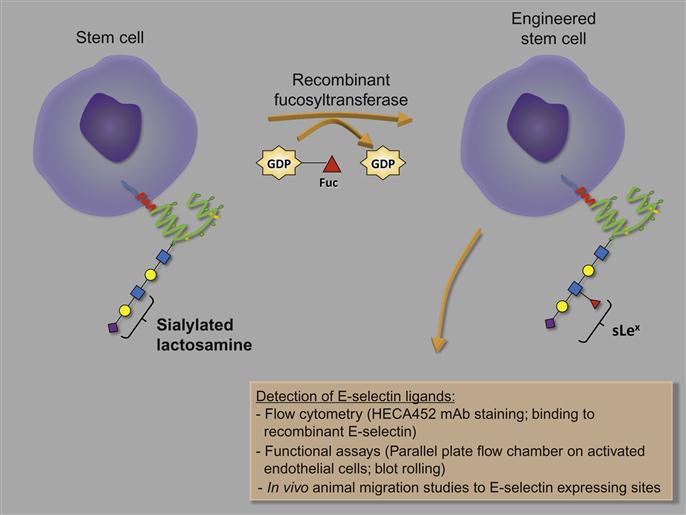
The microvasculature of human and murine bone marrow constitutively expresses E-selectin and, as described earlier, extensive research has determined that HSPC migration to the bone marrow depends on the expression of ligands that bind E-selectin. Both mouse and human HSPCs express a number of E-selectin ligands. Studies to identify the glycoprotein ligands revealed that on human CD34+ HSPCs, at least three different ligands contributed to E-selectin binding. These include the hematopoietic cell E-/L-selectin ligand (HCELL; sLex-decorated glycoform of CD44 that binds E-selectin and L-selectin), cutaneous lymphocyte antigen (CLA; sLex-decorated glycoform of PSGL-1 that binds E-selectin), and a sLex-decorated glycoform of CD43 [96]. In contrast, on mouse HSPCs only CLA and CD43 were identified as ligands suggesting that the modest E-selectin binding detected in mouse HSPCs compared with human HSPCs is due to the absence of HCELL. Indeed, inducing the expression of HCELL on mouse HSPCs leads to significant enhancement of E-selectin binding in mouse HSPCs [96]. HCELL expression was achieved using an innovative tool to custom engineer desirable cell surface glycoforms (i.e., sLex) on therapeutically beneficial cells (i.e., stem cells) using recombinant GTs to transform the cell surface glycans temporarily in order to enhance their migration to specific sites (i.e., E-selectin on bone marrow microvasculature).
Selectin ligands must be α1,3-fucosylated to form the terminal glycan sLex determinant. Often stem cells are inadequately α1,3-fucosylated, this being the case with MSCs [59], HSPCs isolated from cord blood [61,144], as well as with neural stem cells (unpublished data), and so exhibit homing defects. Indeed ex vivo treatment of stem cells (principally human MSCs and HSPCs derived from human cord blood) with FTs, particularly FT-VI and FT-VII, increases cell surface sLex determinants, boosts binding to E-selectin and P-selectin, and enhances homing and engraftment into bone marrow of immunocompromised mice [59,144–146]. By generating a comprehensive set of tools (i.e., GTs) for ex vivo design of sLex structures on the surface of a therapeutic cell, strategies to enhance the migration of these cells for the treatment of diseases can be further anticipated.
For many years, scientists have been interested in understanding and characterizing the complex nature of carbohydrates displayed on cell membranes and the GTs responsible for creating these molecules. Early studies focused on isolating small amounts of GTs from natural sources and using these enzymes in reactions that included radioactive donor sugar nucleotides to rebuild cell surface structures following their removal using glycosidases; the proteins that resulted were then identified by SDS-PAGE [147,148]. These studies also helped to characterize carbohydrate-dependent binding interactions such as those between pathogens and host cells. The use of GTs to manipulate glycans became much more practical with advancements in recombinant DNA technologies that allowed for the expression of proteins and enzymes on a large scale. The potential for creating libraries of recombinant GTs for fine tuning glycans on cells or proteins brings extensive possibilities, such as enhancing the stability of proteins, creating enhanced bioactive/humanized mAbs [149–151], and directing the movement of cells within the body.
The use of recombinant GTs to modify cells ex vivo has been elegantly formulated and discussed by Sackstein [59,79,152–154]. He outlines the guiding principles (summarized in Table 9.1) and methodological details (Figure 9.3) required for the custom engineering of cell surface glycans of a cell population of interest, particularly stem cells, using his “GPS” (glycosyltransferase-programmed stereosubstitution) technology [152,153]. Any GT enzyme can be used to create the glycan of interest using this technique but special attention must be given to ensure that the enzyme assay conditions (storage buffer, reaction buffer) are optimized to maintain cell viability and avoid phenotypic effects (such as the differentiation) on cell(s). One major concern is that many GTs require divalent cations for their catalytic activity [156]. Accordingly, the recombinant human FT-VI enzyme and reaction conditions specifically developed by Sackstein effectively catalyzed α(1,3)-fucosylation at physiologic pH in the absence of divalent cations [59].
