
Bioconjugation Reactions in Living Cells
Development, Advances, and Applications of Glycan-Specific Technologies
Laura Alberch and Kevin J. Yarema, Department of Biomedical Engineering and the Translational Tissue Engineering Center, The Johns Hopkins University, Baltimore, MD, USA
Bioorthogonal ligation reactions, in which synthetic organic chemistry can be accomplished in a physiological milieu including in living cells and animals, have flourished over the past two decades or so with the development of a profusion of coupling partners. This chapter gives an overview of bioorthogonal ligations including the best known example of this approach—the azide–alkyne “click” reaction—but also covers lesser known, more specialized, and recently emerging chemical advances in this field. Although a bioorthogonal ligation approach is proving broadly useful to address many biological and biomedical problems, its use in glycobiology is particularly significant. Accordingly this chapter focuses on examples where this approach is already being used to, or has the potential to, label cell surface glycans for a variety of research (e.g., in vitro and in vivo visualization of glycans) and medical applications (e.g., diagnosis and treatment of cancer and use in tissue engineering and regenerative medicine).
Keywords
Bioorthogonal ligation; cancer therapy; carbohydrates; click chemistry; metabolic oligosaccharide engineering; photoaffinity labeling; Staudinger ligation; stem cell research; tissue engineering
3.1 Introduction
The surfaces of living cells are covered by a layer of complex carbohydrates, collectively known as the glycocalyx (Figure 3.1A), that play critical roles in their interactions with the microenvironment, influence development from conception to adulthood, and are implicated in many diseases. Over the past half century, the study of these sugars has lagged behind other biopolymers (e.g., nucleic acids and proteins) due in part to the complexity of these molecules [1] and in part due to a lack of experimental methods capable of addressing unique challenges that they pose (e.g., their lack of template-driven synthesis precludes methods such as polymer chain reaction that allows the detection and characterization of vanishingly small amounts of nucleic acids) [2]. In recent years, the increased sensitivity of methods such as mass spectrometry [3] coupled with bioinformatics [4,5] and computational tools [6] as well as high-throughput approaches such as carbohydrate [7] and lectin arrays [8,9] has led to major advances in the characterization of complex sugars and their roles in biological systems.
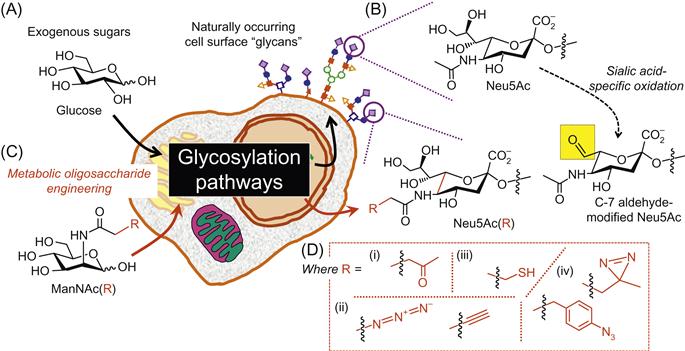
A major limitation of the majority of tools currently available to investigate complex carbohydrates, including the just-mentioned mass spectrometry and array technologies, is that the samples must be isolated from a biological sample for analysis, thus precluding in vivo analysis. The major exception to this limitation is provided by the combined application of bioorthogonal ligation chemistries and metabolic oligosaccharide engineering (MOE) methods, which are well suited even for demanding tasks, such as the in vivo study of the dynamic glycome that has already led to the visualization of glycans in living Caenorhabditis elegans [10] and zebra fish [11] with minimal disturbance to native biological processes. This chapter provides a detailed description of both bioorthogonal ligation reactions (in Section 2) and their utilization in MOE experiments (in Section 3) but first briefly introduces each concept here in the introduction.
A bioorthogonal ligation is defined as a conjugation that occurs in a living system without disturbing native cellular processes. Ideally, they should be highly chemoselective, nontoxic, and form stable products under physiological conditions [12]. The high selectivity of these reactions was initially exploited for drug delivery against Ehrlich ascites carcinoma cells through bioorthogonal aldehyde-based chemistry [13]. Since then, the ability of certain monosaccharides (primarily sialic acid) to be selectively oxidized (Figure 3.1B) has allowed this approach to be extended toward the labeling of cell surface glycans and the imaging of living L6 myoblasts in cell culture [14]. Although this method has long offered a fast and convenient way of measuring sialic acid [15] and more recently additional sugars such as galactose [16] in vitro, it is not applicable for in vivo experiments due to the cytotoxicity of the oxidizing reagents or other chemicals required. Moreover, the detection of naturally occurring, oxidized monosaccharides suffers from the limited repertoire of chemical functional groups available, namely aminoxy [17], hydrazide [14], and thiosemicarbazide [18] groups.
The limited chemical repertoire available naturally for exploitation through bioorthogonal ligation conjugation reactions was overcome through MOE. MOE is a two-step technique where biological macromolecules can be modified by the addition of a synthetic precursor carrying a chemical reporter group (Figure 3.1C). These modified sugars are taken up by the cell, intercept the metabolic pathway without being perceived as being unnatural by the endogenous enzymes, and are finally incorporated in the glycocalyx. In this manner, MOE allows for the noninvasive manipulation of cell surface glycans. This technology has dramatically increased the chemical functional groups available in cell surface glycans for conjugation via bioorthogonal ligation; these groups include ketones, azides, alkynes, thiols, and phenylaryl azides as shown in Figure 3.1D and discussed in detail in review articles [19] as well as in the next section of this chapter in the context of the ability of these functional groups to participate in bioorthogonal ligation reactions.
