
Chapter 8
Evaporative Crystallization
8.1 INTRODUCTION
Increasing the concentration by evaporation or distillation is a common method for increasing supersaturation and inducing crystallization. Since solvent is removed over a finite period of time, it is inherently a semibatch operation. Semicontinuous or continuous operation is also possible. The evaporation or distillation can be run at atmospheric pressure or at reduced pressure when substrate stability is not compatible with the required atmospheric distillation temperature.
One of the primary advantages of evaporative procedures is that they can often be combined with other process operations to reduce equipment requirements and/or time cycles. In addition, it is possible in some cases to complete the crystallization without the addition of a second solvent, thereby avoiding the costs of separation and recovery. Some of the process advantages that may be realized are as follows:
- Combination with a change in solvent and simultaneous crystallization.
- Combination with reaction and removal of a volatile reaction by‐product and simultaneous crystallization.
- Combination with cooling crystallization.
- Combination with antisolvent crystallization.
These operational advantages must be evaluated against the disadvantages that are discussed in the sections to follow. These disadvantages may include
- difficulty in controlling mean particle size and particle size distribution (PSD)
- difficulty in determining the seed point
- unpredictability on scale‐up
- inconsistent batch‐to‐batch performance
8.2 SOLUBILITY DIAGRAMS
Figure 8.1 shows the path of concentration versus time in relation to saturation solubility. With the initial concentration at point A, solvent is removed until the concentration crosses the equilibrium solubility line at point B, where crystallization may occur. In most cases, however, some degree of supersaturation is necessary before crystallization actually starts, depending on the width of the metastable region, the presence or absence of seed, and other factors such as mixing, bubble formation, and impurities level. Once crystallization is initiated, the concentration in solution may follow different pathways. Assuming that crystallization begins at point C, the pathway between points C and E depends on (i) the rate of distillation, (ii) the surface area of the crystals, (iii) the secondary nucleation rate, and (iv) the inherent crystal growth rate. In the absence of crystallization, the concentration would reach point D, the final overall concentration.
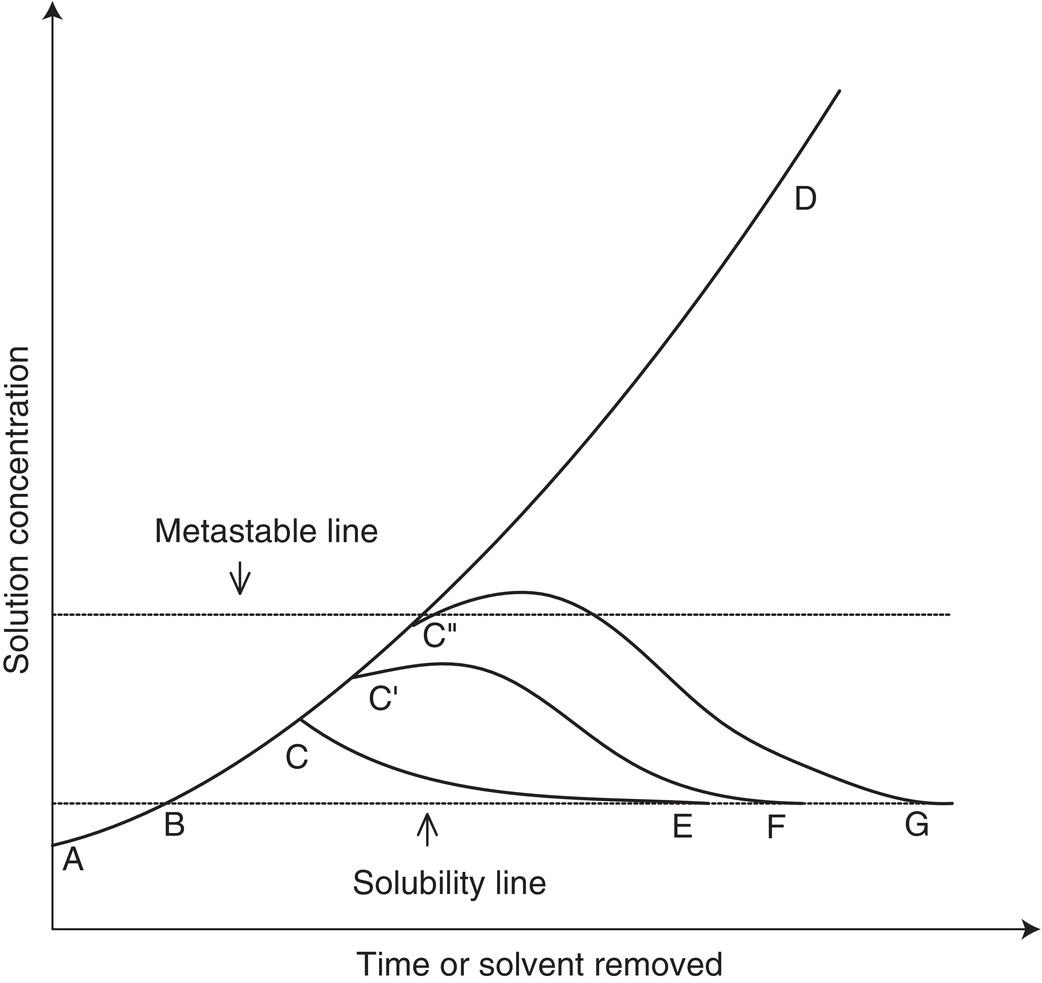
Figure 8.1 Concentration profiles for crystallization by evaporation as a function of time of distillation or amount of solvent removed. A–B–C–E is the preferred pathway for favoring growth.
