
CHAPTER 4
Pericyclic Reactions of Organic Molecules at Semiconductor Surfaces1
KEITH T. WONG AND STACEY F. BENT
4.1 INTRODUCTION
Pericyclic reactions comprise an important class of the chemistry of unsaturated organic molecules. Woodward and Hoffmann first proposed the concept of pericyclic reactions in 1965 [1]. These are reactions that are thought to occur via a concerted process involving a cyclic transition state, without the formation of intermediates. As a result of their concerted nature, pericyclic reactions are known to provide high stereoselectivity [2]. This chapter focuses on a particular type of pericyclic reaction, cycloaddition reactions, in which π electrons from two or more unsaturated reactant molecules form new σ bonds between reactants to create a cyclic product. Due to the aforementioned stereoselectivity and their ability to form new carbon–carbon bonds and new carbon rings, cycloadditions play a central role in organic synthesis [3,4]. The two types of cycloaddition reactions considered in this chapter are the [2+2] cycloaddition and [4+2] cycloaddition, where the reaction is named for the number of π electrons of each reactant molecule. [4+2] cycloaddition is also known as the Diels–Alder reaction.
The Woodward–Hoffmann selection rules describe the reactivity of such cycloadditions. Symmetry analysis of the parity of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), as depicted in Fig. 4.1, indicates that the [2+2] reaction is symmetry-forbidden due to a mismatch in parity whereas the Diels–Alder reaction is symmetry-allowed [5]. In fact, [2+2] cycloadditions in organic chemistry typically require photochemical activation to excite one of the reactants to a state in which the parity of the HOMO and LUMO orbitals that form the new bonds is matched, and the reaction is symmetry-allowed.
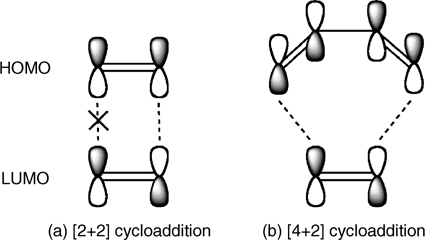
FIGURE 4.1 Schematic depicting the HOMOs and LUMOs of ethylene reacted by (a) [2+2] cycloaddition with another ethylene molecule and (b) [4+2] cycloaddition with 1,3-butadiene. The mismatched parity of the HOMO and LUMO of ethylene leads to [2+2] cycloaddition being symmetry-forbidden according to the Woodward–Hoffmann selection rules.
Diels–Alder reactions, on the other hand, occur readily at room temperature and are commonly used in organic syntheses [2–4].
Since the dimers of the 2×1 reconstructed Group IV semiconductor surfaces (C(100)-(2×1) (diamond), Si(100)-(2×1), and Ge(100)-(2×1)) can be described as having partial π bonds and, thus, alkene character, cycloaddition products may be expected to form on these surfaces. Indeed, numerous unsaturated organic molecules have been found to generate [4+2] or [2+2] cycloaddition products on these surfaces; such reactions are the subject of this chapter. Evidence exists indicating that cycloadditions on semiconductor surfaces do not necessarily proceed via a concerted process with a cyclic transition state; as will be discussed in this chapter, stepwise mechanisms involving short-lived intermediates have been proposed instead [6–9]. Thus, these reactions may not be truly pericyclic, and the term cycloaddition, as it is used in this chapter, is not meant to imply a truly pericyclic mechanism but is convenient nomenclature that highlights the analogies that can be made to organic chemistry. Other pericyclic-like reactions such as 1,3-dipolar cycloaddition and group transfer reactions can occur at semiconductor surfaces; however, these reactions are somewhat less common and less studied than [2+2] and [4+2] cycloadditions at semiconductor surfaces and, thus, will not be addressed in detail here. Additional discussion of pericyclic reactions at semiconductor surfaces can be found in the literature [10–20].
Although it has been the subject of fewer studies than the Si(100)-(2×1) surface, cycloaddition-like reactions are also known to occur on the Si(111)-(7×7) surface, and several examples will be discussed in this chapter. The structure of the Si(111)-(7×7) surface can be described by the widely accepted dimer–adatom–stacking-fault structure [21]. This structure does not contain partial π bonds like the 2×1 reconstructed (100) surfaces; therefore, direct comparison to an alkene cannot be made. However, each unit cell contains seven distinct types of surface atoms. Charge transfer among the dangling bonds at these surface atoms has been theoretically predicted and experimentally observed [22–26]. The resulting uneven distribution of electrons at the surface is believed to enable cycloaddition-like reactions on the Si(111)-(7×7) surface similar to cycloaddition reactions at the buckled dimers of the Si (100)-(2×1) and Ge(100)-(2×1) surfaces.
This chapter first describes the [2+2] cycloadditions of alkenes and alkynes on Si (100)-(2×1)—historically, the first cycloadditions on a semiconductor surface studied. This topic is followed by a discussion of [4+2] reactions of dienes, which adds the possibility of multiple cycloaddition products ([4+2] and [2+2] products). Finally, more recent work on cycloadditions of unsaturated molecules containing N, O, or S heteroatoms is reviewed. In each section, selected adsorbates that highlight particular features of cycloaddition at semiconductor surfaces are discussed. Where possible and informative, comparisons are made between the reactivity on Si(100)-(2×1) and that on Ge(100)-(2×1), C(100)-(2×1), or Si(111)-(7×7).
4.2 [2+2] CYCLOADDITION OF ALKENES AND ALKYNES
Although the [2+2] cycloaddition reaction is symmetry-forbidden according to classic theory (Woodward–Hoffmann selection rules), many molecules have been found to readily form [2+2] cycloaddition products, originally labeled as di-σ or bridge-bonded products, on semiconductor surfaces [11–14,16,18]. This observation highlights some of the important differences between cycloaddition reactions in solution and at semiconductor surfaces.