3.2 Bioorthogonal Chemical Ligation Reactions for Glycan Labeling
This section provides an overview of the main bioorthogonal ligation reactions that have been applied toward the study of surface carbohydrates on living cells starting with a brief mention of aldehydes that can be installed in glycans via chemical oxidation (Figure 3.1B) and then covering the expanded repertoire of functional groups now available through MOE (Figure 3.1D) in greater depth. The advantages and limitations of each existing method are discussed to show how a “chemical biology” strategy has already overcome significant hurdles in developing methods to investigate complex carbohydrates and also to illustrate how opportunities remain to further improve this promising approach.
3.2.1 Aldehyde- and Ketone-Based Bioorthogonal Reactions
3.2.1.1 Aldehyde-Based Reactions
Aldehyde-based ligation has been widely applied toward the quantification of cell surface sialic acids that do not a priori carry a bioorthogonal group for labeling (Figure 3.2). In this method, the vicinal diols in this sugar are selectively oxidized by sodium periodate to provide an aldehyde that—in early colorimetric experiments such as the periodate-resorcinol assay—was reacted with resorcinol and copper and the resulting light absorbance was measured [15,20]. Subsequently, conjugation strategies have been devised where the sugar bearing the aldehyde is subjected to aniline-catalyzed oxime ligation (known as periodate oxidation and aniline-catalyzed oxime ligation (PAL)) to label sialic acid; the aniline catalyst allows the use of low concentrations of aminoxy-biotin at physiological pH [17,21].
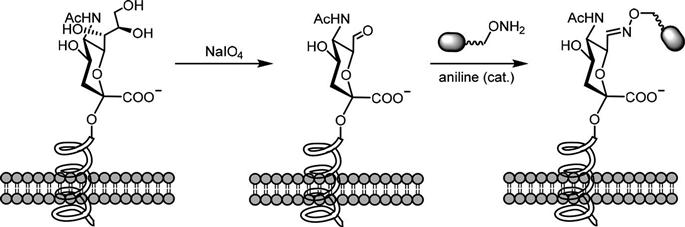
Although used for decades, new developments in a general approach to chemically modify cell surface glycans and then label them (or quantify via colorimetric methods) continue to this day as evidenced by recent reports of new methods to detect sialic acids, for example by using a thiosemicarbazide method [18]. In addition, these techniques are being expanded to include additional monosaccharides, such as galactose [17]. The use of bioorthogonal ligation reactions for glycan labeling, however, has increased much more dramatically through the metabolic incorporation of unnatural sugars that carry reactive functional groups. The rich chemical diversity of these groups (outlined in Figure 3.1D and reviewed extensively elsewhere [19,22–24]) has encouraged the development of a wide range of bioorthogonal chemical ligations. Thus, because of the incorporation of a wide range of functional groups into cell surface glycans via MOE (Figure 3.1B), we are no longer limited to using aldehydes as reactive handles; perhaps ironically however, the first demonstration of the ability of MOE to install bioorthogonal functional groups into glycans was accomplished with a ketone-modified analog [25,26] that had very similar chemical features as nonmetabolically produced aldehydes.
3.2.1.2 Metabolic Incorporation Expands Bioorthogonal Ligation to Include Ketone-Based Reactions
The transition of MOE into the realm of bioorthogonal ligation reactions occurred when the Bertozzi group showed that alkyl “R” groups originally demonstrated by the Reutter group in experiments that pioneered this technology in the early 1990s [27] could be replaced with ketone-modified moieties [25]. Because ketones have chemical reactivity similar to aldehydes, their metabolic incorporation into carbohydrates on the cell surface suffers from many of the limitations of aldehyde-oxidized glycans mentioned above, but as a “proof-of-principle” model the similar chemical reactivity of the two groups allowed the wealth of bioorthogonal ligation established with aldehydes to be applied to MOE rapidly. In particular, cells expressing metabolically ketone-modified sialic acids could be readily conjugated with aminoxy- or hydrazide-conjugated probes (Figure 3.3) [26].
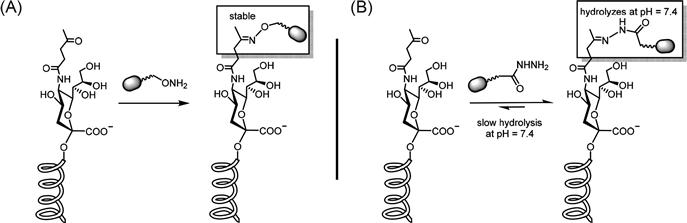
Each reagent used for bioorthogonal ligation of aldehydes and ketones has benefits and drawbacks. For example, hydrazide conjugation reagents are less expensive but the formation of the hydrazone product only occurs readily at subphysiological pH (e.g., pH 5.0) and is reversible (i.e., 100-fold less stable than a reaction between an oxime and a ketone [28,29]) resulting in fewer conjugated molecules. This drawback can be partially alleviated by using excess reagents to shift the equilibrium toward the product. In some cases, the unique features of hydrazide- and aminoxy-conjugated probes can be used in tandem to gain new insights into biological systems; one example is their comparative use to measure kinetic parameters for small-molecule drug delivery by covalent cell surface targeting [29]. Regardless of whether oximes or hydrazides are used for conjugation with ketones or aldehydes, their use in vivo is limited by competition with endogenous keto-metabolites [12]. Moreover, the metabolic incorporation of sugar analogs functionalized with a ketone group, exemplified by Ac4ManLev, is severely restricted in many nonhuman cell lines [19,26] and is rarely if ever as efficient as more recently developed azido-analogs [30], which will be discussed next.