Table 9.1
Guiding Principles Developed by Sackstein [152,153] to Apply GPS Technology to Cells
Strategy for Custom Engineering Cell Surface Glycans
I. Identify target glycoconjugate “acceptor”
II. Use appropriate enzyme(s) that result in stereospecific carbohydrate epitopes
III. Confirm the creation of the target modification biochemically and functionally in vitro and in vivo
Many mammalian proteins have been expressed and prepared as active recombinant proteins [157–159]. A number of different expression systems could be used (bacterial cells, mammalian cell lines, baculovirus-infected insect cells, yeast cells, and even silk worm) to express these GTs but, since several reports have shown that N-glycosylation is necessary, higher order systems are preferred for optimal enzymatic activity; in fact, bacteria-expressed FTs exhibit no detectable activity [160,161]. As bacteria-based systems would not work and production of large amounts of the required enzymes is necessary, Sackstein et al. chose to use the methanotrophic yeast Pichia pastoris expression system. This system offers the advantages of generating a high yield of recombinant proteins and an ability for the proteins to be secreted into the medium which facilitates downstream preparation of the enzyme [162]. Recent efforts have been made to investigate the expression and isolation of GTs from silkworm. This is a clear option for the preparation of recombinant enzymes, which cannot be expressed functionally in bacterial expression systems. The silkworm approach uses pupal or larval individuals of a lepidopteran insect, Bombix mori, and a baculovirus-based vector for exogenous gene expression [163]. This system brings several benefits including optimal folding and posttranslational modifications resulting in proper functionality of the many recombinant proteins, an extremely high yield, great amenability to scaling-up, and a short time period for production of the protein. Although few laboratories currently have the necessary technology, several companies offer a commercial protein expression service, which uses this system.
9.2.3.1 Glycans and Their Importance in Mediating Engraftment
Once HSPCs have used their newly acquired homing molecules to enter the bone marrow, successful transplantation then requires engraftment of the stem cells within their particular niche. Implications for the activity of GTs depend not only on improving the homing of therapeutic cells to their niche but also on the successful engraftment of these cells. Normally HSCs are located within specific regions in the bone marrow that dictate the behavior of these cells. The complex interplay of cells and molecules helps to regulate HSPC quiescence, division, and differentiation. Various cell types have been identified that contribute to the HSPC niche, including various mesenchymal stem cells (CD146+ perivascular MSCs [164], nestin+ MSCs [165], and leptin receptor+ perivascular MSCs [166]), CXCL12 abundant reticular (CAR) cells, and MSC-derived osteoblast lineage cells [167–169]. In addition, endothelial cells form an overlapping HSC niche [166,170]. These niches also appear to be further regulated by local sympathetic nerves [171]. A model has been proposed whereby the “osteoblastic” niche (endosteal surface of bone) harbors and maintains HSCs in a quiescent state, whereas the “vascular” niche (marrow vessels) promotes HSC proliferation and differentiation [65,172,173]. However, many recent studies have challenged this model [174] stating that the osteoblastic niche is actually extensively vascularized [55,175], and that since the most potent and quiescent HSCs prefer to reside in hypoxic [176,177], poorly vascularized regions, these may not reside in the “osteoblastic” niche of the bone marrow. A particularly interesting study has demonstrated that E-selectin, expressed on around 20% of bone marrow endothelial cells (BMECs), directly promotes HSC proliferation and, in the absence of this selectin, HSC quiescence and self-renewal potential increases [65]. These findings suggest that, in addition to directing the migration of HSPCs to the bone marrow, E-selectin—through a currently undefined ligand—also plays a key role in regulating HSC engraftment and fate. Currently, quite which E-selectin ligand(s) on the HSC mediate this proliferation remains unknown as studies focused on knocking down the key E-selectin ligands identified to date do not present the same HSC phenotype [65,96]. Further studies are warranted to identify these ligands.
9.3 Modification of Enzyme Activity Leads to Changes on Cell Surface Structures that Deter Migration and Metastasis
Metastatic colonization, outgrowth at a distant site, starts with the interaction between CTCs and endothelial cells lining lymph and postcapillary vessels. This interaction involves many adhesion molecules and molecular pathways, such as selectins, integrins, intercellular adhesion molecules (ICAMs), and chemokines with their receptors. These molecules work collectively in a process that obeys the multistep paradigm of HSPC recruitment to bone or leukocytes to sites of inflammation (Figure 9.1). Although still debated, “hematopoietic mimicry” or the “homing concept of metastasis” hypothesis states that at Step 1 the circulating cell, moving under high shear force of circulation, uses selectin-mediated low affinity reversible rolling interactions with endothelial cells expressing E-selectin and P-selectin [68,178,179]. This interaction facilitates extravasation of CTCs and determines organ-specific metastasis depending on the tumor type and metastatic environment. Selectins are the receptors for many glycoconjugate selectin ligands.
9.3.1 Selectin–Selectin Ligand Axis in Metastasis
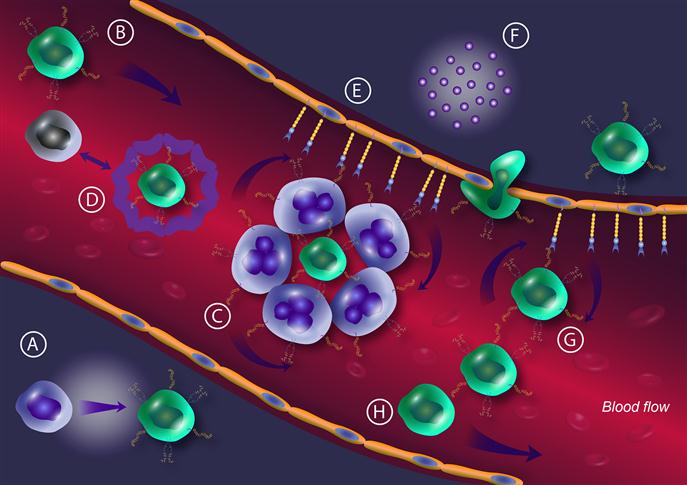
The role of the selectin–selectin ligand axis in promoting cancer metastasis is supported by a number of findings described below and is summarized in Figure 9.4.