Figure 8.1 shows additional concentration pathways (B–C′–F, B–C″–G). Crystallization can begin at different concentrations C′ and C″, depending on the factors mentioned above. Once crystallization is initiated, the degree of supersaturation, as indicated by the departure of the actual concentration profile from the saturation curve (B–E–F–G), that is actually achieved between initial crystallization at C′ or C″ and termination (E, F, or G) is determined by the seed area, inherent crystal growth rate, and secondary nucleation. In addition, oiling out, agglomeration, and/or extensive nucleation can occur if the concentration profile goes beyond the metastable region (above C″). These issues are discussed in Chapter 6 and can occur in any crystallization operation at supersaturations above the metastable region.
8.2.1 Increasing Solubility
Two factors complicate these simple representations. They are the effect of increasing distillation temperature caused by the increasing concentration of substrate and impurities. The impurities, in particular, can dramatically increase the solubility of the substrate. They can also decrease the inherent growth rate by blocking or inhibiting surface incorporation on the growing crystals or by reducing the nucleation rate. These effects are represented as curve H–E in Figure 8.2. As distillation proceeds, the solubility increases. In extreme cases, the crystals once formed could melt as the temperature increases.

Figure 8.2 Concentration profiles for crystallization by evaporation when the solubility of the substrate is increasing because of increasing temperature and/or impurity concentration. The solubility profile is no longer curve H–I but becomes H–E.
Increasing solubility because of increased concentration of impurities will result in a similar equilibrium change, although in some cases, the effect could be much greater. In extreme cases, when the residual solvent concentration is reduced to less than a critical value, the substrate could melt or solidify, depending on the melting point and the impurity effect. This condition is often used in laboratory preparations for convenience in changing solvents and is referred to as concentration to dryness. It is obviously not a scalable operation in a stirred vessel. Specialized tubular evaporators with close‐clearance or scraped‐surface rotors are available for these applications and have been successfully used by the authors for concentration but not for simultaneous crystallization.
The actual concentration curves that could result are shown in curve B–E. Obviously, a yield loss will result due to the increased equilibrium solubility.
8.2.2 Decreasing Solubility
A less common effect of impurities is a decrease in the solubility of the substrate. This effect could be attributed to an impurity that is in high concentration because it will not crystallize under the existing conditions or because the temperature is above its melting point. The solvent capacity for the substrate is thereby decreased, resulting in decreased solubility. In aqueous systems, this effect could be caused by high inorganic salt concentrations and may be referred to as salting out.

Figure 8.3 Concentration profiles for crystallization by evaporation when the batch volume is held constant while an anti‐solvent is being added.
8.2.3 Change in Solvent
A common variation of the evaporative operation is to evaporate the original solvent and charge a second solvent with reduced solubility for the substrate. This can be accomplished in either batch or semicontinuous mode. The solubility will then decrease as distillation proceeds and the system becomes richer in the second solvent. Supersaturation is generated due to the change in equilibrium solubility at different solvent compositions. The temperature will also increase or decrease accordingly, depending upon the boiling points of the solvents.
