
Chapter 6
Critical Issues and Quality by Design
6.1 QUALITY BY DESIGN
A crystallization process for a specific compound will be governed by the compound’s inherent properties for nucleation and growth. Both morphological and polymorphic formation are also species‐specific. The resulting crystal, physical, and chemical attributes will also depend on external properties of the surrounding environment such as impurities, solvents, temperature, and the rate of generation of supersaturation and mixing. For organic compounds, these critical properties vary over exceedingly wide ranges that are caused by their inherent structural and external operational differences. The reader is referred to Chapters 2, 3, 4, and 5 for a discussion of these properties.
Direct scale‐up from an underdeveloped laboratory procedure may result in a different product from the expected one. These differences may include purity, crystal forms, particle size, wide and/or bimodal particle size distribution (PSD), needles or plates with difficult downstream processing properties, and lack of consistent results.
As stated in ICH guidelines Q11, control strategy should ensure that each drug substance’s Critical Quality Attributes (CQAs) are within the appropriate ranges, limit, or distribution to assure drug substance quality. The drug substance specification is one part of a total control strategy, and not all CQAs need to be included in the drug substance specification. CQAs can (i) be included on the specification and confirmed through testing the final drug substance, (ii) be included on the specification and confirmed through upstream controls (e.g. as in Real Time Release Testing), or (iii) not be included on the specification but ensured through upstream controls.
Examples of upstream controls can include:
- In process testing.
- Use of measurements of process parameters and/or in process material attributes that are predictive of a drug substance’s CQA. In some cases, Process Analytical Technology (PAT) can be used to enhance control of the process and maintain output quality.
Regardless of whether a traditional or enhanced process development approach is taken, the use of upstream controls should be based on an evaluation and understanding of the sources of variability of a CQA. Downstream factors that might have an impact on the quality of the drug substance, such as temperature change, moisture level, solvent composition/antisolvent addition rate, and shear/mixing, should be taken into consideration.
When developing a control strategy for crystallization and downstream operations, a manufacturer can consider implementing controls for a specific CQA at single or multiple locations in the process, depending on the risk associated with the CQA and the ability of individual controls to detect a potential problem, for example PAT for in‐line solution concentration or PSD.
The quality of each raw material used in the manufacturing process should be appropriate for its intended use. Raw materials used in operations near the end of the manufacturing process, i.e. active pharmaceutical ingredient (API), have a greater risk of introducing impurities into the drug substance than raw materials used upstream for manufacturing the drug substance intermediates. Therefore, manufacturers should evaluate whether the quality of such materials should be more tightly controlled than similar materials used upstream.
Determining the critical process parameters (CPPs), which affect CQAs, inevitably requires a sound understanding of fundamental principles, such as solubility profiles, solid–liquid phase diagram, crystal growth and nucleation behaviors,. and their dependence on process variables such as solvent types, temperature, and mixing. Additionally, due to the close interaction of these multiple variables, it is highly desirable to apply model‐based statistical design of experiment (DOE) to quantify their interaction and consequence with confidence. Integration of fundamental principles with model‐based DOE tool can be considered to be the “best practice” to achieve QbD.
The purpose of this book in general, and of this chapter in particular, is to highlight the complex interactions between the inherent properties noted above as well as the possibilities for their manipulation to achieve the desired outcome. The objective is to present development, design, and scale‐up guidelines, i.e. quality by design, that can assist in evaluating and manipulating the inherent properties of the compound and its crystallization environment with the goal of achieving a more robust and scalable process from laboratory to commercial manufacturing. This will ensure both consistent manufacturing of high‐quality drugs and favorable approval from the regulatory agents.
Most examples in this book illustrate successful manipulation to achieve particular goals. In some cases, however, the natural growth and/or morphology have/has not been modified sufficiently by the corrective action to achieve success. For difficult situations such as these, downstream problems can be minimized by optimization of process variables (Johnson et al. 1997; Kim et al. 2005).
6.2 BASIC PROPERTIES
6.2.1 Solubility and Crystal Forms
The first set of basic properties of crystallization is solubility and crystal forms phase diagrams. These properties describe the thermodynamic behavior of compound of interests in liquid–solid (or vapor–solid) equilibrium. These equilibrium properties resemble closely to vapor/liquid composition in V–L equilibrium or liquid/liquid composition in L–L equilibrium.
As discussed in Chapter 2, solubility can be a function of multiple variables, including solvents, temperature, counterions (for salt and co‐crystals), and impurities. The solubility difference between the desired molecules and impurities essentially defines the purification efficiency and rejection of these impurities. An example is presented in Section 2.1. Ideally, the solubility of the desired molecule should be low, and undesired impurities, which can be process‐induced or present in the starting materials, should be high. In this scenario, it can maximize the rejection of these impurities from the solid and minimize the loss in the mother liquor of the desired compounds. Since there are many variables which can affect solubilities, it is essential to have a sound understanding of the landscape of solubilities in general and then identify suitable solvent systems, temperature, counterions, etc. based upon the actual values of these properties for a good purification/crystallization process.
If the impurities form solid compound, such as racemic compound, or solid solution with the desired molecules, one option is to find counterions to form salts or co‐crystals which can disrupt the formation of solid compound or solid solution. A classic example is using chiral resolving agents to form diastereomeric salts with the R/S isomers. The diastereoisomeric salts form solid mixture, but the R/S isomer forms racemic compounds. As shown in Example 7.4 of chiral resolution of racemic ibuprofen with S‐lysine. The R‐ibu‐S‐lys‐salt and S‐ibu‐S‐lys salt will simply be different diastereomeric salts with different solubilities, whereas the original R/S ibuprofen forms a racemic compound. If such an approach is not feasible, other purification techniques such as extraction or absorption would be explored for selective removal of these impurities.
The other side of the S–L equilibrium phase is solid state. As expected, different solid/crystal forms, including polymorph and pseudo‐polymorph such as solvates/hydrates, would have different equilibrium solubilities. Also, crystalline solids, in general, have a lower equilibrium solubility and a higher stability than the amorphous solid (sometime also called as subcooled liquid). Since aqueous solubility can be directly related to bioavailability, especially for BSC II and IV class compounds, selection of crystal forms can have direct impact on drug efficacy.
Another complication would be amorphous solid or L–L phase as discussed in Chapter 2. This phenomenon may be less common, but is considered to be an “equilibrium” behavior, which is independent of supersaturation (Section 6.4). Oiling or amorphous solid formation can be true obstacles for crystallization.
6.2.2 Particle Size and Morphology
PSD and morphology are two other unique properties associated with solid state. There are no such properties associated with liquid and gas states.