Many types of metastatic epithelial tumors, including breast [74,180,181], prostate [182,183], colon [184–187], lung [188,189], and pancreatic carcinomas [190], show a substantial ability to roll on endothelial cell layers natively expressing E-selectin. Although this interaction alone is not sufficient to explain the organ specificity of metastasis, it is the initial interaction and the primary adhesion that directs organ-specific metastasis. Lewis lung cancer cells tend to metastasize preferentially to liver where endothelium shows upregulation of E-selectin expression [191,192]. Additionally, prostate and breast carcinomas tend to metastasize to bone, where E-selectin is constitutively expressed and, interestingly, highly metastatic cells show a stronger interaction with bone marrow endothelium compared with weakly metastatic carcinomas [181–183,191,192].
Experiments in mouse models deficient in or overexpressing one of the selectins indicate a direct correlation with selectin expression and metastatic potential. For example, Biancone et al. [193] demonstrated a reversion of metastasis from lung to liver when E-selectin was upregulated in vivo. Another study showed that metastasis is inhibited upon immunoglobulin E-selectin blockade [194]. Moreover, studies in P-selectin and L-selectin deficient mice displayed impaired metastatic potential [195,196]. A growing body of evidence supports the proposal that inflammation potentiates cancer dissemination and progression. Immune cells lying in a tumor microenvironment both at the primary site and at premetastatic sites release proinflammatory cytokines that not only induce the upregulation of selectin expression on endothelial monolayers but also upregulate the enzymes responsible for carbohydrate ligands (GTs) on cancer cells. For instance, E-selectin expression in colorectal tissues is directly proportional to inflammatory reactions at the lesion–environment interface [191,192,197], and this may explain the sensitivity of colon cancer to anti-inflammatory therapy [198].
Selectins support the formation of metastatic emboli. L-selectin and P-selectin expressed on leukocytes and platelets, respectively, bind selectin ligand glycoconjugates and soluble mucins on the surface of CTCs forming metastatic emboli [199]. Metastatic emboli support metastasis in two ways: first, they allow better interaction with E-selectin-expressing endothelium through ligand-expressing leukocytes, and secondly, the coat of platelets is thought to offer a shield against the immune system [195,200].
Carcinogenesis is associated with abnormal GT expression [73] and, hence, typically a shift toward increased sLex/a expression [201–203] (as outlined below). In fact, levels of sLex within tumors correlate with malignancy and patient survival [75,204–208].
Selectin ligand binding, perturbation in GT expression, and subsequent sLex/a expression are correlated with prometastatic modifications such as EMT [209]. EMT is a rapid transformation process involved in embryogenesis, wound healing, and invasiveness of cancer cells, within which a polarized epithelial cell undergoes cellular and biochemical changes to transform into a mesenchymal phenotype. Mesenchymal phenotype is characterized by loss of polarization, increased invasiveness, high motility, elevated resistance to apoptosis, and expression of extracellular matrix components [210–214]. Cancer cells undergoing this transformation, often at the tumor host interface, tend to invade and metastasize to surrounding and distant tissues leading to life-threatening manifestations of cancer progression [215–223]. A study by Sakuma et al. [209] showed that EMT induction in colon cancer cell lines using EGF and bFGF caused elevated expression of sLex and sLea as well as E-selectin binding. These changes were linked to upregulation of ST and FT expression. Pinho et al. [224] reported that there is a negative correlation between sLex and E-cadherin expression in two mammary tumor models. The loss of the latter is a well-known marker for EMT.
