
Molecular Engineering of Cell and Tissue Surfaces with Polymer Thin Films
John T. Wilson1 and Elliot L. Chaikof2, 1Department of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN, USA, 2Department of Surgery, Beth Israel Deaconess Medical Center, Boston, MA; Harvard Medical School, Boston, MA; Wyss Institute for Biological Inspired Engineering of Harvard University, Cambridge, MA, USA
Engineering of polymer thin films has long provided a powerful approach for exquisitely controlling the interfacial properties of diverse materials, and, hence, offers enormous potential as a tool for cell surface engineering. However, most conventional film fabrication methods and polymeric constituents impose a number of barriers, notably cytotoxicity and surface instability, which limit the formation of polymer films on living cellular supports. This chapter focuses on the molecular engineering of polymer films that circumvent some of these major barriers and, in doing so, have facilitated the translation of thin film technology from abiotic substrates to the cell surface. Established and cutting-edge approaches to generating cell surface polymer thin films are discussed and emerging biomedical applications of thin films are reviewed.
Keywords
Polymer thin film; cell surface engineering; polymer monolayer; polyelectrolyte multilayer; layer-by-layer; membrane-anchor; tissue engineering; cell transplantation
13.1 Introduction
From household paints to semiconductors to implantable medical devices, engineered polymer thin films are at the core of many of the influential and emerging technologies that have and continue to play important roles in society and in our daily lives. Polymer films take on many different facets with respect to their physicochemical properties, including thickness, wettability, reactivity, mechanical properties, conductivity, and optical properties, to name only a few. Polymeric thin films can be assembled, deposited or grown on surfaces using a variety of mechanisms including solvent casting, spraying, coating, covalent conjugation, surface initiated polymerization, or self-assembly (i.e., electrostatic interactions, hydrogen bonding). They have been created using an astonishing number of different polymers, including both synthetic and naturally occurring polymers of diverse architecture and properties. The exquisite ability of polymer thin films to control diverse physical and biochemical phenomena at interfaces combined with their enormous utility as surface engineering tools has sparked considerable excitement towards translating polymer thin film technologies for use in cell surface engineering applications. However, unlike conventional inanimate substrates (e.g., glass, ceramic, metallic, or polymeric supports), which are largely passive bystanders of polymer film deposition or growth, cell surfaces present complex and dynamic interfaces capable of chemically and physically restructuring in response to film constituents. Cellular membranes comprise lipids, proteins, and carbohydrates that are essential for maintaining native cell function and phenotype, and, as such, re-engineering these highly dynamic and diverse chemical landscapes with polymer thin films imposes a number of unique challenges and design restrictions which may not normally be encountered in more traditional thin film engineering applications. This chapter will introduce design key principles for engineering living cell and tissue surfaces with polymer thin films, will describe the main classes of cell surface supported polymer films, and will provide notable examples of how such films are being used in biomedical applications.
13.2 General Design Principles and Considerations
As in medicine, “Do no harm,” should be the most important rule in the design of polymer thin films for cell surface engineering applications. Film constituents, solvents, and film deposition methods and conditions must be minimally toxic to cells and should not interfere with cellular functions that are essential to the desired therapeutic application. Therefore, film fabrication methods that use organic solvents, high temperatures, vacuum, or the use of initiators, monomers, catalysts, and polymers with high cellular toxicity are not suitable for cell surface engineering applications. Rather, film formation techniques must be achieved using cell-compatible constituents and film assembly procedures that can be performed under physiological or near-physiological conditions defined by narrow ranges of pH, ionic strength, temperature (e.g., temperature 25−37°C, pH ~7−7.5, osmolarity ~300 mOsM), and mechanical forces. Additionally, to limit toxicity and promote surface deposition, film constituents ideally should not freely pass through the plasma membrane; this is not generally a problem for most hydrophilic macromolecules but can be an issue when using polycations or low molecular weight polymer precursors such as monomers and initiators. Indeed, many initiators and monomers commonly employed in free radical polymerizations exert significant cytotoxicity. Additionally, thin film deposition techniques such as spin coating, spray coating, or chemical vapor deposition are not suitable for cell surface engineering or would require significant modifications from existing standards. For example, chemical vapor deposition (CVD), a popular and widely used approach to generate polymer thin films, is not currently useful for depositing films on cell surfaces due, in part, to the use of vacuum during the film formation. These stringent requirements dramatically reduce the available canon of film constituents and fabrication techniques available to the cell surface engineer. This has spawned a number of highly innovative thin film engineering approaches to circumvent these limitations, many of which will be described in this chapter.
A second major challenge, which, like toxicity, is a common challenge throughout the field of cell surface engineering is the considerable chemical heterogeneity intrinsic to the cell surface. The cell surface is a complex and nonuniform nanostructured material composed of diverse types of lipids, proteins, and carbohydrates, which can vary in relative composition as function of both time and space along the cell surface in a highly cell-type-specific manner. This considerable complexity presents a major challenge for producing uniform films as well as controlling interactions between film components and specific biomacromolecules on the cell surface; interactions that have important implications for regulation of cell function and behavior. The dynamic nature of the cell surface also imposes a major challenge in generating stable polymer thin films. The plasma membrane is a constant state of flux, with lipid and protein components continuously being internalized via endocytosis, degraded, and replaced with fresh molecules [1]. This is a tightly regulated process in which turnover rates, which can vary from hours to weeks, are very specific to a given molecular species [2]. Hence, polymer films formed on cell surfaces are highly prone to intracellular internalization and degradation as a consequence of this process, rendering control of stability, both during and after film formation, a major consideration in film engineering.
Finally, the ultimate objective of most applications of cell surface engineering is application in clinical cell therapy therefore, polymer thin films are subject to an onslaught of mechanical and biochemical forces in the in vivo environment. Polymer films may be exposed to considerable forces as cells are introduced into the bloodstream or migrate through tissues. Depending on the application and cell type, this requires care in generating films that are neither too weak to withstand such forces nor so robust as to hinder cell functions that are dependent on specific mechanical properties of the cell membrane. Additionally, the biochemical environments experienced by cells in vivo, particularly in disease states, can have a dramatic impact on the integrity of the film. Likewise, polymer films can be recognized by the host as a foreign material, triggering inflammation, complement activation thrombosis, and even immunity that can be deleterious to both the engineered cell and the health of the host [3–5]. There exist no universal solutions to these challenges. Films must be engineered considering the intended application and the underlying biology or physiology, the type of cell being used, and the environment in which it will be introduced. The next section of this chapter describes some of the important types of thin films that have successfully met these challenges for a number of important biomedical challenges.
13.3 Cell Surface Engineering with Polymer Thin Films
13.3.1 Covalent Conjugation of Polymer Monolayer Films to Cell Surfaces
The cell surface presents a multitude of reactive groups, including amines, carboxylic acids, and thiols, that are abundant within the natural canon of proteins and carbohydrates present on the cell surface [6,7]. Moreover, through enzymatic, chemical, metabolic, or genetic approaches, additional reactive moieties such as aldehydes, ketones, and azido groups may be introduced to the cell surface milieu [7,8]. As described elsewhere in this book, the development of bioorthogonal and chemoselective conjugation chemistries has revolutionized our ability to reengineer cell surfaces with an enormous diversity of molecules. Accordingly, these reactive sites can serve as anchors for covalent conjugation of appropriately functionalized polymers, representing the most common strategy through which polymer films have been generated on cell surfaces (Figure 13.1A). Among the covalent conjugation chemistries, derivatization of cell surface amines has been the most extensively explored, primarily using polymers functionalized with N-hydroxysuccinimide (NHS) activated esters [9–12], and to a lesser extent benzotriazole carbonate (BTC) [13], cyanuric chloride [14], isocyanate derivatives [15], and aldehydes [16].
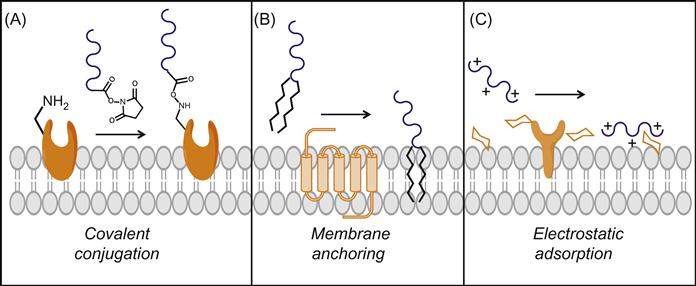
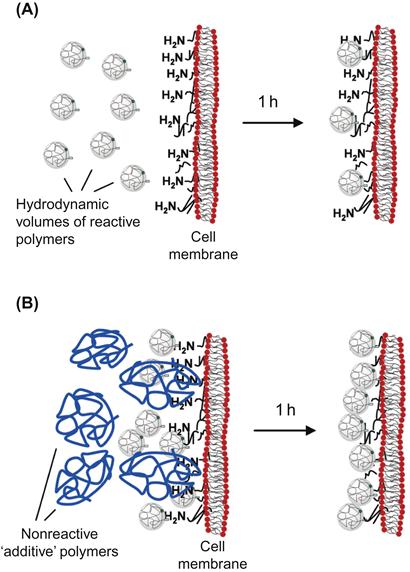
The covalent conjugation of poly(ethylene glycol) (PEG) chains to the cell surface, most commonly to amino groups, represents far and away the most common example of a covalently anchored polymer thin film. PEGylation of cell and tissue surfaces has been utilized for a number of applications, some of which will be described later in this chapter. Accordingly, covalent conjugation of PEG provides an excellent case study for illustrating some of the key challenges and design parameters associated with fabrication of covalently grafted polymer monolayers. In general, hydrophilic and neutral polymers, such as PEG, associate poorly with the cell surface [17]; the same is also true for negatively charged polymers (polyanions), which are electrostatically repelled by the negatively charged cell surface. Consequently, a large stoichiometric excess of reactive PEG is commonly used to increase the surface density of polymer chains immobilized to the cell surface [17]. The need for large molar excess is further exacerbated when using coupling chemistries that involve nucleophilic attack by cell surface amines (e.g., NHS–esters), which are subject to competing ester hydrolysis under aqueous conditions. Although reactive groups (e.g., maleimide) for other cell surface nucleophiles (e.g., thiols) may have slower rates of hydrolysis, the number of reactive groups on the cell surface is usually considerably lower than the number of amines. Not only is the considerable excess of reactive polymer needed is not only a highly inefficient use of reactive polymer, but such high polymer concentrations can exert cytotoxicity. Two approaches to increasing the surface density of chains while minimizing bulk polymer concentration is to perform multiple sequential reactions [18,19] or to utilize reactions that target multiple anchor groups [20,21]. Increased reaction efficiency has also been described using 2-iminothiolane (Traut’s reagent) to convert surface amines to thiol groups to which maleimide-functionalized polymers can subsequently be attached [22]. This approach not only exploits the slower hydrolysis rates of maleimides, conjugation efficiency is further improved by introducing a carbon spacer that renders the resultant thiol more accessible to bulky polymer chains [22]. Recently, a technique termed diffusion modulated macromolecular cell derivatization (DMMCD) has been used to increase the conjugation efficiency of macromolecular species to cell surfaces (Figure 13.2) [17]. By mixing the reactive PEG–NHS with an excess of a nonreactive macromolecule additive such as dextran or hyperbranched polyglycerol (HPG), the surface PEGylation could be increased up to 10-fold. The authors postulate that the synergistic effects of two phenomena can explain this increase in efficiency. First, the diffusion of one type of macromolecule can be enhanced by the presence of a second macromolecule in higher concentration, a process known as enhanced molecular transport. This more rapid diffusion of reactive PEG to the cell surface causes an apparent increase in polymer concentration at the cellular interface, resulting in increased grafting. This enhancement in bulk phase diffusion was postulated to operate synergistically with an increase in the ability of PEG chains to penetrate the glycocalyx, an effect mediated by interactions between the additive macromolecule and the cell surface. Hence, while covalent conjugation protocols must be empirically optimized with respect to polymer type, conjugation chemistry, cell type, and desired application, employing techniques such as DMMCD can widen the window of reaction conditions that may be explored and represents an important tool for improving covalent film deposition on cell surfaces.