4.2.1 Ethylene
Ethylene is the simplest organic molecule that can undergo a [2+2] cycloaddition reaction; it serves as a model for understanding many [2+2] cycloadditions at semiconductor surfaces. Studies in the late 1980s of adsorption of ethylene on Si (100)-(2×1) were among the earliest examples of cycloadditions at a semiconductor surface, although the product was initially labeled as a di-σ product [27,28]. These and later studies of ethylene adsorption on Si(100)-(2×1) using temperature programmed desorption (TPD), scanning tunneling microscopy (STM), scanning tunneling spectroscopy (STS), high-resolution electron energy loss spectroscopy (HREELS), low-energy electron diffraction (LEED), near edge X-ray absorption fine structure (NEXAFS) spectroscopy, ultraviolet photoelectron spectroscopy (UPS), and density functional theory (DFT) calculations indicated that two new Si–C σ bonds are formed, the alkene π bond is broken and the silicon dimer bond remains intact [27–43]. The results show that the product of ethylene reacted at Si(100)-(2×1) can also be labeled as a [2+2] cycloaddition product.
The sticking probabilities of ethylene and other simple alkenes on Si(100)-(2×1) are near unity, indicating facile reaction with the surface [44]. This result is interesting since, as noted previously, the [2+2] cycloaddition reaction is classically symmetry-forbidden and typically occurs slowly in solution [3,5]. To reconcile the different reactivity between the heterogeneous reaction at Si(100)-(2×1) and the homogeneous reaction in solution, low-symmetry stepwise mechanisms with little or no activation barrier(s) were proposed for the reaction on Si(100)-(2×1) of both ethylene and acetylene, which may also undergo [2+2] cycloaddition, as discussed later [7,8]. These mechanisms entail the formation of three-centered π-complex (Fig. 4.2d) and/or diradical (Fig. 4.2c) states as precursors or intermediates. Using a CASSCF(6,6) wave function to model [2+2] cycloaddition of 1,3-cyclohexadiene on Si(100)-(2×1), Choi and Gordon have suggested that the π-complex intermediate is an artifact of using a single-configurational wave function [6], although additional calculations by the same group show a relatively flat potential energy surface associated with dimer buckling [45], which may facilitate initial attack of adsorbates. In agreement with the originally proposed mechanisms, more recent theoretical studies of ethylene adsorption on Si(100)-(2×1) have also found pathways involving a π-complex precursor and diradical intermediate [46–48], and Nagao et al. [48] reported the observation of a π-complex precursor by HREELS at low temperature. Low-symmetry pathways are believed to be possible on the Si(100)-(2×1) surface due to the dynamic tilting of dimers at room temperature, the relative weakness of the dimer π bond and the small (compared to alkenes) separation of the π and π* frontier orbitals. Liu et al. [7] used cis- and trans-dideuteroethylene and the splitting of vs(C–H) and vas(C–H) modes to determine stereoselectivity upon adsorption on Si(100)-(2×1). They find that the symmetry of the original reactant is retained indicating that the symmetry-allowed suprafacial–antarafacial reaction mechanism (Fig. 4.2b) does not occur, whereas nonconcerted (i.e., nonpericyclic), low-symmetry mechanisms involving diradical or π-complex intermediates are possible. The mechanism of [2+2] cycloaddition on Si(100)-(2×1) remains a topic of interest; yet more theoretical studies have recently suggested that a concerted reaction mechanism (similar to that in Fig. 4.2a) may compete with low-symmetry mechanisms [49,50]. The viability of a concerted mechanism has been explained in terms of the crossing of energy bands associated with the surface frontier orbitals through the Fermi level [49] or the presence of crystal wave functions with the appropriate symmetry for bonding interaction with an alkene [50], thus creating a symmetry-allowed pathway.
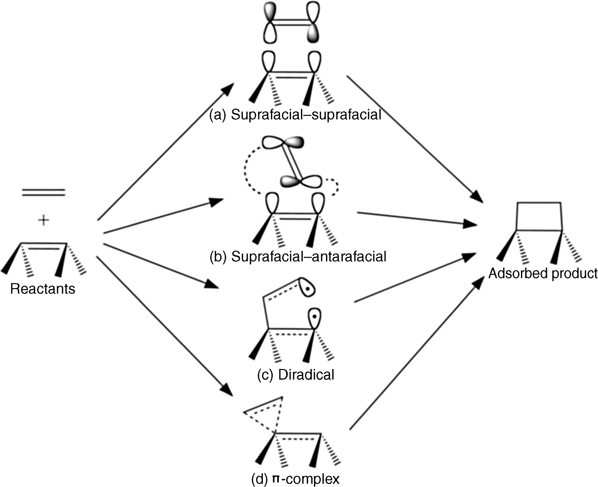
FIGURE 4.2 Schematic representation of commonly discussed reaction pathways for the [2+2] cycloaddition of ethylene on a Group IV (100)-(2×1) surface dimer: (a) concerted suprafacial–suprafacial reaction, which is symmetry-forbidden according to the Woodward–Hoffmann selection rules; (b) concerted suprafacial–antarafacial reaction, which may be sterically hindered; (c) reaction by a diradical intermediate or precursor; and (d) reaction by a π-complex intermediate or precursor. Note that analogous reaction mechanisms are discussed for [2+2] cycloaddition of other unsaturated organic molecules.
Another interesting feature of the [2+2] cycloaddition product of ethylene on Si (100)-(2×1) is its thermodynamic stability. The binding energy is found to be approximately 40 kcal/mol by both experiment and theory [30,33,35,40]. This is somewhat surprising given the significant ring strain, as evidenced by the Si–Si–C bond angle of 78.2°—far from the tetrahedral angle—calculated by Konecny and Doren [30]. Thus, the thermodynamic stability of the [2+2] product must arise primarily from the strength of the two Si–C bonds formed. Indeed, a bond energy of 81.7 kcal/mol has been calculated for the Si–C bond of methylsilane [51]. The strength of the bonds formed with the surface and the resulting overall thermodynamic stability of the ethylene [2+2] product have attracted much interest in the use of [2+2] cycloaddition for functionalizing the Si(100)-(2×1) surface.