9.3.1.1 Selectin Ligands and Metastasis
To date, many selectin ligands are known to be expressed on carcinoma cells, including PSGL-1 [182,225], CD44v [181,185,226–228], podocalyxin-like protein (PCLP) [229], death receptor-3 (DR-3) [186], gangliosides [93], mucin 1 (MUC1) [180,230,231], CD43 [232,233], and Mac-2BP [234] (Figure 9.1). PSGL-1 is the most characterized selectin ligand at the cellular, molecular, and functional levels in addition to being the only ligand currently known to be able to bind all three selectins. PSGL-1 is a type 1 transmembrane sLex/a-bearing mucin-like glycoprotein of 402 or 412 amino acids [235,236]. Moore et al. [237] first identified PSGL-1 as a P-selectin ligand on myeloid cells including neutrophils and HL-60 cells. PSGL-1 is a disulfide homodimer with apparent molecular weights of 240 kD (dimer) and 120 kD (monomer) on SDS-PAGE. The extracellular domain is rich in serine, threonine, and proline, and consists of 15 or 16 decameric repeats extended linearly due to the presence of multiple serine/threonine O-glycosylated sites that straighten the backbone of mucin proteins [238]. Three N-terminal tyrosine amino acids at positions 5, 7, and 10 are arranged in a consensus sequence that favors tyrosine sulfation. The cytoplasmic domain of PSGL-1 has 69 amino acids with signaling functionality but with no consensus sequence for phosphorylation [235]. PSGL-1 is expressed on hematopoietic cells and cancer cells. CD34+ HSPCs and myeloid cells at different states of maturation (except erythrocytes and megakaryocytes) and all peripheral blood leukocytes express PSGL-1 [239,240]. Expression of PSGL-1 on platelets has been reported by Frenette et al., although remains debated [241]. PSGL-1 is also expressed by leukemic cells [242] and carcinoma cells such as colon [225] and prostate cancer cells [182]. PSGL-1 binds to E-selectin in a different way than to L-selectin and P-selectin. Both sialylation and α1,3-fucosylation, that appear on core 2 O-glycan chains, are required for E-selectin binding but sulfation (of either sLex moiety or tyrosine) is not required [243]. The binding to E-selectin of PSGL-1 expressed by CTCs implies a functional role in bone tropism of metastatic cancer cells. Dimitroff et al. demonstrated that PSGL-1, expressed on the surface of the bone-metastatic cell line MDA-PCa-2b, supports rolling on BMECs. This rolling was abolished by using neutralizing antibodies against E-selectin, suggesting that E-selectin/E-selectin ligand interactions mediate this adhesion. Neuraminidase treatment of the MDA-PCa-2b cells also impaired rolling indicating the importance of sialic acid in mediating this interaction and subsequent rolling. Performing a Western blot analysis on the membrane fraction of MDA-PCa-2b for the expression of sLex (detected by HECA-452 mAb) revealed reactivity with CD44 in addition to PSGL-1 [182,183].
Many reports have characterized the binding of CD44 to E-selectin on HSCs, leukocytes, and CTCs. CD44 is known to direct the recruitment of lymphocytes toward inflammatory sites through its interaction with matrix HA (hyaluronin) and endothelial cell expressed E-selectin, and a similar interaction is thought to direct the metastasis of many cancer types [80]. The E-selectin binding form of CD44 is known as HCELL. HCELL is expressed on the surface of HSPCs, human leukemia cells and has recently been located on colon cancer and breast cancer cells. Unlike mucin-like glycoproteins, CD44 bears sLex/a structures on N-linked glycan branches [181,185,226–228,244].
MUC1 is a large heavily glycosylated transmembrane protein with an extracellular region comprising a variable number of tandem repeats that include 20 conserved amino acids (HGVTSAPDTRPAPGSTAPPA) each with five potential sites of O-glycosylation (as underlined). In normal cells, MUC1 is expressed on apical surfaces and glycosylated with core 2 branched O-glycans and lactosamine extensions, whereas in cancer cells MUC1 is expressed on the whole surface and decorated with aberrantly truncated O-glycosylated carbohydrate antigens. MUC1 expressed on breast cancer cells tends to present sialylated and unsialylated core 1 structures instead of core 2 structures. It has long been known that ST expression and activity is higher in breast cancer cells than in normal breast tissue. Thus, the exposure of truncated glycan structures may be due to sialylation of core 1 GalNAc or lactosamine structures leading to a blockade of further extension and expression of T and Tn antigens [245]. The MUC1 interaction with E-selectin was first described in colon cancer cells by Zhang et al. [231]; MUC1 from a colon cancer cell line or colon cancer patient serum was shown to be able to inhibit the binding of leukemic cells to E-selectin-expressing cells. MUC1, expressed by colon cancer cells, was reported to bind E-selectin better than CD43 from the same cells under flow conditions [230]. A recent study by Geng et al. [180] used E-selectin and ICAM-coated microtubes as a model for microvascular endothelium to show that the MUC1-expressing metastatic breast cancer cell line, ZR-75-1, was able to roll and adhere to the walls of the microtube under flow conditions, so implicating MUC1 as a ligand for E-selectin on breast cancer metastatic cells. CD24, another mucin-like glycoprotein, was also identified as an E-selectin and P-selectin ligand in breast cancer cell lines (KS and MCF-7) [246,247].
More recently, a number of novel ligands have been identified as E-selectin ligands on cancer cells including PCLP on colon carcinoma cells [229], Mac-2BP on breast cancer cells [234], and DR-3 on colon cancer [186]. In addition to these glycoproteins, gangliosides have also been shown to play a role as E-selectin ligands in breast cancer cell lines [93]. Mac-2BP is a highly glycosylated protein whose expression is correlated with cancer metastasis. Shirure et al. were able to immunoprecipitate E-selectin ligands from metastatic breast cancer (ZR-75-1) lysate using recombinant E-selectin and identified Mac-2BP as a novel ligand through MS analysis. Gene silencing of Mac-2BP significantly reduced the binding of ZR-75-1 cells to E-selectin-expressing human umbilical vein endothelial cell (HUVEC) cells in flow assays [234]. All these studies highlighted the role of selectin/selectin ligand interaction in supporting metastasis. Since it is the glycan decorations, primarily the expression of sLex/a on these proteins (and lipids), that mediate this interaction of the CTC with the endothelial E-selectin, the contribution of GTs to migration and metastasis-supporting programs such as EMT needs to be better understood.