Another important consideration for covalent grafting of polymer films to cell surfaces is the molecular specificity, or often lack thereof, of the conjugation. As most covalently anchored films exploit native amine or thiol groups, a diverse assortment of cell surface proteins may be modified indiscriminately and to various degrees. This has important implications for not only film properties and performance, but also the resultant cellular behavior and function. For example, conjugation of polymer chains to cell surface proteins involved in cell adhesion or migration (e.g., integrins) may dramatically influence the ability of cells to adhere to substrates or migrate. By contrast, failure to conjugate PEG to key epitopes on red blood cells (RBCs) can result in inefficient masking of antigens from recognition by cognate antibodies [23]. When increased molecular specificity is necessary, metabolic or genetic introduction of nonnaturally occurring reactive groups (e.g., azides) [8] or the use of enzymatic ligation strategies (e.g., biotin ligase) [24] may be harnessed to link polymer chains to specific molecules on the cells’ surface. While such approaches offer exquisite specificity, they are not suitable for all cell types or surface molecules. Improved specificity may also be conferred through control of the molecular weight and architecture of the reactive polymer. Smaller chains or chains that adopt a more compact conformation may more readily access certain cell surface proteins or buried residues. For example, conjugation of 5 kD PEG chains to RBCs has been shown to block antibody binding to D antigens more effectively than 20kD PEG chains, presumably due to the increased access of smaller chains to this particular surface antigen [22]. The molecular specificity of the conjugation can also have important implications for film stability as the turnover rate for surface proteins can vary dramatically [7] and may also be influenced by polymer conjugation [25]. Mass spectroscopy has recently been used to identify the specific cell surface proteins that were linked to polymeric nanoparticles via a thioether bond formed between thiols on the surface of T cells and maleimide-functionalized nanoparticles [26]. Interestingly, the authors found that many of the proteins involving key T cell functions, such as migration and antigen recognition, were not modified, whereas modified proteins were those that tended to accumulate at the immunological synapse. This helped to explain their finding that nanoparticles conjugated to T cells tended to concentrate at the immunological synapse yet did not interfere with migration or antigen recognition. The use of techniques and analyses such as this are critical to understanding patterns of conjugation that have significant impact on both cell behavior and the performance characteristics of the polymer film.
13.3.2 Membrane-Anchored Polymer Thin Films
Covalent chemical manipulation of native cell surface molecules may negatively affect their biological activity and perturb fundamental cell–cell and cell–matrix interactions and signaling processes critical to cell phenotype and survival [23,27,28]. This has spawned interest in noncovalent cell surface modification strategies, the most common of which harnesses passive insertion of a hydrophobic anchor into the phospholipid bilayer of the plasma membrane (Figure 13.1B). Amphiphilic polymers, which contain both hydrophilic and hydrophobic domains, can exploit this entropically favorable process to introduce polymer chains to the cell surface [29]. Amphiphilic polymers of a variety of compositions and architectures, including diblock [28,30], branched [31], and graft copolymers [11,25], can be used to modify cell surfaces, which offers an enormous degree of flexibility in film design. Polymer–phospholipid conjugates represent the most common type of polymer for noncovalent cell surface engineering due to the highly favorable interactions between diacyl lipids and native lipids in the plasma membrane. A key design variable in engineering this class of polymer thin film is the selection of the hydrophobic anchor(s), which determines the efficiency of membrane insertion. In comparing the cell surface density of polysaccharide chains linked to different hydrophobic anchors, a phospholipid (1,2-dipalmitoyl-glycero-3-phosphoethanolamino), a pyrene group, and single octyldodecyl chain, it was found that the degree of polymer incorporation was greatest when using the phospholipid, followed next by the pyrene moiety with the octyldodecyl anchor providing the least incorporation [28]. Liu et al. [30] reported similar findings in demonstrating that the surface density of an oligonucleotide was highest when using a diacyllipid anchor, followed by cholesterol, and, finally, an octyldodecyl anchor. This work also illustrated how chemistry of the hydrophilic segment can also influence membrane insertion efficiency. Introduction of a PEG spacer between the diacyllipid anchor and the oligonucleotide dramatically reduced the degree of insertion, owing to steric repulsion of PEG from the cell surface [30]. Another key consideration in the design of amphiphilic polymers for cell surface engineering applications is to ensure membrane insertion rather than membrane lysis. Many polymer-based surfactants can lyse cell membranes (e.g., Triton X-100), and therefore careful consideration and optimization of polymer design and film deposition conditions (e.g., concentration, time) is critical.
Film stability is perhaps the biggest limitation of polymer films assembled using membrane-anchors, as lipids and integral membrane proteins are in a constant state of flux, continuously being internalized, degraded, and replaced [1,32]. However, conjugation of polymers to lipid anchors may prolong surface residence time of lipids by interfering with natural mechanisms of endocytosis and recycling. For example, the lipid 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine (DSPE) is rapidly internalized into T lymphoblasts within an hour, whereas a PEG–DSPE conjugate could be observed on the surface 3–24 h later and appeared to be released directly into the surrounding culture media as opposed to first being endocytosed and subsequently exocytosed [25]. Such differences in not only in the membrane-bound half-life of polymer films, but also the mechanism of film instability can be an important consideration for some applications. For example, polymer films used for facilitating drug delivery from a cell surface to the surrounding environment (discussed below) could leverage a release mechanism from the cell surface; by contrast, internalization of the film may result in drug degradation in endo/lysosomal compartments [33] or be leveraged to achieve controlled intracellular drug delivery from the surface. Alkyl chain length of polymer–phospholipid conjugates can also influence stability. In comparing PEG–lipid conjugates with diacyl chain lengths of 12, 14, and 16 methylene units, Inui et al. [1,25] found that the stability of lipids on the cell surface increased with increasing alkyl chain length; longer chains, however, were not explored.
An alternative to using polymers with a single lipid anchor is to engineer copolymers bearing multiple hydrophobic moieties to provide multipoint membrane attachment. Graft copolymers comprised of a hydrophilic poly(vinyl alcohol) (PVA) backbone and grafted 14-carbon alkyl chains have recently been used as a facile tool for cell surface engineering [2,11,25,34,35]. By incorporating additional functional groups along the polymer backbone, such as thiols or biotins, such films provide a convenient approach for controlling the chemical composition of the cell surface [3–5,34,36]. Compared to a PEG–lipid conjugate containing the same number of methylene units in the alkyl chains, the PVA–alkyl copolymer remained on the cell surface longer, potentially due to the multiple attachment points [6,7,25]. Though dependent on cell type, coupling chemistry, and polymer properties, polymer thin films assembled through membrane insertion can be comparable to that of covalent modification, as illustrated by a comparative study in which similar stability was demonstrated between surface modification with an NHS–PEG reagent and noncovalent approaches utilizing PEG–lipid and PVA–alkyl polymers. Regardless, most polymers were excluded from the cell surface within 24 h. The short half-life of membrane-anchored polymers is a key consideration in their applicability. Strategies that incorporate crosslinkable sites into the membrane-anchor, such as diacetylene, which upon insertion with cell membranes can be UV polymerized in situ [7,8,37,38] may provide a mechanism for enhancing film stability.
13.3.3 Electrostatic Adsorption of Polymers
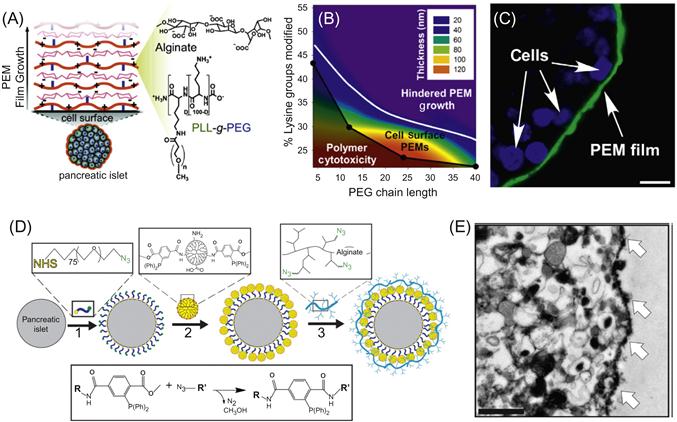
The rich abundance of carbohydrates and glycoproteins renders cell surfaces highly negatively charged, a property that can be harnessed for noncovalently engineering cell surfaces through the use of polycations that interact electrostatically with this negatively charged interface (Figure 13.1C). While monovalent electrostatic interactions with cells are weak due to the high ionic strength of physiological solutions, the multiple positive charges of polycations can promote stable binding via multivalent interactions. However, a significant challenge associated with this facile approach to cell surface engineering has been, and remains, the high cytotoxicity associated with many polycations [9–12,39–42]. Indeed, many polycations commonly used for fabrication of polymer thin films on inanimate surfaces (e.g., silicon wafers and glass slides) can elicit damage to cells by generating pores in the cell membrane resulting in unregulated efflux of molecules across the plasma membrane and ultimately cell death [13,43–45]. Moreover, notwithstanding the toxicity of most polycations, this propensity to rapidly translocate across the membrane in a non-energy-dependent manner, rather than adsorb to the extracellular surface, renders many polycations largely ineffective for cell surface engineering applications. This concept is demonstrated in Figure 13.3A, which shows a confocal micrograph of pancreatic islets incubated with a fluorescently labeled polycation, poly(L-lysine) (PLL). It is obvious in evaluating the images at sufficient magnification that PLL is localized intracellularly, colocalized with the nucleus and distributed broadly within the intracellular space of individual cells within the multi-cellular cluster. Not surprisingly, many such polycations have found utility as carriers in gene delivery applications, due in part to their ability to enhance intracellular uptake [14,33]. By contrast, a poly(L-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) copolymer specifically designed for cell surface applications [15,27,46], is observed predominantly on the extracellular surface (Figure 13.3B). Such a distinction may not be clear with lower magnification imaging and indeed in some published reports it is not; this has been and continues to be a weakness in the field that has confounded interpretation of data. Therefore, careful analysis of cellular localization of film constituents is a vital element of all cell surface engineering endeavors but is perhaps most critical when using polycations which have a propensity for perturbing the integrity of the cell membrane. The toxicity and membrane disruptive properties of polycations is dependent on a number of variables, including the type of positive charge (e.g., primary, secondary, tertiary amine), degree of ionization, concentration, contact time, deposition temperature, molecular weight, charge density, hydrophobicity, and chain conformation [16,39,40,42,45]. Additionally, polycation toxicity can vary widely depending on the cell type studied and the method through which viability is assessed [17,47]. Consequently, there is a considerable amount of disagreement and uncertainty in the literature regarding the use of polycations in cell surface engineering applications. For example, PAH has been reported to be both toxic and nontoxic to primary human pancreatic islets [17,48,49]; similar disparity exists for a number of other polycations.
