
CHAPTER 8
Dative Bonding of Organic Molecules
YOUNG HWAN MIN, HANGIL LEE, DO HWAN KIM, AND SEHUN KIM
8.1 INTRODUCTION
8.1.1 What is Dative Bonding?
A dative bond is a coordinate covalent bond between atoms such that both bonding electrons are contributed by only one of the atoms involved in the bond. In other words, dative bonding occurs when one atom donates an electron pair and the other atom accepts the electron pair, and the atoms share the electrons equally. This bonding mode is distinct from ionic bonding modes. The donating atom is called a Lewis base (an electron pair donor) and the accepting atom is called a Lewis acid (an electron pair acceptor). Formation of a dative bond is, therefore, a Lewis acid/Lewis base reaction or a nucleophilic/electrophilic reaction. One of the most well-known examples of dative bonding is the bond between ammonia (NH3) and boron trifluoride (BF3). The boron of BF3 has only six valence electrons and requires two more electrons to satisfy the octet rule. Due to this deficiency, BF3 can act as a Lewis acid that accepts lone pair electrons. NH3, on the other hand, can act as a Lewis base by donating its two lone pair electrons. Thus, NH3 donates its two lone pair electrons to BF3 to create a dative bond, NH3:BF3.
Interestingly, dative bonding occurs between Lewis bases (or Lewis acids) and reconstructed semiconductor surfaces such as Si(100)-(2×1), Ge(100)-(2×1), and Si(111)-(7×7). Semiconductor surfaces are prepared by cleavage and are reconstructed for stabilization by reducing the number of exposed unstable dangling bonds. In (2×1) reconstructed Si and Ge(100) surfaces, pairs of surface atoms form dimers held together by a strong σ-bond and a weak π-bond, similar to a C=C double bond. The dimers are tilted such that one of the two atoms is tilted up (elevated out of the plane of the substrate surface) and the other is tilted down (recessed below the plane of the substrate surface); the asymmetric dimer is more stable than the symmetric dimer [1]. The tilting of the dimer leads to partial electron transfer from the down atom to the up atom. As a result, the up atom is electron rich and the down atom is electron deficient (see Fig. 8.1) [2]. Because the down atom is electron deficient, it can react with a Lewis base to form a dative bond (see Fig. 8.2a) [3]. In contrast, the electron-rich up atom can donate its electron density to a Lewis acid to form a dative bond (see Fig. 8.2b).
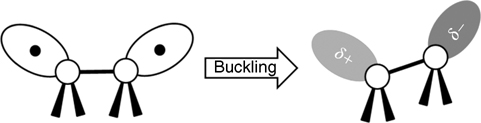
FIGURE 8.1 The tilted dimers of Si(100) and Ge(100) surfaces. The tilting of dimers leads to a partial electron transfer from the buckled-down atom to the buckled-up atom. The dark gray circle indicates the electron-rich orbital, and the light gray circle indicates the electron-deficient orbital.
In the case of Si(111)-(7×7), surface reconstruction reduces the number of unstable dangling bonds from 49 to 19 in each unit cell [4,5]. Upon reconstruction, the rest atom and corner atom have a formal charge of – 1, whereas each adatom has a formal charge of +7/12. The amount of charge transferred from a center adatom to a rest atom is twice that transferred from a corner adatom because each center adatom has two neighboring rest atoms and each corner adatom has only one neighboring rest atom. Due to this difference, the corner adatom has a higher electron density than the center adatom, and as a result, they have different reactivities. The couples formed by an adatom and a neighboring rest atom are zwitterionic in character, similar to the buckled dimers of Si(100)-(2×1) and Ge(100)-(2×1) surfaces.
8.1.2 Periodic Trends in Dative Bond Strength
Two factors determine the dative bond strength between a Lewis base and the electron-deficient down atom of a (100) surface dimer: the size of the HOMO on the Lewis base and the electronegativity (EN) of the electron-donating atom. The strength of a dative bond involving an atom from group V or VI decreases as one moves down the group because the HOMO size increases with increasing atomic mass, reducing the orbital overlap between the HOMO of the Lewis base and the electron-deficient down atom. However, the strength of a dative bond increases as the electronegativity decreases, that is, as one moves down the group, because a less electronegative atom donates electrons more easily to an electron-deficient down atom. Therefore, the strength of a dative bond, as one moves down a group in the periodic table, is determined by the net balance between orbital overlap and electronegativity (see Fig. 8.3).
Previously reported DFT calculations have compared the strengths of dative bonds between the group V Lewis bases dimethylamine (N), dimethylphosphine (P), and dimethylarsine (As) and the down Ge atom of a Ge(100) surface [6]. The calculations indicate that the P–Ge dative bond is slightly stronger than the N–Ge dative bond by 1.1 kcal/mol, whereas the As–Ge dative bond is weaker than the N–Ge dative bond by 4.5 kcal/mol. In the case of P and N, the smaller orbital overlap in P–Ge than in N–Ge is compensated by the lower electronegativity of P (2.19) than of N (3.04). This compensation results in approximately equal N–Ge and P–Ge dative bond strengths. However, the electronegativities of P (2.19) and As (2.18) are similar. Therefore, orbital overlap dominates the formation of a dative bond, and the smaller orbital overlap in As–Ge results in a weaker dative bond than is observed in P–Ge.
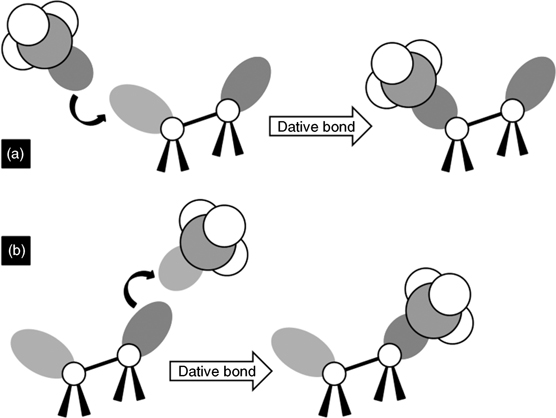
FIGURE 8.2 (a) The electron-deficient down atom can react with a Lewis base to form a dative bond. (b) The electron-rich up atom can react with a Lewis acid to form a dative bond. The dark gray circles indicate electron-rich orbitals, and the light gray circles indicate electron-deficient orbitals.
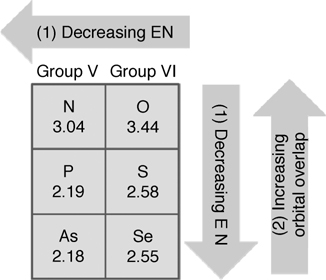
FIGURE 8.3 The overall effects of (1) electronegativity and (2) orbital overlap determine the dative bond strength. As the electronegativity decreases and the orbital overlap increases, the dative bond strength increases. The numbers under the elements indicate the Pauling electronegativity of each element.
In the case of group VI, the electronegativities of oxygen, sulfur, and selenium are 3.44, 2.58, and 2.55, respectively. Previously reported IR studies show that diethyl ether (O–Ge) and diethyl sulfide (S–Ge) adsorb via a dative bond on Ge(100)-(2×1) surfaces with bond strengths of 17.9 and 23.8 kcal/mol, respectively [7]. This indicates that the S–Ge dative bond is stronger than the O–Ge dative bond by 5.9 kcal/mol. DFT calculations of the strengths of dative bonds involving diethyl ether, diethyl sulfide, and diethyl selenide support these experimental data [6]. A comparison of dative bond energies (O, S, Se) shows that the dative bond strength increases as one moves down the group VI. This suggests that the electronegativity effects dominate group VI dative bonds.
The electronegativity effects are applicable along a period as well as down a group. Comparison of methanol and trimethylamine, both of which feature sp3 hybridization on the electron-donor atom, indicates that oxygen forms a weaker dative bond (18.5 kcal/mol) than nitrogen (23.4 kcal/mol) with the down atom of a Si dimer (as shown in Fig. 8.4), in agreement with the relative electronegativities. Hybridization of the HOMO on the electron-donating atom also influences the dative bond strength. As the p character of the hybridized HOMO increases, orbital overlap between the HOMO and the down atom of the surface dimer increases. As a result, the dative bond energy increases [12]. Thus, sp3 hybridized atoms can donate more charge to form a more stable dative bond than sp hybridized atoms. As shown in Fig. 8.4, methanol (sp3) and trimethylamine (sp3) form stronger dative bonds than acetone (sp2) and acetonitrile (sp), respectively.
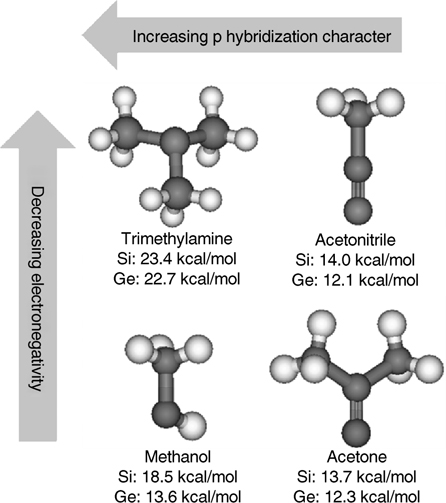
FIGURE 8.4 Theoretically calculated dative bond energies for trimethylamine [3], acetonitrile [8], methanol [9], and acetone [10] on Si(100)-(2×1) and Ge(100)-(2×1). The calculation for methanol on a Ge(100) surface was described in Ref. 11. As electronegativity decreases and π hybridization character increases, the dative bond strength increases on both surfaces. Gray: hydrogen; black: C, N of trimetylamine and acetonitrile, and C, O of methanol and acetone.
8.1.3 Examples of Dative Bonding: Ammonia and Phosphine on Si(100) and Ge(100)
Although group V hydrides are not organic molecules, reactions of these compounds with Si(100) and Ge(100) surfaces are good examples of dative bond formation as well as proton transfer. Two examples are ammonia and phosphine, which may act as Lewis bases. Adsorption of these molecules on group IV semiconductor surfaces has been extensively studied.
Musgrave and coworkers suggested, based on DFT calculation results, that molecular adsorption of ammonia on Si(100)-(2×1) or Ge(100)-(2×1) occurs by formation of a dative bond with the down atom of the surface dimer [13,14]. Formation of the molecularly adsorbed state is barrierless. The barrier separating the adsorbed state from the dissociated state was found to be 24 kcal/mol for Si (100) [13]. The transition state energy is lower than the energy of the reactants. As a result, dissociative adsorption is not activated with respect to the reactants. In summary, ammonia predominantly dissociates on Si(100) to form NH2 and H species at 300 K. In addition to the dissociated species, a fraction of molecular NH3 species may be present on Si(100). The fraction of dative-bonded species increases at lower temperatures. Increasing ammonia coverage favors dissociative adsorption. In contrast, the calculated activation barrier for the dissociation of NH3 on Ge(100)-(2×1) is higher than the barrier for reversible desorption of the dative-bonded state [14]. The transition state energy is higher than the reactant energies. Although N–H dissociation is thermodynamically favored on Ge(100), the dissociation process is slow compared with the reversible desorption of NH3 from the adsorbed state; the dissociation barrier is higher than the reactant energies, whereas molecular adsorption is barrierless. Thus, ammonia adsorbs molecularly and does not undergo dissociation on Ge(100) surfaces. Ammonia adsorbed onto Ge(100) forms a stable molecularly adsorbed species.
Phosphine (PH3) behaves similar to ammonia and prefers dissociative adsorption on Si(100) surfaces. Radny and coworkers investigated the adsorption of phosphine on Si(100) and found that most adsorbed phosphine molecules dissociate to form PH2+H at room temperature [15,16]. The dative bond between phosphine and Si(100) has a binding energy of 14 kcal/mol, 12 kcal/mol less stable than that formed by ammonia (26 kcal/mol). The transition state associated with proton transfer from phosphine is approximately 16 kcal/mol higher than the energy of the dative-bonded state, a smaller barrier than the barrier to proton transfer from ammonia (24 kcal/mol). The thermodynamic stability of the dissociative structure with respect to the dative-bonded state is higher for phosphine adsorbed on Si(100) (32 kcal/mol) than for ammonia adsorbed on Si(100) (22 kcal/mol). Meanwhile, molecular adsorption on Ge(100) was found to prevail at room temperature [17] but comprehensive theoretical results are not yet available.