Different crystal forms can have different crystal morphology, due to inherent arrangement of molecules within the crystal structure. Crystals may be one‐dimensional such as needles, two‐dimensional such as plate, or three‐dimensional such as rock. Different crystal forms may have different crystal morphology and it may easily serve as a quick index for differentiation of crystal forms.
Particle size, even for the same crystal form, can vary widely from submicron to few hundred microns for pharmaceutical solids. Particle size is strongly affected by multiple variables in the crystallization process, i.e. initial seed, supersaturation, nucleation, crystal growth, mixing intensity which are rate dependent kinetics, and mixing which can be scale‐dependent.
6.3 SEED
The influence of seeding on crystallization is often critical to control a process. The importance of an appropriate seeding strategy cannot be overemphasized. While some systems will nucleate spontaneously under some level of supersaturation, a robust control of a process, especially upon scale‐up, that depends on spontaneous nucleation can be subject to extreme process variation for several reasons, including:
- The presence or absence of seed particles and/or foreign particles from a previous batch.
- Differences in concentration of impurities from batch to batch that affect the nucleation rate.
- Differences in the rate of generation of supersaturation from batch to batch.
- Differences in the concentration of solute from batch to batch.
- Differences in mixing scale‐up.
- Local conditions in the crystallizer such as wall temperature, anti‐solvent addition point, and evaporation rate.
The importance of seeding has been underscored by several authors including Mersmann (2001, pp. 410ff.), Mullin (2001, pp. 197), Myerson (2001, pp. 240–241), Tung (2013) and is observed in many processes by the authors of this book, such as Examples 7.1, 7.2, 9.3, 10.1, and others
6.3.1 Determination of Seed Form, Size, and Quantity
At the conclusion of a crystallization operation, the crystal form, number, and size of the product crystals will be determined by a number of factors:
- Seed added or generated at the outset.
- Nuclei generated during the operation (e.g. by energy input of mixing, local regions of high supersaturation).
- Fragments of crystals generated by attrition during the operation.
- Ostwald ripening, growth dispersion, and size‐dependent growth.
Of these critical factors, the one that is most subjected to predetermination and control is the seed added or generated at the outset which is also called in situ seed. The following section offers guidelines on effective seeding strategy and methods.
6.3.1.1 Seed Crystal Form and Quantity
It is a typical requirement that crystal form of seed should be the same as desired crystal form of final product after crystallization. Seed with the desired crystal form will facilitate the formation of desired crystal form and minimize the formation of undesired crystal form during crystallization. Conversion of crystal form during crystallization should be avoided as it essentially falls back into the non‐seed scenario. For anhydrates or desolvates which are generated after drying, one approach is to slurry some of these crystals in the crystallization solvent first to convert them back to hydrates or solvates for use as seed.
Four levels of seeding can be described, depending on the purpose of seed addition as follows:
- Pinch, to hopefully avoid oiling out and/or uncontrolled nucleation, crashing, or snowing out. It may be satisfactory in the laboratory but is rarely effective or reliable on scale‐up.
- Small (<1%), to hopefully aid in more controlled nucleation but not adequate to achieve primary growth on scale‐up. It is subject to additional nucleation and bimodal distribution of the product. However, this approach can be adopted in the in situ seed generation approach, which generates massive seeding for the subsequent crystallization operation, thus maintaining the benefit of massive seeding but minimizing the amount of external seed used.
- Large (5–10%), to improve the probability of growth with the possibility of preventing further nucleation and bimodal distribution.
- Massive (>10%), to provide maximum opportunity for all growth. (See Examples 7.6 and 13.6 on resolution of optical isomers.)
Seed can be critical in the control of enantiomer separation and selection between polymorphs and hydrates/solvates. In these applications, the seed will maximize the growth of the desired product where nucleation of undesired isomer/polymorph/hydrate/solvate are minimized/prevented.
6.3.1.2 Estimation of Seed Size and Quantity
The impact of seed size and quantities on final product particle size increase if an all‐growth process were calculated (Table 4.3) by a simple relationship. Just a note that validity of this calculation depends on an all‐growth process. Nucleation—which is virtually always present to some degree — will result in a reduction in the actual particle size of the product as well as an increase in total particles per unit volume. This increase can come in the form of a bimodal distribution made up of growth on seed and nucleated smaller crystals.
6.3.1.3 Seeding Procedures
There are several sources of seed crystals that may be used with varying degrees of effectiveness. Each seeding application requires analysis to determine which of the following sources will be most satisfactory for the operation in question. These sources and methods of preparation include the following:
- Seed from the previous batch—added separately or as heel recycle; effective if the physical and chemical attributes and stability remain satisfactorily constant or can be normalized by intermediate treatment.
- Seed prepared in a batch for that purpose and used in many batches; effective provided that the seed so prepared has the necessary physical and chemical attributes and stability.
- Seed prepared as above and then (dry) milled to achieve a mean particle size and PSD as required; effective for achieving increased control of physical attributes.
- Seed generated in situ from the current batch or heel recycling, both with wet milling, to control the mean particle size and PSD; one of the most effective seeding procedures when applicable (see Example 10.1).
The issues involved in the necessary record keeping, good manufacturing practice (GMP), and regulatory control that are associated with a seeded process in the pharmaceutical industry are sometimes cited as reasons not to seed. In response to an article endorsing this premise (Pessler 1997), the following rebuttal by C.B. Rosas (1997, personal communication) summarizes his and the authors’ views on this topic.
Not only is timely seeding an excellent way to do away with batch‐to‐batch vagaries in the width and sensitivity of the metastable, supersaturated region, but the avoidance of nucleation is often crucial for achieving crystallization elegance—a requisite for enhanced rejection of impurities. To trade away such a powerful tool for the sake of trivially lessened inconveniences in record keeping in a GMP plant is most unsound as an operating principle. For those frequent and difficult purification tasks that crystallization from solution does so well, seed early, seed often and, above all, seed always.
6.3.2 Effectiveness of Seeding
The effectiveness of seeding is linked to several key factors, including the following:
- timing of seed addition
- condition of the seed surface
- method of seed addition
- rate of generation of supersaturation
6.3.2.1 Seeding Point
The obvious problem of adding the seed before reaching saturation is that of dissolution of some or all of the seed, and the problem of adding the seed after reaching saturation is that of already experiencing nucleation. These difficulties are greatly exaggerated in cooling crystallization of solute with steep solubility dependence on temperature, as described in Example 7.1. In this example, suitable timing of addition of seed was considered to be unachievable in the manufacturing operation and an alternate strategy utilizing seed heel recycling, as described in the example, was utilized.
This problem can also be severe in evaporative methods particularly at reduced pressure, as is common. For these and other reasons described in Chapter 8, evaporative methods are considered to be the most difficult for predictable control of mean particle size and PSD on scale‐up.