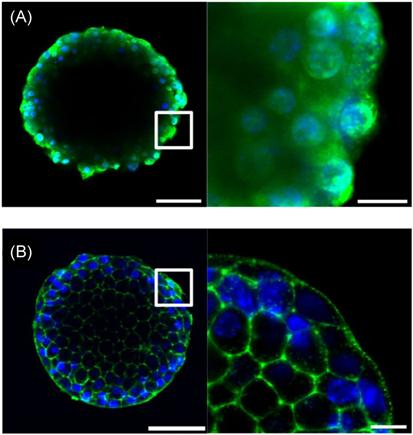
The cytotoxicity of polycations therefore requires careful selection of not only the appropriate polycation properties, but also deposition conditions. Low concentrations, short contact times, and low temperatures can be utilized to allow polycations to be deposited on cell surfaces while minimizing toxic effects. Additionally, naturally derived polycations such as chitosan and protamine tend to exhibit lower toxicity profiles and, therefore, have been used in a number of cell surface engineering applications [16,18,19,50,51]. Another approach has been to re-engineer polycation properties to render them less toxic and membrane destabilizing. While not explicit in their seminal work, Hubbell and coworkers first described that the grafting of PEG to a polycation, specifically PLL, can mitigate cytotoxicity and enable adsorption of polymer chains on the cell surface through electrostatic interactions [20,21,52]. Through adsorption of PLL-g-PEG copolymers, they were able to noncovalently introduce PEG chains to the surface of RBCs to prevent lectin-induced hemagglutination. This concept has recently been extended with the goal of further understanding the relationship between the degree of PEG grafting and PEG chain length and polycation cytotoxicity [22,27,46]. In this work, a library of 24 PLLMW-g[D]-PEGn (PMWPn[D]) graft copolymers of variable PLL backbone molecular weights (PMW) was synthesized, where MW is the PLL molecular weight in kDa, Pn is the PEG graft length, where n is the number of monomeric repeats, and D is the degree of grafting, defined as the percentage of backbone lysine groups grafted to PEG chains. These polymers were incubated with pancreatic islets, multicellular aggregates that were focus of these investigations, and cell viability was subsequently assessed. It was found that polycation toxicity decreased as charge density was reduced but, interestingly, at a fixed molar concentration and degree of grafting, increasing the PEG graft length decreased cytotoxicity, suggesting synergism between the degree of grafting and PEG chain length in reducing toxicity. Significantly, for each length of PEG chain used, a critical degree of grafting, Dc, could be identified where copolymers did not exert significant toxicity under the conditions tested, and Dc was found to decrease exponentially with increasing PEG chain length. Confocal microscopy of pancreatic islets incubated with fluorescently labeled PLL and PMWPn[Dc] revealed very distinct contrasts in behavior (Figure 13.3). PLL, which is highly toxic, translocated across the plasma membrane and was localized within the cytoplasm of individual cells, whereas labeled PMWPn[Dc] copolymers adsorbed to the apical surface of individual cells within pancreatic islets, indicating maintenance of cell membrane integrity and minimal endocytosis of copolymers. Such contrasting behavior suggests that conjugation of PEG chains to PLL inhibits the capacity of PLL to cross the cell membrane, most likely through inhibition of membrane pore formation, consistent with observed reductions in toxicity.
PLL-g-PEG copolymers engineered for enhanced cytocompatibility through control of structural variables can also be used as “cell surface active” polymeric carriers for ligands and reactive groups [22,27]. This technology, termed PAINTS (Polycations Assembled at Interfaces for Noncovalent Tissue Surfacing), utilizes copolymers bearing terminally functionalized PEG grafts to molecularly paint cell surfaces with a diversity of chemical functional groups. PEG grafts were terminally functionalized with biotin, hydrazide, and azide moieties, to selectively capture streptavidin-, aldehyde-, and cyclooctyne-labeled molecules, (e.g. carbohydrates and peptides) respectively, on cell surfaces [17,27,53]. The introduction of hydrazide groups, which are a useful handle for immobilizing oligosaccharides and glycoconjugates, is particularly notable since covalent generation of hydrazide groups on cell surfaces is not feasible using the commonly employed NHS–ester reagents. Additionally, this approach facilitates presentation of cell surface azido groups within minutes, providing a facile and rapid alternative to metabolic engineering approaches for chemically targeting cell surfaces via copper-free click chemistry, albeit with reduced molecular specificity. For example, a “one-pot” modification of cell surfaces with bioactive peptides could be achieved through concomitant adsorption of an azide-functionalized copolymer and chemoselective ligation of a cyclooctyne-modified peptide. Co-adsorption of differentially functionalized PLL-g-PEG copolymers was also demonstrated for simultaneous display of multiple functional groups on cell surfaces, and in principle, a library of copolymers bearing a diverse array of small molecules (e.g., peptides, oligosaccharides, other reactive groups) could be used combinatorially to achieve exquisite control over the composition of cell surfaces in a single step. Cell surface engineering using PLL-g-PEG copolymers also offers a number of advantages over conventional covalent approaches, particularly NHS–ester coupling. In contrast to NHS–ester conjugates, PLL-g-PEG copolymers allow cells to be functionalized in aqueous solvents containing primary amines (e.g., cell culture media) and may be dissolved well in advance of application without the hydrolysis associated with NHS–esters. Using a biotin-functionalized polymer as a model, adsorbed PLL-g-PEG copolymers generated surface densities of functional groups similar to that obtained by treatment with an NHS–PEG(biotin) reagent. However, pancreatic islets treated with the NHS–ester reagent adopted an altered morphology, possibly a result of impaired cell–cell adhesion due to covalent modification of surface proteins. However, the relative stability of this approach in relation to covalent strategies has not yet been evaluated.
13.3.4 Multilayer Polymer Thin Films
Over the past two decades, layer-by-layer (LbL) assembly of polymer thin films has emerged as one of the most versatile and facile bottom-up approaches for engineering surfaces of defined biological and physicochemical properties [23,54–60]. The premise behind the assembly of multilayer thin films is quite simple (Figure 13.4): polymer A binds to a surface via a number of possible interactions (electrostatic, hydrogen bonding, covalent conjugation), unbound polymer is then washed away, and the surface is then incubated with a second polymer, polymer B, that has an affinity for the bound polymer A. This completes the assembly of a polymer bilayer, and this process can be repeated indefinitely to form more complex multilayered assemblies. Through incorporation of enzymes and other proteins, nucleic acids, liposomes, biologically active nanoparticles, polymers functionalized with bioactive motifs, and guest–host supramolecular complexes, LbL films provide an immensely versatile platform for controlling the biochemical properties of surfaces. Moreover, film thickness, permeability, mechanical properties, and surface chemistry may be tailored through film design, providing additional mechanisms for manipulating biophysical phenomenon at interfaces. Relative to polymer monolayers, LbL films offer considerably more control over film properties, enabling a wider range of possible applications. For example, through control of film thickness and/or permeability, LbL films can be used to regulate the diffusion of molecules to and from the cell surface. Additionally, LbL films provide a three-dimensional scaffold for the presentation or controlled release of bioactive molecules and/or drug carriers and facilitate significantly higher drug loading than polymer monolayers. Moreover, LbL films may provide a route for engineering significantly more stable cell surface modifications since LbL films are anchored multivalently to the cell surface, and become much thicker than the lipid bilayer as successive layers are deposited and effectively “glued” together. This creates a cell–material interface with a very high aspect ratio from the perspective of the cell, which has been shown to inhibit endocytosis [8,61,62]. Furthermore, the mechanical properties of the film may be modulated [24,46] to render it difficult for cells to endocytose the film and its associated cargo [22,63]. Therefore, multilayer polymer thin films offer an enormous opportunity to expand the molecular repertoire of available cell surface modifications beyond what is currently possible with genetic and metabolic approaches or covalent and noncovalent chemistries.
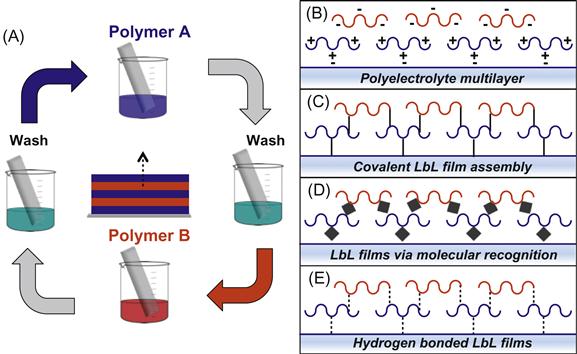
However, until recently, this powerful surface engineering technique has been largely inaccessible to living cell surfaces, due largely to the cytotoxicity inherent to conventional LbL film constituents and/or architectures. Polyelectrolyte multilayer (PEM) films (Figure 13.4B), which are assembled through alternating deposition of polycations and polyanions [7,54], represent the most common and versatile LbL film architecture. Here, a polycation is first deposited onto the negatively charged cell surface, followed by a polyanion, until the desired number of layers is obtained. Cells are washed after each polymer deposition step to eliminate unbound polyelectrolyte. While many (though not all) polyanions are highly cytocompatible, the well-documented toxicity elicited by most polycations [25,39–42] (see above) in direct contact with the cell membrane poses a significant hurdle in employing PEMs in cell surface engineering. Nonetheless, through careful selection of polycations and assembly routines, a number of reports have described the assembly of PEM films on mammalian cells or cellular aggregates without significantly compromising cell viability. Owing to its reduced cytotoxicity relative to synthetic polycations, chitosan, a naturally derived polysaccharide, has been commonly employed as the polycation component in the assembly of PEM films. For example, Rajagopalan et al. [26,50] reported use of chitosan in the assembly of PEMs on hepatocytes, Pickup and colleagues [23,27,28,51] have described the assembly of chitosan/alginate films on MIN6 cells, and Tabrizian and coworkers [29,64] have utilized phosphorylcholine-modified chitosan for coating RBCs with PEM films. Hence, chitosan may be an attractive choice of polycation for generating PEMs on cell surfaces, though it should be noted that dissolution of chitosan requires slightly acidic conditions (~pH 6.0–6.5), which may not be suitable for some cell types. Another approach to minimize polycation cytotoxicity has been to deposit an initial basement layer onto the cell surface to serve as a molecular spacer between polycations and the cell surface. For example, a cationic lipid conjugate was first inserted into cell membranes to enable deposition of a polyanionic alginate spacer layer prior to polycation deposition [28,30,65]. While such approaches may reduce direct contact between the polycation and the cell membrane, the capacity of polycations to diffuse within PEM films [31,66] may still permit direct interaction with the cell surface.