All of the above control factors are issues, as well as the local concentration gradients at the point of addition of the second solvent (also referred to as the anti‐solvent). This key point will be discussed in Chapter 9. Also discussed in that chapter is the method of seed introduction as a slurry with the anti‐solvent. This method can be useful in this case as well.
A change in solvent can be operated at essentially constant volume by balancing the distillation rate with the feed rate of the second solvent being added. In this case, the solution concentration profile will resemble closely that of cooling crystallization, as shown in Figure 8.3. The amount of the original solvent remaining is shown as the abscissa. The evaporation is originated at point A and the anti‐solvent addition is started at the same time at a rate to maintain constant volume. By choosing the correct rate of evaporation and by seeding at point B, the concentration can adhere closely to the equilibrium saturation and end at point D. If the evaporation rate is too fast or seeding is delayed, the concentration can exceed the metastable limit (point C) and the concentration will then follow (C → D). Examples are given below to highlight the impact of distillation on supersaturation and PSD.
8.3 FACTORS AFFECTING NUCLEATION AND GROWTH
One of the primary difficulties with distillation as a method of inducing crystallization is control of nucleation. The factors affecting nucleation are the potentially large local gradients in both concentration and temperature that are induced by the generation of vapor at the heating surfaces and at the liquid–vapor interface as the vapor bubbles leave the liquid surface. Local concentration can be greater than the limits of the metastable region, thereby causing increased local nucleation rates. The result on the ultimate mean particle size and PSD can be much less predictable. Both of these results can be affected by the distillation rate, wall temperature, vapor disengagement, and vapor bubble distribution as well as by the factors discussed above.
Distillation is analogous to the cooling rate in creating supersaturation and may be controlled by similar methods to match the evaporation rate with the surface area available for growth. However, the wall and vapor disengagement effects on local concentration can cause excessive nucleation such that a predictable growth rate may not be achieved. Other factors that are difficult to control are as follows:
- Build‐up of product scale at the evaporation surface and above the surface on the heated wall (see Section 8.5.1).
- Decomposition of product in the scale due to local overheating.
- Occlusion of impurities and/or solvent in the crystals due to a local rapid nucleation/growth rate.
- Poor bulk mixing caused by increasing slurry concentration.
- Foaming (can be increased by the presence of fine particles).
Seed, as highlighted in examples of this chapter and discussed in earlier chapters, however, can effectively reduce the risk of nucleation and enhance the crystal growth. Thus, evaporation remains a feasible option for crystallization if properly seeded and designed.
8.4 SCALE‐UP
All of the noted problems with control of nucleation and growth are exacerbated on scale‐up, thereby increasing the risk of an unpredictable PSD and other physical attributes. The causes of these scale‐up problems include:
- Reduced surface area to volume ratio for heating.
- Reduced relative distillation rate and/or higher jacket/coil temperature.
- Reduced vapor disengagement area relative to volume—increased foaming.
- Increased mixing turnover time.
- Change in secondary nucleation due to changes in mixing speed, shear, micro‐ and macro‐mixing.
These factors may have conflicting effects on PSD. They are difficult to quantify and are system specific, depending primarily on the crystallization characteristics of the substrate. Therefore, laboratory results can vary greatly from pilot scale results and from pilot scale to manufacturing scale.
The net result of these control issues is that crystallization by evaporation of solvent may be less stringent for intermediates when control of PSD and other physical factors may not be a significant issue (with the possible exception of impurity rejection and/or downstream operations such as filtration). However, for final bulk drug products where control of these attributes is critical, this method has proven to be challenging and requires proper considerations of seeding and supersaturation control.
8.5 EQUIPMENT
Evaporative crystallization can be carried out in a wide variety of equipment for both semi‐batch and continuous operation. Discussions of these systems may be found in several references including Myerson (2002, chapter 10) and Mullin (2001, chapter 7). They are utilized in some areas of the chemical industry that require high production rates that can be achieved with these specialized designs.
Our experience in the pharmaceutical industry has been confined to semi‐batch operation in standard stirred vessels, as illustrated in Figure 8.4. These vessels may be operated at atmospheric pressure or more commonly under reduced pressure, for which vacuum pumps are the most common vacuum source.
As indicated in the discussions above, these applications may be subject to difficult control issues on scale‐up. Impeller type and power input need to be carefully specified in providing adequate circulation without breaking crystals under excessive shear and/or creating excess nucleation, as discussed in Chapter 5.