Unlike on Si(100)-(2×1) where ethylene is believed to adsorb in a single configuration (on top of a single Si dimer) [52,53], ethylene is found to form at least two molecular adsorption states on Ge(100)-(2×1) [54–56]. The first experimental studies of ethylene adsorption on Ge(100)-(2×1) performed by Lal et al. [56] using multiple internal reflection Fourier transform infrared spectroscopy (MIR-FTIR) and TPD were consistent with the formation of a [2+2] cycloaddition product. However, time- and coverage-dependent behavior and the presence of two molecular desorption peaks in TPD measurements indicate the existence of at least two molecular adsorption states [56]. TPD and angle-resolved UPS experiments by Fink et al. [54] attributed the minor peak in TPD measurements to adsorption at step sites. STM images, such as that shown in Fig. 4.3, later showed the presence of two distinct bonding geometries at room temperature, which are attributed to a [2+2] product across a single dimer (on-top configuration; shown in Fig. 4.4a) and two ethylene molecules bridging between two dimers in the same row (pair end-bridge configuration; shown in Fig. 4.4c) [55]. Calculations for acetylene on Ge (100)-(2×1) show that the pair end-bridge configuration is most stable [57,58], suggesting that the same may be the case for ethylene; however, according to theoretical calculations of ethylene on Ge(100)-(2×1), the on-top [2+2] product is most stable [59]. Though there is debate over the exact adsorption states of ethylene on Ge(100)-(2×1), this system clearly demonstrates several important concepts: (1) similar but not necessarily identical reaction products are observed for cycloadditions at the Group IV semiconductor surfaces; (2) interdimer reactions can be important even for small molecules such as ethylene; and (3) multiple reaction products can form even for the simplest cycloaddition systems.

FIGURE 4.3 Filled state STM image of a Ge(100)-(2×1) surface exposed to a low dose of ethylene. Two distinct bonding configurations can be distinguished and are labeled A and B. The authors attributed feature A to on-top adsorption and feature B to pair end-bridge adsorption. A 10×10 nm2 area is imaged at −1.8V sample bias and 100 pA tunneling current. Reprinted with permission from Ref. 55. Copyright 2004 American Chemical Society.
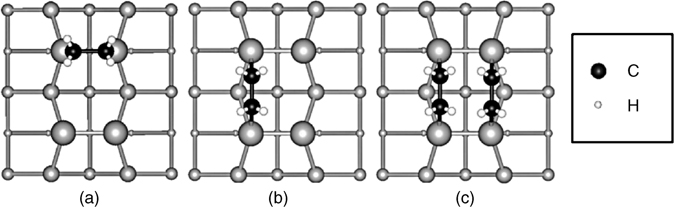
FIGURE 4.4 Top view of adsorption configurations of ethylene on a Group IV (100)-(2×1) surface: (a) on-top; (b) end-bridge; and (c) pair end-bridge. Surface dimer atoms are represented by the largest gray circles, and subsurface atoms are represented by increasingly small gray circles. Adapted with permission from Ref. 55. Copyright 2004 American Chemical Society.
Finally, ethylene is found to desorb molecularly from the Ge(100)-(2×1) surface, whereas decomposition occurs on Si(100)-(2×1) [14,18,54–56]. Similarly, reversible adsorption on Ge(100)-(2×1) versus decomposition on Si(100)-(2×1) has been observed for other systems including the [2+2] product of acetylene and the [4+2] product of 1,3-butadiene [18,51]. This difference in stability can be attributed to the difference in the strength of bonds formed with the surface. The bond energy of the Ge–C bond in methylgermane was calculated to be 72.7 kcal/mol, which is 9 kcal/mol less than the Si–C bond energy in methylsilane (81.7 kcal/mol) [51]. Thus, the binding energy of adsorbates on Ge is expected to be significantly less than on Si, allowing for molecular desorption to occur upon heating before decomposition. This result highlights the important influence the surface can have on the stability of adsorbates. On the basis of this concept, it has been suggested that reactions at the Si surface are more often under kinetic control, whereas reactions at the Ge surface are more often under thermodynamic control since less favorable adsorption products may have such low binding energies on Ge that they readily desorb [16].
4.2.2 Acetylene
The behavior of acetylene, C2H2, at Si(100)-(2×1) and Ge(100)-(2×1) mimics that of ethylene to some degree, although its triple bond adds the possibility of additional tetra-σ bonded products in which an acetylene molecule undergoes two [2+2]-like cycloadditions to form four σ bonds with the surface (two per carbon atom; shown in Fig. 4.5c). Like ethylene adsorption on Ge(100)-(2×1), the bonding configuration of acetylene on Si(100)-(2×1) has been debated at length in the literature with tetra-σ, end-bridge [2+2] and on-top [2+2] products (Fig. 4.5) all suggested [8,30,32,35,37,38,43,60–85]. It is generally agreed that multiple of these products form in appreciable quantity at room temperature (perhaps with different coverage dependences), thus reinforcing the assertions made earlier that cycloadditions across multiple dimers can be important and even simple molecules may form multiple products at the surface.
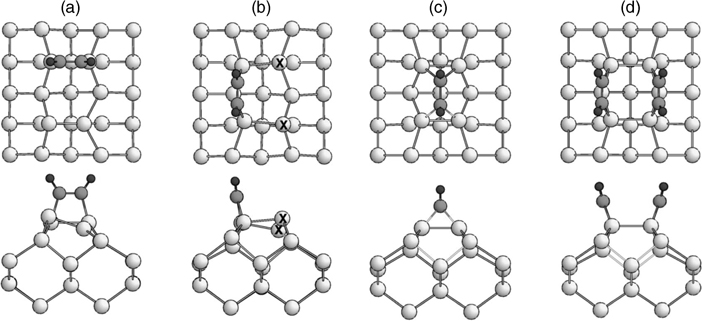
FIGURE 4.5 Top and side views of optimized structures of acetylene on Si(100)-(2×1): (a) on-top; (b) end-bridge; (c) tetra- σ; and (d) pair end-bridge. Light gray, medium gray, and dark gray circles correspond to silicon, carbon, and hydrogen, respectively. Note that tetra- σ configurations with the acetylene molecule perpendicular (as shown) and parallel (not shown) to the surface dimer have both been proposed. Figure adapted from Ref. 84. Copyright 2004 by The American Physical Society.