9.3.1.2 Perturbation in the Expression of GTs in Metastasis
It is widely accepted that abnormalities in glycosylation are closely associated with malignant transformation and progression [201–203]. Accordingly, highly metastatic carcinomas (e.g., colon, breast, and prostate) exhibit high levels of sLex/a expression [74,180–183,185,186,225]. sLex/a biosynthesis involves the concerted action of four GTs [68] and, hence, any dysregulation in GT expression and function has a direct impact on selectin-mediated metastasis. For instance, C2GnT is overexpressed in colorectal adenocarcinoma [248,249]. Lung carcinoma cells were found to acquire a colonizing phenotype upon FT-VII overexpression [188,189,250] and, in colon carcinoma, FucT-III gene silencing caused downregulation of sLex expression and subsequent inhibition of tumor proliferation and metastasis to the liver [251,252]. Matsuura et al. [74] investigated the expression of FTs and STs in breast cancer samples and cell lines and found that FT-VI and -III are significantly elevated, and that by expressing these FTs in breast cancer cells normally negative for sLex and sLea expression (MCF-7), rolling events are amplified on IL-1β activated HUVECs. One decade later, Barthel et al. reported similar results, i.e., elevated expression of FucT-III and -VI in the metastatic prostate cancer cell line, MDA-PCa-2b, compared with nonmetastatic cell lines. When PC3 cells, a prostate cancer cell line that is normally negative for E-selectin binding, were transfected with FucT-III, -VI or -VII, the cells gained sLex expression and the ability to roll on E-selectin-expressing endothelial cells and to metastasize to bone [71,201].
Moreover, it was demonstrated that the most highly expressed ST in patient samples of breast carcinoma was ST3Gal-III. This ST is responsible for sLea biosynthesis and its overexpression was associated with shorter overall survival [253,254]. Correspondingly, ST expression in metastasizing mammary tumors is higher compared with levels in nonmetastasizing tumors [255]. A recent report has indicated that induction of EMT, a prometastatic transformation program, was accompanied by elevation in levels of ST3Gal-I/-III/-IV and FT-III which are responsible for terminal capping of lactosamine units with sialic acid and fucose residues to form sLex/a moieties on colon cancer cells [209]. In combination, these studies indicate the fundamental role of GTs as modifiers of surface glycome features in supporting cancer metastasis and also reveal the potential significance of GT inhibitors as basic research and therapeutic tools.
9.3.2 Metabolic Inhibition of GTs
In common with all glycans, sLex/a is not directly encoded by the genome and its upregulation is achieved through overexpression of GTs responsible for its biosynthesis. The tetrasaccharide sLex/a requires four GT enzymes to catalyze its biosynthesis: N-acetylglucosaminyltransferase, β-galactosyltransferase, α-fucosyltransferase, and α-sialyltransferase. A detailed biosynthesis pathway of sLex/a and an explanation of the differences in glycan structure recognized by the three different selectins are outlined in Figure 9.2. Glycoside bond synthesis by the Leloir pathway involves the transfer of a sugar from an activated sugar nucleotide donor, nucleoside diphospho sugar (NDP sugar), or a nucleoside monophospho sugar (NMP sugar), to the hydroxyl group of an aglycone acceptor, catalyzed by the action of a GT enzyme. GT action may either have an overall retaining or inverting effect on the configuration at the anomeric center of the donor sugar [156]. sLex/a acts as a recognition marker for selectin and is involved in diverse biological processes including cell–cell adhesion, cell growth, cell differentiation, morphogenesis, leukocyte trafficking, and tumor dissemination [46,256]. In addition, aberration of glycan structure to sLex/a and expression occurs in various diseases including inflammatory disorders and tumor metastasis [68,204,257]. Consequently, GT inhibitors as therapeutic tools are promising in targeting diseases associated with surface glycoproteins such as inflammatory diseases and cancer metastasis. Alternatively, inhibitors of GTs provide valuable information about the role of glycans and glycoconjugates in biological processes through functional experiments in which the glycosylation of surface markers is perturbed and the cell surface glycoproteome is altered. Inhibitors are also used to study the active sites of GT enzymes and the molecular mechanisms of their action. Due to their biological and therapeutic significance, numerous inhibitors have been developed to target GTs.