8.2 DATIVE BONDING OF LEWIS BASES (NUCLEOPHILIC)
8.2.1 Aliphatic Amines
8.2.1.1 Primary, Secondary, and Tertiary Amines on Si(100) and Ge(100)
The reaction of aliphatic amines with Si(100)-(2×1) surfaces involves dative bonding, as shown in Fig. 8.5. Dative bond formation occurs without an energy barrier through donation of the lone pair electrons from the nitrogen atom to the electron-deficient down Si atom. Quaternary ammonium ions may be stabilized by the electron-donating alkyl groups on the amine, unlike ammonia. On the other hand, the up Si atom of the dimer is electron rich and acts a nucleophilic surface site. Nucleophilic attack by the up Si atom may occur, resulting in a N–H dissociative product.
The most important factor that influences the surface reactions of aliphatic amines on Si(100) is the presence of a N–H bond in the reacting amine molecule. Primary and secondary amines, both of which contain at least one N–H bond, can undergo proton transfer whereas tertiary amines cannot. The reactivity of cyclic aliphatic amines is similar to that of noncyclic amines.
Mui et al. investigated the bonding of aliphatic amines on Si(100)-(2×1) via multiple internal reflection Fourier transform infrared (MIR-FTIR) spectroscopy and density functional theory (DFT) calculations [3,18]. Two simple primary and secondary amines, methylamine and dimethylamine, were found to undergo N–H dissociative adsorption on Si(100)-(2×1) via dative-bonded precursor states in a manner similar to that observed for ammonia on Si(100)-(2×1). The presence of a v(Si–H) stretching mode near 2070 cm–1 in the MIR-FTIR spectrum provides strong evidence for the presence of the dissociated states of methylamine and dimethylamine (see Fig. 8.6b and d). In addition, strong v(– C–H) stretching modes, observed at 2803 and 2787 cm–1, indicate that a lone pair is present in the adsorption product. These modes, known as Bohlmann bands, originate from interactions between the nitrogen lone pair and the C–H σ-orbitals positioned trans-periplanar to the lone pair. Moreover, only a weak or no N–H stretching mode is observed at 3043 cm–1 for methylamine and dimethylamine, respectively. The spectral data support the presence of a N–H dissociative product.
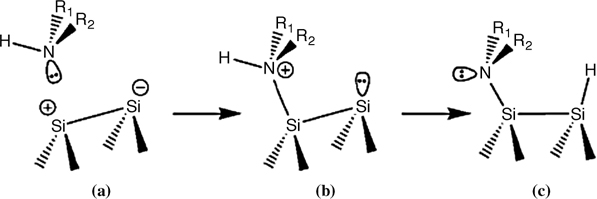
FIGURE 8.5 An illustration of the reaction of an amine with Si(100)-(2×1) surface (from Ref. 3). Reprinted with permission from Mui, C. et al., J. Am. Chem. Soc., 124, 4027. Copyright (2002) American Chemical Society.
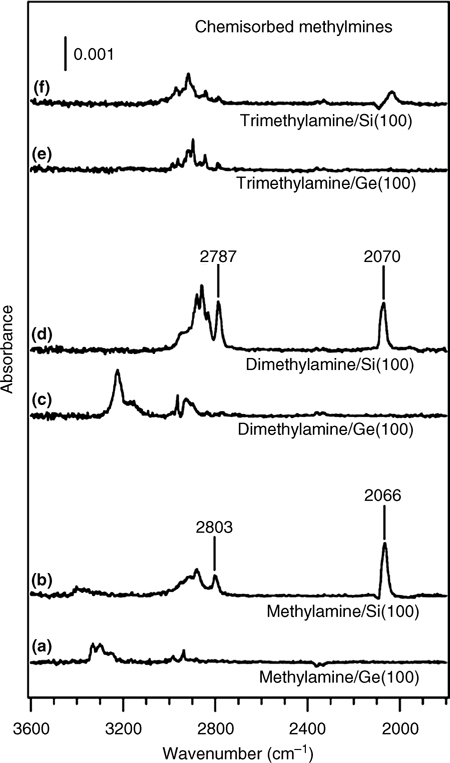
FIGURE 8.6 Infrared spectra of saturated coverage of methylamine, dimethylamine, or trimethylamine on Si(100)-(2×1) and Ge(100)-(2×1) surfaces at room temperature (from Ref. 3). Reprinted with permission from Mui, C. et al., J. Am. Chem. Soc., 124, 4027. Copyright (2002) American Chemical Society.
The reaction pathway for dimethylamine on Si(100)-(2×1) (see Fig. 8.7) shows that this system is under kinetic control [3,19]. Although the binding energy for the methyl dissociative product is higher than that for the hydrogen dissociative product, the barrier for N–C bond cleavage is high, 19.3 kcal/mol above the vacuum level. In contrast, the transition state for N–H cleavage is 9.3 kcal/mol below the entry level. The barrier for N–C cleavage is too high to be overcome at room temperature, explaining the experimental result that methylamine and dimethylamine undergo only N–H dissociation.
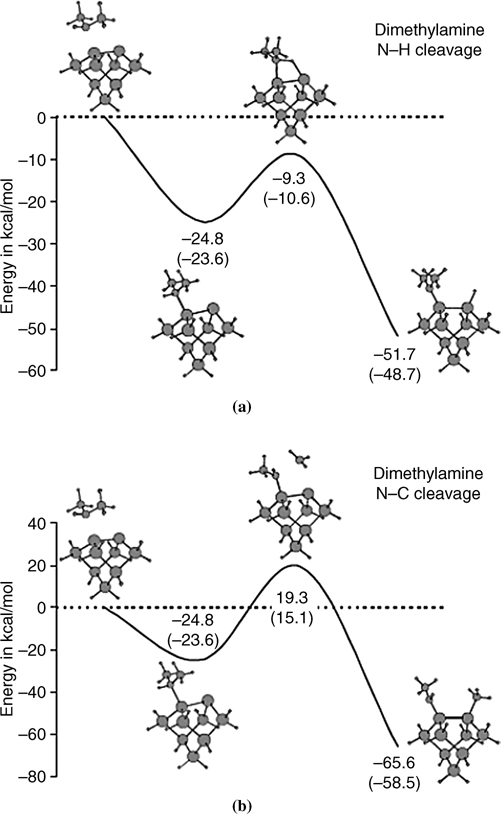
FIGURE 8.7 Theoretically calculated potential energy surfaces for (a) N–H dissociation and (b) N–C dissociation of dimethylamine on Si(100)-(2×1). All energies are given with respect to the vacuum level in kcal/mol (from Ref. 18). Reprinted with permission from Mui, C. et al., J. Chem. Phys. 114, 10170. Copyright (2001) American Institute of Physics.
The difference between activation barriers of the two pathways can be understood by considering the transition state geometries [18]. The N–C bond must be stretched to accommodate front-side nucleophilic attack by the electron-rich up Si atom. The N–C and the Si–C bond lengths in this transition state are stretched by 36% and 63%, respectively, relative to the corresponding N–C and Si–C bonds in the reactant and the product. As a result, the energy of the transition state is high and the N–CH3 dissociation process has a relatively high activation barrier [18]. These results indicate that the selectivity of N–H cleavage over N–CH3 dissociation for aliphatic amines on Si(100)-(2×1) surfaces is due to kinetic effects rather than thermodynamic effects. Similar kinetic selectivity holds for other aliphatic amines, such as methylamine [18] and pyrrolidine [19], on Si.
Trimethylamine is a typical tertiary aliphatic amine and contains no N–H bonds to assist with dissociative adsorption. Figure 8.8 shows the N 1s XPS spectrum obtained after exposing a Si(100) surface to tetramethylamine at 300 K, followed by annealing at higher temperatures [20]. These data show that two N 1s photoelectron peaks are present near 402.2 and 398.9 eV at room temperature. The peak of N 1s for typical amines is around 398.5 eV. The peak at 402.2 eV represents a dative-bonded trimethylamine and contributes 85% of the total peak area. The lower energy peak at 398.9 eV is from a minor surface species, possibly a N–C dissociative product. When trimethylamine adsorbs onto Si(100)-(2×1) at 190 K, only one N 1s peak at 402.2 eV is observed, suggesting that the reaction pathway to N–C dissociation must overcome a high activation barrier. Thus, most of the trimethylamine molecules adsorb molecularly via dative bonding.
Dative bonding of trimethylamine on Si(100) was confirmed through MIR-FTIR analysis by Mui et al. [3]. Trimethylamine does not exhibit a significant peak in the v(Si–H) stretching region. For multilayer adsorption of trimethylamine on Si(100) at 100 K, the observed C–H stretching vibrations below 2800 cm–1 are assigned to the vibrations of CH bonds positioned trans-periplanar to the nitrogen lone pair of the amine. These bands are known as Bohlmann bands and indicate the presence of a nitrogen lone pair [21]. This peak was very weak for trimethylamine adsorbed onto Si(100) at 300 K, suggesting loss of the lone pair and formation of a dative-bonded product. The formation of a dative bond between the lone pair of the nitrogen atom and the electron-deficient down atom of a Si dimer is exothermic with an adsorption energy of 23.4 kcal/mol. Cleavage of the N–C bond from the dative-bonded state requires crossing the transition state barrier, located 21.7 kcal/mol above the vacuum level. The N–C dissociative product energy is 62.3 kcal/mol below the vacuum level. Although the N–C dissociative product is thermodynamically stable, the high activation barrier makes the dissociation process kinetically unfavorable.
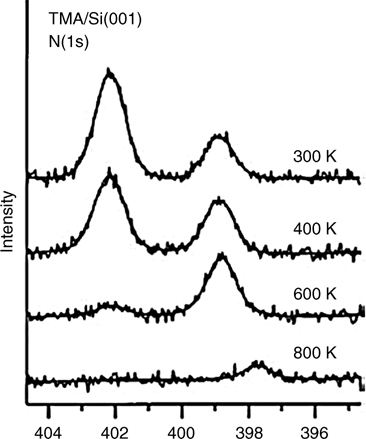
FIGURE 8.8 XPS spectral data for Si(100)-(2×1) surfaces exposed to trimethylamine, at room temperature and after annealing. Both spectra show two distinct binding energies for the N 1s core electrons (from Ref. 20). Reprinted with permission from Cao, X. et al., J. Am. Chem. Soc. 123, 10988. Copyright (2001) American Chemical Society.
Mui et al. studied a series of alkylamines on Ge(100)-(2×1) surfaces and found that N–H cleavage does not occur for any of the alkylamines [3]. A distinct absorption peak in the N–H stretching region indicates the retention of N–H bonds. The difference between the chemical reactivities of the Ge(100)-(2×1) and Si (100)-(2×1) surfaces toward N–H dissociation is related to a decrease in the electron density as one moves down a group in the periodic table. The proton transfer process is facilitated by electron donation from the electron-rich up atom of a dimer. Thus, the dimer atom with largest electron density will most easily attack the proton of a N–H bond. Because the electron density on the nucleophilic Ge dimer atom is less than its Si counterpart, the N–H bond must be stretched to acquire sufficient electron density to form a Ge–H bond. The additional stretching would induce additional strain, increasing the activation energy. The adsorption of trimethylamine on either Ge(100) or Si(100) results in a datively bonded product. Trimethylamine has no N–H bonds but contains nitrogen lone pair electrons that can interact with the electrophilic down atom of a tilted dimer to form a dative bond.
8.2.1.2 Cyclic Aliphatic Amines on Si(100) and Ge(100)
Pyrrolidine is a secondary amine like dimethylamine, whereas N-methylpyrrolidine is analogous to trimethylamine (see Fig. 8.9). The main concern about the reaction of these amines on (100) semiconductor surfaces is whether the final product is N dative bonded or if it proceeds to a N–H dissociated structure. For comparison, pyrroline and N-methyl-3-pyrroline have an additional reactive site at the C=C double bond. Thus, reactions may occur competitively either at the nitrogen atom or at the C=C double bond.