8.5.1 Heat Transfer
In addition to these standard crystallization issues, evaporative crystallizers must have adequate circulation at the wall surface and maintain sufficient temperature difference between the jacket fluid and batch fluid for heat transfer to achieve satisfactory distillation rates. This issue can be complicated by restrictions on wall temperature imposed by the temperature instability of certain organic compounds. These problems can be severe when the crystallizing material forms a crust on the vessel wall, where it can decompose as well as reduce heat flux.
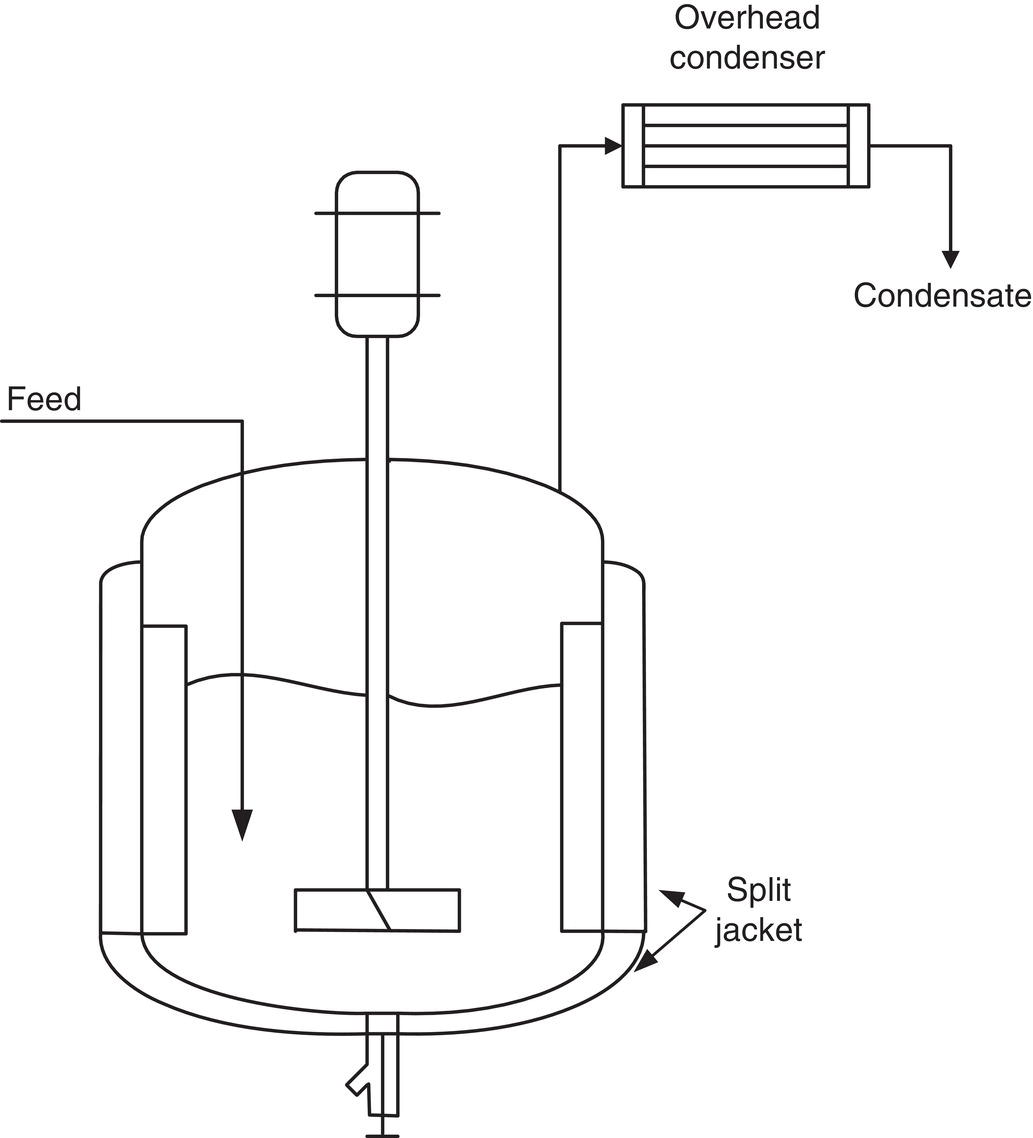
Figure 8.4 A standard jacketed stirred tank with baffles and an overhead condenser for evaporative crystallization. During distillation, a second stream of solvent can be charged to maintain a constant batch volume in the vessel.