For anti‐solvent addition methods, the seed can be added to a small part of the anti‐solvent, which is then added as slurry as the saturation point is approached. Although some of the seed may dissolve, this technique is considered to be more reliable than adding the seed all at once.
For reactive crystallization, the solubility of the product is usually low and seed can be added before the operation is initiated without concern for dissolution. However, since this method generally produces the smallest particles and is subject to agglomeration, the effectiveness of seeding is critically dependent on providing sufficient surface area for growth at the outset to prevent nucleation at the high local supersaturation conditions at the point of addition and throughout the resulting slurry. The reader is referred to Examples 10.1 and 10.2 for descriptions of operations in which nucleation was minimized in favor of growth.
6.3.2.2 Instrumentation for Seeding Timing
Online instrumentation can be a critical aid in detecting the correct seeding point (Zhou et al. 2006). Measurement of solution concentration by, e.g. Fourier transform infrared (FTIR), near infrared (NIR), UV/visible or Raman spectroscopy, can be very effective in determining the seeding point. After seeding, in order to determine whether or not seeding was effective, particle count and size distribution measurement can be made by an in situ particle size and counting instrument. Sampling and laboratory measurement can be used, but changes that can occur in the sample and the time delay can often introduce unreliability.
Depending on the outcome of the particle number and size determination, corrective measures may be initiated in the event that the seed is dissolved or there was excessive nucleation. Reseeding can correct the former and reheating to dissolve excess nuclei may be applicable in the latter.
6.3.2.3 Seed Aging and Annealing
Many crystallization operations can benefit from a seed age in which the temperature is held constant or the anti‐solvent addition is temporarily halted while the seed crystals grow and deplete the supersaturation before continuing. This procedure can help in normalizing the PSD resulting from the initial steps of seeding and supersaturation generation. The use of online instrumentation can again be very effective in determining when the seed age is complete and the operation can continue.
In some cases, a crystallization procedure is unsatisfactory for producing satisfactory seed crystals for subsequent use. This is particularly true for reactive crystallization because of the fine particles normally produced by this method. Since no amount of fine particle seed is satisfactory for significant growth in succeeding batches, the next possibility is to increase the particle size of the seed. To accomplish this, it is necessary to grow seeds in a separate operation from the reactive crystallization. Other applications may also require separate preparation of grown seed. The following procedure is one possible method.
In a separate operation, starting with fine needles, these crystals are subjected to many heat/cool treatments with sonication/wet milling between cycles. A suitable solvent is required in which the solubility approximately doubles on heating (e.g. a temperature increase of 20–50°C). During heating, the finer crystals will dissolve. During slow cooling, growth on the remaining crystals may be achieved. Sonication/wet milling after cooling can break the needles lengthwise, creating more particles. In subsequent cooling cycles, the diameter of the needles, the slowest growth dimension, can slowly increase eventually producing three‐dimensional crystals.
This procedure is applicable to other shapes. Crystal breakage by sonication/wet milling may result in other types of crystal cleavage but three‐dimensional growth may also be achieved in these cases.
The needles shown in Figure 10.7a were subjected to this heat–cool treatment resulting in the large three‐dimensional crystals shown in Figure 10.7a. Success in growing seed crystals both prepares seed for larger batches and establishes that this compound will actually grow. It is then necessary to determine if it will grow at a practical rate in the actual reactive crystallization system.
Several authors indicate the importance of the condition of the surface of seed crystals, and some qualitative suggestions can be made. These include (i) making a seed slurry to help condition the surface by dissolution of shards and providing some surface activation (which is also a good method of seed addition) and (ii) avoiding the use of dried seed. Milled seed may be necessary for particle size control, but milled seed should either be reslurried or the milling should be done by wet‐milling methods.
6.3.2.4 Continuous Operation
The most effective seeding is achieved in semicontinuous and continuous crystallizations by the nature of the operations themselves, in which the seed is always present and in large quantity. Although common in large industrial operations, these techniques have found more limited application in the pharmaceutical industry. As observed, there are more pharmaceutical applications using the in situ wet seed generation approach (Chapter 4) which has the advantages of mass seeding as in the continuous crystallization, but without the burden of large industrial operations.
There are excellent examples as detailed in Examples 7.6 and 13.6, on the continuous resolution of optical isomers in fluid bed crystallizers, and in Example 10.1, which presents a semicontinuous method of utilizing seed heel recycle in reactive crystallization to achieve primary growth. Also, Example 9.7 represents a semicontinuous method utilizing in situ generated seed in anti‐solvent crystallization. Because they are primary growth processes, these applications require reduction in seed crystal size to prevent oversized crystals from limiting the surface area. The methods documented in these examples include sonication and wet milling.
6.3.2.5 Method of Seed Addition
Seeding with dry powder seed through a vessel head nozzle, while widely practiced in the past, is now limited because of safety and exposure considerations. Slurry seed additions by pump or from seed tanks are preferred and are superior to powder addition for the reasons discusse 9.7d above. The advantages of retaining the seed in the system as utilized in continuous operation or in heel recycling are also indicated above. Needless to say, for in situ generated seed it is no longer an issue.
6.4 SUPERSATURATION
6.4.1 Generation of Supersaturation
As described in Chapter 2, there are multiple variables which can affect the solubility, including temperature, solvent composition, and counterions. Therefore, by varying and controlling these variables, it can generate and control the supersaturation. Additionally, by evaporating the solvents, the batch concentration can increase and become supersaturated. Figure 6.1 displays these four basic techniques to generate the supersaturation and Table 6.1 further highlights some unique characteristics associated with each method.
In Figure 6.1, the solid line represents the solubility of compound of interests. The dotted line represents batch concentration. The dotted arrows represent the direction of change of process variables and the solid arrow represents the supersaturation.
For the cooling crystallization, the batch concentration remains constant. But as temperature decreases, the solubility decreases. Therefore, the batch concentration will exceed the solubility and become supersaturated. For the anti‐solvent crystallization, the batch concentration will be diluted upon the addition of anti‐solvent. However, the solubility decreases faster than the batch concentration. Therefore, the batch concentration will exceed the solubility and become supersaturated. For the reactive crystallization, similarly, the batch concentration will decrease due to dilution. However, the desired compound will react with the counterions to form salts/co‐crystals which have much lower solubility than the free molecule, and the batch becomes supersaturated. For the evaporative crystallization, the batch concentration actually increases due to solvent evaporation. But the solubility remains basically unchanged. Therefore, batch becomes supersaturated.

Figure 6.1 Four types of supersaturation generation.