3.2.2 Azide- and Alkyne-Based Bioorthogonal Reactions
A major advance in the use of bioorthogonal ligation reactions to investigate glycans came when the Bertozzi group showed that azide groups could be metabolically incorporated into cell surface carbohydrates [30]. As discussed in more detail below, the cell surface azides were first subject to a modified Staudinger ligation reaction and later to the well-known copper-catalyzed “click” reaction and recently also to strain-promoted cycloaddition reactions. It should be noted that the discussion below is focused on using MOE to install the azide group onto the cell surface, but in many cases the complementary coupling partner for click chemistries (e.g., alkynes [31,32]) can also be metabolically incorporated into glycans thereby providing this methodology with great flexibility and versatility.
3.2.2.1 Classical and Modified Staudinger Ligation Reactions
A benign chemoselective ligation was developed by the Bertozzi group based on the Staudinger ligation where an azide reacts with a phosphine (Figure 3.4) [30,33]. While the classical Staudinger reaction reduces azides to amines, the intermediate aza-ylide readily hydrolyzes to the phosphine oxide and an amide in the presence of water making it unsuitable for use in physiological conditions. The Bertozzi group envisioned a modification of this reaction as a potential method for bioconjugation by installing a methyl ester to the aryl phosphine, which acts as an electrophilic trap. In this manner, the aza-ylide reacts with the ester moiety faster than hydrolysis of the aza-ylide, resulting in a stable amide bond for the ligation of bioorthogonal probes [12]. The carboxylic acid group allows for facile attachment of any biological probe. This ligation strategy was first used for proteins [34] and protein-associated lipids [35] and subsequently has been used extensively for the chemoselective targeting of cell surface glycans [36–38]. Another step forward was in the development of the “traceless” Staudinger ligation, where the ester [39] or thioester [40] attached to the aza-ylide intermediate is displaced and hydrolysis of the resulting intermediate affords an amide bond and liberates the aryl phosphine oxide.
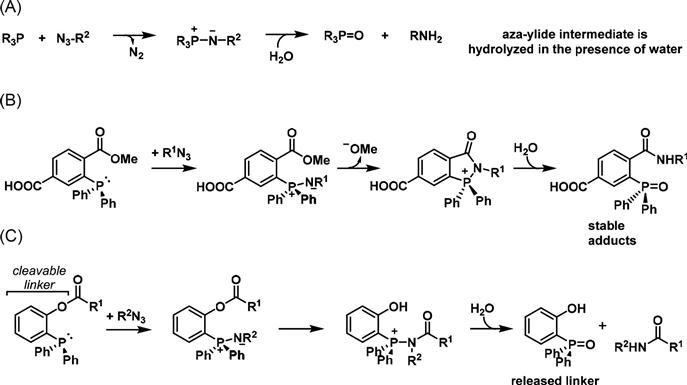
In contrast to the hydrazone ligation used with ketone-modified MOE analogs, the efficiency of the modified Staudinger ligation does not vary with pH. Moreover, the azide tag is abiotic and its small size allows the use of azido sugars to undergo metabolic incorporation more efficiently than their keto counterparts. However, the use of this reaction has been limited by the oxygen sensitivity of phosphines [41]. The slow reaction kinetics of phosphine reagents require high concentrations for in vivo experiments, which results in an elevated background signal when using fluorogenic probes. This problem can be alleviated by the increased sensitivity observed when a luciferin-conjugated phosphine designed to release luciferin upon Staudinger ligation is used. Once luciferin diffuses into the cell, it reacts with luciferase and is converted to oxyluciferin with the release of a photon of visible light [36]. This is an example of bioluminescence imaging, where the visible light generated upon reaction with luciferase can be detected by a highly sensitive CCD camera [42].
3.2.2.2 Click Reactions: The Copper-Catalyzed [3+2] Azide–Alkyne Cycloaddition
The most common and simplest procedure for the conjugation of cells to biomolecules via an azide functional group is through a click reaction. The term “click chemistry” was coined by Barry Sharpless to describe any reaction which would follow a set of criteria such as high yields, high stereospecificity, physiologically stable, does not require chromatographic separation, forms only benign by-products, and can be carried out in water [43]. The most widely used click reaction is the copper-catalyzed [3+2] azide–alkyne cycloaddition, often abbreviated as CuAAC reaction (Figure 3.5). Its great success is rooted in the fact that the azide and the triazole product are essentially inert to most components in a biological environment. In addition, the azide is small in size and usually is ignored by endogenous enzymes. The CuAAC-reaction mechanistic details have been thoroughly studied and reviewed [44,45]. Its applications are countless, as recently reviewed in depth [46], and so we will focus on its recent advances in bioconjugation of living cells.