Comparatively few studies have been published regarding acetylene adsorption on Ge(100)-(2×1). Theoretical calculations initially suggested that the on-top [2+2] product should be observed [58,86]. The presence of both the on-top di-σ and tetra-σ products was suggested by STM studies [87]; however, these results were later reinterpreted following additional DFT calculations to show that acetylene adsorbs on Ge(100)-(2×1) in on-top di-σ and pair end-bridge configurations, similar to ethylene [57]. Also like ethylene, acetylene binds to Ge(100)-(2×1) more weakly than to Si(100)-(2×1) according to calculations [88]. This is confirmed experimentally, as mentioned previously, by the dominance of dissociative desorption of acetylene from Si(100)-(2×1) [61] versus molecular desorption from Ge(100)-(2×1) [87], which again highlights the difference in thermodynamic stability typically observed for cycloaddition products on the Si versus Ge surface.
4.2.3 Cis- and Trans-2-Butene
Issues of stereoselectivity or stereospecificity are further explored by the reactions of cis- and trans-2-butene at Si(100)-(2×1). The structures of cis-and trans-2-butene are equivalent to the aforementioned molecules cis- and trans-dideuteroethylene, respectively, with the substitution of methyl groups in the place of deuterium. Thus, the 2-butene stereoisomers can similarly be used to shed light on the mechanism of [2+2] cycloaddition at Si(100)-(2×1) with the additional advantage that different adsorption configurations can be probed by STM. Indeed, Lopinski et al. [9,89] showed that two binding configurations—one observed for cis-2-butene and the other for trans-2-butene—can be differentiated using STM. However, they found that 2–3% of both stereoisomers isomerized upon adsorption to the clean surface (not at defect sites) [9]. An example of a trans-2-butene molecule isomerized to cis-2-butene upon adsorption is circled in Fig. 4.6. In Fig. 4.6, the dimer rows are imaged as gray features running diagonally across the image, and the paired light-colored protrusions correspond to the methyl groups on both ends of adsorbed 2-butene molecules. Most of the adsorbed 2-butene molecules are angled slightly with respect to the underlying dimers, consistent with adsorption of trans-2-butene without isomerization, but the circled feature is parallel to the dimer direction, indicating isomerization to cis-2-butene. Since earlier work had indicated that the trans adsorbate is 4 kcal/mol more stable than the cis adsorbate [90], equal isomerization of both isomers indicates the process is under kinetic rather than thermodynamic control. Their results add to the previous studies of dideuteroethylene [7] by showing that adsorption by [2+2] cycloaddition at Si(100)-(2×1) is highly stereoselective but not truly stereospecific. They conclude that the mechanism must proceed in a stepwise fashion on a timescale of picoseconds in order to allow for isomerization by rotation about the C–C bond [9].
Interestingly, Madachik and Teplyakov [91] report that 2,3-dimethyl-2-butene does not react with the Si(100)-(2×1) surface at room temperature. No signs of chemisorption are present in the IR spectra after large doses at room temperature or heating multilayers adsorbed at 100K to room temperature. In agreement with the experimental results, the authors' DFT calculations indicate that formation of the expected [2+2] cycloaddition product is kinetically unfavorable compared to desorption from the two π-complex precursor states considered. Steric hindrance due to the additional methyl groups (as compared to 2-butene) is suggested as the reason for the large barrier to [2+2] cycloaddition. Due to its lack of reactivity with the Si(100)-(2×1) surface, it is suggested that 2,3-dimethyl-2-butene could be used as an inert carrier gas or precursor ligand.

FIGURE 4.6 Empty state STM image of a Si(100)-(2×1) surface exposed to trans-2-butene. Most trans-2-butene molecules lead to paired light-colored protrusions angled with respect to the dimers, but the circled feature shows a pair of protrusions that is aligned with the underlying dimers, which corresponds to a trans-2-butene molecule that isomerized to cis-2-butene upon adsorption. A 75×75 Å area is imaged at a sample bias of 2 V and tunneling current of 40 pA. Reprinted with permission from Ref. 9. Copyright 2000 American Chemical Society.
4.2.4 Cyclopentene
Cyclopentene has served as a model system for exploring ordering of [2+2] cycloaddition products at semiconductor surfaces. Due to the nature of the [2+2] cycloaddition product, which contains two directional bonds to the surface per molecule, Hamers et al. [11] suggest that [2+2] cycloaddition provides a means to control both translational and orientational ordering of adsorbates at the interface. Indeed, the STM images in Fig. 4.7 demonstrate such ordering of cyclopentene adsorbed on Si(100)-(2×1). These images and others obtained by the same authors show the formation of a highly ordered monolayer with each cyclopentene molecule aligned over a Si dimer [92–95]. FTIR spectroscopy and X-ray photoelectron spectroscopy (XPS) confirm that cyclopentene adsorbs to form a [2+2] cycloaddition product [92,93,95–97], and anisotropy in the C–H stretching region of the FTIR spectra suggests that the ordering observed by STM is present over macroscopic (centimeter) length scales [92,93,95]. The formation of ordered monolayers has also been demonstrated by STM for 1,5-cyclooctadiene [98] and 1,3,5,7-cyclooctatetraene [99], which form single- and dual-[2+2] cycloaddition products on Si(100)-(2×1), respectively. Later analysis by photoemission spectroscopy (PES) and NEXAFS indicated the presence of some dual-[4+2] products in the reaction of 1,3,5,7-cyclooctatetraene with Si(100)-(2×1) [100]. Nonetheless, both 1,3-cyclooctadiene and 1,3,5,7-cyclooctatetraene provide examples of generating a functionalized semiconductor surface with ordered arrays of unreacted alkenes that may be apt for further functionalization [101].