Another approach to designing PEM films for cell surface engineering has recently been reported by the team of Wilson and Chaikof [11,25,46] (Figure 13.5A–C). To circumvent the molecular hurdle of polycation cytotoxicity, they exploited PEG-dependent conformational changes in polycation (PLL) structure to design cytocompatible polycations for direct cell surface assembly of PEM films with tunable physicochemical and biochemical properties. A major challenge in designing films using PLL-g-PEG copolymers is a complex interplay between copolymer structural variables that influence cell viability and PEM growth in opposing manners. For example, decreasing polycation charge density generally attenuates cytotoxicity [28,39], while insufficiently charged species may be incapable of participating in film growth [30,67,68]. Similarly, PLL-g-PEG copolymers bearing long PEG chains can create steric barriers to protein adsorption and molecular recognition and similarly may hinder electrostatic interactions necessary to drive film assembly [30,69]. By mapping the relationships between copolymer properties and both cell viability and PEM film growth (using alginate as the polyanion), they were able to unveil a narrow window, where films of diverse and unique composition, thickness, and mechanical properties could be generated without compromising cell viability or function (Figure 13.5B). Additionally, through use of functionalized polyelectrolytes, a diversity of reactive groups could be incorporated into films, providing a strategy for further tailoring film properties and integrating bioactive molecules. By elucidating the effect of key film design variables (e.g., polycation structure, layer number) on both film properties and cellular viability/function, this work serves as a model for the rational design of cell surface supported multilayer thin films.
While PEM films represent arguably the most versatile architecture, the polycation toxicity challenge has also spawned efforts to design new architectures that are less dependent on electrostatic interactions and instead seek to utilize covalent bonds, molecular recognition, and hydrogen bonding to drive film growth. Covalently assembled films (Figure 13.4C) utilize the toolbox of bioorthogonal and chemoselective chemistries described briefly previously in this chapter and elsewhere in this book to link polymer layers both to the cell surface and to each other. Iwata and coworkers [1,32,36] described the assembly of a multilayer PVA film via covalent disulfide exchange reactions. To generate the initial PVA layer, the cell surface was modified noncovalently with maleimide groups using a maleimide-functionalized phospholipid and thiol-modified PVA was subsequently bound covalently via thiol-ene reaction. Through alternating deposition of thiol-modified and pyridyl disulfide-functionalized PVA, covalently crosslinked thin films of PVA could be assembled. Similarly, covalent films could be generated through alternating deposition of thiol- and maleimide-functionalized polymers (e.g., alginate, PEG, and lipid-based micelles). Significantly, these films could be grown to be approximately 30 μm thick, substantially thicker than most LbL assemblies and were shown to be stable on the surface of pancreatic islets for at least 30 days, representing a very stable surface modification [25,70]. The design of covalently assembled films is also exemplified by the recent work of Stabler and coworkers [33,71] who generated covalently crosslinked LbL films on the surface of viable and functional pancreatic islets using the chemoselective Staudinger ligation to link polymers functionalized with azide and phosphine groups (Figure 13.5D–E). Specifically, they utilized alternating deposition of a hyperbranched alginate bearing azide groups and poly(amidoamine) (PAMAM) dendrimers functionalized with methyl-2-(diphenylphosphino)terephthalate moieties. This not only provided a mechanism for orthogonal covalent crosslinking but also a strategy through which to integrate azide- or phosphine-functionalized molecules into the film. Interestingly, since PAMAM and alginate are polycationic and polyanionic, respectively, this allowed for generation of hybrid PEM–covalent films as well as comparison of films assembled via both mechanisms. Similar to the approach taken by Wilson and Chaikof [46], the degree of positive charge of phosphine-modified PAMAM dendrimers was attenuated via reaction of residual primary amines with glutaric anhydride (GA). Indeed, decreasing the PAMAM positive charge reduced toxicity and enabled deposition of an initial phosphine-rich layer on the cell surface, which served as the foundation for covalent immobilization of azide-functionalized hyperbranched alginate. Through repeated alternating deposition of azide–alginate and PAMAM–phosphine polymers, multilayer thin films could thereby be grown. Deposition of six bilayers generated films approximately 70–100 nm thick on the cell surface as measured by TEM (Figure 13.5E); this represents one of the more rigorous attempts to characterize film properties on the cell surface, a significant technical challenge. However, the amount of alginate deposited on the surface and the uniformity of the coating tended to decrease as the PAMAM cationic charge was decreased, prompting the investigators to fabricate films assembled completely through covalent interactions. This could be achieved by first introducing azide groups to the cell surface using a heterobifunctional NHS–PEG–azide, followed by covalent LbL assembly of a net neutral phosphine-modified PAMAM dendrimer and azide-modified hyperbranched alginate. Their results demonstrate that covalent cross-linking of LbL films could significantly enhance film stability, though films assembled purely via covalent interactions demonstrated a higher degree of surface inhomogeneity. This was attributed to incomplete and nonuniform coverage of the initial PEG-azide layer via NHS coupling relative to the more complete coverage achieved via electrostatic deposition of the PAMAM dendrimer. This highlights the need to balance the surface density and uniformity of the basement layer with film stability and toxicity, and also suggests that hybrid films assembled via multiple mechanisms may provide an ideal combination of these two important variables.
Cytocompatible LbL films can also be generated on cell surfaces using molecular recognition between two cognate-binding partners to drive film assembly (Figure 13.4D). Film growth is initiated using polymers functionalized with motifs that bind endogenous surface molecules or by first introducing an exogenous group (e.g., biotin) to the cell surface. Through alternating deposition of polymers or biomacromolecules that specifically bind to one another film growth can be achieved. This is perhaps best exemplified through the use of the biotin–streptavidin interaction, which represents one of the strongest noncovalent interactions known in nature. Wilson et al. [48] utilized electrostatic adsorption of cytocompatible poly(L-lysine)-g-poly(ethylene glycol)(biotin) (PPB) copolymers to create a biotin-rich cellular interface. This enabled binding of streptavidin; since streptavidin has four binding sites for biotin, a fraction remains unoccupied, which facilitated the binding of another layer of PPB and the regeneration of the biotin-rich interface. Therefore, through alternating deposition of streptavidin and PPB, PEG-rich multilayer films could be assembled on cell surface without compromising cell viability. Films can also be fabricated using natural biopolymers and/or biomacromolecules with complementary binding affinities. An elegant example of this concept was described by Akashi and coworkers [72] who generated thin films comprised of fibronectin and gelatin on the surface of adherent fibroblasts. Although both film components are negatively charged, they can be used to generate LbL films through binding of gelatin to fibronectin via a collagen-binding domain.
LbL assembly driven by hydrogen bonds has emerged as a powerful technique for generating films under physiologic conditions using polymers that carry no charge and, consequently, have recently been explored as an alternative to PEM films for cell surface engineering (Figure 13.4E). Originally developed for surface engineering and nanoencapsulation of yeast cells [73], hydrogen-bonded LbL films assembled through alternating deposition of tannic acid (TA) and poly(N-vinylpyrrolidone) (PVPON) have recently been translated for mammalian cell surface engineering [74]. Film growth is initiated through deposition of PVPON which bears pyrrolidone rings that contain a proton-accepting carbonyl group to facilitate hydrogen bonding with carbonyl, amide, and hydroxyl groups on the cell surface. The TA layer can subsequently be deposited through hydrogen bonding between PVPON carbonyl groups and hydroxyl groups on TA. This deposition cycle can be repeated to generate films entirely stabilized by hydrogen-bonding interactions. Moreover, TA possesses antioxidant properties and is capable of scavenging free radicals. Hence, these films possess intrinsically anti-inflammatory properties, as evidenced by reduced pro-inflammatory cytokine production when incubated with activated macrophages.
Despite the enormous potential offered by LbL films for cell surface engineering, a number of challenges and design considerations must be addressed when creating such films. As with any surface modification, film stability is a key consideration. While LbL films offer potential for generating more stable films, particularly the covalently assembled films described above, they are nonetheless susceptible to endocytosis and chemical and/or enzymatic degradation [51,71,74]. Moreover, film stability must also be accounted for during the film assembly process, as deposited layers may be endocytosed prior to addition of subsequent layers. Minimizing polymer incubation times and assembling films at reduced temperatures, where endocytosis is inhibited, can help ensure complete film assembly prior to returning cells to physiological temperatures. This, of course, must be balanced with the sensitivity of cells to lower temperatures over the time of the coating process. The coating process itself also represents a significant practical challenge in LbL film assembly, as the numerous deposition and wash steps are both time consuming and laborious. Most approaches have utilized centrifugation to separate cells from coating and wash solutions, a process that can mechanically damage cells, is time consuming and subject to human variability. Methods for automating assembly of LbL films on cells and tissues have been explored and are currently in development [75]. Automated methods will not only save time and increase the efficiency of the coating process, but also allow for high throughput optimization of film design and process variables.