The possible restriction on jacket temperature can preclude the use of a common jacket service such as pressure steam. An alternative is hot water or a heat transfer fluid at a temperature compatible with the decomposition limits. Both of these liquids result in decreased jacket side heat transfer coefficients compared to those for steam. Another possible alternative is steam under vacuum, which can be readily achieved with a vacuum pump and can be used to limit jacket temperatures to ~60°C while maintaining good wall heat transfer rates.
8.5.1.1 Split Jacket Vessels
Perhaps the most important and most readily achievable equipment design to minimize wall decomposition is the use of jacket services on the bottom section of the vessel only. Such services can be provided either by a lower jacket only or, more commonly, by a split jacket to provide for full services in multipurpose vessels. Decomposition can be especially severe on the wall above the boiling surface, where any liquid may evaporate and leave the residual compound to dry and be exposed to the maximum temperature in the jacket (baking).
The best defense against these possibly severe problems of product loss and contamination with impurities is a design and an operation that preclude heat supply near or above the boiling surface at all times. This precaution may be especially important as the batch volume decreases and baking on the wall may be severe.
Split jacket designs are available for virtually all vessels, and the additional cost may be offset by the savings realized in preventing decomposition at the upper wall. In addition, by limiting exposure at the critical liquid–vapor interface, the temperature of the heating fluid may be increased at the submerged heating surface and the additional driving force may compensate for the decrease in overall heat transfer surface area.
8.5.1.2 External Heat Exchangers and Internal Coils
In some cases, the required distillation rate can be achieved by forced circulation through a heat exchanger, as shown in Figure 8.5. This design can achieve higher heat input at a minimum temperature difference by providing more surface area than the vessel jacket and higher heat transfer coefficients. However, the crystals are subject to additional shear stresses in the pump and possible encrustation on the heat exchanger tube surfaces, thereby limiting this design to robust systems.
Internal coils can also be used to increase heat transfer by supplying additional surface area. However, the coil surfaces may be subject to encrustation and may be difficult to clean.
8.5.1.3 Alternatives to Evaporation
In cases of extreme temperature instability, evaporative methods of concentration have not been feasible because distillation temperatures at even the lowest feasible pressures are still too high (e.g. distilling water at 25°C may be possible but this temperature may be still too high for the extended time cycle required). Alternative means of concentration are reverse osmosis for aqueous systems and some membrane systems with molecular weight cutoffs compatible with the compound in question. Obviously, these systems preclude crystallization during operation. An example of an antibiotic with severe temperature restrictions is presented in Example 13.1.