FIGURE 4.7 STM images of (a) a clean vicinal Si(100)-(2×1) surface (4° miscut toward the ![]() direction) showing terraces on the surface with 8–12 dimers in each row and (b) the same surface after exposure to cyclopentene. Figure adapted with permission from Ref. 95. Copyright 1998 Elsevier.
direction) showing terraces on the surface with 8–12 dimers in each row and (b) the same surface after exposure to cyclopentene. Figure adapted with permission from Ref. 95. Copyright 1998 Elsevier.
Cyclopentene adsorption has also been studied on Ge(100)-(2×1) and C(100)-(2×1), thus providing for useful comparison among the Group IV semiconductor surfaces. The lack of high-frequency alkene C–H stretching modes in the FTIR spectra indicates the formation of a [2+2] cycloaddition product on all three surfaces [92,102,103]. Hovis et al. [102] found the interesting result that the sticking coefficients are approximately 1,0.1 and 10−3 on Si(100)-(2×1), Ge(100)-(2×1), and C(100)-(2×1), respectively. They proposed several explanations for this trend. First, the sticking coefficients correlate negatively with the π–π* splitting of the surfaces. Large π–π* splitting is expected to slow the rate of the symmetry-forbidden suprafacial–suprafacial reaction mechanism. Second, the relative strength of the C dimer π bond compared to that of Si and Ge may slow the reaction rate on C(100)-(2×1). Finally, the geometry of the surface dimers may impact the reaction rate. Tilting out of plane of the Si and Ge dimers enables a low-symmetry pathway with little or no activation barrier [102,104] equivalent to π-complex mechanisms that have been suggested for other adsorbates [7,8,46–48]. The clean C(100)-(2×1) surface does not exhibit the same dimer tilting, and calculations show that the presence of an alkene does not induce tilting [102]. The lack of dimer tilt results in a much higher activation barrier and is suggested as the primary explanation for the much lower sticking coefficient on C(100)-(2×1) [11]. The Si (100)-(2×1) and Ge(100)-(2×1) surfaces are both comprised of similarly tilted dimers yet the sticking coefficients of cyclopentene on these surfaces differ by about an order of magnitude. Lee et al. [103] suggest that the slightly larger dimer bond length on the Ge surface (about 0.2 Å longer than on Si) may increase the energy of the transition state between the π-complex intermediate and the product, leading to lower sticking probability. Thus, cyclopentene provides an excellent example for exploring the effects of surface properties on reactivity of adsorbates by [2+2] cycloaddition.
4.2.5 [2+2]-Like Cycloaddition on Si(111)-(7×7)
Adsorption of ethylene [105] and acetylene [106] on Si(111)-(7×7) were first investigated beginning in the 1980s. As with early studies of these molecules on Si(100)-(2×1) around the same time, the primary adsorption products were initially termed di-σ products. Later studies made the analogy to [2+2] cycloaddition and such products are now sometimes referred to as [2+2]-like cycloaddition products. [2+2]-like cycloaddition products have also been suggested for adsorption on Si(111)-(7×7) of other nonconjugated unsaturated hydrocarbons such as cyclopentene, cyclohexene, and 1,4-cyclohexadiene [107,108], but most research has focused on ethylene and acetylene.
Early studies by Yoshinobu et al. [105,106] used HREELS to show that both ethylene and acetylene primarily adsorb without dissociating and that they rehybridize to near sp3 and between sp2 and sp3 hybridizations, respectively. They suggested that both molecules adsorb to form primarily di-σ bonded products (i.e., [2+2]-like products) on the basis of these results. A study of ethylene adsorption on Si(111)-(7×7) by STM and STS suggested that ethylene molecules bridge adjacent adatom–rest atom pairs [109]. Furthermore, it was shown that adsorption of an ethylene molecule changes the local density of states at neighboring surface atoms in addition to the surface atoms to which it bonds, but no changes to the surface reconstruction were observed. Later study by XPS agreed with the assignment of the primary product to a di-σ bonded product and was able to differentiate a minor product attributed to dissociative adsorption [110]. By comparison to ethylene adsorption on Si(100)-(2×1), the authors estimate that the maximum coverage of ethylene on Si(111)-(7×7) is several times higher than would be possible with binding only at adatom–rest atom bridge sites. Based on this result and the LEED pattern, which is reported to change from 7×7 to 7×1 with increasing ethylene exposure, it is suggested that a rearrangement of the surface atoms does occur. A more recent DFT study found a barrierless diradical reaction pathway with no intermediate or transition states for the adsorption of ethylene on Si(111)-(7×7) [111]. Reaction by such a pathway should occur readily, and the authors suggest that significant reconstruction of the surface should not be induced by this reaction. The binding energy of ethylene on Si(111)-(7×7) is calculated to be 47.9 kcal/mol—slightly larger than values typically obtained for ethylene adsorption on Si (100)-(2×1).
Acetylene adsorption on Si(111)-(7×7) has been the focus of more studies than that of ethylene. Formation of a di-σ bonded product, or [2+2]-like cycloaddition product, upon acetylene adsorption on Si(111)-(7×7) was supported by early theoretical studies, but it was suggested that restructuring of surface atoms accompanies adsorption in order for the spacing between dangling bonds of surface Si atoms to be close enough to interact with acetylene [112,113]. STM results again agreed with the di-σ product, but no restructuring of the surface was observed [114]. MacPherson et al. [115] attempted to resolve the discrepancy between theory and experiment by suggesting that easily resolved features of the 7×7 unit cell (e.g., corner holes and dimer walls) remain even though restructuring within the unit cell that cannot be resolved by STM occurs. Later studies using XPS, X-ray absorption spectroscopy (XAS), UPS, and HREELS were consistent with the di-σ model bridging an adatom–rest atom pair [116,117]. Changes in the angular dependence of the HREELS spectrum with increasing exposure suggested that a different adsorption geometry may dominate at higher coverage, possibly due to restructuring of the surface [117]. Calculations indicated that an upright adsorption geometry (in which acetylene is bound to the surface through only one carbon atom) may also explain the HREELS angular dependence [118]. A pathway similar to some suggested for [2+2] cycloaddition on Si(100)-(2×1) involving two diradical intermediate states and three transitions states was calculated for adsorption of acetylene to form a di-σ bonded product on Si(111)-(7×7) [111].