Table 6.1 Characteristics of generation of supersaturation.
| Methods | Characteristics |
|---|---|
| Cooling | Solubility slope is normally higher at higher temperatures. Slower cooling rate at higher temperatures may be required. Crystallization kinetics are normally slower in comparison to anti‐solvent or reactive crystallization. Therefore, it can be less mixing sensitive. |
| Anti‐solvent | Solubility can be nonlinear. More solubility data would be needed to design a crystallization process. Upon addition of anti‐solvent, it is likely to create high local supersaturation, which leads to rapid local nucleation. |
| Reactive | Solubilities between free molecule and salt/co‐crystals can be very different. Upon addition of counterions, it may create high local supersaturation. Crystallization kinetics can be fast under high local supersaturation. |
| Evaporative | Can be integrated with other techniques, including cooling, anti‐solvent, or reactive, for improvement of recovery. There is a possibility of foaming under rapid evaporation. |
The seed size and amount are critically linked with the release of supersaturation. Thus the use of increased seed amounts affords an increase in rate of supersaturation generation by providing more surface for growth while avoiding nucleation. Addition and cooling rates will be less sensitive but can still be influential. Seeding is essential, but is not the only consideration for successful operation.
These interacting process parameters are illustrated in Figure 6.2, where supersaturation is plotted against an average solution concentration that would be experienced during addition over the indicated time interval (each point represents an average—one point per run, not a sequence of points in one run). The amount of seed is shown as a parameter in allowing an increased addition rate. This concept is also valid for anti‐solvent addition time and reactive reagent addition time.

Figure 6.2 Effect of time of addition of an anti‐solvent or reagent on supersaturation with seed level as the parameter.
6.4.2 Oiling Out, Agglomeration/Aggregation
The most important property of a compound from a crystallization point of view may be its inherent growth characteristics. The critical question is: will it grow? Additionally, in an actual crystallization operation, in order to be crystalline, nucleation must occur and the resulting nuclei are then assumed to grow to some limit in the nucleation phase. The question here is whether, after reaching this nucleation limit, further conditions of growth will or will not result in additional growth. While the authors have found that most compounds will continue to grow to some limit, depending on many process and inherent factors, there are some that do not exhibit significant growth beyond the 5–10 micron range.
A substantial‐sized subgroup of those compounds which do not exhibit typical crystal growth, in which a repeated lattice grouping or crystal structure, is so difficult to achieve that the compound resembles a liquid as it emerges from solution. This is the phenomenon of oiling out. It is perhaps helpful to begin this discussion with the consideration of oiling out since it may be the first event in the pathway of crystallization or, in extreme cases, the operation may end with an oil, gel, or intractable gum or tar (Bonnet et al. 2002).
6.4.2.1 Oiling Out
Oiling out, beyond as an equilibrium phenomenon as discussed in Section 6.2.1, can be a kinetic phenomenon in crystallization by any of the methods of creating supersaturation and becomes increasingly possible under several conditions, including:
- high supersaturation
- rapid generation of supersaturation
- high levels of impurities
- presence of crystallization inhibitors even at low levels
- absence of seed
- inadequate mixing (high local supersaturation)
Kinetically, when supersaturation is achieved rapidly such that the concentration is beyond the upper metastable limit—as can often be the case in a nucleation‐based process—the substrate is forced to separate into a second phase by the creation of the resulting high solution concentration. However, crystallization is delayed by a slow crystallization rate. This combination may result in the creation of a nonstructured oil or possibly an amorphous solid. The rates of phase separation and nucleation are relative to each other such that “slow nucleation” implies only that nucleation was not fast enough to create discrete particles before oil separation.
Transition of an oil or an amorphous solid or a crystal can then occur. However, this type of operation can be difficult to control, and scale‐up is treacherous because the oil droplets may coalesce into masses and/or form gum balls and increase in size to intolerable levels.
Oiling out may be minimized or eliminated by control of supersaturation and seeding (Deneau and Steele 2005), as discussed below. Seeding has been proven to be essential to prevent oiling out in some systems because, although the oil may not be the thermodynamically stable phase, the transformation to crystals may be sufficiently slow and uncontrolled to cause severe processing problems, as discussed above.
The initial formation of an oil, gel, gum, or amorphous, solid conforms to the Ostwald step rule discussed in Myerson (2001, p. 39). This rule states that in any process, the state that is initially obtained is not the most stable state but rather the least stable state that is closest in terms of free energy change to the original one. It has been postulated that the initial state of crystallization processes is amorphous clusters and that the difference in time constants for the transformation to more stable crystalline states (nuclei) is a key determining factor in the course of the crystallization. It is difficult to distinguish between cluster formation, nucleation, agglomeration, and growth in the early stages (Mersmann 2001, p. 235). The following possibilities can be recognized qualitatively as determined by the time constants and the physical chemistry of the specific compound and system.
As is well known, some compounds have never been crystallized, and phase separation results in a stable oil or an amorphous solid. The search for solvents and conditions, or the introduction of foreign particle seeds (e.g. by scratching a glass test tube) to induce crystal formation for a new compound, becomes a matter of trial and error. Combinatorial techniques continue to be developed that can aid in this evaluation. A critical factor for success may be removal of impurities to achieve a very high level of purity, because the effect of even very low levels of impurities on homogeneous nucleation will not be known at this stage.
High supersaturation can lead to very small (nano‐sized) particles and result in agglomeration and gel formation. This phenomenon has been discussed by Mersmann (2001, p. 295) and Mullin (2001, p. 317). Although difficult to define or predict, one mechanism of phase separation leading to agglomeration may be a combination of gelling, to produce a colloidal system, followed by oiling out and nucleation in which the oil serves as a bridge between developing nuclei. Operation at high global or local supersaturation ratios is expected to promote this effect. Mixing can also be a key factor. Although increased mixing may result in breakup of agglomerates in some cases, it may also cause an increased rate of formation by impact between “particles.” The effect of mixing on agglomeration of particles smaller than the Kolmogoroff length scale has been termed by Smoluchowski (1918) as “orthokinetic.” This would predominate in a stirred vessel. The other term used is “perikinetic,” pertaining to Brownian motion in a static fluid and when particles are in the submicron size range.
6.4.2.2 Agglomeration and Aggregation
The distinction between agglomeration and aggregation has been described differently by various authors. Both have received further study by several investigators, including Myerson (2001, pp. 110–111, 146), Mullin (2001, pp. 316ff.), and Mersmann (2001, pp. 235ff, 527). In agreement with these authors, the differences between them are not significant and agglomeration will be the term used in this discussion.