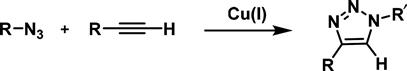
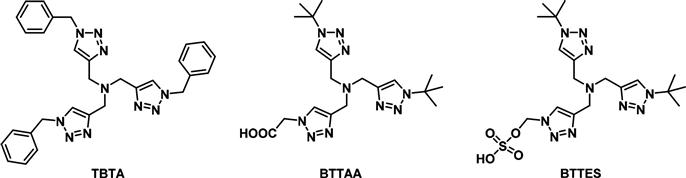
A major limitation to the use of the CuAAC reaction in living systems is the cytotoxicity imparted by the copper catalyst, which is required to speed up the rate of reaction at levels useful for biological systems. The high concentrations of copper catalyst can be reduced by the addition of Cu(I) stabilizing ligands (Figure 3.6). These ligands mainly function by protecting the copper catalyst from oxidation by oxygen reactive species and by preventing the formation of copper clusters, thus increasing the effective concentration of copper. Although tris-(benzyltriazolylmethyl)amine (TBTA) has been the most widely used ligand [47,48], others have been designed with modifications at the triazole side chain to improve their physical properties and kinetics. For example, the acetic acid group in (2-[4-{(bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]-amino)methyl}-1H-1,2,3-triazol-1-yl]acetic acid) (BTTAA) improves its water solubility as it becomes acetate at physiological pH, while the two tert-butyl groups prevent the polymerization of copper acetylides [49]. Introducing a highly polar sulfate group as in the case of 2-[4-{(bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino)-methyl}-1H-1,2,3-triazol-1-yl]ethyl hydrogen sulfate (BTTES) minimizes the cell membrane permeability of the coordinated copper [50]. The development of these ligands has encouraged the revisiting of the CuAAC for use in cell surface labeling, as it is still the fastest bioorthogonal reaction and it is compatible with both azide- and alkyne-tagged carbohydrates.
3.2.2.3 Strain-Promoted Azide–Alkyne Cycloaddition Reactions
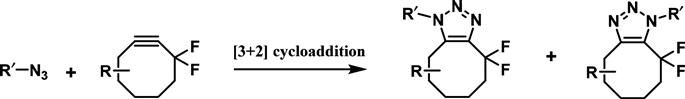
An ingenious alternative to the use of copper in the azide–alkyne cycloaddition is to increase the reactivity of the alkyne (lowering LUMO energy) by constraining it in a ring. This strategy, known as a strain-promoted azide–alkyne cycloaddition (SPAAC), can be used under physiological conditions without the need of a catalyst, and consequently it is often referred to as the copper-free click reaction (Figure 3.7).
Novel cyclooctynes are constantly being developed (Figure 3.8). Their design has been assisted by computational models and theoretical studies that explain and predict the enhanced reactivity of novel cyclooctynes [51,52]. An important design strategy is centered in decreasing the electron density around the triple bond and thus lowering the LUMO energy, either by incorporation of propargylic electron withdrawing fluorine groups (DIFO) [53] or by installing two adjacent aromatic rings which allow the delocalization of the alkyne π-system (DIBO) [54]. The fast reaction kinetics of these highly strained biarylazacylooctynones with azides allow for their use in visualizing metabolically labeled glycans [55,56].
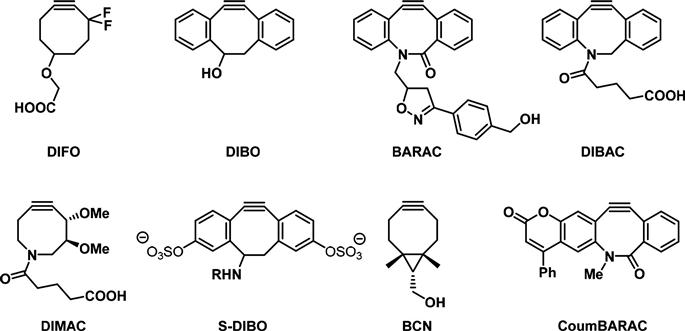
Other modifications to the cyclooctynes focus on increasing their solubility in water, a common problem encountered with the first generation of cyclooctyne derivatives. Their poor hydrophilicity resulted in high nonspecific binding to serum proteins and promoted sequestration by membranes thereby reducing bioavailable concentrations [57]. BARAC, DIBAC, and DIMAC possess a heteroatom in the eight-membered ring, which greatly improves aqueous solubility and pharmacokinetics [57]. Incorporation of sulfated groups such as in S-DIBO renders the compounds completely water soluble, while retaining excellent reaction kinetics. Moreover, unlike other biarylcyclooctynes, S-DIBO does not pass through the cell membrane and so exclusively labels extracellular glycoconjugates further decreasing the background signal [58]. Novel bicyclo–[6.1.0]nonyne, which has been applied for imaging of glycans on the cell surface of melanoma MV3 cells, offers the advantage that it can be prepared in only two synthetic steps [59].
While the use of benign cyclooctynes has provided an advance in the field of biological imaging in vivo, challenges with non-specificity and sensitivity remain. For example, nonspecific biotinylation, presumably by reaction of the alkyne moiety with cysteine residues, reduces the sensitivity of detection [60,61]. Protection of the peptidylcysteines by treatment with iodoacetamide prior to labeling results in a strong reduction of nonspecific labeling [62]. In an alternative approach, a cyclooctyne probe that becomes fluorogenic upon reaction with an azide, CoumBARAC, was developed by the Bertozzi laboratory [63,64]. Although this compound paved the way toward the development of other such probes, the low-fluorescence quantum yields of CoumBARAC have so far limited its use for biological imaging applications. The design of fluorogenic probes that are activated upon triazole formation would prove useful in the dynamic imaging of the glycome in situations (e.g., in living animals) where it is not possible to wash away unbound probes.