Figure 8.10 shows the infrared spectra of Si(100)-(2×1) and Ge(100)-(2×1) surfaces after the surface is saturated with pyrrolidine [19]. The appearance of a Si–H stretching mode at 2070 cm–1 or a Ge–H mode at 1904 cm–1 suggests that N–H or C–H dissociative products are formed. Complete loss of the N–H stretching mode at 3190 cm–1 indicates cleavage of the N–H bond on Si(100). In contrast, a significant v(N–H) mode is observed after saturating doses of pyrrolidine on Ge(100). The presence of a Bohlmann band at 2780 cm–1 for pyrrolidine adsorbed onto Si(100) indicates that the lone pair is not involved in dative bonding. The absence of a Bohlmann band on Ge(100) suggests formation of a dative bond with the lone pair electrons. These spectra strongly suggest that pyrrolidine undergoes N–H dissociation on Si(100) surfaces, whereas both dative bonding and N–H dissociative products form on Ge(100).
The reaction product of pyrrolidine adsorbed onto Ge(100) depends on the adsorbate coverage. The infrared spectra shown in Fig. 8.11 reveal that molecular adsorption through dative bonding is the only product at low coverage, whereas N–H dissociative adsorption also occurs at high coverage [19]. The different behavior of pyrrolidine on Si or Ge surfaces can be explained in terms of kinetic effects. The dative-bonded products form without crossing an activation barrier in each case. On Ge (100), the barrier to N–H dissociative adsorption is 21.9 kcal/mol and the transition state is only 1kcal/mol below the energy of the reactants. On Si(100) surfaces, the barrier is 13.3 kcal/mol and the transition state is 11.9 kcal below the reactant energy. These differences explain why some pyrrolidine molecules are trapped in a dative-bonded state on Ge(100). The presence of neighboring dative-bonded species lowers the barrier for N–H dissociation and favors the dissociative products at high coverage. The chemistry of pyrrolidine binding to Si(111)-(7×7) surfaces is characterized by two types of dative bonding (β1 and β2) at 85 K [22]. In the β1 state, one pyrrolidine molecule engages in both electron acceptance by the adatom and electron donation by the rest atom simultaneously through dative bonding and hydrogen bonding, respectively. In the second dative-bonded state (β2), the adsorbed pyrrolidine is directly bonded to the silicon surface adatom via a N–Si (adatom) dative bond. The presence of a N–H dissociative product at higher temperatures could not be clearly determined.
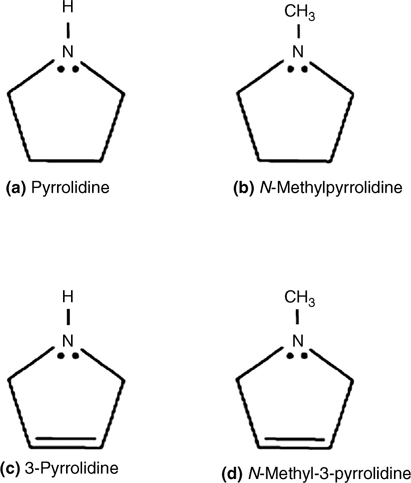
FIGURE 8.9 Examples of five-membered cyclic aliphatic amines.
N-Methylpyrrolidine forms a stable dative bond at room temperature on both Si(100) and Ge(100) surfaces [19]. The saturation coverage is significantly higher on the Ge(100) than on the Si(100) surface, probably due to steric effects. Ge(100) has a slightly larger spacing between adjacent dimers in each row than does Si(100): 4.00 Å versus 3.84 Å. Thus, the steric repulsion between adjacent adsorbates may be weaker, and a higher coverage may be attained for Ge(100). 3-Pyrroline primarily undergoes molecular adsorption through Ge–N dative bonding on Ge (100) [19], whereas N–H dissociation is favored on Si(100) along with [2+2] cycloaddition as a minor product [4]. 3-Pyrroline exhibits coverage-dependent adsorption on Ge(100), similar to pyrrolidine. Some of the dative-bonded species undergo N–H dissociation at higher coverage. N-Methyl-3-pyrroline adsorbs through Ge–N dative bonding on Ge(100) surfaces, as expected from the high barrier to cleavage of the N–CH3 bond [22].
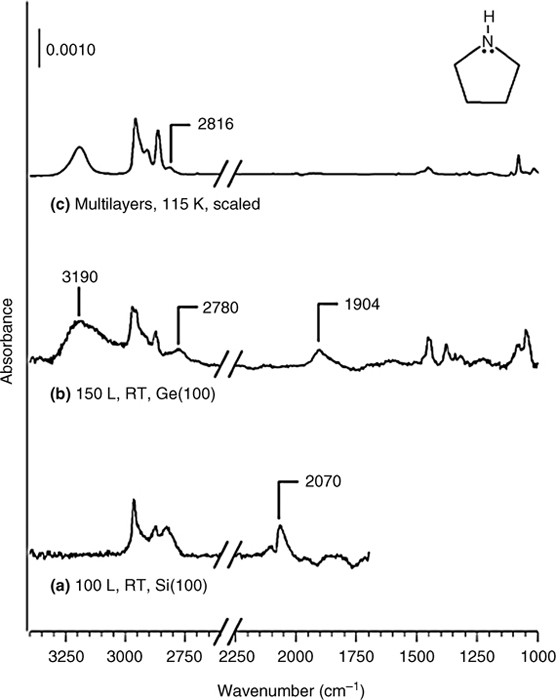
FIGURE 8.10 Infrared spectra of pyrrolidine on Si(100)-(2×1) and Ge(100)-(2×1) surfaces: (a) 100L at 298 K on Si(100); (b) 150 L at 298 K on Ge(100); (c) multilayers (scaled to fit the figure) at 117 K (from Ref. 19). Reprinted with permission from Wang, G. T. et al., J. Phys. Chem. B, 107, 4982. Copyright (2003) American Chemical Society.
8.2.1.3 Ethylenediamine on Ge(100)
Ethylenediamine is a bifunctional molecule with two amino end groups. Either one or two of the amino groups may be involved in ethylenediamine surface reactions. The reaction can be performed through N dative bonding or N–H dissociation. The selectivity for forming each of the possible products is important for exerting appropriate control during multilayer organic functionalization.
FIGURE 8.11 Infrared spectra of pyrrolidine on a Ge(100)-(2×1) surface as a function of exposure: (a) 1.5 L at 298 K; (b) 15 L, in ratio with 1.5 L at 298 K; (c) 150 L, in ratio with 15 L at 298 K (from Ref. 19). Reprinted with permission from Wang, G. T. et al., J. Phys. Chem. B, 107, 4982. Copyright (2003) American Chemical Society.
Both amino end groups were found to undergo dissociative adsorption on Ge(100) surfaces at low coverage, and two N–Ge covalent bonds form with two down Ge atoms in adjacent dimer rows [23]. Although ethylenediamine is a primary amine, it behaves differently from other primary amines. The formation of an interdimer row bridging structure affects the reaction pathway, leading to dual N–H dissociative products. The calculated potential energy surfaces (see Fig. 8.12) support the experimental results that indicate dual N–H dissociation reactions. Initially, ethylenediamine adsorbs in a dual dative-bonded precursor state, in which both amine lone pairs are donated to the electrophilic dimer atoms of neighboring dimers, a state which lies 43.7 kcal/mol below the energy of the reactants. From the dual dative bond, it is now possible for ethylenediamine to transfer a hydrogen atom to each surface dimer, thus creating a dual N–H dissociation product. The transition states associated with transfer of the first and second hydrogen atoms are –10.4 and –16.5 kcal/mol below the energy of the reactants, respectively. As the coverage increases, N–H dissociation is inhibited, and formation of a single Ge–N dative-bonded structure dominates. Changes in the electronic structure at the surface due to neighboring adsorbates could account for the inhibition of the N–H dissociation reaction at higher coverage.
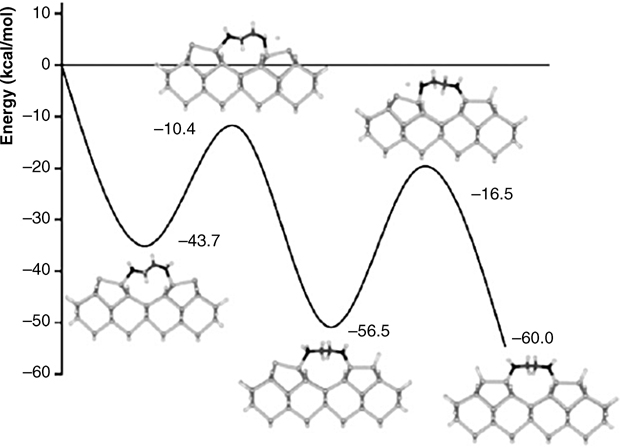
FIGURE 8.12 Calculated potential energy surface for the interdimer N–H dissociation of ethylenediamine on a Ge(100) surface (from Ref. 23). Reprinted with permission from Kim, A. et al., J. Phys. Chem. B, 109, 19817. Copyright (2005) American Chemical Society.
The adsorption product at high coverage, a single dative-bonded ethylenediamine on Ge(100), was utilized for the controlled formation of multiple organic layers on a Ge(100) surface. One of the ultimate goals of organic functionalization is the appropriate control of the deposition of multiple organic layers so that precisely tailored surfaces may be prepared with a variety of useful functionalities. A multilayer organic functionalization system was built on Ge(100)-(2×1) using a combination of ethylenediamine and 1,4-phenylene diisocyanate under vacuum conditions to form urea linkages [24]. The idealized scheme for producing the multilayer film is illustrated in Fig. 8.13.
8.2.2 Aromatic Amines
Aromatic amines may be classified into two different categories. In the first group, nitrogen atoms are external to the aromatic phenyl ring. In this case, the presence of the phenyl ring may affect the reactivity of the amine on semiconductor surfaces. Reaction through the phenyl ring via [4+2] or [2+2] cycloaddition is not competitive with the reaction through the amine group, mainly due to the aromaticity of the phenyl ring. The second group includes heteroaromatic amines, in which some of the carbon atoms in the aromatic ring are substituted with nitrogen atoms. The concepts of aromaticity and resonance stabilization are applied to the bonding of heteroaromatic molecules on Si(100)-(2×1) or Ge(100)-(2×1). Pyrrole, pyridine, pyrazine, and s-triazine have all been investigated experimentally and/or theoretically on semiconductor surfaces. Beyond possessing a large resonance stabilization energy and nonbonding electrons, many heteroaromatic molecules have significant dipole moments due to resonance and induction effects. This leads to an asymmetric charge distribution in the cyclic π-system and can activate new reaction pathways and produce new surface species not observed for benzene. The participation of the lone pair electrons of the nitrogen atom in an aromatic π-electron system is an important factor for determining whether the dative-bonded state is the major product.
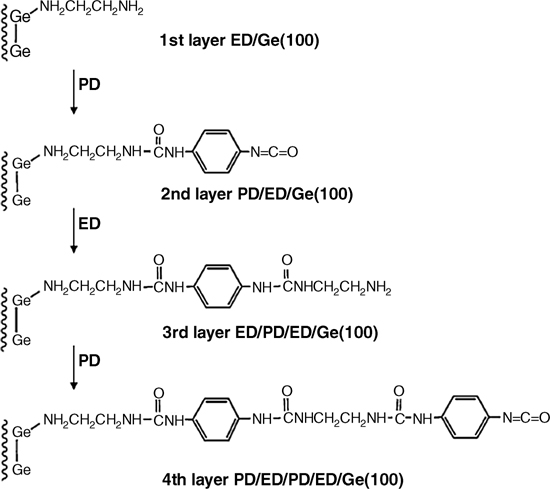
FIGURE 8.13 Schematic illustration of the synthetic route for the layer-by-layer growth of polyurea films on a Ge(100)-(2×1) surface (from Ref. 24). Reprinted with permission from Kim, A. et al., J. Am. Chem. Soc., 127, 6123. Copyright (2005) American Chemical Society.