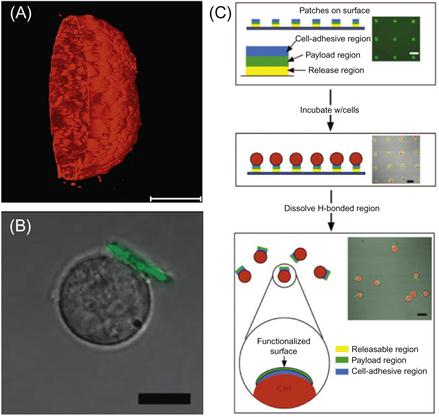
The direct assembly of LbL films on cell and tissue surfaces results in the formation of a thin coating that conforms to the geometry of the surface (Figure 13.6A). While this is favorable in some applications, such as generating barriers to diffusion of molecules to the cell surface, it can be disadvantageous in certain applications if the film interferes with cell–cell and cell–matrix interactions that are essential for desired cellular function. The team of Irvine and Rubner [76] has devised an elegant solution to this challenge that enables cell surface engineering with LbL films that does not completely occlude the cell surface. Rather than using the cell surface as the foundation upon which to generate films, they have focused on generation of LbL films that can be attached to cells postassembly, generating a so-called cell backpack (Figure 13.6B,C). In this approach, films are assembled onto a photolithographic patterned surface to create distinct patches of LbL film. The film is comprised of a temperature and pH-responsive basement region that allows for triggered release of the film from the substrate, a central payload region, and an outer cell-adhesive region that contains hyaluronic acid (HA), a ligand recognized by CD44 receptors on the surface of lymphocytes. Cells are then seeded onto the array of patches where they can bind to the patches via receptor–ligand interactions. Upon a change in temperature or pH, the PEM patches, now connected to cells, are released from the underlying substrate yielding a suspension of cells modified with a “backpack” that may contain diverse functional payloads. Significantly, cells maintained their native ability to migrate, a phenomenon dependent on molecular interactions between the cell surface and underlying substrate. This approach may prove particularly advantageous in applications where anisotropic cell surface modification is desired and also allows films to be assembled under nonphysiologic conditions using constituents that may be toxic in soluble form but are not when deposited on a surface. This approach greatly expands the possible options available for using LbL films in cell surface engineering applications where conformal cell coating is not required.
13.4 Biomedical Applications
13.4.1 Molecular Camouflage and Nano-Encapsulation
Cell- and tissue-based therapies continue to emerge as promising strategies for disease treatment. Early examples include blood transfusion and organ transplantation, now mainstays of modern medicine. Substantial advancement in areas such as adoptive T cell transfer [77], engineered induced pluripotent stem (iPS) cells [78], and mesenchymal stem cells (MSCs) [79] therapies has led to numerous promising clinical trials that continue to ignite excitement for the burgeoning field of cell-based therapy. However, in many instances, organs, cells, and tissues are derived from a nonautologous source, resulting in recognition and destruction of graft by the host immune repertoire that target epitopes present on the foreign cell surface and vice versa (i.e., destruction of host tissue by foreign immune cells). Therefore, cell and organ transplantation often necessitates the use of immunosuppressive drugs to prevent the destruction and rejection of the transplanted tissue by the host immune system. While considerable advancements have been made over the past two decades in the development of immunosuppressive drug therapy, systemic immunosuppression requires life-long treatment and continues to impose a number of adverse side effects. This challenge fueled one of the earliest applications of cell surface engineering: the use of polymer thin films to effectively hide or “camouflage” transplanted cells from detection by the immune system. To accomplish this, polymer thin films on the cell surface must provide a physical barrier between the cellular and molecular components of the immune system, including T cells, antibodies, and complement proteins, and their targets on the transplanted cell surface [80] (Figure 13.7). At the same time, polymer films must allow the diffusion of small molecules such as oxygen, glucose, and ions as well as the transport and binding of other molecules essential to cell viability and desired function. Therefore, in designing such films, it is essential to consider what cell functionalities are essential for a given application and which may be compromised at the benefit of gaining increased immune protection. RBC transfusion and islet transplantation are two prominent examples where the immune protection conferred by polymeric barriers can outweigh any deleterious effects on cell function.

There are two general classes of barrier: steric barriers, which prevent or inhibit molecular interactions as a result of the spatial structure of the film; and permselective barriers, which exclude diffusion of molecules on the basis of molecular size, charge, and/or physical configuration. The most common approach utilized in RBC transfusion and islet encapsulation has been to generate steric barriers via immobilization of PEG chains to the cell or tissue surface to prevent molecular recognition between cell surface receptors and soluble ligands. PEG is a hydrated, flexible polymer chain due to repeating, highly mobile, ether units, which creates a large hydrodynamic volume that allows the polymer chain to act as a polymer exclusion layer on the cell surface. PEG can be synthesized with a range of molecular weights and architectures (linear, branched, and star). These films have mostly been formed through covalent coupling of PEG to amine groups on the cell surface or by direct insertion of PEG–lipid conjugates into the cell membrane, although other approaches have been used including PEG-based copolymers designed to adsorb to cell surfaces [48,52,81,82].
The most common use of PEG in cell surface engineering has been in the pursuit of the “universal” RBC for blood transfusion. Although the discovery of ABO blood group antigens on RBCs has revolutionized the categorization and matched transfusion of blood, patients that receive multiple transfusions develop alloimmunity against minor antigens on RBCs that consequently prohibits identification of appropriate blood donors [83]. Moreover, limitations in donor blood supply, particularly for group O type, continue to drive interest in strategies to generate universal RBCs [23]. To prevent immune rejection of RBCs, surface-bound PEG chains must efficiently mask blood group antigens (e.g., A, B, Rh) from recognition by cognate antibodies in circulation; even a small amount of residual antibody-binding activity can have deleterious effects on RBC survival [23]. Additionally, surface PEGylation must not dramatically hinder the functionality or circulation time of RBCs nor interfere with natural splenic RBC clearance mechanisms [23,83]. Toward addressing this considerable challenge, investigations have focused on inhibiting antibody recognition of RBCs by optimizing the mechanism of PEG immobilization (i.e., conjugation chemistry), surface grafting density, molecular weight, and polymer architecture. Due to the relative lack of molecular specificity of PEGylation reagents and the diversity of antigens present on the RBC surface, there exists a complex and incompletely understood interplay between these variables in achieving complete masking of blood antigens without compromising in vivo survival and circulation properties. This is exemplified by studies demonstrating that only RBCs PEGylated at and below grafting concentrations 0.4 mM of 5 kDa PEG and 2 mM of 20 kDa PEG survived in mice, despite having normal in vitro cell structure, function, and viability [84].
The surface density of PEG and the efficiency of antigen masking does not necessarily correlate with the solution concentration of reactive PEG, which is confined by an upper limit where RBC morphology and function are compromised [23,85]. Hence, largely empirical approaches are commonly utilized to determine optimal conjugation conditions. For example, for a 5 kD cyanuric chloride-functionalized PEG, it was found that a maximal 15 mg/ml PEG concentration, pH of 8.7, temperature of 14°C, and reaction time of 30 min were most suitable for RBCs [85]. Likewise, the dependence on PEG molecular weight and architecture in masking RBC antigens is not entirely intuitive. Several groups have demonstrated that PEG chains of different molecular weight are more effective at masking certain blood group antigens; for example, 20 kD PEG has been shown to mask A antigens better than 5 kD PEG, whereas D antigens were more efficiently blocked by 5 kD PEG chains [22]. Similarly, in comparing linear PEG chains to highly branched polyglycerol (HPG), PEG chains formed larger exclusion zones and, consequently, masked major blood antigens better than HPGs, though the more compact HPG structure was found to be more effective than PEG at protecting certain minor antigens [84]. As the inhibition of antibody binding is dependent on steric blockade of cognate antigen epitopes, this result may reflect differences in the accessibility of amino groups within specific blood antigens to different sized PEG chains. Therefore, using combinations of molecular weight PEG can lead to more effective antigen masking. Additionally, while amine-reactive chemistries have been most commonly explored, some evidence suggests that the maleimide linkage may be more stable under certain environments encountered during transfusion, although further study is necessary to more thoroughly characterize the properties of RBCs modified via different chemistries in vivo [86].
Optimization of PEG conjugation chemistries has yielded a number of preclinical studies in animal models that illustrate both the promise and challenges of RBC PEGylation. Scott et al. [14] were among the first to evaluate PEGylated RBCs in vivo, using cyanuric chloride coupling to conjugate methoxy poly(ethylene glycol) (mPEG) to RBC surface amines. PEGylated RBCs lost their ABO group reactivity and were more resistant to phagocytosis by monocytes in vitro [14], but, significantly, appeared to be functionally normal in vivo [87]. However, the concentration of reactive mPEG used to achieve normal in vivo RBC survival (0.4 mM) was not found to efficiently block antigen binding in vitro; conversely, the high concentrations used to mask antigens in vitro lead to poor in vivo survival. The group also demonstrated that PEGylated sheep RBCs exhibited significantly prolonged survival when transfused into mice, with ~90% less antibody produced in response to mPEG-coated RBCs, illustrating the promise of using PEG coatings to improve transfusion across species. In a study comparing the conjugation of 2, 5, and 20 kD mPEG coupled using cyanuric chloride, BTC, and NHS ester amine-reactive chemistries, they found that the 20 kD mPEG chains to provide better immunocamouflage and, using a 20 kD BTC-functionalized mPEG, reported normal survival of RBCs in mice [13]. Recently, Chen et al. have also demonstrated no ill effects of RBC PEGylation on the recipient microcirculation, as indicated by no changes in hematocrit, hemoglobin concentration, blood vessel diameter, blood flow velocities, and the interstitial partial oxygen pressure (pO2) before, during, and after the injections of PEG–RBCs coated with a 20 kD mPEG [12]. While RBCs have been the major target, PEGylation of other blood cell types has also been explored. mPEG modification of peripheral blood mononuclear cells effectively decreased antibody recognition and inhibited T cell activation [88]. Similarly, PEGylated donor T cells prevented allorecognition and initiation of graft-versus-host disease in vivo [89], and PEGylation of allografts prolonged survival in a bone marrow transplantation model [90]. Platelet concentrates have also been modified by PEG to prevent bacteria binding and biofilm formation [91]. Attempts have even been made to perfuse entire hearts with reactive PEG ex vivo to attenuate hyperacute xenograft rejection upon transplantation [92].
Despite promising in vivo reports throughout the last two decades, generation of universal RBCs through surface PEGylation has yet to achieve clinical realization. In addition to the continued challenge of obtaining complete antigen masking, the discovery of anti-PEG antibodies that can shorten the survival and circulation time of PEGylated RBCs poses another significant barrier to the use of PEG in the engineering of “universal” RBCs [23]. This has prompted recent investigation into other surface conjugated polymers. Kizhakkedathu and colleagues [84,93,94] have recently explored NHS activated HPG as an alternative to PEG. Like PEG, HPG is highly hydrated and biocompatible but is more compact and contains multiple hydroxyl groups for linking therapeutic molecules. Through appropriate control of HBG molecular weight and grafting concentrations, HBG-functionalized RBCs demonstrated normal in vivo circulation times and splenic clearance mechanisms [93]. Though PEG grafting provided better shielding and protection of ABO antigens from antibody recognition than HPG polymers [84], HBGs provided superior protection against some minor antigens [84,93], which is important in chronic blood transfusion where alloantibodies to minor antigens develop. Importantly, HPG-grafted RBCs do not appear to be immunogenic, as a similar RBC circulation profile was observed upon repeated administration in mice over several months. Covalent conjugation of polyethyloxazoline has also been explored as a PEG alternative [95], and though polyethyloxazoline grafting better preserved RBC morphology, mPEG was superior for masking blood group antigens.