4.3 [4+2] CYCLOADDITION OF DIENES
The [4+2] cycloaddition, or Diels–Alder, reaction between a conjugated diene and alkene (the alkene is referred to in this sense as the dienophile) is well known to occur readily in solution. In 1997, Konecny and Doren predicted on the basis of DFT calculations that the dimers of the Si(100)-(2×1) surface could act as a dienophile and react with a diene with minimal activation barrier [119]. Their calculations for adsorption of 1,3-cyclohexadiene on an Si9H12 cluster representing a single surface dimer result in a large 54.0 kcal/mol binding energy for the [4+2] product and no significant activation barrier. Moreover, reaction of one double bond of 1,3-cyclohexadiene to yield a [2+2] product is calculated to be 15.2 kcal/mol less exothermic. The difference in stability is attributed primarily to the difference in ring strain; Si–Si–C bond angles of approximately 78° and 97° are predicted for the [2+2] and [4+2] products, respectively, yielding higher ring strain for the [2+2] product. [4+2] cycloadditions on Ge(100)-(2×1) and C(100)-(2×1) were subsequently investigated theoretically and shown to be similarly favorable [51,120]. Besides the greater product stability and minimal activation barrier predicted for [4+2] cycloaddition, this reaction inherently leaves an alkene moiety on the surface. It was suggested that this could be the starting point for further functionalization [119]; however, the alkene moieties at the surface have proven to have low reactivity, and work to date on layer-by-layer growth of organic films has focused instead on other chemistries [121–142]. Since conjugated dienes are capable of forming both [2+2] and [4+2] cycloaddition products, they present a more complicated chemistry than simple alkenes and alkynes because competition and selectivity among multiple reaction channels is often important.
4.3.1 1,3-Butadiene and 2,3-Dimethyl-1,3-Butadiene
Following Konecny and Doren's theoretical predictions, the first experimental evidence of [4+2] reactions at the Si(100)-(2×1) surface was provided by Teplyakov et al. [143]. In this and a later [144] study by the same authors, adsorption of 1,3-butadiene and 2,3-dimethyl-1,3-butadiene were studied by MIR-FTIR, NEXAFS, and TPD. Lack of absorption in the Si–H stretching region of the IR spectra for both molecules and presence of features attributed to both single bonds and nonconjugated double bonds in the NEXAFS spectrum of 2,3-dimethyl-1,3-butadiene indicate that these molecules do not react by C–H dissociation at room temperature. Figure 4.8 shows the IR spectra of chemisorbed and physisorbed 1,3-butadiene and two of its deuterated derivatives. Vibrational modes consistent with a terminal =CH2 group near 3080 cm−1 are present in the low-temperature multilayer IR spectra (Fig. 4.8d and f), and are clearly identified as they shift out of the plotted range upon substitution of deuterium for the terminal hydrogens (Fig. 4.8e). No absorption near 3080 cm−1 is discernable upon adsorption at room temperature (Fig. 4.8a–c). Olefinic and aliphatic C–H stretches, however, are clearly observed at room temperature near 2990 cm−1 and 2900 cm−1 (Fig. 4.8a), respectively, and can again be identified by comparison to the deuterated derivatives (Fig. 4.8b and c). NEXAFS spectra of 2,3-dimethyl-1,3-butadiene are shown in Fig. 4.9. There is a reduction in the number of π* transitions from four in the physisorbed spectra (Fig. 4.9a and b) to only one in the room temperature spectrum (Fig. 4.9c). Additionally, the energy of the single π* transition at room temperature is consistent with that of monounsaturated compounds. Both IR and NEXAFS results are consistent with the formation of a [4+2] product at the surface since this product contains a C=C group within the ring rather than terminal =CH2 groups (as indicated by IR results), and the conjugation that leads to multiple π* transitions in the NEXAFS spectra at low temperature is no longer present in the [4+2] product.
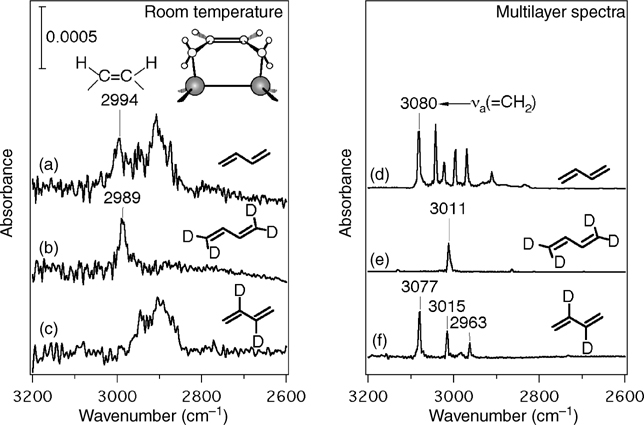
FIGURE 4.8 Infrared spectra showing the C–H stretching region of 1,3-butadiene adsorbed on Si(100)-(2×1): (a) 1 L (1 L=10−6 Torr · s) of 1,3-butadiene adsorbed at 300K; (b) 1L of 1,3-butadiene-1,1,4,4-d4 adsorbed at 300K; (c) 1000 L of 1,3,-butadiene-2,3-d2 adsorbed at 300K; (d) multilayers of 1,3-butadiene (1000 L) at 100K; (e) multilayers of 1,3-butadiene-1,1,4,4-d4 (1000 L) at 100K; and (f) multilayers of 1,3-butadiene-2,3-d2 (1000 L) at 100K. Adapted with permission from Ref. 144. Copyright 1998 American Institute of Physics.