8.2.2.1 Aniline on Si(100) and Ge(100)
The reaction of aniline (C6H5NH2) with semiconductor surfaces may occur either at the amine group or at the phenyl ring. Figure 8.14 shows an FTIR spectrum of a Si(001) surface exposed to 10 L aniline at 300 K [4]. A comparison of the spectra of surface-adsorbed aniline (Fig. 8.14a) and liquid aniline (Fig. 8.14b) reveals the spectral changes resulting from adsorption of aniline onto Si(001). The presence of a strong Si–H stretching vibration at 2072 cm–1 shows that adsorption is accompanied by cleavage of either N–H or C–H bonds. Meanwhile, the C–H spectral region and the ring-stretching combination band at 3217 cm–1 remain nearly unchanged. No significant absorption is detected in the 2600–3000 cm–1 region, in which alkane-like C–H vibrations are typically observed. These observations suggest that the benzene ring in aniline remains unperturbed. The two N–H modes at 3356 and 3431 cm–1 (Fig. 8.14b) of liquid aniline collapse into a single N–H mode at 3356 cm–1 (Fig. 8.14a) for the surface-adsorbed aniline, which strongly suggests cleavage of one of the N–H bonds.
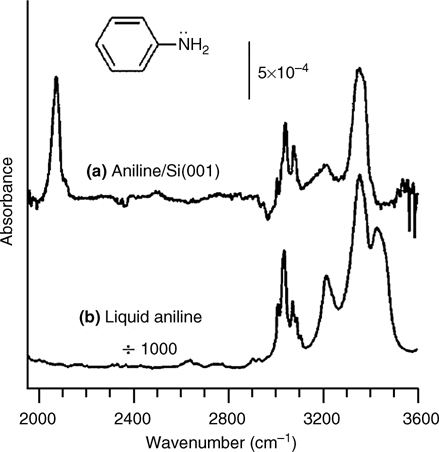
FIGURE 8.14 FTIR spectra of aniline. (a) Spectrum of a Si(001) surface exposed to 10 L of aniline at 300 K. (b) Spectrum of neat liquid aniline (from Ref. 4). Reprinted with permission from Cao, X. et al., J. Phys. Chem. B, 105, 3759. Copyright (2001) American Chemical Society.
The N 1s XPS data in Fig. 8.15 show a single peak at a binding energy of 398.9 eV [4]. This energy is almost identical with the energy associated with binding of primary amines on Si(100) surfaces, which was demonstrated to undergo N–H dissociation as discussed above. The similarity in N 1s binding energy between aniline and primary amine compounds suggests that aniline also binds to Si(100) surfaces via N–H dissociative adsorption.
Thus, it was concluded that aniline exclusively undergoes dissociative adsorption to form Si–N–C and Si–H bonds, as depicted in Fig. 8.16. The reaction is similar to the adsorption of the primary aliphatic amine, methylamine, onto Si(100).
It is notable that the reaction between aniline and Si(100) proceeds exclusively along one of the two or more adsorption modes available: Si–N–C linkage or a benzene-like direct ring interaction. Reaction through the π-electrons of the phenyl ring results in loss of aromaticity and is not thermodynamically favorable. No reports have yet described the adsorption of aniline on Ge(100); however, considering the dative bonding of aliphatic primary amines on Ge(100) and the reactivity of the phenyl ring of aniline, aniline is predicted to adsorb onto Ge(100) through N–Ge dative bonding.
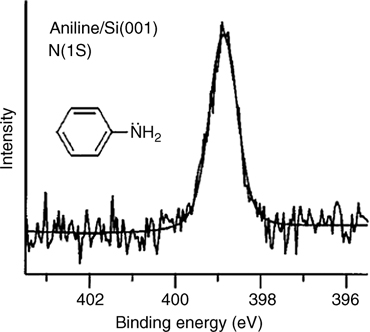
FIGURE 8.15 XPS N 1s spectrum of a Si(001) sample exposed to 5 L of aniline at 300 K (from Ref. 4). Reprinted with permission from Cao, X. et al., J. Phys. Chem. B, 105, 3759. Copyright (2001) American Chemical Society.
8.2.2.2 Five-Membered Heteroaromatic Amines: Pyrrole on Si(100) and Ge(100)
Pyrrole is a five-membered cyclic aromatic compound with one nitrogen atom. In this molecule, the two lone pair electrons of the nitrogen atom and the four π-electrons form a conjugated π-electron system. High-resolution electron energy loss spectroscopy (HREELS) and IR spectroscopic studies reveal that pyrrole on Si(100) and Ge(100) undergoes dissociative adsorption through either N–H cleavage (major product) or C–H cleavage (minor product) [4,19,25]. The retention of aromatic v(C– H) stretching modes near 3100 cm– 1 and the growth of v(Si–H) stretching modes at 2107 cm–1 in HREELS support the notion that the dominant surface species is a N–H dissociative product on Si (100) [25]. The adsorption peak corresponding to v(Si–H) at 2093 cm–1 in the IR spectrum also supports either N–Hor C–H cleavage [4]. Few or no absorption bands are observed in the 2800–3000 cm–1 region normally associated with saturated C–H vibrational modes, excluding the possible cycloaddition through a C=C double bond in the pyrrole ring. The presence of a v(N–H) stretching mode near 3400 cm–1 suggests that both C–H and N–H dissociation are possible at room temperature. Comparing the relative intensities of the Si–H and C–H stretching peaks, the C–H dissociative adsorption product may be assigned as a minor product. On the other hand, the IR spectrum for pyrrole on Ge(100)-(2×1) has a significant peak at 1988 cm–1 [19], similar to the peak observed for absorption on Si(100): (a) a significant peak corresponding to the v(Ge–H) mode at 1988 cm–1 indicates that Ge–H bonds are present at the surface; (b) the retention of a low-amplitude v(N–H) mode at 3403 cm–1; and (c) the absence of sp3 v(C–H) modes in the region 2800–3000 cm–1. These observations suggest that pyrrole also undergoes dissociative adsorption on the Ge(100) surface, and cycloaddition does not occur.
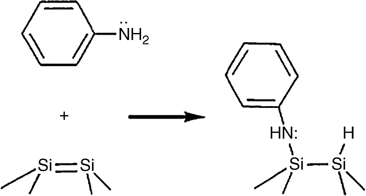
FIGURE 8.16 Bonding configuration of aniline on the Si(100) surface (from Ref. 4).
Dissociative adsorption would likely occur via a dative-bonded precursor state [19]. The aromatic nature of pyrrole enables dative bonding at either the ring carbons or the nitrogen heteroatom. A dative bond precursor through the nitrogen lone pair of the pyrrole accompanies loss of aromaticity, and the transition state is unstable relative to the energy of reactants. Thus, this pathway must overcome a high activation barrier for N–H dissociation. The other type of dative bonding, via a ring carbon, is possible because of resonance stabilization by the ring. The precursor state may give way to N–H dissociation to form a pyrrolyl radical, followed by isomerization to produce a N–H dissociated state. Otherwise, a C–H dissociation product may form from the latter type of dative bonding, analogous to electrophilic aromatic substitution. This reaction is specific to aromatic systems and occurs when an electrophilic down atom of a dimer attacks a ring carbon to cleave the aromatic C–H bond, and the resultant carbocation is stabilized through resonance. In the reaction of pyrrole with Si(100)-(2×1), the N–H dissociative product dominates. This product supposedly forms via the lower energy α- C–H dative bonding.
The adsorption of pyrrole on Ge(100) results in a mixture of dissociation products, whereas other heterocyclic aromatic compounds, such as pyridine and pyrimidine, adsorb selectively through nitrogen dative bonds to the Ge(100)-(2×1) surface. The main product of adsorption of pyrrole on Ge(100) is a N–H dissociation product [19], the same adsorption product on Si(100) surfaces. Evidence for the minor electrophilic aromatic substitution product of C–H bond cleavage is also observed. The major factor that distinguishes these bonding species from those of other aromatic heterocyclic compounds adsorbed onto Ge(100) is the delocalization of the nitrogen lone pair in the pyrrole over the ring as part of the aromatic π-system. Consequently, the N dative-bonded state is relatively unstable due to loss of resonance energy. Thus, initial adsorption through dative bonding is unfavorable, and electrophilic aromatic substitution may compete as a reaction pathway such that the nitrogen atom is not a reaction site. The reaction pathways for N–H dissociation in Fig. 8.17 compare the two possible intermediate structures, which involve either Ge–N or Ge–C dative bonding [19].
The adsorption of pyrrole on Si(111)-(7×7) is similar to that on Si(100). The disappearance of the N–H stretching band upon adsorption at room temperature, along with the appearance of Si–H vibrational peaks, clearly indicates that pyrrole chemisorbs dissociatively on Si(111)-(7×7) [26]. The N–H dissociative product is calculated to be 24–31 kcal/mol more stable than the 2,5-dihydropyrrole-like adduct formed via a [4+2]-like addition, implying that the retention of aromaticity is an important factor in the reaction pathway. Furthermore, pyrrolyl binding at an adatom is sterically more favorable than attachment to a lower lying rest atom, in which the ring may experience repulsive interactions with the neighboring adatoms.
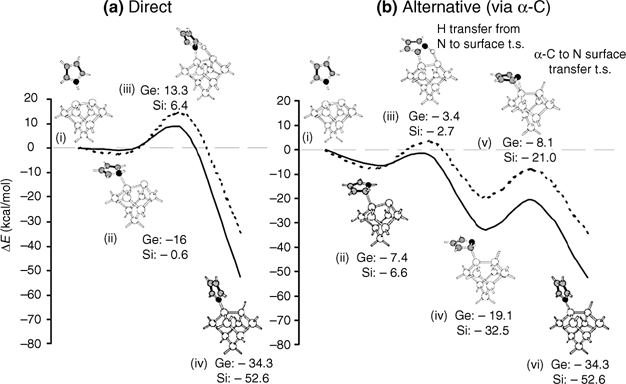
FIGURE 8.17 Reaction pathways for N–H dissociation in pyrrole adsorbed onto Si(100)-(2 × 1) and Ge(100)-(2 × 1) surfaces: (a) direct pathway; (b) alternative pathway (from Ref. 19). Reprinted with permission from Wang, G. T. et al., J. Phys. Chem. B, 107, 4982. Copyright (2003) American Chemical Society.
Protection of the pyrrole nitrogen with a methyl group reverses the reactivity [27]. Because C–N bonds have a much higher dissociation barrier than N–H bonds, N-methylpyrrole reacts exclusively through both [2+2]-like and [4+2]-like additions.
8.2.2.3 Six-Membered Heteroaromatic Amines
Pyridine, pyrazine, and s-triazine are six-membered heterocyclic amines with different number of nitrogen atoms in the ring. Two reaction modes compete during adsorption of these molecules on Si(100)-(2×1): adsorption may occur through the lone pairs of the nitrogen atom or through the π-electrons of the double bond [28]. DFT studies suggest that all three molecules adsorb through N dative bonding with the buckled-down silicon atom of the silicon surface dimer [29]. In addition, [4+2] cycloaddition occurs, similar to the adsorption of benzene. Reaction between the remaining double bond and the adjacent Si dimer may follow via cycloaddition, leading to a tetra-σ-bonded state called a “tight-bridged product.”
Pyridine on Si(100) and Ge(100)
The lone pair electrons on the N atom of pyridine are not directly involved in aromatic π-conjugation, and instead are donated to form a dative bond with the electron-deficient atom of a surface dimer. Two [4+2] cycloaddition configurations may be attained from pyridine. Adducts may have either N–Si/C–Si bonds or C–Si/C–Si bonds. In fact, it is found that pyridine forms both the dative-bonded product and the [4+2] cycloaddition-like product on Si(100) and the nearly completely dative-bonded product on Ge(100) [30–32].
The N 1s XPS spectrum shown in Fig. 8.18 indicates the presence of two adsorption states for pyridine adsorbed onto Si(100) [30]. Observation of two N 1s binding energies at 398.8 and 401.8 eV implies the presence of at least two types of species. The higher binding energy state (401.8 eV) corresponds to dative bonding between the nitrogen atom of the pyridine and the electron-deficient surface Si atom. Dative bonding is dominant at 110 K. An increase in temperature leads to a gradual decrease in the intensity of the dative bonding peak and growth of the 398.8 eV peak. Increasing the temperature to 350 K causes all pyridine molecules remaining on the surface to yield a N 1s peak at 398.8 eV.