3.2.3 Thiol-Based Chemistry
Although bioconjugation with thiol reagents is the most well known for labeling cysteine residues on proteins, this approach is also becoming useful for labeling unnatural sugars synthetically modified with a thiol group on the cell surface. Because most native surface thiols are complexed with metals, form disulfide bonds, or are in membrane proteins shielded by the glycocalyx, the metabolic incorporation of thiol-containing sugar analogs (Ac5ManNTGc) allows for chemoselective labeling at the outermost periphery of the cell surface [65]. A major advantage of thiol-based ligation strategies is the plethora of commercially available reagents for labeling these functional groups [66]. One option, maleimide-functionalized probes (Figure 3.9), offer the advantage that they are highly efficient at physiological pH, unreactive with tyrosine and hystidine side chains, and are not light sensitive. Alternatively, the strong coordinate–covalent interaction between thiols and gold allows for cell surface thiols to be directly attached to gold surfaces, which has a strong impact on stem cells [67] and can even influence cancer cells to dramatically alter their behavior [68,69].
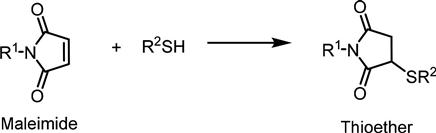
The oxidative nature surrounding the cell surface under in vitro culture conditions, which can also occur in vivo, results in approximately 90% of thiols from modified sialic acid forming cis-disulfides [65], and these must be reduced to free thiols to maximize their labeling in bioorthogonal conjugation reactions. For this purpose, treatment with tris-(carboxyethyl)-phosphine (TCEP) is preferred over dithiothreitol, as the former does not contain any thiols and so does not require washing before reacting with the maleimide reagent [70]. The use of TCEP in sufficiently high concentrations nevertheless remains toxic to cells limiting the use of this reagent in biological systems.
3.2.4 Photoactivated Ligation Reactions
The demonstration that functional groups suited for photoaffinity labeling (PAL) such as phenylaryl azides [71] and diazirines [72,73] could be metabolically incorporated into cell surface glycans further expanded the potential of bioorthogonal ligation reactions for the investigation and understanding of complex biological carbohydrates. In PAL, UV light converts the inert photoreactive crosslinker to a highly reactive carbene or nitrene, which forms a covalent bond with its target molecule at predetermined points during the experiment, for example, after binding of the photoreactive ligand to a receptor as exemplified by the Paulson’s group’s efforts to identify the in situ binding partners for CD22 [71]. When using this strategy, it is important that the photoreactive group of choice causes minimal perturbation to the binding affinity of the ligand to its receptor and its functionality. When this criterion is met, the ability of PAL strategies to form robust covalent bonds to capture noncovalent interactions [74] comprises a powerful tool to capture biologically relevant binding events in their cellular context.
Two types of carbenes can be generated upon irradiation: a singlet carbene, where both electrons are in a single orbital and a triplet carbene, where each electron occupies a separate orbital. Understanding the ground-state carbene multiplicity is important as it dictates the reactivity of the unpaired electron and thus the coupling efficiency of the carbene. The carbene’s electronic distribution is determined by the steric and inductive effects exerted by each substituent. Although a full explanation is beyond the scope of this book (but is reviewed elsewhere [75–77]), in brief it is well established that singlet carbenes are stabilized by σ-electron withdrawing groups and π-electron donating groups that are delocalized into the empty p-orbital (Figure 3.10). For example, fluorine is often incorporated due to its high electronegativity (σ-direction) and its ability to donate its lone pair of electrons into the p-orbital, stabilizing the singlet carbene.
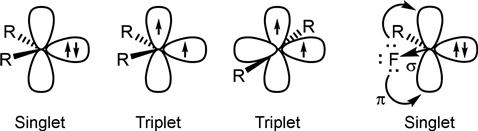
An ideal photoactivatable chemical group for bioconjugation has several requirements including that: (i) it is activated at a long UV wavelength to minimize damage to other biomolecules, (ii) it is similar in structure to the parent molecule that its biological activity is not significantly altered, (iii) it forms a stable covalent bond with the receptor upon activation, and (iv) it generates short-lived intermediates to minimize nonspecific binding. Largely based on these requirements, the three most popular photoactivatable groups used in bioconjugate labeling are benzophenones, arylazides, and diazirines [78]. Each group has advantages and limitations and these will be highlighted in further sections.
3.2.4.1 Benzophenones
Benzophenones generate triplet carbenes upon irradiation (Figure 3.11). Although they require prolonged exposure, which often leads to nonspecific labeling, the long UV range for activation and their inertness to solvent have made them attractive photoactivatible crosslinkers [79,80]. However, their bulkiness may prohibit their use for certain biological imaging applications [81].
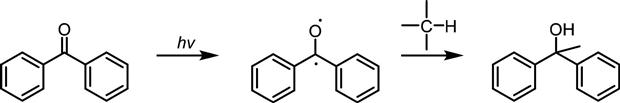
 H insertion into carbenes generated from benzophenones.