To further characterize the adsorption of 2,3-dimethyl-1,3-butadiene on Si(100)-(2×1), Teplyakov et al. [144] used NEXAFS to determine the angle between the transition moment of the π* orbital and the surface normal. Their experimentally determined value of 41 ± 2° differs slightly from Konecny and Doren's theoretical prediction of 30°. The discrepancy may result from interactions with adjacent dimers not captured by the single-dimer theoretical model, surface defects present experimentally or the formation of side products with different adsorption geometries. Hovis et al. [145] investigated the latter possibility by analyzing STM images of Si(100)-(2×1) exposed to 2,3-dimethyl-1,3-butadiene. Their images clearly show the presence of two products: a primary product accounting for about 80% of adsorbates was assigned to the [4+2] product and a side product accounting for the remaining adsorbates was assigned to a [2+2] product involving reaction of only one of the C=C double bonds. However, it has also been suggested by Doren that a [4+2] product bridging neighboring dimers may be the source of the side product seen in STM images [12,16]. The assignment by Hovis et al. [145] of the side product to a [2+2] product was supported by the presence of a small C–H stretch in the IR spectrum at 3093 cm−1—in the region expected for terminal =CH2. Given the significantly greater thermodynamic stability (about 15 kcal/mol for cyclohexadiene [119]) of the [4+2] product over the [2+2] product, the results of Hovis et al. suggest that the reaction of 2,3-dimethyl-1,3-butadiene is under kinetic control and that, despite being classically symmetry-forbidden, the [2+2] reaction at the Si surface competes to some extent with the [4+2] reaction for this molecule. This result implies that obtaining high selectivity between [2+2] and [4+2] products, as is typical in solution organic chemistry, may be difficult to achieve at semiconductor surfaces.
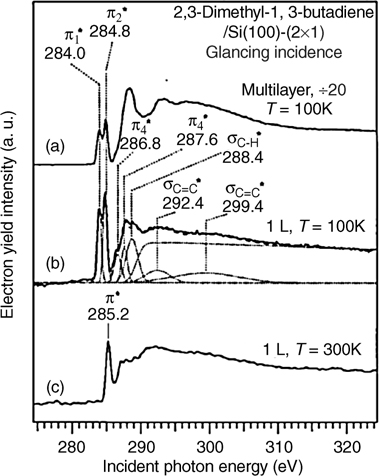
FIGURE 4.9 NEXAFS spectra of 2,3-dimethyl-1,3-butadiene adsorbed on Si(100)-(2×1): (a) multilayers of 2,3-dimethyl-1,3-butadiene physisorbed at 100K; (b) submonolayer (1 L exposure) of 2,3-dimethyl-1,3-butadiene physisorbed at 100K; and (c) submonolayer (1L exposure) of 2,3-dimethyl-1,3-butadiene chemisorbed at room temperature. Spectra were obtained at glancing incidence of the incoming photon flux. Reprinted with permission from Ref. 144. Copyright 1998 American Institute of Physics.
The adsorption of 1,3-butadiene has also been studied on the analogous Ge [51,146] and diamond [120,147] surfaces. The IR spectrum of 1,3-butadiene on Ge (100)-(2×1) closely resembles that on Si(100)-(2×1); hence, it was concluded that the [4+2] product is also the dominant species on Ge [146]. The spectral features of C(100)-(2×1) differ noticeably from those of the other two surfaces [147]. In particular, the peaks are shifted up in frequency from their positions on the other surfaces. This shift was hypothesized to result from different electron donation effects on the various surfaces or the smaller bond length of the dimers on the diamond surface leading to greater ring strain. Nonetheless, isotopic studies led to the conclusion that 1,3-butadiene predominantly forms the [4+2] product on the C(100)-(2×1) surface as well [147]. It has also been noted that ~1–2 orders of magnitude larger doses of 1,3-butadiene are required to saturate the C(100)-(2×1) surface than either the Si(100)-(2×1) or Ge(100)-(2×1) surfaces [12]. This result is in agreement with calculated activation barriers [120] and largely mimics the result noted previously for the sticking coefficient of cyclopentene on these surfaces. Of the proposed explanations for the trend in [2+2] reactivity toward cyclopentene—based on differing π–π* splitting, dimer π bond strength, and dimer tilt—only the dimer bond strength is expected to impact the reactivity of the surface toward a diene since the [4+2] cycloaddition reaction is symmetry-allowed [12].
Also of interest is the differing thermal chemistry exhibited by 1,3-butadiene on the Si(100)-(2×1) and Ge(100)-(2×1) surfaces. Upon heating, 1,3-butadiene on Si(100)-(2×1) primarily decomposes to form surface carbon and Si–H species [144]. On Ge(100)-(2×1), however, molecular desorption is the predominant pathway upon thermal annealing [146]. Again, this is similar to results presented earlier for ethylene and acetylene on these surfaces, and the difference has been attributed to the approximately 10 kcal/mol weaker Ge–C bond than Si–C bond. Molecular desorption of 1,3-butadiene from Ge(100)-(2×1) indicates the occurrence of the reverse of the Diels–Alder reaction, that is, the retro-Diels–Alder reaction. It has been suggested that this reversibility provides a possible means for spatially controlled modification by using photo- or electron-induced reaction to selectively react another precursor with the adsorbed dienes, followed by removal of unreacted dienes by retro-Diels–Alder reaction.
4.3.2 1,3-Cyclohexadiene
1,3-Cyclohexadiene was the subject of the first theoretical prediction of [4+2] cycloaddition on Si(100)-(2×1) [119] as well as several later experimental and theoretical investigations [6,30,145,148]. STM images obtained by Hovis et al. [145] show the presence of three surface products accounting for 55%, 35%, and 10% of adsorbates. Likewise, at least four peaks are discernable by infrared spectroscopy in the alkene C–H stretching region. The three products observed by STM were attributed to [4+2] cycloaddition (55%), [2+2] cycloaddition (35%) and an unidentified product (10%) based on the shape of the observed protrusions and their alignment with the underlying dimers. IR and NEXAFS spectroscopy data consistent with the presence of a mix of [4+2] and [2+2] products was later provided by Kong et al. [148]. Given the theoretical predictions of significantly greater thermodynamic stability (~15 kcal/mol) of the [4+2] product [6,30], which would suggest several orders of magnitude higher abundance of the [4+2] product if the reaction were under thermodynamic control, the observed product distribution implies that the reaction of 1,3-cyclohexadiene is under kinetic control on the Si(100)-(2×1) surface. The same phenomenon has been noted for several other previously discussed adsorbates. Later STM images obtained by Teague and Boland [149,150] distinguish multiple bonding configurations for both [2+2] and [4+2] products, as shown in Fig. 4.10. In addition to intradimer products for both cycloaddition reactions (Fig. 4.10a, d, and e), interdimer products bridging dimers in the same row (Fig. 4.10b) or adjacent rows (Fig. 4.10c) are identified for [4+2] cycloaddition. DFT calculations by the same group agree with the finding that the reaction of 1,3-cyclohexadiene is under kinetic control on Si(100)-(2×1) [151].