The other peak at 398.8 eV was identified as a butterfly-like [4+2] product by HREELS (see Fig. 8.19) and DFT calculations [30]. The observation of Si–C (615 cm– 1) and Si–N (495cm–1) peaks, together with the coexistence of C(sp2)–H (3045 cm–1) and C(sp3)–H (2895 cm–1) stretching modes, suggests formation of a di-σ-bonded pyridine on Si(100) at 350 K. The suggested configuration was a butterflylike [4+2] product with di-σ-bonding between the N/C atoms of the pyridine molecule and Si dimer atoms, similarly to the bonding of benzene on the same surface.
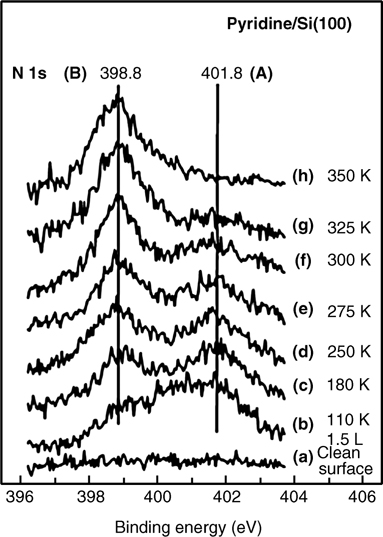
FIGURE 8.18 The N 1s core level spectra for pyridine adsorbed onto Si(100) as a function of temperature (from Ref. 30). Reprinted with permission from Tao, F. et al., J. Phys. Chem. B, 107, 6384. Copyright (2003) American Chemical Society.
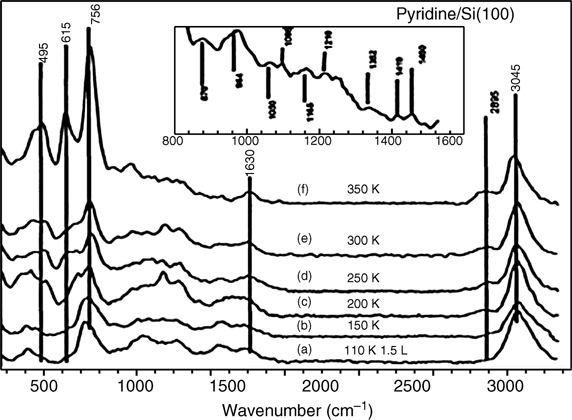
FIGURE 8.19 HREELS spectra for pyridine adsorbed onto Si(100) as a function of temperature (110–350 K). The inset shows an enlarged view of spectrum (f) in the region 830–1530 cm–1 (from Ref. 30). Reprinted with permission from Tao, F. et al., J. Phys. Chem. B, 107, 6384. Copyright (2003) American Chemical Society.
STM measurements identified two interesting features [31]. The feature shown in Fig. 8.20b is a two-dimer footprint in which a bright feature, centered over one of the dimers, represents a tight-bridged configuration. Gradual conversion of the dative-bonded configuration (see Fig. 8.20a) into a tight-bridged species (see Fig. 8.20b) is observed at low coverage. The barrier for conversion is estimated from the observed rate of conversion to be 0.9 eV. The di-σ-bonded adduct may further proceed to a tight-bridged species via appropriate choice of adsorbate coverage and temperature, among other factors. The formation of a tight-bridged configuration requires two surface dimers. Thus, they may be invisible under certain coverage conditions.
An XPS study of pyridine on Si(111)-(7×7) at 110 K also shows two features in the N 1s edge: one at 401.8 eV (a dative-bonded state) and another at 398.8 eV (the monolayer coverage) [33]. Based on the vibrational studies, the second feature is assigned to the [4+2] adduct with Si–N and Si–C bonds. The dative-bonded state is converted into a cycloadduct with increasing temperature, similar to the behavior observed on Si(100).
In contrast, pyridine adsorbs selectively through a Ge–N dative bond on the Ge(100) surface. Furthermore, STM images show formation of a highly ordered monolayer at the surface with coverage of 0.25 ML, as shown in Fig. 8.21 [32]. The pyridine overlayer forms a c(4 × 2) structure in which the molecules bind to the down atoms of every other dimer to minimize repulsive interactions between pyridine molecules.
Theoretical calculations show that the dative-bonded adduct is more stable than the other possible reaction products on Ge. The calculations for the most stable dative-bonded structure indicate that (1) the Ge–N bond length (2.04 Å) is significantly longer than the covalent bond length (1.70 Å) such that the pyridine molecule datively binds to the down Ge atom; (2) the Ge dimers are rearranged into a c(4×2) structure; (3) the adsorbed pyridine molecules retain their aromaticity; and (4) the plane of the aromatic ring is tilted with respect to the surface (see Fig. 8.22).
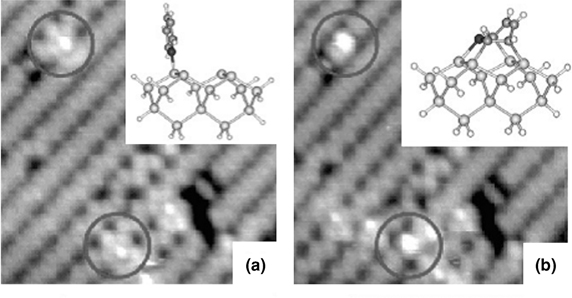
FIGURE 8.20 Occupied state STM images (7×7nm2, –2 V, 40 pA) during dosing of pyridine on Si(100). In (a), two dative-bonded adducts are highlighted and are observed to relax to the tight-bridged configuration in (b). The insets show model bonding geometries for the dative and tight-bridged configurations, respectively (from Ref. 31). Reprinted with permission from Miwa, J. A. et al., J. Phys. Chem. B, 109, 20055. Copyright (2005) American Chemical Society.

FIGURE 8.21 Sequence of filled state STM images (10×10 nm2, 0.1 nA) of (a) a clean Ge (100) surface (–1.8 V) and 0.25 ML pyridine adsorbed onto the surface at bias voltages (Vs) of (b) –1.0 V, (c) –1.8 V, and (d) –2.4 V. Arrow indicates a pyridine molecule (from Ref. 32). Reprinted with permission from Cho, Y. E. et al., J. Am. Chem. Soc., 125, 7514. Copyright (2003) American Chemical Society.
The electronic character of the dative-bonded structure can be clearly described through the charge density difference Δρ(r) before and after adsorption [34]. The Δρ(r) contours corresponding to the clean Ge(100) surface show a pronounced maximum (red color) in the buckled dimer region, indicating bonding between the two Ge atoms. Above the up Ge atoms, the existence of an electron-rich region may be explained by charge transfer from the buckled-down atom to the buckled-up atom. Figure 8.22b shows a pronounced maximum (indicated in red) between the down Ge atom and the pyridine molecule. The maximum resides closer to the pyridine molecule, represents a nitrogen lone pair, and, thus, indicates a dative bond.
Comparison of pyridine adsorption on Ge(100) with that on Si(100) reveals significant differences. The tight-bridged-type configuration readily available on Si(100)-(2×1) cannot form on Ge(100)-(2×1) because of the apparent negative adsorption energy predicted by DFT calculations [35]. This energetic difference causes pyridine to attach to a single Ge atom by dative bonding to form a very stable c(4×2) pattern observed by STM [32]. The difference between the Ge(100) and the Si(100) surfaces mainly arises from the fact that the strength of the covalent bond between C and Ge, formed by a cycloaddition-type reaction, is weaker than that between C and Si. Indeed, the Ge–C bond strength was found to be 8.9 kcal/mol less than its silicon analogue. Thus, on Ge(100), the dative-bonded product is favored such that the aromaticity of the pyridine ring is retained after adsorption onto the surface.
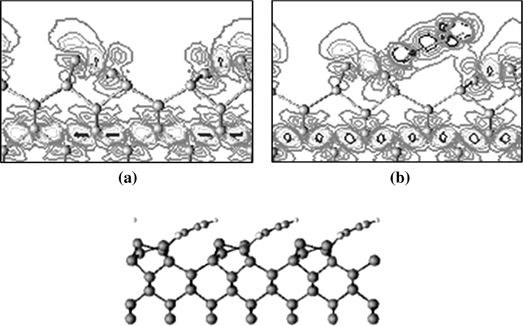
FIGURE 8.22 Plot of the charge density difference between the total valence charge density and the superposition of atomic valence charge densities for (a) clean Ge(100)-c(4×2) and (b) pyridine/Ge(100)-c(4×2). Side view of the optimized dative-bonded structure (from Ref. 34). Reprinted with permission from Hong, S. et al., J. Phys. Chem. B, 108, 15229. Copyright (2004) American Chemical Society.
Pyrimidine on Ge(100)
Pyrimidine (1,3-diazine, C4H4N2) is a six-membered aromatic molecule with four carbon atoms and two nitrogen atoms, each with a lone pair of electrons. Pyrimidine is one of the important parent molecules for synthesizing nucleic acid bases. The structure of pyrimidine-adsorbed semiconductor (100) surfaces can assist an understanding of the interaction between biomolecules, such as amino acids, nucleic acids, or DNA, and semiconductor (100) surfaces.
Lee et al. studied the adsorption of pyrimidine on Ge(100) using STM and found that pyrimidine undergoes molecular adsorption through dative bonding on Ge(100) [36]. For coverage of pyrimidine up to 0.25 ML, a well-ordered c(4×2) structure results from states that appear as oval-shaped protrusions in the STM micrographs (see Fig. 8.23). The oval-shaped protrusions, surrounded by six neighboring up atoms of the buckled-up Ge dimers, are located between the Ge dimer rows. The STM image indicates that pyrimidine molecules are located between adjacent dimer rows.
Through DTF calculations, Lee et al. found that the adsorbed pyrimidine molecules are tilted by 40° with respect to the Ge surface and form bridges between the down Ge atoms of neighboring Ge dimer rows via double Ge–N dative bonding without loss of aromaticity (see Fig. 8.24) [36]. At higher coverage (0.5 ML), the pyrimidine overlayer converts to a p(2×2) structure. This high-coverage overlayer is attributed to pyrimidine in a single dative-bonded configuration, a state that is stable only in the presence of a continuous pyrimidine flux.

FIGURE 8.23 (a) A filled state STM image (15×15 nm2, Vs = –1.8 V, It=0.1 nA) of a clean Ge(100) surface. (b) A filled state STM image (Vs = –1.6 V) of pyrimidine on Ge(100) at a coverage of 0.01 ML. The arrow indicates an adsorbed pyrimidine molecule. STM images of pyrimidine on Ge(100) at various bias voltages: (c) –2.0 V, (d) –1.6V, (e) –1.0 V, (f) +1.4 V, (g) +1.0 V, and (h) +0.8 V (from Ref. 36). Reprinted with permission from Lee, J. Y. et al., J. Phys. Chem. B, 109, 348. Copyright (2005) American Chemical Society.
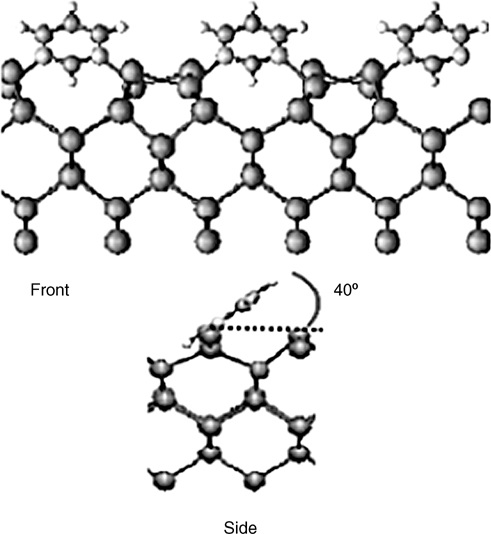
FIGURE 8.24 Front and side views of the adsorption structure of pyrimidine on Ge(100) (from Ref. 36). Reprinted with permission from Lee, J. Y. et al., J. Phys. Chem. B, 109, 348. Copyright (2005) American Chemical Society.