9.3.2.1 GT Inhibitors and Primers
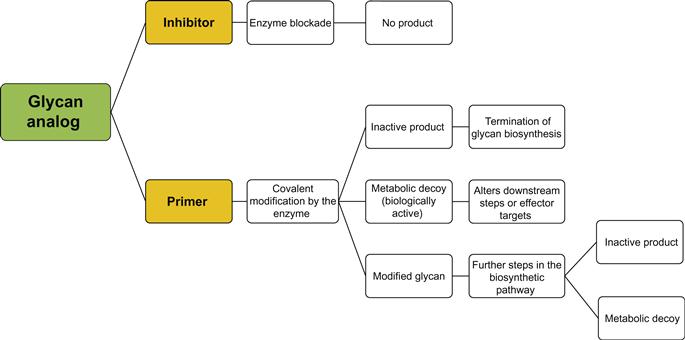
Natural GT inhibitors include papulacandin B [258], polyoxin D [259], tunicamycin [260], castanospermine [261], swainsonine [262], deoxymannojirimycin [263], and N-butyldeoxynojirimycin [264]. Natural inhibitors may cause extensive modifications in glycoconjugate structures such that it is difficult to study or control a specific interaction or enzyme; this introduces the necessity for the design and synthesis of selective agents. Synthetic GT inhibitors can be divided into two classes (refer to Figure 9.5). The first comprises substrate analogs which cause dead-end inhibition whereby a modified substrate is used to mimic one or more of the enzyme substrate(s) in order to block the enzyme or compete with natural substrates and terminate or delay glycan chain initiation or elongation. This class is further separated into three classes: donor substrate analogs (mimetics) of the donor sugar, nucleoside diphosphate, nucleoside monophosphate, or a diphosphate moiety. Because the mammalian system has only nine sugar nucleotide donors, the selectivity of GTs is thought to be associated with the identification of the acceptor rather than with the donor substrate. Within the group of donor analogs, diphosphate mimetics are thought to confer even less selectivity than sugar-modified donor analogs. In order to fulfill the selectivity requirement of GT inhibitors, acceptor mimetics have been developed. Acceptor substrate analogs compete with natural acceptors or block the active site of the enzyme to prevent the glycosyl transfer to the natural acceptor. Acceptor analogs usually have modifications at the hydroxyl group. The group may be deoxygenated, derivatized, or replaced with a halogen or other functionalities. Bisubstrate analogs have two covalently linked motifs mimicking both the acceptor and donor substrates. These analogs are also referred to as transition state analogs because they mimic the transition state of the reaction, and they are thought to be the most active and selective inhibitors of GTs. The second class includes GT primers and metabolic decoys. In contrast to the dead-end GT inhibitors, GT primers prime the enzyme activity using irrelevant substrates leading to the consumption of free active enzyme and the production of irrelevant products. Products of the priming reaction may be either inactive or metabolic decoys, or further modified with downstream enzymes to produce metabolic decoys capable of inhibiting a target pathway or ligand. GT synthetic inhibitors will be discussed in more detail in the following section. For simplicity, the first class will be designated as “inhibitors” and the second class as “primers.”
9.3.2.2 GT Substrate-Analogous Inhibitors
As this chapter is focused on selectin ligand prototype sLex/a, this section in turn will discuss inhibitors against the key GTs involved in its biosynthesis (Figure 9.2) and with which sLex/a expression and/or selectin ligand binding were reported to be affected: C2GnT, FT, and ST.
C2GnT inhibitors: C2GnT (β(1,3)-galactosyl-O-glycosyl-glycoprotein β(1,6)-N-acetylglucosaminyltransferase; EC 2.4.1.102) is an inverting GT that catalyzes the transfer of a GlcNAc residue from UDP-αGlcNAc to β(1,6) linkage on core 1 structure (Galβ1,3GalNAcα-O-pp) to form core 2 branching, one of the major mucin-type O-glycan core structures [265]. An inhibitor to C2GnT GT was developed based on a deoxygenated analog of the enzyme acceptor (Galβ1,3,6-deoxy-GalNAcα-O(CH2)8COOCH3) which produced moderate inhibition compared with the corresponding acceptor (Galβ1,3GalNAcα-O(CH2)8COOCH3) in vitro [266]. A recent study showed that 5-thio-GlcNAc could act as a donor substrate analog against the C2GnT enzyme. This inhibitor is active in vitro and the first to be reported as active in vivo. Although it caused a decrease in levels of cellular O-GlcNAc, the surface glycan profile did not change. In vivo activity is thought to be due to peracetylation of 5T-GlcNAc, which overcomes the problem of poor diffusion of charged analogs. The peracetylated prodrug is converted to the 5T-GlcNAc by the intracellular estrases and, in turn, is converted to UDP-5T-GlcNAc through the salvage pathway [267].
FUT inhibitors: α1,3-fucosyltransferase (FucT-IV, -V, -VI, -VII; EC 2.4.1.152) is an inverting enzyme that catalyzes the transfer of the fucosyl moiety from GDP-fucose to the 3-OH of group of N-acetyllactosamine to give a Lewis X trisaccharide. α1,3/4-fucosyltransferase (EC 2.4.1.65) transfers a fucose sugar from GDP-fucose to the 4-OH of Galβ1,3GlcNAc moiety to give a Lewis A trisaccharide. FTs can act on sialylated LacNAc to give sLex/a tetrasaccharides [268].
As for 5T-GlcNAc, the same research group developed 5-thio-fucose and evaluated its activity against sLex expression and selectin binding activity. Peracetylated 5T-Fuc was taken up by the relevant cells and converted through a salvage pathway into a sugar nucleotide donor analog, GDP-5T-Fuc that, in turn, inhibited the addition of fucose by the FT enzyme. 5T-Fuc was able to inhibit sLex expression in HepG2 cells and impaired adhesion of the cells to selectin-coated surfaces, and activated endothelial monolayer cells [269]. A cell-permeable acetylated analog of a fucose bearing a fluorine atom near the endocyclic oxygen, 2-fluoro-fucose (2F-Fuc), was reported to exhibit global inhibition of FTs. After being deacetylated intracellularly, this fluorinated analog is converted to the corresponding nucleotide sugar substrate GDP-2-fluoro-Fuc by the salvage pathway and is able to inhibit sLex formation in a human myeloid cell line and, subsequently, to hinder E-selectin- and P-selectin-mediated rolling on coated surfaces [270]. A novel study investigated the oral bioavailability of 2F-Fuc in a murine model. 2F-Fuc inhibited protein fucosylation, neutrophil sLex expression, and selectin-mediated adhesion during oral administration with no apparent toxicity in mice over the course of 3 weeks. Inhibitory activities were reversed upon discontinuing the drug administration. 2F-Fuc was also found to inhibit tumor outgrowth in colon and renal cancer metastasis mouse models after oral administration. This study supports the development and potential use of clinically effective GT inhibitors [179,180] in the treatment of cancer and prevention of metastasis.