One mechanism for agglomerate growth is the collision of growing nuclei followed by “cementing” together from continuing growth between two or more crystals. Although simultaneous collision of more than two particles is not statistically important, the addition of a large number of nuclei to an original two‐crystal agglomerate can readily occur by ongoing collisions, leading to very large agglomerates. Aggregation is weak bonding of colloidal particles. Aggregates are relatively easily separated
Several investigators have developed models for the effectiveness of collisions that lead to agglomeration including Nyvlt et al. (1985) and Sohnel and Garside (1992). This complex interaction of hydrodynamics and crystallization physical chemistry is difficult to predict or describe but can be critical to the successful operation and scale‐up of a crystallization process. In particular, for reactive crystallization in which high supersaturation levels are inherently present, agglomeration is very likely to occur as the precipitate forms. Careful control may be necessary to avoid extensive agglomeration, as outlined below and in Examples 10.1 and 10.2 for reactive crystallization.
The difficulties that can result from agglomeration include:
- entrapment of solvent and/or impurities in the crystal mass
- reduced effective surface area for true growth
- subsequent breakup of agglomerates into small crystals that were captured during nucleation without an opportunity for growth
- difficulties in downstream processing because of these small crystals
For these reasons, agglomeration is generally to be avoided. The use of additives (Myerson 2001, p. 146) may be considered for minimizing agglomeration. However, the use of additives in the pharmaceutical industry—particularly for final products—is generally not done for regulatory reasons barring extreme need.
There are operations, however, which may intentionally generate agglomerates for a particular purpose. These operations are described as flocculation and/or coagulation. However, a discussion of the purposeful generation of these clusters is beyond the scope of this section.
6.4.2.3 Minimization of Agglomeration
As indicated above, the primary process variables that can be manipulated to minimize agglomeration are
- operation within the metastable region
- controlled rate of supersaturation generation
- removal of crystallization inhibitors from the feed stream
- appropriately high seed levels
- mixing conditions to achieve particle suspension
These factors are all discussed in the applicable sections of this book and are the same as those recommended for achieving growth.
Both the formation and disruption of agglomerates are functions of mixing conditions and local shear. The reader is referred to the detailed treatment by Mersmann and Braun (Mersmann 2001, pp. 235ff.) in their chapter on “Agglomeration” for a comprehensive analysis of the forces involved in these phenomena. This discussion includes the distinction between attrition and disruption, which are both functions of mixing. Disruption refers to breakup of agglomerates that formed under conditions of low supersaturation and can be broken because the binding forces are small. Agglomerates that are formed under conditions of high supersaturation are much stronger and not subject to disruption by mixing. Attrition refers to breakup of large primary crystals that were formed by growth at low supersaturation and is a function of local shear and crystal lattice energy.
6.4.3 Nucleation
In the absence of control of supersaturation, nucleation will usually predominate. Nyvlt et al. (1985, p. 36) notes that “the primary requirement in control of the crystallization process is thus control of the number of crystals formed.” As discussed below, this control may be difficult to achieve on any scale and may be particularly difficult to reproduce on scale‐up, often because of mixing issues.
A specific crystallization can be dominated by either nucleation or growth. Which of these does dominate depends on the methods of control of critical variables, the amount of seed, the size distribution and surface qualities of the seed, and the environment in which supersaturation is created, as well as the specific nucleation and growth rate properties of the compound.
Both nucleation and growth almost always proceed simultaneously. In general, nucleation will dominate when supersaturation, either local or global, is close to or greater than the upper limit of the metastable region. Growth can dominate at low supersaturation and in the presence of a sufficient crystal surface area.
Three different mechanisms for nucleation may occur in any crystallization operation, as discussed by Mersmann (2001, pp. 45ff.): homogeneous primary, heterogeneous primary, and activated secondary. Industrial crystallizers are usually operated under conditions of the last two simultaneously. For simplicity, nucleation as used in this discussion refers to any or all three of the mechanisms that may be important in a particular crystallization.
A nucleation‐dominated process may be chosen in order to produce fine particles for a product attribute such as bioavailability for a pharmaceutical with low water solubility. The reader is referred to Examples 9.5 and 9.6 for discussion of a process for creating fine particles. Nucleation‐dominated processes may be difficult to operate in stirred vessels for the reasons as outlined below and may require intense in‐line mixing, as presented in the examples.
6.4.3.1 Nucleation Issues
In some cases, a process that is dominated by nucleation can result in an acceptable process outcome. The process may appear satisfactory in a laboratory‐scale operation. However, there are several potential problems with this type of process for laboratory and pilot plant operation or when scaling up for manufacturing, including:
- fine crystals and/or wide PSD
- high surface area
- low bulk density
- the risk of large batch‐to‐batch variation
- occlusion of solvent and impurities
- agglomeration/aggregation
- lack of control of hydrates, solvates, and polymorphs
In addition, scale‐up of a nucleation‐dominated process is difficult to predict, unless the generation of supersaturation is well controlled.
A principal reason for the difficulty in controlling local supersaturation and nucleation is the differences in the local energy dissipation rates within a stirred vessel. But under high mixing intensity with in‐line mixer, the issues of variability of metastability of a supersaturated solution for nucleation can be minimized.
With an extremely high nucleation rate, for example, one with a characteristic time in milliseconds, the degree of control required to prevent formation of the large number of nuclei, may not be achievable in a stirred vessel, in part, because of the inherent properties of the compound and/or because of mixing scale‐up issues (the most common being the geographical distribution and magnitude variation of the energy dissipation rate). Extreme examples of this are found in ionic reactions to precipitate inorganic salts. Organic acid–base reactions can also generate nuclei at high rates. An in‐line type of crystallization approach may be required for anti‐solvent and/or reactive crystallization, both of which may be dominated by nucleation in the absence of strategies to promote growth. The processes referred to above for creating fine particles (Examples 9.5–9.7) are carried out using in‐line mixer/crystallizers.
Nucleation must also be minimized by tight control of supersaturation in processes involving resolution of optical isomers. In some cases, nucleation must be avoided to prevent the formation of undesired polymorphs.
6.4.3.2 Nucleation Rate
As discussed in Chapter 4, the nucleation rate is both species‐specific and a function of the supersaturation ratio. The relation between nucleation rate, growth rate, and particle size as a function of the supersaturation ratio is illustrated qualitatively in Figure 6.3. The actual rate and supersaturation characteristics, such as metastable zone width, are system specific and can vary over wide ranges. In typical stirred vessels, it has been observed that the nucleation rate may vary from milliseconds to hours, and the metastable zone width may vary from less than 1 mg/ml to tens of mg/ml.
In addition, for a specific compound, the nucleation rate is also dependent on the solvent(s) system, impurities, and mixing. These factors combine to cause the difficulties that are often encountered in controlling a nucleation‐based crystallization process, especially on scale‐up.