Pyrazine on Si(100) and Si(111)
Pyrazine is expected to adsorb through either N dative bonding or [4+2] cycloaddition on Si(100) and Si(111). The [4+2] product may have either two C–Si bonds or two N–Si bonds. The formation of N dative bonding is favored only at low temperatures [27]. The binding energy for the dative-bonded pyrazine (–18.6 kcal/mol) is lower than that of pyridine (–27.0 kcal/mol), reflecting the smaller electrostatic interactions between nonpolar pyrazine and electron-deficient down Si atoms. More polar pyridine molecules have stronger attractions with Si atoms. Moreover, the electron density at the nitrogen atom in pyridine is higher than that in pyrazine. Thus, pyrazine is a poor electron donor, and dative bonding through the nitrogen atom is unfavorable at room temperature. At elevated temperatures, the thermodynamically most favored [4+2] cycloaddition may occur to form a 1,4-N,N-dihydropyrazine-like state [37,38]. Chemisorption does not cause significant shifts in the stretching frequency of C–H, indicating that all four C atoms of the molecule are not rehybridized. In addition, a new peak at 1617 cm– 1, attributed to the unconjugated C=C double bond, is observed. Furthermore, the characteristic vibrational modes of the aromatic ring at 1330–1547 cm–1 are absent in the HREELS spectra of chemisorbed pyrazine, indicating the disruption of aromaticity in the pyrazine. The formation of a [4+2] product is accompanied by loss of aromaticity. The reaction of pyrazine with the Si (111)-(7×7) surface results in formation of a [4+2] product in which both nitrogen atoms of the pyrazine form covalent bonds with the adatom–rest atom pair on the Si(111)-(7×7) surface. Such a reaction pathway produces two unconjugated C=C bonds [39].
8.2.3 O-Containing Molecules
8.2.3.1 Alcohols on Si(100) and Ge(100)
Methanol (CH3OH) is a simple O-containing molecule that is used as an oxidant and precursor in the formation of oxide layers. Adsorption of methanol on semiconductor surfaces has been extensively studied [40–45]. Previous experimental and theoretical results revealed that methanol undergoes dissociative adsorption on Si(100) and Ge(100) surfaces at room temperature.
Figure 8.25 illustrates the probable reaction pathways of methanol adsorption onto Ge(100). First, methanol may enter a molecular adsorption state as a result of Ge–O dative bonding between the O lone pair electrons and the electrophilic down Ge atom of a dimer (see Fig. 8.25a). This surface reaction occurs via a nucleophilic/ electrophilic reaction. If the molecular adsorption state is unstable, the molecule subsequently undergoes proton transfer via O–H bond dissociation (see Fig. 8.25b) or methyl group transfer via C–O bond dissociation (see Fig. 8.25c).
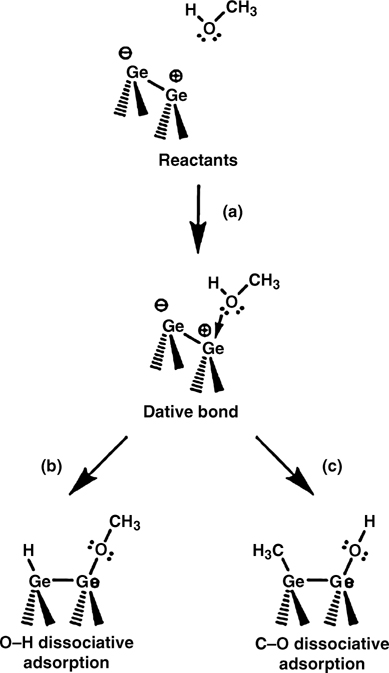
FIGURE 8.25 Schematic illustration of the possible reactions of methanol on Ge(100) surfaces: (a) Lewis acid-base interaction via Ge–O dative bonding; (b) proton transfer via O–H bond dissociation; and (c) methyl group transfer via C–O bond dissociation (from Ref. 44). Reprinted with permission from Bae, S.-S. et al., J. Phys. Chem. C, 111, 15013. Copyright (2007) American Chemical Society.
Kim et al. studied the adsorption of methanol on Ge(100) using STM and DFT calculations [11,44]. At low coverage, high-resolution experimental STM (see Fig. 8.26a) shows that methanol undergoes O–H bond dissociative adsorption on a single Ge–Ge dimer. As the methanol coverage increases to saturation, dissociative adsorption of methanol results in formation of dimer row-based chain-like arrays. DFT calculations (see Fig. 8.26b) show that at room temperature, O–H bond dissociative adsorption is kinetically more favorable than C–O bond dissociative adsorption, although the final product of C–O bond dissociative adsorption is more stable geometrically and thermodynamically. STM experiments and DFT calculations together suggested that the adsorption structure of methanol on Ge(100) at room temperature has a H–Ge–Ge–OCH3 geometry as a result of O–H dissociative adsorption onto a single dimer.
Kachian and Bent described the adsorption structure of ethanol on Ge(100) [7]. Figure 8.27 displays the IR spectra for the interdimer O–H dissociated product and the intradimer O–H dissociative product for ethanol on a Ge(100) surface. As shown in this figure, it is difficult to identify the difference between inter- and intradimer O–H dissociated products. Thus, DFT calculations are needed to determine the most favorable adsorption structure.
8.2.3.2 Ketones on Si(100) and Ge(100)
Armstrong et al. reported that the majority of surface adducts formed by acetone, acetaldehyde, and biacetyl at low temperatures are [2+2] C=O cycloaddition products, as illustrated in Fig. 8.28a for the case of acetone [46,47]. Barriocanal and Doren identified a barrierless pathway that passes through a dative-bonded precursor state for the [2+2] C=O cycloaddition product of glyoxal [48]. On the other hand, Wang et al. were not able to find a barrierless pathway for the reaction of acetone on Si(100)-(2×1), and they propose that steric interactions of the methyl groups with the surface likely play a role in hindering the reaction of acetone [10]. The conjugated double bonds in glyoxal may weaken the C=O bond and reduce or remove any activation barrier. Using IR spectroscopic results, Wang et al. [10] showed that acetone becomes trapped in a dative-bonded well at low temperatures on Ge(100)-(2×1), as evidenced by a 70–90 cm–1 red shift observed in the m(C=O) stretching mode.
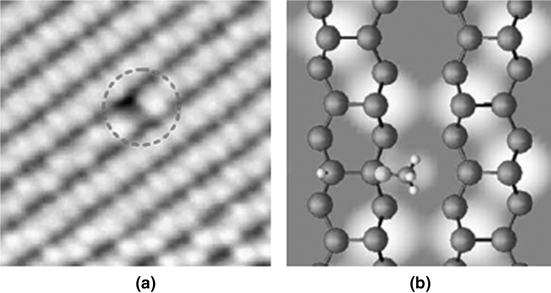
FIGURE 8.26 (a) The experimental filled state STM image (Vs = –1.6 V); (b) the corresponding theoretical schematic illustration of OH dissociative adsorption, with the OH group parallel to the Ge dimer row (from Ref. 44). Reprinted with permission from Bae, S.-S. et al., J. Phys. Chem. C, 111, 15013. Copyright (2007) American Chemical Society.
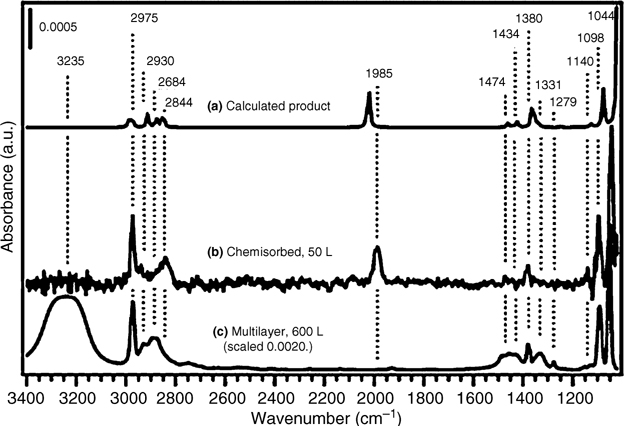
FIGURE 8.27 (a) Calculated IR spectrum of the intradimer O–H dissociation product of ethanol across a Ge dimer on the two–dimer intrarow Ge15H16 cluster. (b) The average of two IR spectra taken following saturation exposure of ethanol to Ge(100)-(2×1) at 310 K. (c) An IR spectrum of a multilayer of ethanol on Ge(100)-(2×1), taken at 140 K (intensity scaled). The peak labels correspond to chemisorbed and multilayer spectra. The product used to calculate the IR spectrum displayed in part (a) was geometrically optimized without geometric constraints (from Ref. 7). Reprinted with permission from Kachian, J. S. et al., J. Am. Chem. Soc., 131, 7005. Copyright (2009) American Chemical Society.
We also consider the α-CH dissociation product, as depicted in Fig. 8.28b, which was first identified on a semiconductor surface by Wang et al. in their study of acetone on Ge(100)-(2×1) [10]. As shown in Fig. 8.29, the IR spectrum revealed absorption peaks near 1638 and 1967 cm–1 corresponding to v(C=C) and v(Ge–H) stretching modes, respectively. Wang et al. postulated that the nucleophilic dimer atom more easily abstracts a hydrogen atom once acetone datively bonds through one of the oxygen lone pairs to form the final product illustrated in Fig. 8.28b.
8.2.3.3 Carboxyl Acids on Si(100) and Ge(100)
Several research groups have previously investigated carboxylic acids on Si(100)-(2×1) and have provided convincing evidence that the major reaction pathway proceeds via O–H dissociation [49,50]. On the basis of the observed energy difference between the v(C=O) and v(C–O) peaks in the HREELS spectra, Tanaka et al. concluded that formic acid forms a monodentate O–H dissociation product on Si(100)-(2×1) at room temperature. More recently, Lopez et al. [51] reported an O–H dissociation product on Si(100)-(2×1), following a reaction with benzoic acid or the multifunctional molecules glycine and 4-aminobenzoic acid, respectively. Hwang et al. [52] investigated the reaction of vinyl acetic acid on Si(100)-(2×1) using low-energy electron diffraction (LEED), X-ray photoelectron spectroscopy (XPS), and synchrotron radiation photoemission spectroscopy (SRPES).
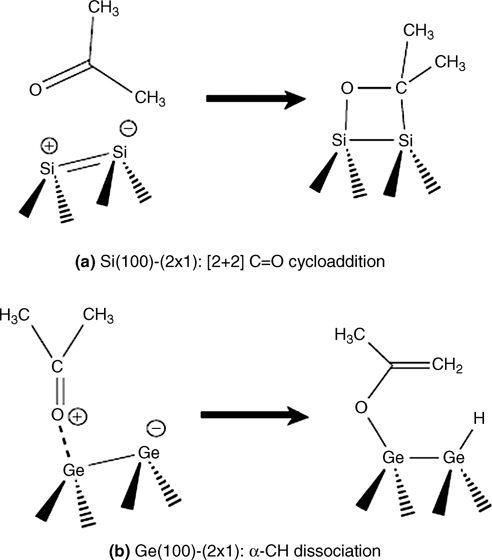
FIGURE 8.28 The major products of acetone adsorption onto Si(100)-(2×1) or Ge(100)-(2×1) are different. (a) [2+2] C=O cycloaddition is kinetically favored and is observed on Si(100)-(2×1) at low temperatures. (b) An α-CH dissociation product is thermodynamically favored and is observed on Ge(100)-(2×1) at room temperature.
Bent and coworkers reported that the major reaction pathway of carboxylic acids on Si(100) was via O–H dissociation [53]. Although O–H dissociation is reported to be the major pathway on Si, different or additional surface reactions may potentially be observed for acetic acid adsorption.
Figure 8.30 shows the IR spectra corresponding to the reaction of acetic acid on Ge(100)-(2×1) as a function of coverage. Infrared results from a coverage-dependent study of acetic acid adsorbed onto Ge(100)-(2×1) at room temperature or multilayers adsorbed at low temperatures are shown in Fig. 8.30a and b, respectively. The key peaks of the multilayer spectrum, appearing at 1697, 1440, 1414, 1360, and 1302 cm–1, are in excellent agreement with previously published low-temperature spectra of molecular acetic acid [54]. The strong peak at 1697 cm– 1 is attributed to a v(C=O) stretching mode. A comparison with Haurie and Novak [55] suggests that the peaks at 1440 and 1360 cm–1 may be assigned to asymmetric and symmetric δ(CH3) bending modes, respectively, whereas the mode at 1414 cm–1 may be assigned to a δ(OH) bending mode. The intense absorption feature at 1302 cm–1 may be attributed to a skeletal stretching vibration with the strongest contribution attributed to the C–O single bond [54,56]. They conclusively confirmed, based on IR spectral studies, that carboxylic acids undergo O–H dissociation at 310 K, and the product is both kinetically and thermodynamically favored.