ST inhibitors: The α2,3-sialyltransferase enzyme (ST3Gal) is a retaining enzyme that catalyzes the transfer of sialic acid from cytidine-5′-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac) to the terminal Galβ1,4GlcNAc (EC 2.4.99.6) or Galβ1,3GlcNAc. Donor-analogous substrates were developed to mimic the common donor substrate, CMP-Neu5Ac, by replacing the anomeric carbon-linked oxygen atom with an ethyl bridge. This analog caused significant inhibition as it probably prevented the nucleophilic attack by the enzyme basic group [271]. Recently, using a cell-permeable acetylated analog of a sialic acid bearing a fluorine atom near the endocyclic oxygen, a global inhibition of ST activity was reported. After being deacetylated intracellularly, this fluorinated analog is converted to the corresponding nucleotide sugar substrate, CMP-2-fluoro-NeuAc, by the salvage pathway. This analog is then able to inhibit sLex formation in a human myeloid cell line as well as E-selectin- and P-selectin-mediated rolling on coated surfaces. It is thought that the observed inhibition is due to both enzyme and feedback inhibition of de novo synthesis of CMP-Neu5Ac by the salvage pathway and, to date, this is the only α2,3-sialyltransferase analog that has been found to produce inhibitory effects in vivo [270,272]. Acceptor-analogous inhibitors were reported to inhibit rat liver α2,3-sialyltransferase activity in vitro using a trisaccharide analog with a modified 3′-OH. The modifications include deoxygenation, fluorination, amination, or methoxy derivatization [273]. The 3′-deoxy lactosamine derivative was also reported, but with no inhibition activity in vitro [266]. Bisubstrate analogs containing the donor CMP-Neu5Ac mimetic and galactose, lactose or lactosamine acceptor have been developed and found to produce weak inhibitory effects against α2,3-sialyltransferase in the case of galactose- and lactose-containing acceptors, whereas they produced potent effects in the case of lactosamine-containing analogs [271,274].
9.3.2.3 GT Primers and Metabolic Decoys
Traditional GT inhibitors are analogs with alterations in specific functionalities that affect the thermodynamics and kinetics of the reaction. These analogs can still bind to the enzyme, but cannot undergo covalent modifications to give reaction products (dead-end inhibition). Conversely, priming activity is achieved when the modified analog binds the active enzyme and undergoes covalent modification to yield modified soluble glycan. Modified glycans can be inactive and thus inhibitory by only the consumption of the active enzyme in an extraneous pathway diverting glycan synthesis from endogenous glycoconjugates (see Figure 9.5). Otherwise, the modified glycan can be biologically active so that it acts as a metabolic decoy, or further modified by the biosynthesis pathway enzymes to a metabolic decoy that can inhibit the glycoconjugate formation or block an effector target. Many of the well-known inhibitors are inactive in vivo probably due to poor permeation into cells and subcellular compartments such as the Golgi apparatus where most glycan formation takes place. Primers can circumvent this problem by conjugating the substrate to a hydrophobic aglycone that enables passive diffusion through the lipophilic cell membrane without affecting the binding kinetics to the target enzyme. Conjugating specific aglycones may also facilitate the isolation of reaction products for further study or create probes for the study of active sites. Also, heterogeneity between cell types in glycosylation patterns can be studied through analysis of the primed products. The monosaccharide glycoside paranitrophenol α-GalNAc (pNp α-GalNAc) was used to inhibit mucin glycosylation through priming modified lactosamine formation and extension in colon cancer and lymphoid tumor cells [275,276]. PNp α-GalNAc is an acceptor analog for β1,3-galactosyltransferase that transfers a Gal residue from UDP-Gal to GalNAc to form core 1 structure. The core 1 structure (Gal β1,3GalNAc) acts as a molecular scaffold for mucin decorations with glycan side chains (i.e., core 2 branching, lactosamine extension, and sLex/a formation). PNp α-GalNAc could enter viable cells, probably due to the hydrophobic aryl group, and consume glycosylation machinery from mucin decoration toward the priming reaction. The aryl group was also used to isolate and secrete the priming reaction products Galβ1,3GalNAcα-pNp and Galβ1,3(Galβ1,4GlcNAcβ-6)GalNAcα-pNp, and to study the glycosylation patterns in these cells. Two independent groups who used GalNAcα-O-benzyl as a primer for such pathways [277–279] also reported similar results. Likewise, several hydrophobic glycosides of GlcNAc, GlcNAcα-O-benzyl, GlcNAcβ-O-benzyl, GlcNAcβ-O-phenyl, and GlcNAcβ-O-pnp could act as primers for polylactosamine chain synthesis [280]. More recently, fluorinated peracetylated GalNAc was reported to be incorporated in selectin ligands expressed by human promyelocytic leukemia cells (HL-60) suggesting a metabolic priming activity and inhibition of glycan chain elongation through the blockade of carbon 4 with a fluorine atom, so preventing the addition of either fucose or galactose sugars. Addition of 4F-GalNAc to the culture medium caused a significant decrease in sLex expression and leukocyte adhesion in vivo [281]. Another study confirmed the activity of 4F-GlcNAc in inhibiting sLex expression and selectin binding. However, this research showed that there was no incorporation of 4F-GlcNAc into the growing glycan chain, suggesting that the main mechanism of 4F-GlcNAc activity lies in interference with UDP-GlcNAc biosynthesis and not in the priming and incorporation of 4F-GlcNAc [282].