H insertion into carbenes generated from benzophenones.3.2.4.2 Arylazides
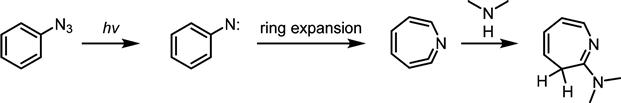
Arylazide-modified sugars can be metabolically incorporated into glycans of interest, and upon exposure to UV light, the arylazide generates a nitrene that can insert into C![]() H and N
H and N![]() H sites of a neighboring protein. While an advantage of using arylazide-modified sugars is their ease of preparation, a drawback is that they are activated at short UV wavelengths (254 nm) that have the potential to damage other biomolecules, such as nucleic acids that have a strong absorbance at this wavelength. Moreover, simple arylazides are rarely used as photoreactive crosslinkers because they rapidly undergo ring expansion to dehydroazepenes, which tend to react with nucleophiles such as amines and thiols, rather than C
H sites of a neighboring protein. While an advantage of using arylazide-modified sugars is their ease of preparation, a drawback is that they are activated at short UV wavelengths (254 nm) that have the potential to damage other biomolecules, such as nucleic acids that have a strong absorbance at this wavelength. Moreover, simple arylazides are rarely used as photoreactive crosslinkers because they rapidly undergo ring expansion to dehydroazepenes, which tend to react with nucleophiles such as amines and thiols, rather than C![]() H insertion (Figure 3.12) [82,83].
H insertion (Figure 3.12) [82,83].
Substitution of the phenyl ring with electron withdrawing groups prevents rearrangement and allows these nitrenes to undergo C![]() H insertion with high efficiency (Figure 3.13) [84].
H insertion with high efficiency (Figure 3.13) [84].
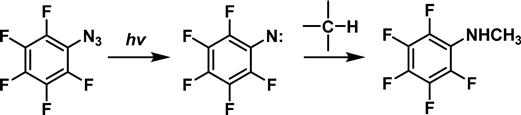
A limitation of using arylazides for photoaffinity labeling is that the relatively large phenyl group can interfere with the biological function of the original compound. Moreover, in the cell, the arylazides can be reduced to anilines by endogenous thiols; interestingly, Chang’s group has taken this side reaction as an opportunity to detect intracellular hydrogen sulfide levels [85,86].
3.2.4.3 Diazirines
While traditionally, arylazides were the most widely used photoreactive group, diazirines are growing in popularity, and increasingly the photoreactive group of choice. Like benzophenones, diazirines have the advantage that they are activated at longer UV range (330–370 nm), which minimizes damage to other biomolecules present in a cell. In addition, the small size of the diazirine makes it particularly suited for PAL because it causes the least structure perturbation compared to the original substrate. Finally, the small size of this group has allowed sugars bearing a diazirine at the N-acyl position to be metabolically incorporated on the cell surface to capture carbohydrate–protein interactions [72].
3.3 Bioorthogonal Ligation Reactions: Exploitation in MOE-Based Applications
The previous section of this chapter outlined emerging strategies to exploit chemical functional groups that now can be installed on the cell surface via MOE by using bioorthogonal reaction chemistries. For these spectacular “chemical biology” advances ultimately to be meaningful, however, the hope is that they will find use in solving real-world biological or medical problems. This section addresses this issue, first by a general discussion of nuances of analog incorporation into various glycoforms that ultimately determine biological or medical significance, and then by giving several specific examples of biological problems (e.g., imaging) or medical conditions (e.g., treatment of degenerative disease or cancer) where a combination of MOE and bioorthogonal ligation chemistries appear to be particularly promising.
3.3.1 MOE Extends Beyond N-Acyl-Modified Sialic Acid
The examples illustrated in Figure 3.1, which are largely used to illuminate the bioorthogonal chemistries discussed in Section 2 of this chapter, where chemical modifications focused on the N-acyl position of sialic acid, which comprised the original in vivo demonstration of MOE [27] and which remains the best studied example of this technology to date [19,24]. In recent years, however, additional sites of modification on sialic acid, for example, the C-7 position accessed through modifications to the C-4 position of ManNAc [87], have been successfully functionalized with chemical reporter groups. In other cases, modifications at the C-6 position of ManNAc to express C-9-modified sialic acid are not tolerated because one of the steps during its biosynthetic conversion to sialic acid involves phosphorylation at the C-6 position. Instead, the C-9-modified sialic acid can be administered directly to the cells [88].
The metabolic incorporation of sialic acid and its precursor ManNAc has been the most extensively studied, as sialic acid is conveniently located at the outermost periphery where it is readily accessible for binding with exogenous substances, such as labeling reagents, lectins, and toxins [89]. Moreover, sialyltransferases tolerate a wide range of modified substrates [22] ranging from compounds with reactive chemical handles for imaging the glycome [10] to alkyl-modified analogs that regulate cellular processes during embryonic neural development [90] or the neurite outgrowth of PC12 cells [91]. Despite the focus on sialic acid in this report, we wish to emphasize that over the past decade, there has been an increasing focus on expanding MOE technologies to additional monosaccharide vehicles, which have been successfully demonstrated to date for GlcNAc, GalNAc, and fucose analogs [19]. By exploiting these additional monosaccharides, we believe that a vast realm of currently unexplored glycan-based biological activities can be fruitfully investigated in the future. In the remainder of this report, however, we offer only a small sampling of how a combination of MOE and bioorthogonal ligation chemistries have already contributed to biology and medicine.