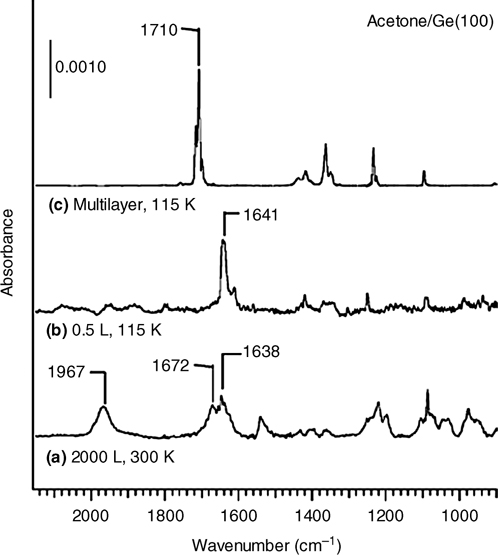
FIGURE 8.29 Infrared spectra of acetone adsorbed onto a Ge(100)-(2×1) surface: (a) 2000 L at 300 K, (b) 0.5 L at 115 K, and (c) multilayers (scaled) at 115 K. Stretching modes at 1967 and 1672/1638 cm–1 are evidence of v(Ge–H) and v(C=C) stretching modes, respectively, and indicate an α-CH dissociation product. At low temperatures, the presence of a red-shifted v(C=O) mode at 1641 cm–1 indicates a dative bond species. Figure reprinted with permission from Ref. 10, copyright 2001 American Chemical Society. Reprinted with permission from Wang, G. T. et al., J. Phys. Chem. B, 105, 12559. Copyright (2001) American Chemical Society.
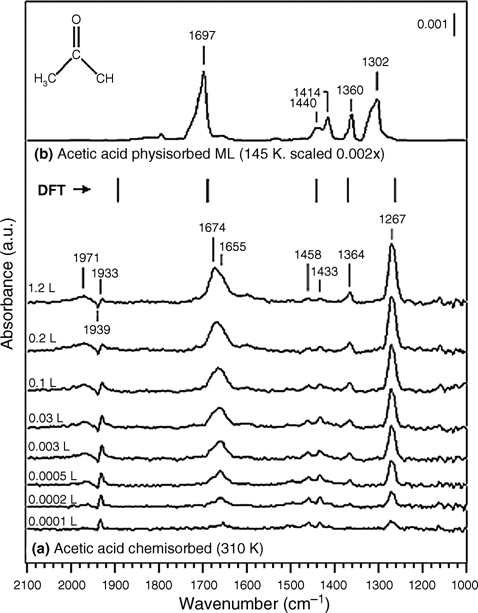
FIGURE 8.30 (a) Coverage-dependent infrared spectra for the reaction of acetic acid on Ge(100)-(2×1) at 310K, and (b) acetic acid multilayers adsorbed at 145 K (scaled). The vertical lines mark the theoretically calculated vibrational frequencies of the O–H dissociation product (scaled by 0.96 for the B3LYP/6–31G* level of theory [53]. Reprinted with permission from Filler, M. A. et al., J. Am. Chem. Soc., 128, 770. Copyright (2006) American Chemical Society.
8.2.4 S-Containing Molecules
8.2.4.1 Thiophene on Si(100) and Ge(100)
A heteroaromatic thiophene molecule contains a sulfur atom and an aromatic ring. Thus, it can adsorb on Si and Ge(100) surfaces in a variety of ways, such as via [4+2] cycloaddition, [2+2] cycloaddition, or Lewis acid-base reaction. Lu et al. reported the adsorption of thiophene (C4H4S) on a reconstructed Si(100)-(2×1) surface using a hybrid density functional B3LYP method in combination with the cluster model approach [57]. Two chemisorption mechanisms, [4+2] or [2+2] cycloaddition of C4H4S onto a surface dimer site, have been examined. The calculations revealed that the former process is barrierless and favored over the latter process, which requires an activation energy of 2.6 kcal/mol. The di-σ-bonded surface species formed by the [4+2] cycloaddition-type chemisorption can undergo further [2+2] cycloaddition with a neighboring Si=Si dimer site. As a result, on Si(100)-(2×1) surfaces, only cycloaddition (chemisorption) can occur.
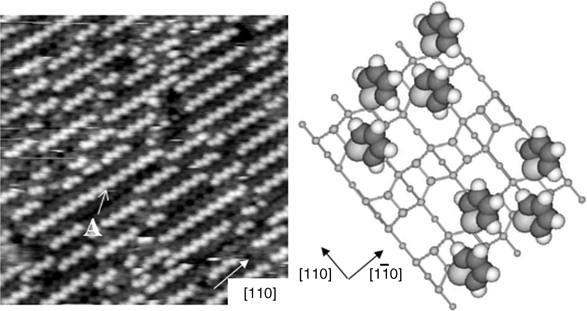
FIGURE 8.31 (left panel) Filled state STM images (20 × 20 nm2, Vs = –2.0 V, It=0.1 nA) of a Ge(100) surface when exposed to 2L (0.13 ML) of thiophene. (right panel) Schematic diagram for dative bonding structure of thiophene on Ge(100) system. Each light gray, dark gray, and white ball indicates sulfur (S), carbon (C), and hydrogen (H), respectively [58]. Reprinted with permission from Jeon, S. M. et al., J. Am. Chem. Soc., 128, 6296. Copyright (2006) American Chemical Society.
In contrast, Jeon et al. described the adsorption of C4H4S on a Ge(100)-(2×1) surface using STM, DFT calculations, and photoemission spectroscopy. They conclude that C4H4S on Ge(100) can be concurrently adsorbed through dative bonding and [4+2] cycloaddition at a coverage of < 0.25 ML [58]. The initial deposition of thiophene on Ge(100)-(2×1) proceeds via dative bonding between thiophene and the Ge(100)-(2×1) surface through the sulfur atom. Figure 8.31 shows that the thiophene molecules preferentially form one-dimensional molecular chains (feature A: dative bonding feature) if a clean Ge(100) surface is exposed to 2 L of thiophene.
DFT calculations of the C4H4S/Ge9H12 system, shown in Fig. 8.32, agree with the theoretical results for thiophene on Si(100) [57] in that the [4+2] cycloaddition reaction product is predicted to be the most thermodynamically favorable product. The Ge–S dative bonding configuration was found to be the least stable structure from a thermodynamic perspective, although this reaction may have the lowest activation barrier, making it the most favorable from a kinetic perspective. The sulfur atom in thiophene is sp3-hybridized, and therefore, the lone pair electrons are tilted away from the aromatic ring plane. As a consequence, the ring plane of the adsorbed thiophene is tilted with respect to the surface when it is bound to Ge(100) via Ge–S dative bonding.
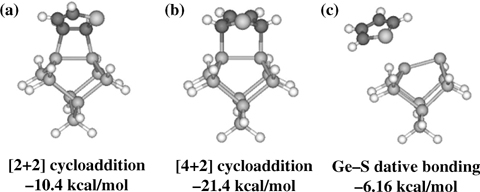
FIGURE 8.32 Local minima for the C4H4S/Ge9H12 system calculated at the B3LYP/ LACVP** level of theory. The values indicate the adsorption energies with respect to free thiophene and Ge9H12.
For thiophene adsorbed onto Ge(100)-(2×1), Ge–S dative bonding can occur, although [4+2] cycloaddition is more energetically favorable.
8.3 DATIVE BONDING OF LEWIS ACIDS (ELECTROPHILIC)
As mentioned in previous sections, the formation of datively bonded Lewis bases with electron-deficient atoms on semiconductor surfaces has been intensively studied. In contrast, relatively few examples of the formation of dative bonds between Lewis acids and electron-rich atoms on semiconductor surfaces are available.
Konecny and Doren investigated the adsorption of BH3 on Si(100) using nonlocal DFT with a cluster model for the surface. They predicted that a Si–B bond would form via nucleophilic attack on the boron, leaving BH2 and H fragments bound to the surface [59]. Yoshinobu and coworkers studied the adsorption of BF3 (a Lewis acid) on Si(100)-c(4×2) using high-resolution Si 2p photoelectron spectroscopy [60]. They found that BF3 dissociates to form Si–F and Si–BF2 species on Si(100), and the dissociated species (BF2 and F) adsorb predominantly on the up atoms of Si(100) dimers. Cao and Hamers reported that the adsorption of trimethylamine (a Lewis base) and BF3 (a Lewis acid) on a Si(100) surface leads to formation of a novel surface-mediated donor-acceptor complex with the structure TMA–Si–Si–BF3 [61]. They found that the sequential adsorption of TMA and BF3 leads to a dative bond between BF3 (a Lewis acid) and the electron-rich up atom of a single Si(100) dimer.
Aluminum trichloride (AlCl3) is a Lewis acid that forms donor-acceptor complexes with electron-rich molecules. Interestingly, Jung et al. studied the adsorption structures of AlCl3 on Ge(100) using STM and high-resolution photoelectron spectroscopy. They found that AlCl3 molecules adsorb onto Ge(100) surfaces via a cycloaddition-like reaction to form di-σ-bonds in which the AlCl3 molecules interact with the surface dimers of Ge(100) [62]. These results are quite different from those of NH3 adsorbed onto Ge(100), in which a single dative bond forms. Recently, an STM investigation by Kim et al. revealed that vinylferrocene, an organometallic molecule, adsorbs onto Ge(100) surfaces through Ge–Fe dative bonding between the electron-deficient Fe atom of the vinylferrocene and the electron-rich up atoms of Ge(100) dimers (see Fig. 8.33) [63]. They also predicted that there would be an interaction between the electron-rich cyclopentadiene (Cp) rings and the electron-deficient down Ge atoms at both ends of the adsorbate. An STM image of vinylferrocene adsorbed onto Ge(100) displays dumbbell-shaped fuzzy features (dotted circles), consisting of two coupled bright protrusions on one side of a dimer row, attributed to adsorbed vinylferrocene molecules. As indicated in an enlarged STM image of this feature (see Fig. 8.33b), the distance between the top positions of coupled protrusions is 3.6 Å, in agreement with both experimental and theoretical values of 3.3 Å for the distance between the Cp rings in a ferrocene molecule [64]. Therefore, it is reasonable that the bright dumbbell-shaped protrusions are associated with the coupled Cp rings of a vinylferrocene molecule. An atom-resolved STM investigation indicated that the center of the dumbbell-shaped protrusion, the position of the iron atom, is exactly located on top of an up Ge atom (see Fig. 8.33c). Thus, formation of a dative bond between the Fe and the up Ge atom is expected. The formal charge of the iron atom in the vinylferrocene molecule is considered to be positive [65], and the electron-rich buckled-up Ge atom is predicted to donate electrons to an iron atom.
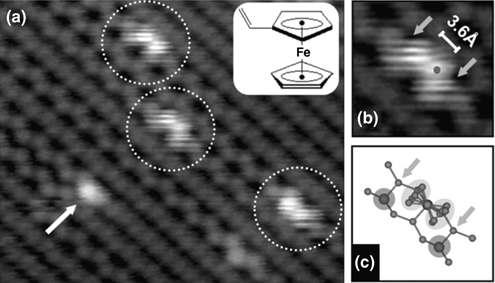
FIGURE 8.33 (a) A filled state STM image of a vinylferrocene adsorbed onto a Ge(100) surface, acquired at room temperature (Vs = –1.1 V, It=0.1 nA). The dotted circles and arrow indicate parallel vinylferrocenes and tilted vinylferrocenes, respectively. (b, c) An enlarged image and a proposed schematic representation of the adsorption structure of the parallel vinylferrocene. Arrows indicate the positions of down Ge atoms.