The variation in supersaturation range, as indicated by the variability in width of the metastable region, can play a critical role in the operability of a process. An example of this type of variability is described in Example 13.1. In this process for the crystallization of an antibiotic, a high degree of supersaturation can be achieved and maintained for several minutes (S ~ 15) in an all‐aqueous system allowing completion of sterile filtration at this high degree of supersaturation (high concentration). The subsequent addition of 3% acetone, however, effectively increases the nucleation rate and narrows the width of the metastable region (as defined in Section 2.2). Crystallization is then completed by the addition of sufficient acetone to achieve the desired yield.
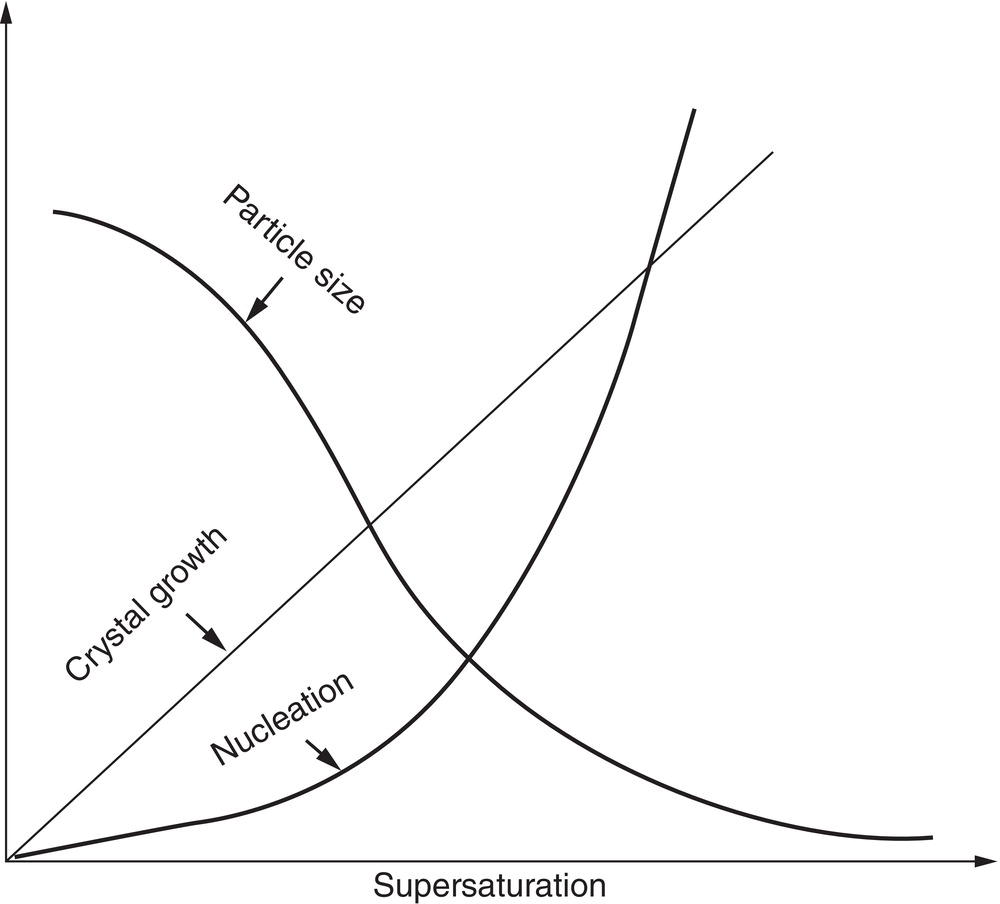
Figure 6.3 Effect of supersaturation on nucleation, growth, and nucleate particle size.
As noted above, nucleation rate differences can be extreme within the same crystallization process. With small changes, a nucleation‐based process can be made to run predictably when the nucleation rate is low (characteristic nucleation time is tens of minutes or hours), and can be essentially an all‐growth process. At the other extreme, nucleation control can be virtually impossible when high rates are encountered (characteristic time of seconds or less).
By increasing the mixing intensity or energy dissipate rate per unit volume by orders of magnitudes (see Table 5.2) using in‐line mixer such as impinging jet, rotor–stator homogenizer, the variability of metastable zone can be significantly reduced and results become more consistent (Examples 9.5–9.7). As a result, the in‐line mixer is considered to be leader for controlling the nucleation, especially. for generation of seed in situ.
6.4.4 Crystal Growth
When crystal growth is desired, tight control of several variables is almost always required. The outcome is species dependent, however, and in some cases growth may not be achievable because of the inherent growth rate or morphology. Nucleation followed by growth on a large number of nuclei may then predominate, thereby limiting the ultimate particle size, as discussed in Section 5.5. As noted in many examples in this book, the type of crystallization equipment selected can play a key role in determining the success or failure of the operation.
Processes that require and/or benefit from operation under conditions in which growth predominates include:
- required product purity
- large three‐dimensional crystals for easy downstream operations: filtration, washing, and drying
- predictable dry solid flow characteristics
- control of polymorphs
- resolution of optical isomers
6.4.4.1 Growth Issues
A growth‐dominated process may have several advantages, including:
- larger particle size and lower surface area
- higher bulk density
- improved solvent/impurity rejection
- improved control of hydration, solvation, and polymorphs
- decreased sensitivity to mixing
- improved reproducibility on scale‐up
- adaptability to continuous operation
All scales of operation are dominated by the effects of supersaturation, although the outcome is usually more critical on scale‐up to pilot plant and plant operations. The local and global supersaturation ratios that are experienced over the course of a crystallization operation are critical because they determine the balance between nucleation and growth, not only at the onset of crystallization, but throughout the course of a batch or semi‐batch operation. This balance, in turn, determines the resulting physical properties and, in many cases, the distribution of chemical impurities between the crystals and the liquors.
6.4.4.2 Growth Rate and Growth Characteristics
Inherent crystal growth rates are system specific and can vary over several orders of magnitude for different compounds. Measurement techniques for growth rate are presented by Myerson (2001, pp. 58ff.) and Mersmann (2001, pp. 220ff.). A laboratory procedure for measuring the growth rate in a fluidized bed—chosen to minimize nucleation—is presented in Chapter 4. Readers can also read Example 7.2, which discusses crystal growth impact on particle size.
The factors influencing the crystal growth rate of a specific compound are discussed by Myerson (2001, pp. 93ff.). In addition to molecular structure and the solvent system, the growth rate can be greatly modified by the presence of dissolved impurities that may either compete for growth sites or block these sites. As with the nucleation rate, these differences can be so extreme as to make growth impracticably slow, or at the opposite extreme, the rate may be sufficient to achieve an essentially all‐growth process with careful control of supersaturation and growth area. In practice, it has been observed that the release rate of supersaturation by crystal growth can vary from less than a second to several days.