As covalent grafting of polymer chains can damage RBCs through modification of integral membrane proteins, resulting in sequestration of RBCs by the immune system, noncovalent approaches have been explored as an alternative to covalent grafting of PEG and other polymer chains. Elbert and Hubbell [52] were among the first to utilize a noncovalent strategy, using PLL-g-PEG copolymers that adsorbed electrostatically to RBC surfaces. By controlling the length of the PLL backbone and the PEG grafting ratio, they created an adsorbed film that was able to prevent lectin-induced hemagglutination; however, camouflage of specific RBC surface antigens was not explored. More recently a conceptually similar approach has been explored by Grandfils and coworkers [96] using poly(2-dimethylamino ethylmethacrylate) (PDMAEMA) and PDMAEMA–PEG copolymers, though a low efficiency of blood group antigen masking was observed for all polymers evaluated.
Tabrizian and colleagues [64,97] have recently taken a different polymer film engineering approach to generating a universal RBC. Instead relying on a single polymer layer to form a steric barrier between RBC surface antigens and antibodies, they instead assembled a PEM thin film on the surface of RBCs. Key to their design was selection of a polyelectrolyte pair, particularly the polycation, and a film architecture that did not result in RBC lysis or loss of function while also providing a barrier to antibody binding. Moreover, an optimal balance between solution cell density and polyelectrolyte concentration had to be determined to avoid cell aggregation during the film buildup process due to cross-bridge formation. To achieve this, the group assembled a film consisting of an inner protective shell made of four bilayers of chitosan-graft-phosphorylcholine (CH-PC)/alginate (AL) to prevent RBC lysis and an outer shell composed of two bilayers of alginate/PLL-g-PEG terminated with a negatively charged alginate layer to repel proteins from the RBC surface. This film could be assembled without RBC lysis and did not comprise RBC oxygen uptake. Significantly, a multilayered shell composed of 13 biocompatible polyelectrolyte layers of (AL/CH-PC)4−(AL/PLL-PEG)2-AL was found to dramatically inhibit the binding of antibodies against A, B, D, and Rh RBC surface antigens, as measured in an agglutination assay. While LbL films had previously been deposited on fixed RBC templates [98], this work represents the first to develop an LbL film that can be assembled on functional RBCs. It remains to be seen how LbL-coated RBCs will function in vivo, particularly given the possibility that other key RBC properties may have been altered as a result of film assembly, for example, RBC mechanical properties or accessibility of the essential RBC membrane protein CD47 [84]. While covalent conjugation of PEG is still the gold standard in RBC camouflage technology, there remains a significant need for the development of new polymer thin-film-based approaches to enhance RBC transfusion.
The replacement of compromised insulin production through transplantation of pancreatic islets has long been conceived as a promising therapy for the treatment of diabetes [99]. However, immune recognition and destruction of transplanted cells remains a major limitation to the widespread clinical realization of islet transplantation [80]. Host reaction to transplanted islets can be generally categorized into two intertwined phases. The first phase, which happens immediately upon transplantation, results in activation of the coagulation cascade and induction of an inflammatory response initiated by islet-surface-derived factors [99]. Recruitment of immune cells and production of proinflammatory cytokines leads to nonspecific destruction of a large fraction of injected cell mass within days of transplantation. The second phase of the response is mediated by adaptive immunity and the generation of antibodies and T cells that recognize foreign antigens on the islet surface, resulting in destruction of the graft [100]. Both phases of the response are largely driven by molecular recognition of islet-surface molecules by cells (e.g., macrophages, cytotoxic T cells) and molecules (e.g., complement, antibodies) of the host immune system, and, hence, molecular camouflage of these cell surface mediators using polymer thin films has emerged as a promising strategy for improving the outcome of islet transplantation.
Similar to surface modification of RBCs, optimized chemistries and conditions have been developed that allow PEG chains to be displayed on the surface of islets without compromising viability or function. These strategies have primarily utilized amine-reactive PEG [10,15,18,19,101–104] (e.g., NHS) or passive membrane insertion of PEG–lipid conjugates [16,105]. The efficacy of islet PEGylation in attenuating immune responses and improving islet function has been evaluated in a number of animals of both allo- and xenotransplantation. Byun and colleagues [10] have demonstrated that covalent conjugation of PEG (5 kD)–propionic acid to islets extended islet survival from 5 to 11 days in a rat allograft model. Histological evaluation demonstrated the presence of the PEG on islets 30 days after transplantation, a significant finding considering the rapid turnover of many cell surface molecules. While PEGylation prevented graft infiltration by host immune cells, it did not prevent host cell recruitment to the graft site [10,18]. Though the PEG film appeared to impede recognition of the islet by immune cells, it did not appear to inhibit the release of antigens and immunostimulatory compounds (e.g., chemokines, cytokines), resulting in lymphocyte recruitment and localized production of toxic mediators. Hence, it was postulated that islet PEGylation could synergize with systemic administration of immunosuppressive drugs, enabling prolonged islet survival while reducing the dose, and side effects, of immunosupressive drug required. Indeed, in a model of allotransplantation, the combination of islet PEGylation with low-dose cyclosporin A extended islet survival time of PEGylated islets from 12 to more than 100 days, the lifetime of the rats used in this study [10]. More recently, the group has developed an innovative approach through the use of 6- and 8-arm PEG stars terminally functionalized with catechol groups, which react with islet-surface amines [101]. Here, in a more challenging xenograft model of islet transplantation, conjugation of the PEG stars did not significantly enhance islet survival relative to unmodified islets (~11 days), but doubled survival (21 days) when administered in combination with the immunosupressive drug Tacrolimus. Survival could be extended further (>50 days) using a combination of PEGylation, Tacrolimus, and anti-CD154 mAb. Similarly, Dong et al. [102] demonstrated that islet PEGylation resulted in long-term (>100 days) normoglycemia in 30% of recipients, whereas no long-term normoglycemia was observed for mice receiving uncoated islets. This effect could be extended to 57% by tethering nanoparticles loaded with the immunomodulatory drug leukemia inhibitory factor (LIF) to PEG chains. Currently, islet PEGylation is not able to completely protect islets from the host immune system, a consequence of both the relatively short stability of the modification compared to the life span of the graft as well as the complex immunological mechanisms underlying islet destruction [80]. Nonetheless, the synergy between PEGylation and low-dose immunosuppression holds considerable promise for improving the outcome of islet transplantation while reducing the potentially severe side effects of immunosuppressive drugs. In this regard, cell surface PEGylation should be considered to be part of the therapeutic arsenal when designing combination immunosuppressive drug regimens.
While the relatively short residence time of most monolayer polymer thin films on cell surfaces limits their efficacy in completely preventing immune rejection of islets over the desired life span of the graft, the surface residence time of such thin films may be more appropriate to camouflage early cellular and molecular recognition events that lead to deleterious inflammatory responses [99]. For example, Contreras and coworkers [103,104] have used islet-surface PEGylation in a xenogenic model of intraportal islet transplantation, where xenoreactive natural antibodies and complement act rapidly to destroy the islet graft. Within the immediate posttransplant period (2 weeks), animals that received islets treated with PEG presented significantly better control of glucose than animals receiving nonmodified islets. PEG with a molecular weight of 5 kD performed slightly better than 2 kD PEG, and capping surface-grafted PEG with albumin proved most efficacious, which was attributed to the enhanced capacity of thicker films and/or larger steric barriers. More recently, Teramura and Iwata [105] have utilized a noncovalent approach to islet-surface PEGylation using lipid-modified PEG in both allograft and autograft [106] models of islet transplantation. In both reports, surface engineering of islets with PEG resulted in improved islet engraftment, extending the duration of graft survival and permitting infusion of a fewer number of islets to achieve normoglycemia, mostly likely through inhibition of early inflammatory responses. However, as is the case in adaptive immunity to transplanted islets, the presence of a molecular blockade may not be sufficient to completely abrogate responses, which may also be mediated by the release of chemoattractants and proinflammatory compounds from the islet [99]. Therefore, molecular camouflage has been combined with surface immobilization of active regulators of inflammatory and thrombotic responses. For example, PEGylation has been combined with surface immobilization of heparin, thrombomodulin (TM), and urokinase, all active regulators of the coagulation cascade [9,16,34,107].
While islet PEGylation has clearly demonstrated some encouraging results, it remains unclear whether or not surface-grafted PEG will ultimately remain stable enough to provide protection for the anticipated lifetime of a human islet graft. Moreover, data suggests that the efficacy of PEGylation is at least partially limited by the lack of a defined pore structure and dependence on a steric exclusion effect in order to provide an immunoprotective barrier. Inspired largely by the concept of islet microencapsulation, which provides a higher level of immunoprotection than PEGylation but is limited by high transplant volumes and diffusional limitations [80], several groups have recently began to explore the possibility of constructing nano-thin films of controlled permeability and surface chemistry directly on the surface of pancreatic islets via LbL polymer self-assembly (discussed above), effectively creating conformal, permselective membranes of nanoscale thickness. Wilson and Chaikof [46,48] were the first to describe in vivo investigations into the ability of LbL films to protect islets from host inflammatory and immune responses. In their most recent report [46], islets coated with an eight bilayer PEM film comprised of PLL-g-PEG and alginate showed a trend toward improved engraftment in vivo in a murine allograft model but failed to improve rates of conversion to normoglycemia to a statistically significant level. While this study marked the first of its kind in that viable cells coated with PEM films were shown to function in an in vivo environment, the data suggest that the films provided an insufficient barrier for protection against early mediators of islet destruction. To date, no other in vivo investigations into the use of LbL-coated islets, or other cell types, have been reported. Instead, other investigators have focused on evaluating the barrier capacity of the film in vitro by assessing the ability of the film to prevent binding of fluorescently labeled ligands for islet-surface targets. For example, Pickup and colleagues [51] demonstrated reduced penetration of an antibody against class I major histocompatibility complex upon coating MIN6 cell spheroids with chitosan/alginate PEM films. The assembly of LbL films on cell surfaces is a nascent area of research, with most efforts focusing on developing new film types and deposition strategies to circumvent the attendant fabrication and toxicity challenges; however, tools for evaluating cell surface film permeability and barrier capacity, among other properties, are in their infancy.