8.4 SUMMARY
The chemical reactions of organic compounds with semiconductor surfaces such as Si(100)-(2×1), Si(111)-(7×7), and Ge(100)-(2×1) are summarized in Table 8.1. Additional surface reconstructions may occur after the initial reactions. Cycloaddition and dissociative adsorption compete with dative bonding. With respect to aliphatic amines, both primary and secondary amine molecules dissociatively adsorb, whereas tertiary amines molecularly adsorb onto Si(100). In contrast, most amines absorb mainly through dative bonding on Ge(100) surfaces, except for ethylenediamine and pyrrole. O-containing molecules such as alcohol, ketone, and carboxylic acids on Si(100)-(2×1) and Ge(100)-(2×1) mainly undergo dissociative reaction through a dative-bonded intermediate state. S-containing molecules such as thiophene adsorb on Si(100)-(2 × 1) and Ge(100)-(2 × 1) through dative bonding and cycloaddition reaction.
TABLE 8.1 Reactions Between Organic Molecules and Si(100), Si(lll), and Ge(100) Surfaces


REFERENCES
1. Kang, H. C. Surf. Interface Anal. 1999, 28, 92.
2. Lui, Q.; Hoffmann, R. J. Am. Chem. Soc. 1995, 117, 4082.
3. Mui, C.; Han, J. H.; Wang, G. T.; Musgrave, C. B.; Bent, S. F. J. Am. Chem. Soc. 2002, 124, 4027.
4. Cao, X.; Coulter, S. K.; Ellison, M. D.; Liu, H.; Hamers, R. J. J. Phys. Chem. B 2001, 105, 3759.
5. Waltenburg, H. N.; Yates, J. T., Jr., Chem. Rev. 1995, 95, 1589.
6. Kachian, J. S.; Wong, K. T.; Bent, S. F. Acc. Chem. Res. 2010, 43, 346.
7. Kachian, J. S.; Bent, S. F. J. Am. Chem. Soc. 2009, 131, 7005.
8. Filler, M. A.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Am. Chem. Soc. 2003, 125, 4928.
9. Lu, X.; Zhang, Q.; Lin, M. C. Phys. Chem. Chem. Phys. 2001, 3, 2156.
10. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 2001, 105, 12559.
11. Kim, D. H.; Bae, S.-S.; Hong, S.; Kim, S. Surf. Sci. 2010, 604, 129.
12. Bent, H. A. Chem. Rev. 1961, 61, 275.
13. Widjaja, Y.; Mysinger, M. M.; Musgrave, C. B. J. Phys. Chem. B 2000, 104, 2527.
14. Mui, C.; Musgrave, C. B. Langmuir 2005, 21, 5230.
15. Warschkow, O.; Wilson, H. F.; Marks, N. A; Schofield, S. R.; Curson, N. J.; Smith, P. V.; Radny, M. W.; McKenzie, D. R.; Simmons, M. Y. Phys. Rev. B 2005, 72, 125328.
16. Schofield, S. R.; Curson, N. J.; Warschkow, O.; Marks, N. A; Wilson, H. F.; Simmons, M. Y.; Smith, P. V.; Radny, M. W.; McKenzie, D. R.; Clark, R. G. J. Phys. Chem. B 2006, 110, 3173.
17. Tsai, H.-W.; Lin, D.-S. Surf. Sci. 2001, 482, 654.
18. Mui, C.; Wang, G. T.; Bent, S. F.; Musgrave, C. B. J. Chem. Phys. 2001, 114, 10170.
19. Wang, G. T.; Mui, C.; Tannaci, J. F.; Filler, M. A.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 2003, 107, 4982.
20. Cao, X.; Hamers, R. J. J. Am. Chem. Soc. 2001, 123, 10988.
21. McKean, D. C.; Ellis, I. A. J. Mol. Struct. 1975, 29, 81.
22. Tao, F.; Cai, Y.; Ning, Y.; Xu, G.; Bernasek, S. L. J. Phys. Chem. C 2008, 112, 15474.
23. Kim, A.; Filler, M. A.; Kim, S.; Bent, S. F. J. Phys. Chem. B 2005, 109, 19817.
24. Kim, A.; Filler, M. A.; Kim, S.; Bent, S. F. J. Am. Chem. Soc. 2005, 127, 6123.
25. Qiao, M. H.; Cao, Y.; Deng, J. F.; Xu, G. Q. Chem. Phys. Lett. 2000, 325, 508.
26. Yuan, Z. L.; Chen, X. F.; Wang, Z. H.; Yong, K. S.; Cao, Y.; Xu, G. Q. J. Chem. Phys. 2003, 119, 10389.
27. Tao, F.; Yuan, Z. L.; Chen, X. F.; Qiao, M. H.; Wang, Z. H.; Dai, Y. J.; Huang, H. G.; Cao, Y.; Xu, G. Q. Phys. Rev. B 2003, 67, 245406.
28. Lu, X.; Lin, M. C. Int. Rev. Phys. Chem. 2002, 21, 137.
29. Lu, X.; Xu, X.; Wu, J.; Wang, N.; Zhang, Q. New J. Chem. 2002, 26, 160.
30. Tao, F.; Qiao, M. H.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2003, 107, 6384.
31. Miwa, J. A.; Eves, B. J.; Rosei, F.; Lopinski, G. P. J. Phys. Chem. B 2005, 109, 20055.
32. Cho, Y. E.; Maeng, J. Y.; Kim, S.; Hong, S. J. Am. Chem. Soc. 2003, 125, 7514.
33. Tao, F.; Lai, Y. H.; Xu, G. Q. Langmuir 2004, 20, 366.
34. Hong, S.; Cho, Y. E.; Maeng, J. Y.; Kim, S. J. Phys. Chem. B 2004, 108, 15229.
35. Kim, H.-J.; Cho, J.-H. J. Chem. Phys. 2004, 120, 8222.
36. Lee, J. Y.; Jung, S. J.; Hong, S.; Kim, S. J. Phys. Chem. B 2005, 109, 348.
37. Huang, H. G.; Huang, J. Y.; Ning, Y. S.; Xu, G. Q. J. Chem. Phys. 2004, 121, 4820.
38. Shimomura, M.; Ichikawa, D.; Fukuda, Y.; Abukawa, T.; Aoyama, T.; Kono, S. Phys. Rev. B 2005, 72, 033303.
39. Huang, H. G.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2004, 108, 12560.
40. Kanaya, H.; Usuda, K.; Yamada, K. Appl. Phys. Lett. 1995, 67, 682.
41. Horie, T.; Takakuwa, Y.; Miyamoto, N. Jpn. J. Appl. Phys. 1994, 33, 4684.
42. Kato, T.; Kang, S.-Y.; Xu, X.; Yamabe, T. J. Phys. Chem. B 2001, 105, 10340.
43. Zhang, L.; Carman, A. J.; Casey, S. M. J. Phys. Chem. B 2003, 107, 8424.
44. Bae, S.-S.; Kim, D. H.; Kim, A.; Jung, S. J.; Hong, S.; Kim, S. J. Phys. Chem. C 2007, 111, 15013.
45. Carbone, M.; Meloni, S.; Caminiti, R. Phys. Rev. B 2007, 76, 085332.
46. Armstrong, J. L.; Pylant, E. D.; White, J. M. J. Vac. Sci. Technol. A 1998, 16, 123.
47. Armstrong, J. L.; White, J. M.; Langell, M. J. Vac. Sci. Technol. A 1997, 15, 1146.
48. Barriocanal, J. A.; Doren, D. J. J. Am. Chem. Soc. 2001, 123, 7340.
49. Tanaka, S.; Onchi, M.; Nishijima, M. J. Chem. Phys. 1989, 91, 2712.
50. Lopez, A.; Heller, T.; Bitzer, T.; Richardson, N. V. Chem. Phys. 2002, 277,1.
51. Lopez, A.; Bitzer, T.; Heller, T.; Richardson, N. V. Surf. Sci. 2001, 480, 65.
52. Hwang, H.-N.; Baik, J. Y.; An, K.-S.; Lee, S. S.; Kim, Y.; Hwang, C. C.; Kim, B. J. Phys. Chem. B 2004, 108, 8379.
53. Filler, M. A.; Deventer, J. A. V.; Keung, A. J.; Bent, S. F. J. Am. Chem. Soc. 2006, 128, 770.
54. Teragni, P.; Masetti, G.; Zerbi, G. Chem. Phys. 1978, 28, 55.
55. Haurie, M.; Novak, A. Spectrochim. Acta 1965, 21, 1217.
56. Srivastava, T. N.; Onyszchuk, M. Can. J. Chem. 1963, 41, 1244.
57. Lu, X.; Xu, X.; Wang, N.; Zhang, Q.; Lin, M. C. J. Phys. Chem. B 2001, 105, 10069.
58. Jeon, S. M.; Jung, S. J.; Lim, D. K.; Kim, H.-D.; Lee, H.; Kim, S. J. Am. Chem. Soc. 2006, 128, 6296.
59. Konecny, R.; Doren, D. J. J. Phys. Chem. B 1997, 101, 10983.
60. Machida, S.; Nagao, M.; Yamamoto, S.; Kakefuda, Y.; Mukai, K.; Yamashita, Y.; Yoshinobu, J. Surf. Sci. 2003, 532–535, 716.
61. Cao, X.; Hamers, R. J. J. Phys. Chem. B 2002, 106, 1840.
62. Jung, S. J.; Youn, Y.-S.; Lee, H.; Kim, K.-J.; Kim, B. S; Kim, S. J. Am. Chem. Soc. 2008, 130, 3288.
63. Kim, Y. B; Jung, S. J.; Min, Y. H.; Kim, S. J. Am. Chem. Soc. 2010, 132, 12782.
64. Koch, H.; J∅rgensen, P.; Helgaker, T. J. Chem. Phys. 1996, 104, 9528.
65. Haaland, A. Acc. Chem. Res. 1979, 12, 415.
66. Cao, X.; Hamers, R. J. J. Vac. Sci. Technol. B 2002, 20, 1614.
67. Carman, A. J.; Zhang, L.; Liswood, J. L.; Casey, S. M. J. Phys. Chem. B 2003, 107, 5491.
68. Bitzer, T.; Alkunshalie, T.; Richardson, N. V. Surf. Sci. 1996, 368, 202.
69. Bitzer, T.; Richardson, N. V. Appl. Phys. Lett. 1997, 71, 662.
70. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 2001, 105, 3295.
71. Liu, H.; Hamers, R. J. Surf. Sci. 1998, 416, 354.
72. Romero, A. H.; Sbraccia, C.; Silvestrelli, P. L. J. Chem. Phys. 2004, 120, 9745.
73. Cantele, G.; Trani, F.; Ninno, D.; Cossi, M.; Barone, V. J. Phys.: Condens. Matter 2006, 18, 2349.
74. Cao, X.; Hamers, R. J. Surf. Sci. 2003, 523, 241.
75. Tomimoto, H.; Sumii, R.; Shirota, N.; Yagi, S.; Taniguchi, M.; Sekitani, T.; Tanaka, K. J. Vac. Sci. Technol. B 2000, 18, 2335.
76. Kim, D. H.; Choi, D. S.; Kim, A.; Bae, S.-S.; Hong, S.; Kim, S. J. Phys. Chem. B 2006, 110, 7938.
77. Silvestrelli, P. L. Surf. Sci. 2004, 552, 17.
78. Zhou, J.-G.; Hagelberg, F. Int. J. Quantum Chem. 2005, 105, 359.
79. Kim, J.; Kim, K.; Yong, K. J. Vac. Sci. Technol. A 2002, 20, 1582.
80. Kim, J. W.; Carbone, M.; Tallarida, M.; Dil, J. H.; Horn, K.; Casaletto, M. P.; Flammini, R.; Piancastelli, M. N. Surf Sci. 2004, 559, 179.
81. Casaletto, M. P.; Carbone, M.; Piancastelli, M. N.; Horn, K.; Weiss, K.; Zanoni, R. Surf. Sci. 2005, 582, 42.
Functionalization of Semiconductor Surfaces, First Edition
Edited by Franklin (Feng) Tao and Steven L. Bernasek.
© 2012 John Wiley & Sons, Inc. Published 2012 by John Wiley & Sons, Inc.