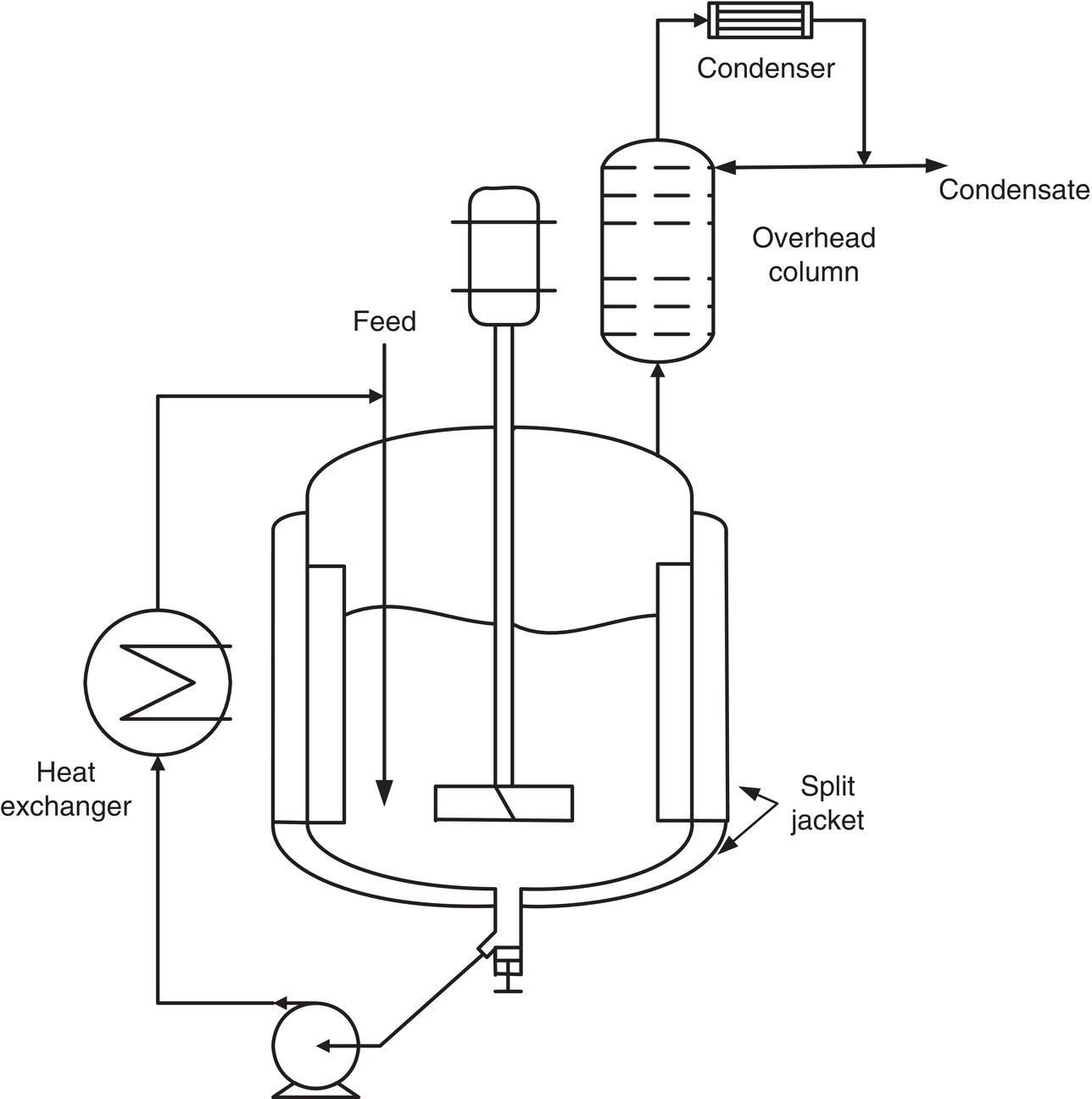
Figure 8.5 A standard jacketed stirred tank for evaporative crystallization with baffles, an overhead condenser, and an external heat exchanger to improve heat transfer. Similarly, during distillation, a second stream of solvent can be charged to maintain a constant batch volume in the vessel. Fractionation is a viable option for aiding solvent change and/or recovery.
8.5.2 Overconcentration
The potentially most damaging operational error is overconcentration. As the solids’ concentration in solution and/or in the crystal slurry increases, the viscosity and temperature can increase. This condition can most likely occur as the residual volume reaches the region of the impeller and perhaps—in extreme cases—below the impeller. Possible consequences when a critical concentration is reached include
- solidification of the entire mass
- decomposition of the substrate
- damage to the mixing components
In extreme cases, the temperature could increase to the temperature of the heating fluid and initiate a decomposition exotherm in the residual mass, causing a catastrophic failure of the vessel. Measures to prevent such events include:
- Determination of the decomposition initiation temperature.
- Limiting heating fluid temperatures to a safe margin below this temperature.
- Provision of control measures to detect the approach to a critical temperature, concentration, and/or volume limit.
- Provision of shutdown procedures if this temperature, concentration, and/or volume are reached (by cooling but not breaking the vacuum).
8.5.3 Combination of Evaporation and Cooling
In addition to the common combination of evaporation and anti‐solvent addition, evaporation is often combined with cooling to provide maximum yield. In some cases, the substrate does not crystallize during concentration, either because the solubility at the higher distillation temperature is greater than the solution concentration or because of a wide metastable zone allowing high supersaturation. In these cases, the issues discussed in Chapter 7 on cooling become important when the concentration is complete and cooling is initiated. Cooling a solution that is highly supersaturated can readily result in oiling out, agglomeration, and/or crash‐out.
In most cases, crystallization occurs during concentration and the precautions outlined in this chapter are relevant.
8.6 PROCESS DESIGN AND EXAMPLES
Design of a crystallization requires consideration of the impact of many of the above factors on the resultant chemical purification and physical properties. With proper control of seed and supersaturation, as well as V–L and L–L relationship, the following examples illustrate evaporative approach was applied to crystallization challenges.