3.3.2 Imaging of Living Cells
Cell surface glycans comprise a fingerprint of a cell and are specifically recognized by carbohydrate-binding proteins generically known as lectins. The dynamic display of cellular glycans is constantly changing throughout the cell cycle and abnormal changes can be indicative of disease. Imaging these glycans through bioorthogonal chemical ligations provides information on the cell’s state. The biosynthesis of glycans is not directly encoded by the genome and so the imaging techniques that rely on genetic reporters for proteins and DNA are not applicable. The imaging of the cell surface is further complicated by the high heterogeneity of glycans structures, differing in linkage, composition, branching, and anomericity. While the use of lectins and antibodies has been used extensively to image glycoconjugates, they require prior isolation of the glycan from the cells. Moreover, they are not suitable for in vivo dynamic imaging due to their low-binding affinity and tissue impermeability. Toward this end, bioorthogonal chemical reporters have been developed to image glycans on the cell surface and the advantages and limitations of each existing method have been described above.
The labeling of cell surface glycans is dominated by the SPAAC due to the benign nature and faster reaction kinetics of this reaction compared to the modified Staudinger ligation. However, the development of copper ligands which reduce catalyst loading has encouraged the revisiting of the CuAAC for use in biological reactions, as it is still the fastest bioorthogonal reaction and is compatible with both azide- and alkyne-tagged biomolecules [49].
The small size and inertness of an azide are ideal for MOE experiments, and once expressed on the cell surface it can be reacted with other biomolecules via the Staudinger ligation, the SPAAC, or the CuAAC. The promiscuity of the enzymes involved in the hexosamine pathway allows for the efficient enzymatic conversion of azide-modified precursors to SialNAz, GlcNAz, and GalNAz. However, this is not the case for fucose-modified analogs since 90% of GDP fucose utilized by the cell originates from GDP mannose through the de novo biosynthetic pathway. Intercepting the fucose salvage pathway using enzymatically modified azide or alkyne GDP-fucose analogs significantly improves its metabolic uptake [50].
While metabolic incorporation of Ac4ManNAz exclusively yields the azide-modified sialic acid, it is more difficult to achieve precise pathway targeting with analogs such as Ac4GlcNAz because a portion of this compound, or its metabolites, is reversibly converted by GlcNAc-2-epimerase into Ac4GalNAz. This problem can be avoided by treating the cells with the alkyne-functionalized GlcNAc analog (Ac4GlcNAlk), which is not recognized by the enzyme and thus is a more specific reporter of O-GlcNAc modification. Moreover, the metabolic efficiency of Ac4GlcNAlk is comparable to that of Ac4GlcNAz [92], and the use of azide-detection tags has improved sensitivity over the reverse copper-catalyzed click chemistry orientation due to decreased background signal [93,94].
Photocrosslinking has been used successfully for the identification of cis-ligands of CD22, a glycoprotein that regulates B-cell signaling. Metabolic incorporation of the 9-arylazide derivative of sialic acid (9AAzNeuAc) in B cells followed by irradiation established that CD22 mostly binds to sialic acids on cis-ligands of neighboring CD22 resulting in homomultimeric complexes [71,95]. This carbohydrate-mediated interaction has also been demonstrated by installing a diazirine at the N-acyl side chain of mannosamine (Ac4-ManDAz) and sialic acid (Ac5-5-SiaDAz). N-acyl modification is preferred over the 9-position of sialic acid, as the latter can interfere with biological processes [96]. Metabolic incorporation of an arylazide at the N-acyl position of sialic acid was extremely poor [97], while substitution with a diazirine led to high levels of expressed Ac5-5-SiaDAz [72]. Ideally, the sialic acid possesses both a diazirine moiety for the capture of carbohydrate–protein interactions and another functional group for conjugation with a chemical probe. Toward this end, bifunctional sialic acids have been developed (9AzSiaDAz) and incorporated into CHO, HeLa, Daudi, and K20 cells [98].
3.3.3 In vivo Labeling/Imaging
With the continuing success of in vitro imaging and the wide variety of bioorthogonal ligation strategies, the in vivo labeling of living organism has been made possible. Zebra fish and C. elegans are ideal models for imaging the glycome as they are transparent, their development is well studied, and exogenous substances are easily incorporated. Opaque organisms such as mice can be visualized using far-red fluorescence imaging and single-photon emission computed tomography. The MOE approach has been successfully extended towards the in vivo imaging of cell surface sialylated [99], fucosylated [100], and polylactosamine [101] glycans in zebra fish by metabolic incorporation of the appropriate azide-modified sugar precursor.
Using a multicolored labeling strategy, the mucin-type O-glycans in zebra fish [11] and C. elegans [10] were imaged from embryo to adult stage by inoculating GalNAz, followed by copper-free click chemistry of the azide with a DIFO reagent conjugated to red and green fluorescent probes. In this manner, it was shown that the biosynthesis of mucin-type O-glycans for these two models is highly dynamic and tissue dependent. This methodology was extended toward the imaging of sialic acids in zebra fish by incubation with Ac4ManNAz [99].
A limitation of MOE is that the metabolic incorporation of Ac4ManNAz results in Sia5Az into both N- and O-linked glycans. However, it was found that Ac3-4-azido-ManNAc is selectively expressed as Sia7Az in O-glycosylated proteins, and this analog was used for the in vivo imaging of the midbrain and hindbrain regions in zebra fish, which contain high levels of O-glycosylated proteins [87].