13.4.2 Polymer Thin Films for Enhancing Cell and Drug Delivery
Cell-based therapies have recently found utility in the treatment of numerous pathologies, including heart and vascular disease, stroke and spinal injury, musculoskeletal disorders, cancer, and diabetes. The field of cell surface engineering has enabled modification of cell surfaces with an enormous diversity of biologic drugs, as well as drug delivery vehicles, that bestow the cell with new biochemical functionalities to improve therapeutic performance (Figure 13.7). For example, cell surfaces can be engineered with specific targeting moieties to facilitate cell homing to an injured area [31,108,109], enzymes that can enhance localized production of therapeutic products [9,20,107], and nanoparticle carriers for sustained release of drugs that can improve cell function and survival [26,102,110]. In some instances, the local production or release of drugs from the cell surface has been shown to be superior to parental drug administration [110], which may be limited by systemic toxicity, inadequate circulation time, and lack of targeting. In many such cell surface engineering applications, polymer thin films play an essential role in enabling immobilization of exogenous bioactive species.
As in many other applications described herein, surface immobilized PEG serves as the most common and notable example of a polymer thin film that enables the introduction of exogenous bioactive molecules to the cell surface. The high biocompatible and water solubility of PEG render it an attractive choice as a linker and spacer between the cell surface and the tethered species. Moreover, the increasing commercial availability of heterobifunctional PEG molecules with diverse reactive groups has increased the accessibility of cell surface engineering with PEG-based linkers. Work by Chaikof and coworkers serves as a good illustration for the use of functionalized PEG films for immobilization of biological active biomacromolecules. Their team developed a two-step process for covalently conjugating recombinant TM, an important inhibitor of thrombosis and inflammation, to the surface of pancreatic islets in a chemo- and bio-orthogonal manner [9]. The group first introduced triphenylphosphine moieties to the cell surface through use of a heterobifunctional NHS–PEG–triphenylphosphine linker. This enabled subsequent chemoselective Staudinger ligation of a recombinant TM construct engineered with a C-terminal azido (N3) group. This process could be achieved without compromising the viability or function of the islet. By increasing the surface density of TM on the islet surface, the thrombin-mediated conversion of protein C to activated protein C (APC) was dramatically enhanced. Similarly, Iwata and colleagues have utilized lipid–PEG conjugates with a terminal maleimide group for covalent anchoring of thiol-modified TM, urokinase, a thrombolytic enzyme, and complement receptor 1 [107,111]. The team has also utilized graft copolymers comprised of a hydrophilic PVA backbone functionalized with both pendant 14-carbon alkyl chains and maleimide groups and used this carrier for conjugation of similar biomacromolecules [34]. Given the deleterious effects of thrombosis and inflammation on islet survival in the immediate posttransplant period, arming islet surfaces with an arsenal of anticoagulant and anti-inflammatory molecules offers a promising strategy for improving clinical outcomes of islet transplantation.
Heterobifunctional PEG has also been an essential tool for engineering cell surfaces with nanoparticles for controlled drug release. Recently, Dong et al. [102] utilized an NHS–PEG–biotin compound to first introduce biotin groups on islet surfaces, allowing subsequent immobilization of ~100 nm PLGA nanoparticles coated with avidin molecules. The PLGA particles were designed to enable efficient loading and controlled release of LIF, a cytokine that can improve islet engraftment through reduction of inflammation, induction of immunological tolerance, and promotion of islet beta cell survival and growth. They postulated that islet viability, insulin secretion, and in vivo function could be enhanced through local and sustained of LIF to both islet cells and the surrounding milieu. Indeed, they demonstrated that while islet PEGylation resulted in long-term (>100 days) normoglycemia in 30% of recipients, this effect could be extended to 57% by tethering LIF-loaded nanoparticles to the cell surface. This work is predated by the seminal work of Irvine and colleagues, who tethered drug-loaded nanoparticles to the surface of T cells and hematopoietic stem cells to improve in vivo survival. In this study, however, nanoparticles were tethered directly to cell surface thiols without the use of a polymer film as a linker.
Holden and colleagues have recently demonstrated another manner by which cell surface polymer films can potentially be used for drug delivery from cell surfaces [112]. In a proof-of-concept study, the group covalently tethered PEGylated poly(amido amine) (PAMAM) dendrimers, a common polymeric drug carrier, to the surface of macrophages. To do so, they first generated cell surface aldehydes via mild sodium periodate treatment. Resultant aldehydes were then coupled to amine groups on the dendrimer via Schiff base formation, a bond that was subsequently reduced to improve stability using sodium cyanoborohydride. However, as conceded in their report, surface modification of highly endocytotic cells such as macrophages and dendritic cells poses a significant challenge for stable surface modification. Toward addressing this challenge, the team of Rubner and Mitragotri utilized the “cell backpack approach” described earlier in this chapter to functionalize macrophage surfaces with patches of LbL polymer films that were largely resistant to phagocytosis [63]. Because macrophages struggle to phagocytose very flat and disk-shaped particles [61,62], such as the LbL patches used, stable cell surface modification can be achieved. To exemplify the potential utility of polymer films for drug delivery, PEM patches were loaded with a layer containing bovine serum albumin as a model biologic payload, which could be released from the film over several hours. Owing to the versatility of such PEM films in loading diverse therapeutic agents, this strategy offers great potential for cell surface engineering of highly phagocytic cells, such as dendritic cells currently used in cancer immunotherapy [113]; however, the ability of therapeutically relevant cells modified in this manner to function in vivo remains to be demonstrated. Though both of the aforementioned studies stopped short of loading the polymer film with therapeutically relevant drugs, by harnessing the ability of macrophages to migrate to infected sites, damaged tissue, and tumors, these reports establish an important foundation for using drug-loaded polymer films to empower cell-mediated delivery of therapeutics.
While some cell-based therapies can be directly injected into the target tissue (e.g., pancreatic islet transplantation) or have the intrinsic capacity to locate pathologic tissue (e.g., adoptive T cell transfer), many therapeutic cell products inefficiently home to the desired site because the infused cells lack key receptors required for homing. This results in only a small fraction of the infused cells, as well as any associated drug cargo, arriving at the site of interest; hence, engineering cell surfaces with molecules to improve the efficiency and specificity of targeting is an area of active investigation [31,108,109]. In this application, polymers have also played an important role (Figure 13.7). Sarkar et al. introduced the selectin ligand sialyl Lewisx (sLex) to the surface of MSCs via a three-step process, first covalently conjugating biotin to MSCs via NHS coupling, followed by immobilization with streptavidin, and lastly displaying a biotinylated tetravalent sLex construct with a poly-(acrylamide) polymer linker [108]. By maximizing the surface density of sLex, the MSC homing to a site of inflammation in vivo via selectin-mediated rolling as well as subsequent extravasation through the endothelium was significantly enhanced. Here, not only did the poly(acrylamide) linker provide a facile strategy for immobilization of sLex, the authors also suggest that the polymer helped space the sLex groups at a distance from the cell surface comparable to that of native ligands, providing a sufficient length to bypass the steric and electrostatic repulsion provided by the endothelial glycocalyx. More recently, Jeong et al. utilized an HPG functionalized with both octadecyl chains and a peptide sequence that targeted vascular endothelial adhesion molecule (VCAM) overexpressed by inflamed blood vessels [31]. This multimodal polymer efficiently incorporated with the surface of MSCs through insertion of the octadecyl chains into the lipid bilayer, resulting in a surface-bound HPG polymer monolayer displaying the targeting peptides on the surface in a single step and without any covalent modification. By engineering the cell surface with this novel “cell-guidance” molecule, MSCs more efficiently adhered to inflamed endothelial cells in an in vitro flow camber model. While the in vivo homing capabilities of this technology have not been assessed, these works serve as excellent illustrations of how multifunctional polymers, specifically engineered to interact with the cell surface, and carry biologically relevant molecule, can provide a versatile and facile approach for re-engineering the molecular landscape of cell and tissue surfaces.
13.4.3 Tissue Engineering
The ability to organize individual cells in a spatially controlled manner, particularly in three dimensions, offers great potential for de novo engineering of tissues, for creating in vitro models of pathology, and for modulating the cellular composition of a target tissue. Polymer thin films have played an important role toward this end (Figure 13.7). A relatively simple but important application has been to use cells modified with polymeric thin films to generate 3D cell aggregates [114–119]. In these applications, the polymer film serves as “glue” that helps hold the cells together, a synthetic extracellular matrix of sorts that drives cellular assembly, and provides initial structural support. The use of simple polymer films to assemble cell aggregates is exemplified by the work of Yu and coworkers who have utilized covalent linkages between hydrazide-functionalized PEI [114] or cationic dendrimers [115] and cells engineered with surface aldehydes through mild periodate treatment. Here, the polycation serves to bring the cells together through electrostatic crossbridge formation, allowing hydrazone bonds to subsequently be formed between the cells. Of note, the polymer concentrations and contact times were optimized in these studies to minimize potential toxicity. This linkage was reported only to last for about 5–7 days after which time the aggregate structure was maintained by natural cell–cell and cell–matrix interactions. This strategy has been used to dramatically accelerate the formation of spheroids comprised of carcinoma cell line for use as a 3D cell culture model for in vitro drug penetration studies [116]. More recently, the group has utilized an entirely noncovalent approach using oleyl–PEG conjugated to a polypropylenimine hexadecaamine dendrimer [117]. Here, the positively charged dendrimer serves to concentrate the polymer on the surface such that when cells are brought into close contact with each other through mechanical means (e.g., centrifugation, micromanipulation), they may be linked together through insertion of the hydrophobic oleyl groups into adjacent cell membranes. The ability to link cells into complex patterns was demonstrated using optical tweezers to spatially organize engineered cells; though perhaps not a scalable strategy for tissue engineering applications, it serves to demonstrate the enormous potential of this approach in controlling tissue architecture and may prove useful in understanding cell–cell interactions in hierarchical cellular architectures.
Gartner and Bertozzi [118] have also described a versatile and elegant approach to generating 3D cellular assemblies. By covalently modifying cell surfaces with oligonucleotide sequences through Staudinger ligation or copper-free click chemistry, cells engineered with complementary DNA strands self-assembled in a highly specific manner to create heterotopic 3D microtissues of defined composition and connectivity. Cell connectivity via DNA hybridization could be reversed through melting of DNA sequences at elevated temperatures or through addition of DNase, providing simple and convenient mechanisms for eliminating extracellular DNA after tissue assembly. As a simple but telling demonstration of tissue engineering using this approach, a murine B-cell line that is dependent on the cytokine IL-3 for survival was linked via DNA hybridization to CHO cells engineered to express IL-3. Only when the B cells were linked to CHO cells were they able to survive, demonstrating the formation of a paracrine signaling network in three dimensions. In contrast to approaches utilizing specific receptor–ligand interactions (e.g., biotin–avidin) or orthogonally reactive chemistries, this approach to tissue self-assembly is particularly powerful since the number of complementary DNA sequences that may be employed to drive assembly is practically unlimited. This provides a combinatorial strategy that, in principle, could allow precise control of the arrangement of a diversity of modified cell types in a single step. Moreover, the group has also demonstrated the ability to use this approach for templating cells on substrates in a controlled manner [120,121], offering further potential for this technology in engineering tissues from scaffolds of more complex geometries.