Supersaturation has an important effect on the growth rate, as shown in Figure 6.4. As can be seen, the increased growth rate that can be achieved at higher supersaturation may come at the expense of increased nucleation, leading to broader PSD and possibly a bimodal distribution.
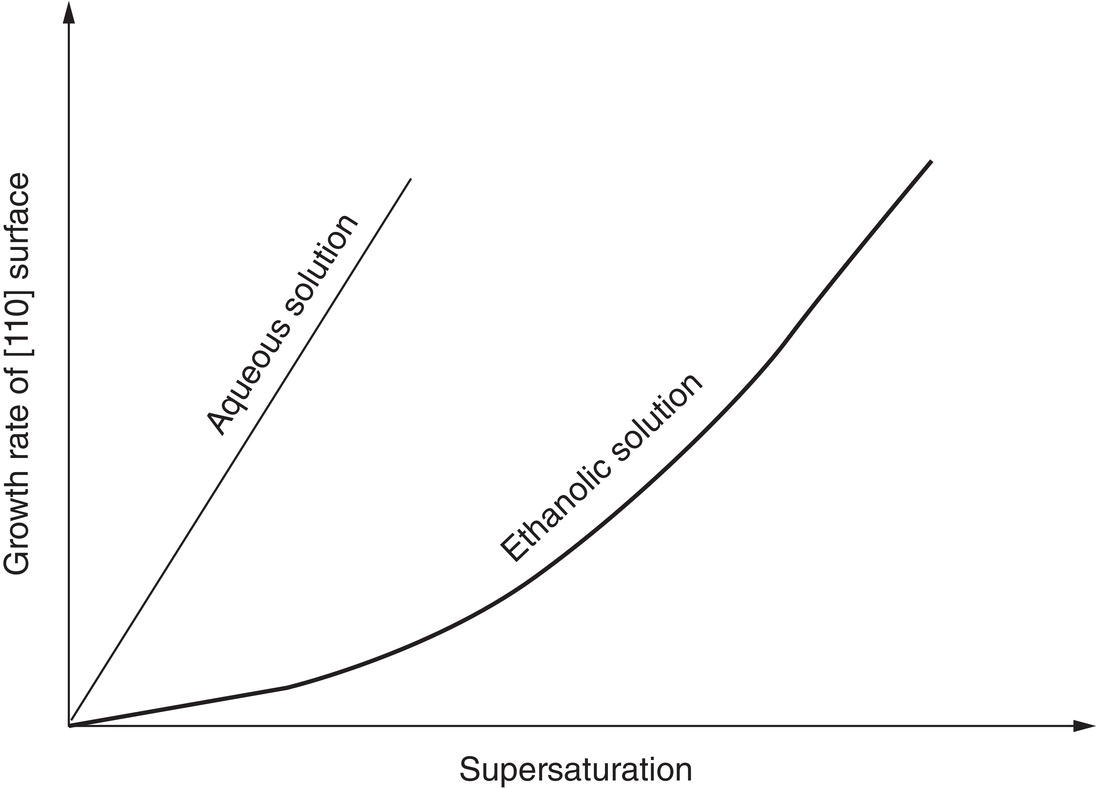
Figure 6.4 Comparison of the growth rate of hexamethylene tetramine crystals as a function of supersaturation in aqueous solution and in ethanol solution.
Source: Reproduced with permission from Davey et al. (1982).
The effect of supersaturation also can depend on the solvent system, as shown in Figure 6.4 for hexamethylene tetramine (Davey et al. 1982).
In the design of a crystallization process, therefore, the balance that is achieved between nucleation and growth rates is critical to particle size under the operational constraints of equipment and facilities. The supersaturation ratio can be controlled to limit nucleation in order for growth to predominate. This becomes increasingly difficult at lower inherent growth rates, since it will extend the batch time cycle substantially and because the nucleation rate becomes more critical at lower growth rates.
Solvent and impurity effects must also be considered. Solvent effects are important and may play a key role in inclusions and in affecting the width of the metastable zone, as discussed in Example 13.1. However, variations in impurity composition can have more influence and can dominate the course of crystallization in many ways.
Impurities can:
- • slow the nucleation rate, leading to high supersaturation and create oiling (see below)
- • retard or stop growth
- • co‐crystallize and/or form solid solutions (see Chapter 2)
Experimentation is required to evaluate these effects. The most useful experiments utilize spiking with known impurities when they can be isolated for this use. However, as is often the case, the number and possibly the low concentration of impurities often make this impractical. An experimentally simpler method is to recrystallize the compound with and without spiking of the mother liquors obtained from the process isolation. Differences in nucleation and growth may readily be observed by comparing photomicrographs of the resulting crystals. Both size and shape can be expected to be affected. If no significant differences are observed, the impurities from the process may not cause any nucleation or growth changes, and the inherent properties of the compound may be assumed to prevail.
Ideal steps in determining growth potential include the following:
- purification to the highest possible extent (using chromatography if necessary),
- selection of a solvent with solubility <50 g/l and some dependence on temperature,
- preparation of a clear solution with low supersaturation,
- aging of this solution with minimal or no mixing in the presence of some seeds, and/or
- subjecting the solution to heat/cool cycles (some fines dissolve during each heating cycle, and some growth may occur on slow cooling).
This procedure may show that growth is possible. A growth rate can then be determined by various methods, including the fluid bed method described in Chapter 4 using the crystals from the heat/cool experiments as seed.
6.5 MIXING AND SCALE—SELECTION OF EQUIPMENT AND OPERATING PROCEDURES
In addition to the basic properties and kinetics, mixing can be considered the third pillar to achieve the quality‐by‐design of crystallization and to deliver the desired quality crystals. As detailed in Chapter 5, mixing factors include mixing time, mixing intensity, and mixing distribution/scale‐up and different types of equipment have different mixing characteristics.
6.5.1 Stirred Vessels
Stirred vessels are the common process equipment in pharmaceutical industries, including crystallization. Its size can vary several orders of magnitude from hundreds of milliliter to thousands of liters. In this book, stirred vessels would be considered as the first reference standard equipment in designing the crystallization process. The overall geometry of stirred vessels consists mainly a cylindrical‐shape tank with overhead stirrer as detailed in Section 5.6. In the laboratory, round‐bottom flask may also be used to as an alternative to stirred vessels, but is not recommended. The batch is typically contained within the vessels for the batch operation.