Another challenge in labeling glycans in vivo is that the typical biotin–streptavidin system used for detection in living cells has limited vessel permeability due to the large size of streptavidin. As a result, this approach cannot be applied for the imaging of normal glycans and is largely restricted to the imaging of tumor glycans. Usually, glycans in optically opaque organisms such as mice are labeled in vivo but are analyzed ex vivo. Toward this end, Brindle’s group has developed fluorescently labeled tetrazine reagents which are small enough to access normal tissue and have lower levels of nonspecific retention. The tetrazene moiety undergoes a Diels–Alder reaction with an alkene appended to the cyclooctyne probe and its fluorescence measured [102].
3.3.4 Tissue Engineering, Stem Cell Research, and Regenerative Medicine
The oxidative nature around the glycocalyx allows for thioglycolyl sugars to readily form disulfide bonds with adjacent thiol-containing cells, which results in cell–cell clustering. These adhesive properties imparted by the thiol group can be extended to attachment of the cell to matrix materials, which is particularly challenging [103]. The strong gold–thiol interaction and the small spacing of the sulfur anchors on gold (4.97 Å) make thiol-bearing sialic acids ideal for attachment to gold substrates. Alternatively, the analog-treated cells can be anchored to maleimide-functionalized glass surfaces. The physical properties of the biomaterial, such as its elasticity and topography, can modulate the morphology of stem cells, which in turn directs the stem cell differentiation into specific lineages [104]. Yarema’s group demonstrated that treating hEBD stem cells with Ac5ManNTGc induced differentiation to a neuronal cell linage [67]. When Jurkat cells were grown on gold-coated nanofibers, they developed a distinctive “spreading” morphology which was exclusive to sialosides of N-linked glycoproteins [69]. Extending this methodology from 2D- to 3D-scaffolds (gold- or maleimide-coated nanofibers) significantly increased cell adhesion (66–81%) compared to the 2D films (23–35%) [68].
The early experiments with thiol-modified sialic acids highlight the role of these sugars on adhesion processes involved in stem cell fate. In follow-up studies, a more sophisticated understanding of the underlying mechanism for these compounds, for example, through modulation of potassium channel activity [105], is being gained. From a more practical point of view, the thiol analogs illustrate an important aspect of nonnatural monosaccharide residues installed in the cell surface via MOE. Specifically, the thioglycolyl sialic acids, similar to other sialosides installed via MOE such as ketone groups [26], are slowly removed from the cell surface over time thus allowing thiol-dependent responses to be reversed in some cases. In other cases, such as stem cell differentiation, however, it appears that thiol-analog-driven biological responses are irreversible. Nevertheless, the levels of surface display can be maintained by continuous addition of the analog precursor Ac5ManNTGc while the removal of the synthetic sugars can be accelerated by the addition of the natural sialic acid precursor ManNAc [26,65]. The removal of nonnatural sugars from the cell surface is potentially important for tissue engineering applications of MOE to avoid possible deleterious immune responses triggered by the nonnatural glycan.
3.3.5 Cancer Therapy
It is well established that the composition of cell surface glycans in diseased cells, especially cancer cells [106,107], is different than those of healthy cells. For example, elevated levels of sialic acid [108] and tri- and tetra-antennary N-glycans [109–111] are an indication of cancer. Some of the earliest MOE experiments showed that a tagged sialic acid can be introduced to the cancer cell by MOE and quantified for diagnostic purposes [112] or used as a handle for delivery of toxins for cancer therapy [113]. An ongoing challenge in using MOE for cancer therapy, however, is the selective targeting of cancer cells over healthy cells. This problem is partially alleviated because sialic acid is overexpressed in metastatic cells and so they are most affected by any modifications in sialic acid. A strategy to increase the tissue-specific delivery of Ac3ManNAz was to install a peptide recognized by the prostate-specific antigen (PSA) protease at the O-6 position of the sugar. In this manner, the caged sugar could only be released upon reaction with the PSA secreted by prostate tumor tissue [114]. Another approach to selectively deliver azido sugars to cancer cells is by encapsulating them in ligand-targeted liposomes [115]. Controlled release of cancer prodrug Bu4ManNAc over a 10-plus day period was achieved by encapsulating the sugar in polyethylene glycol-sebacic acid [116] resulting in a substantial improvement compared to the otherwise twice daily administration required for in vivo studies [117].
3.4 Concluding Comments
This chapter provides an overview of bioorthogonal ligation reactions that—when applied to the study of glycans—were first used to label selectively oxidized sialic acids. The development of MOE, which allows a profusion of nonnatural chemical functional groups suitable for various bioorthogonal ligation approaches to be incorporated into glycans in living cells and animals, provided a major impetus to this field. Although the development of underlying chemistry-based technologies continues, some of the most exciting recent developments in the bioorthogonal ligation field include the burgeoning use of this approach to confront pressing biomedical research and clinical challenges. The introduction to these issues provided by this chapter is designed to provide a foundation for the interested reader to independently investigate the application of bioorthogonal ligation to glycobiology in more depth and hopefully begin making experimental contributions to this exciting field.
Acknowledgments
Funding for this contribution was provided by the National Institutes of Health (R01CA112314 and P01HL107153).