Disaccharide glycosides can also be used as primers for GTs. GlcNAcβ1,3Galβ-O-naphthalenemethanol (GlcNAcβ1,3Galβ-O-NM) and GlcNAcβ1,4Galβ-O-NM inhibited sLex expression in leukemia, colon cancer, and lung cancer models, and subsequently reduced E-selectin-dependent cell adhesion to activated endothelial cells [283–286]. Furthermore, it was shown that the treatment of colon and lung adenocarcinoma with the peracetylated disaccharide primer (Ac)6GlcNAcβ1,3Galβ-O-NM reduced the potential for lung colonization in immunodeficient mice. Acetylated derivatives are thought to be more readily bioavailable to cells and, hence, more active in vivo as the polar hydroxyl groups are masked. Acetyl groups are then removed by the action of intracellular and membrane bound esterases [287]. 4-deoxy derivative of (Ac)6GlcNAcβ1,3Galβ-O-NM acted as an acceptor-analogous substrate against β1,4-galactosyltransferase and inhibited experimental tumor metastasis by Lewis lung carcinoma in vivo [288].
The development of a diverse group of GT inhibitors as modulators of surface selectin glycoconjugates holds great promise for the treatment of metastatic tumors and inflammatory disorders. Although this field is several decades old, most of the inhibitors known lack activity in vivo due to their polar nature and underprivileged cell permeability. Recently, several reports have indicated that this problem can be avoided by using the prodrug technology whereby peracetylated derivatives show enhanced uptake. Peracetylated prodrugs are easily deacetylated intracellularly to give the active form of the drug. Another approach is to use bulky hydrophobic aglycones to confer hydrophobic properties on the inhibitor candidate. Nevertheless the development of GT inhibitors with acceptable bioavailability remains a goal in the development of clinically effective antimetastasis therapeutic agents. On the other hand, since both physiological and pathological cell players share the sLex decoration, increased efforts should be directed toward to the development of selective agents that target-specific GT isoforms that are differentially expressed in CTCs but not in normal immune cells.
9.4 Use of Enzymes to Improve the Therapeutic Function of Cells Through Modification of the Cell Surface
Other types of enzymes have been used or are potentially available for ex vivo enzymatic treatment of the cell surface with the aim of therapeutic applications. One such example is the transient proteolytic treatment of MSCs to improve the efficiency of delivery to the target sites [289,290]. When a systemic infusion is conducted for the delivery of MSCs, one major obstacle is the natural tendency of the injected cells to accumulate initially in the lung, and this may lower the efficiency of delivery of the MSCs to the target sites. Use of pronase rather than the standard use of trypsin for the preparation of the cell suspension results in increased lung clearance of MSCs and a better delivery to the site of inflammation, without any apparent functional defect, presumably due to the enhanced cleavage of cell surface proteins involved in adhesion and migration.
In another example, O-sialoglycoprotein endopeptidase (OSGE) treatment has been shown to enhance the functionality of aged CD4 T cells [291]. In humans and mice, the function of the antibody-mediated immune system becomes limited with age, there being lowered clonal diversity of antibodies and inadequate formation of long-term memory [292], due in part to age-related defects in CD4 T cells, especially a malfunction in the interaction between T cells and antigen presenting cells (APCs) [293]. The treatment of aged CD4 cells with OSGE ex vivo restored the age-dependent loss of function, possibly through a mechanism that removes glycoproteins resulting in increased access to the CD28 molecules for interaction with APCs [291].
9.5 Conclusion
This chapter has discussed in detail that the adhesive recruitment of both therapeutic stem cells as well as detrimental metastatic cancer cells are initiated by glycoprotein–selectin interactions facilitating cell rolling on endothelium. The formation of the key glycan responsible for mediating these interactions, sLex, is controlled by the precise activity of a family of ER/Golgi localized enzymes, the GTs. Through the identification and manipulation of these enzymes, migration can be effectively controlled either to enhance or inhibit it. In applications dealing with enabling a remedial cell to migrate to a predetermined site in vivo, the sLex structure needs to be created by the sequential activity of GTs under conditions that do not alter the viability, metabolism, or physiology of that cell, such as through the application of “GPS” technology. Multiple therapeutic avenues can then be sought including the guiding of stem cells or cell-based drug delivery systems to selectin expressing endothelial cells intrinsic to autoimmune or inflammatory diseases. In contrast, for applications where the focus is to inhibit migration/metastasis of detrimental cancer cells or immune cells in inflammatory disease, this interaction between selectins and glycans can be perturbed by inhibiting the activity of these enzymes. Since every family of GT has numerous members, identifying unique enzymes that are responsible for contributing to sLex formation in these harmful cells, but not in “normal” cells, and engineering selective inhibitors to these unique GTs, the migration of the harmful cells could potentially be targeted. Most of the inhibitors known to date lack this specificity and in addition lack activity in vivo due to their polar nature and underprivileged cell permeability. Although recent approaches have helped to overcome membrane permeabilization issues, the development of inhibitors with acceptable bioavailability and specificity remains a goal in the development of clinically effective antimetastasis therapeutic agents.