For stirred vessels, typically, mixing time is on the order fraction of minute to minutes from laboratory to production scale. This time frame in general is shorter than the time constant of crystal growth or nucleation rate of many pharmaceutical compounds. Therefore, mixing time is not a concern. For compounds which have faster kinetics or shorter time constant, especially under high supersaturation, stirred vessels would not provide adequate mixing time for content uniformity. In this situation, in‐line mixer can be an excellent add‐on to achieve the rapid (micro‐scale) mixing time.
6.5.2 In‐line Mixers
The second reference standard equipment is in‐line mixer which focuses primarily on micro‐scale mixing. For in‐line mixer, there can be different configurations, such as impinging jet, mixing elbow/tee, vortex mixer, or rotor–stator homogenizer. For the in‐line mixer, the batch and feed streams flow together into the mixer, rapidly mixed in the mixer, and flows out of the mixer. In‐line mixer plays an important role in the crystallization process, because of its simplicity and its complementary mixing characteristics in comparison to stirred vessels.
In‐line mixer can be an excellent add‐on for the stirred tank vessel to achieve the rapid (micro‐scale) mixing time. Under high mixing intensity, the mixing time in the in‐line mixer can be reduced significantly to less than a fraction of second or milliseconds. This magnitude of time frame is faster than the time constant of crystal growth or nucleation for almost every crystallization cases known to the authors.
Since in‐line mixer can operate under much higher mixing intensity than that of stirred vessels, in‐line mixers will in‐parallel create higher degree of nucleation and/or particle attrition than stirred vessels. Thus, in‐line mixers become the natural choice to generate massive seed in situ at the beginning of crystallization. Once the massive seed is generated, needless to say, the stirred vessels become the choice to grow the bulk of the batches on these in situ seeds under much gentle mixing intensity. At that time, the mixing intensity in the in‐line mixer can be reduced or bypassed in the recirculation loop to minimize nucleation as appropriate.
6.5.3 Fluidized Bed
The third and unique type of equipment is fluidized bed crystallizer. Fluidized bed crystallizer is essentially a tubular crystallizer. The crystals are fluidized within the crystallizer without exiting the fluidizer. The liquid is fed into the bottom of fluidizer and exited from the top of the fluidizer. Fluidized bed crystallizer is less common in the pharmaceutical industry, but it has been used successfully in the past. Regardless, fluidized bed design offers the ideal mixing environment to maximize crystal growth with minimum nucleation. It achieves the best chemical purity, i.e. rejection of undesired racemic isomers up to 50% (or more), and best physical solid property, i.e. growing crystals of unimodal PSD with narrow aspect ratio.
It is well‐known that mixing in stirred vessels can be highly nonuniform. A small portion of the batch surrounding the agitator blade can be rapidly mixed, whereas the bulk portion of the batch, especially the top portion, is sluggishly mixed at probably <1/10 of the mixing rate relative to the rapidly mixed zone around the agitator blade. To minimize this nonuniform mixing situation, it would be desirable to “pump” the portion of the batch in the rapidly mixed zone near the agitator blade to the slowly mixed zone outside the agitator blade region, and vice versa. An external recirculation loop, which can pump the batch out of the stirred vessel and pump the batch back into the stirred vessel, will greatly facilitate this need. The inlet and outlet of the streams to/from the stirred vessels should be properly located apart to minimize the risk of bypass. With the integration of in‐line mixer and the stirred vessels, the mixing pattern in the stirred tank vessel will be acting closer to the “fluidized bed crystallizer.” The integrated configuration can serve for the needs of massive seeding (via in‐line mixer to generate in situ seed at the beginning of crystallization) for maximizing crystal growth, gentle mixing (via stirred vessels during the bulk of crystallization) to minimize the nucleation, and recirculation of the batch through recirculation loop to ensure mixing uniformity (with in‐line mixer operated at reduced mixing intensity or bypass the in‐line mixer).
In authors’ opinion, integration of in‐line mixers and stirred vessels can serve as “the best practice” for crystallization setup, with necessary adjustment and tuning to fit specific compound characteristics and project needs.
6.6 STRATEGIC CONSIDERATIONS FOR CRYSTALLIZATION PROCESS DEVELOPMENT
Below is a brief set of guidelines for factors to consider and proposed activity, when developing a new crystallization process for a pharmaceutical compound.
| Investigation Screening | Investigation action |
|---|---|
| Laboratory observations of solid generated: oily/gummy, amorphous, or crystalline solid |
|
Note: Different techniques may be applied for polymorph or salt/co‐crystal screening. However, slurry approach with sonication/wet milling is the preferred technique of the authors. Further, impurity may have strong influence on oil/amorphous solid/crystal formation.
| Solubility, metastability, and crystallization trials |
|
Note: These studies can be iterative due to many unknown factors that can influence the outcome of crystallization. For example, the trials may show good solid purity but with low recovery, or different crystal forms are generated in different solvent systems unexpectedly.
| Laboratory process research and development | Developmental action |
|---|---|
| Set objectives for chemical and physical attributes |
|
| Experimental apparatus |
|
| Design of experiment (DOE) and analysis |
|
| Crystallization optimization |
|
Note: Preferred crystal morphology for growth, good filtration, and, downstream qualities would be three‐dimensional; whether the basic shape is a needle, plate, or cube.
| Scale‐up | Pilot plant/manufacturing |
|---|---|
| Development of laboratory‐scale experiment results can be used for scale‐up |
|
| Test in pilot plant and scale‐up to manufacturing | From pilot plant results, determine variables—seed, agitator speed, cooling/addition rate, seed amount and timing, effect of point of addition, etc.—which contribute to differences in chemical purity and physical attributes. |
Note: Continue to update the DOE multivariable analysis results and compare the difference, if any, between the lab and scale‐up batches. The difference of results could reflect the impact of equipment at different scale and manufacturing sites. For example, at larger scale, the PSD may become wider or there is more caking on the crystal wall observed. The agitator tip speed may need to be decreased or increased accordingly.
6.7 SUMMARY OF CRITICAL ISSUES
The complex interactions between solid properties, seed, supersaturation, and mixing are critical to all crystallization operations. These properties vary over large ranges because of the complexity and variety of molecular structures. Crystallization conditions for a particular compound are therefore highly species‐dependent and difficult to predict without experimentation.
Nevertheless, some guidelines can be useful, after some critical properties are determined experimentally, and can be used in the development of effective procedures for specific goals. These guidelines may be summarized briefly as follows for processes in which growth is required:
- Seed as much as allowed, i.e. massive seeding, using crystals of desired form.
- Create and maintain the minimum amount of supersaturation throughout the crystallization.
- Provide effective mixing (see Chapter 5).
All of the examples in this book are intended to provide information on the utilization of these guidelines in practice.