The formation of cellular aggregates has also been driven through the use of patches of multilayer thin films assembled using the previously described “cell backpack” approach. To utilize the LbL patches for this application, Swiston et al. [119] mixed cells with a suspension of PEM patches that had been released from the underlying substrate. The cell surface linked to the patches through receptor–ligand interactions, here, CD44 on B cell surfaces and HA deposited on the surface of the PEM patch. In this scenario, backpacks, displaying multiple ligands on their surface, are free to associate with multiple cell membranes simultaneously, thereby linking multiple cells together. By controlling the dimensions of the backpack and the ratio of cells to backpacks, the size of cell aggregates could be tailored. This approach to generating self-assembling cell aggregates may be particularly advantageous over strategies that use a polymer monolayer in that the PEM patches provide a versatile method for incorporating a diversity of bioactive molecules that can be released from the film after aggregate assembly, delivering key chemical cues to direct the fate of the aggregate.
LbL films have also recently emerged as a strategy for creating layered assemblies of cells for tissue engineering applications. Rajagopalan et al. [50] assembled cell-adhesive chitosan/DNA PEM films on adherent cell monolayers in order to promote the adhesion and growth of a second layer of cells, the film effectively serving as a glue between layers in a multilayered cellular construct. This approach is limited, however, by the use of chitosan as a PEM film constituent, owing to its cytotoxicity and low solubility in neutral buffers. Akashi and coworkers [122] later reported a conceptually similar approach to generate 3D cellular multilayers but employed LbL films comprised of the extracellular matrix proteins fibronectin and gelatin, which interact with each other through biorecognition rather than electrostatic interactions. Using this approach, they were able to generate assemblies comprised of four cell layers, both homo- and heterotopic, that could be subsequently removed from the underlying substrate to which cell were initially adhered, providing a strategy for engineering cell sheets. Given the conformal nature of films assembled through LbL assembly, these approaches could reasonably be applied to generating cellular multilayers on more geometrically complex substrates as well, which might further expand the potential of this approach in tissue engineering applications.
In addition to de novo assembly of cellular assemblies, investigators have also used thin-film-based approaches to integrate exogenous cell types with naturally occurring cell aggregates, most notably pancreatic islets. This has been exemplified by the work of Iwata and colleagues who have utilized a number of different polymer thin-film-based approaches to encapsulate pancreatic islets with a layer of living cells. This not only creates both a physical barrier between the islet and the surrounding environment but also a biochemical barrier generated by factors expressed or secreted by the immobilized cell. The group has utilized phospholipid–PEG(biotin) to introduce biotin groups to the cell surface, facilitating subsequent immobilization of streptavidin-coated HEK293 cells [35]. They have also utilized an approach similar to Gartner and Bertozzi wherein polyDNA–PEG–lipid conjugates were used to noncovalently engineer cell surfaces with specific DNA sequences. By incubating poly(adenine) functionalized HEK293 cells with poly(thymine) functionalized islets, HEK293 cells could be specifically immobilized on the islet surface through DNA hybridization [123].
13.4.4 In vivo Modification of Cells and Tissues with Polymer Thin Films
While cell-based therapy and cellularized tissue engineered constructs offer incredible potential for treating a number of diseases, challenges such as cell sourcing, cell and tissue preservation and banking, and notably, high cost remain potential barriers to the clinical translation of these technologies. Hence, in situ fabrication of thin films in living organisms using specifically engineered polymers or film deposition strategies may provide a solution to some of these translational challenges as well as open new opportunities in drug delivery and tissue engineering (Figure 13.7). Elbert and Hubbell [124] were the first to demonstrate in vivo surface remodeling of tissue surfaces with polymer thin films. Building upon their previous work using PLL-g-PEG copolymers to noncovalently modify cell and tissue surfaces with PEG brushes in vitro, they postulated that self-assembly of polymers with a tissue-binding domain (PLL) and a tissue repelling domain (PEG) could be used to prevent formation of surgical adhesions in a rat model. In this model, an ischemic injury is made to the uterine horn that untreated results in fibrous adhesions to other organs in the peritoneal cavity. By applying a solution of PLL-g-PEG to both the injury site and to the organs in the vicinity of the uterine horn, the incidence of surgical adhesion could be reduced by 88% owing to disruption of cell–tissue adhesive interactions as well as inhibition of fibrin formation. This work highlights a number of potential advantages of using such a noncovalent approach for in vivo tissue remodeling, most notably that the modification was achieved nearly instantaneously, no leaving groups or other reaction byproducts were generated, and the modification could be accomplished in a complex biochemical milieu comprised of proteins and carbohydrates that might otherwise compete with covalent methods to modify the cell surface.
In vivo cell surface engineering has also been used to improve the efficacy of therapeutics. Irvine and coworkers [30] have recently described a noncovalent approach for in situ engineering of tissues with oligonucleotides, specifically CpG oligodeoxynucleotide, a single-stranded DNA stimulator of the immune system that is being explored as an adjuvant for vaccination and as a cancer immunotherapeutic. Toward this end, they conjugated CpG to a diacyl lipid and postulated that the amphiphilic construct would enable spontaneous in vivo cell surface engineering via membrane insertion of the diacyl chains. They found that CpG–diacyllipid conjugate was retained at an intratumoral injection site significantly longer than unmodified CpG, a relatively small and hydrophilic macromolecule that rapidly diffused from the injection site. The authors hypothesized that cell surface engineering with CpG provided a mechanism for prolonging residence time in the tissue, creating a depot that enhanced localized immune attack of the tumor. While the insertion of the conjugate into cell membranes in vivo was not explicitly demonstrated, and it is conceivable that micellization of CpG–lipid conjugates might also effect biodistribution kinetics. This study serves as an excellent example of the potential benefit that can be achieved through in vivo engineering of cells and tissues with therapeutic agents.
Integration of implantable and injectable biomaterials and tissue engineered scaffolds with the surrounding host tissue remains a major challenge to the field and an active area of research. To address this challenge, several groups have developed implantable polymeric materials that covalently react with surrounding cells and tissue surfaces. These covalent strategies must utilize chemistries that do not rely on toxic catalysts or generate harmful by-products and reactive groups must be sufficiently stable to allow for aqueous solubilization prior to injection. Elisseeff and coworkers [125,126] have described the synthesis of a multifunctional condrotin sulfate (CS) bearing both acrylate and aldehyde groups for use in cartilage repair applications. A solution of the polymer can be applied to a defect in cartilage, where aldehyde groups on the polymer bind to proteins on cells and extracellular matrix through Schiff base formation. The excess polymer is then washed away, generating a CS thin film functionalized with acrylate groups on the tissue surface. Acrylate groups at the tissue surface could then be covalently integrated with an injectable acrylate-based hydrogel formed via in situ photoinitiated polymerization. Thus, the polymer thin film serves as a bridge between the injured tissue surface and the injectable biomaterial scaffold, promoting tissue integration at the interface. Similarly, Brubaker et al. [127] recently utilized a four-arm PEG bearing terminal catechol groups to effectively glue pancreatic islets to tissue surfaces. Oxidation of the catechol-functionalized polymer solution with sodium periodate induces rapid hydrogel formation with o-quinone groups generated during the process capable of covalently coupling with primary amines at cell surfaces. The group leveraged this strategy to apply a thin polymer film that covered pancreatic islets deposited on the epididymal fat pad and external surfaces of the liver. By mixing the sodium periodate and polymer solutions immediately before deposition, the polymer gelled in situ while covalently coupling with both islets and tissue surfaces, creating a stable interface between two. This allowed islets to be immobilized in anatomic positions not possible without the use of such an adhesive. Significantly, the film elicited minimal acute or chronic inflammatory responses and permitted normoglycemic recovery and islet revascularization.
13.5 Conclusion
Over the past century, engineered polymer thin films have had an enormous technological impact, and their use as functional coatings has enabled the realization of a plethora of products and devices that now are mainstays of daily life. However, most polymer thin film technologies, even those intended for coating of medical devices, were not developed for cell surface engineering applications, where the health and survival of the cellular support is of utmost importance. Recent advances in polymer and organic chemistry, combined with an improving understanding of cell–polymer interactions, are now allowing the rich surface engineering toolbox afforded by polymer thin films to be translated to living cellular interfaces. This has opened up a number of new and exciting areas in the field of cell surface engineering with significant potential for clinical impact. Notably, the recent advances in engineering of cell surfaces with LbL thin films is ushering in a new wave of cell encapsulation and molecular camouflage research. For the past several decades, the field has largely been dominated at two ends of the length scale: large microcapsules, which provided maximal protection but imposed mass transfer restrictions, and surface immobilized polymer chains, which did not add to the transplant volume, but lacked permselectivity. Many of the LbL films currently in the early phases of development may provide the solution to this conundrum. Moreover, LbL films, particularly covalently crosslinked LbL films, are also well poised to circumvent the stability challenge associated with many cell surface engineering approaches. Rapid growth and development in the field of cell-based therapeutics has also identified a need for the delivery of drugs to support the survival and function of transplanted cells. This merger of cellular therapy and drug delivery has spawned highly innovative research focused on engineering of cell surfaces with nanoparticle vehicles and thin films for local delivery and controlled release. While polymer films have thus far taken only a modest role in this area, with some of the more prominent reports described here, the inevitable need to increase drug loading and control drug release profiles is likely to invoke the use of polymer films as linkers or scaffolds in the near future. For some applications, LbL films, such as the cell backpack, may be uniquely suited to deliver large amounts of cargo in a highly controlled manner. For others, it may be necessary to use a diversity of linker chemistries and immobilization methods to maximize immobilization of enzymes or nanoparticle delivery vehicles. Finally, while most cell surface engineering endeavors are performed ex vivo and under controlled conditions, the emerging field of regenerative medicine and tissue engineering has led to the development of injectable polymers that can integrate, both covalently and noncovalently, with the surface of cells and tissue. In vivo cell surface engineering strategies are also beginning to merge with the drug delivery paradigms of controlled release depots and targeted drug delivery, where immobilization of drugs or drug carriers on cell surfaces can enhance local retention time, improve uptake at a desired site, or modulate the pharmacokinetics of the drug. In all of these in vivo applications, polymers will likely play an important role in mediating the interaction with the target tissue or otherwise improving the therapeutic efficacy. Looking forward, engineered polymer thin films will undoubtedly continue to play a major role in the development, improvement, and optimization technologies that will define this century. The innovation and research over the past several decades now allows cell surface engineering to be added to this list of technologies.