Due to the greater stability of the [4+2] products, annealing may be able to skew the product distribution in favor of the [4+2] products. Upon heating above approximately 400K, C–H stretching modes in the IR spectra are found to decrease in intensity without noticeable change in shape, concurrent with an increase in Si–H stretching modes, as shown in Fig. 4.11 [145,148]. These IR results as well as TPD spectra suggest that 1,3-cyclohexadiene primarily desorbs or decomposes before isomerization can occur, in agreement with the large activation barrier (>40 kcal/mol) calculated for isomerization between [4+2] and [2+2] products [6].
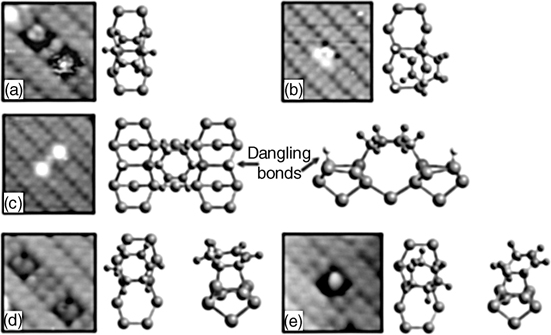
FIGURE 4.10 High-resolution empty state STM images showing multiple distinct adsorption configurations of 1,3-cyclohexadiene on Si(100)-(2×1) at 300K and models of the corresponding proposed bonding configuration. The features in the STM images are proposed to correspond to: (a) [4+2] cycloaddition on a single dimer; (b) [4+2] cycloaddition bridging two dimers in the same row; (c) [4+2] cycloaddition bridging two dimers in adjacent rows; and (d) and (e) two conformations of the [2+2] cycloaddition product. Approximately a 4×4 nm area is imaged at a tip bias of −1.3 V. Adapted with permission from Ref. 149. Copyright 2003 American Chemical Society.
4.3.3 Cyclopentadiene
An interesting study of cyclopentadiene and dicyclopentadiene adsorption on Si(100)-(2×1) was carried out by Wang et al. [152]. Dicyclopentadiene is formed by the spontaneous dimerization of cyclopentadiene by Diels–Alder reaction in solution. The spectral features in the C–H stretching region of chemisorbed cyclopentadiene at room temperature align well with calculated frequencies for the [4+2] product. The authors concluded that cyclopentadiene yields primarily the [4+2] product although the presence of a small amount of [2+2] side product could not be ruled out.
Dicyclopentadiene was also studied as it was hypothesized that the surface might catalyze the retro-Diels–Alder reaction (yielding two cyclopentadiene monomers per dicyclopentadiene dimer), as the formation of two [4+2] adducts (of cyclopentadiene) is thermodynamically much more favorable than formation of a single [2+2] adduct (of dicyclopentadiene). If this were the case, the IR spectrum after exposing the surface to dicyclopentadiene should be nearly, if not exactly, identical to that of cyclopentadiene. In fact, the spectra of chemisorbed cyclopentadiene and dicyclopentadiene chemisorbed on Si(100)-(2×1) at room temperature, shown in Fig. 4.12, differ significantly in both peak positions and intensities in the C–H stretching region, and chemisorbed dicyclopentadiene was instead assigned to a [2+2] product. The authors' calculations show a thermodynamic favorability of the surface-catalyzed retro-Diels–Alder reaction resulting in [4+2] cycloaddition of cyclopentadiene of nearly 46 kcal/mol over the [2+2] cycloaddition of intact dicyclopentadiene; thus, the experimental evidence indicates that kinetics controls the outcome of reaction. Though they do not calculate an activation barrier for surface-catalyzed retro-Diels–Alder reaction, it is noted that the energy barrier in the gas phase (33.7 kcal/mol) provides an upper bound on the energy barrier for the reaction catalyzed by the surface. Nonetheless, the energy barrier at the surface may still be much greater than that of the [2+2] reaction, which, as discussed earlier, occurs readily at the Si(100)-(2×1) surface. Moreover, geometric constraints for a transition state involving interaction of a dicyclopentadiene molecule with two surface dimers may yield a low pre-exponential factor for the surface-catalyzed reaction.
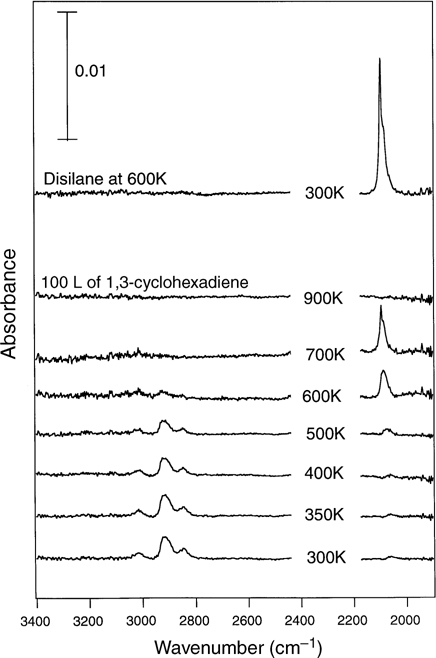
FIGURE 4.11 IR spectra for an annealing series of a Si(100)-(2×1) surface exposed to 100 L of 1,3-cyclohexadiene. The top spectrum corresponds to a well-ordered monohydride-terminated surface resulting from the exposure of Si(100)-(2×1) to disilane at 600K. Reprinted with permission from Ref. 148. Copyright 2000 American Chemical Society.
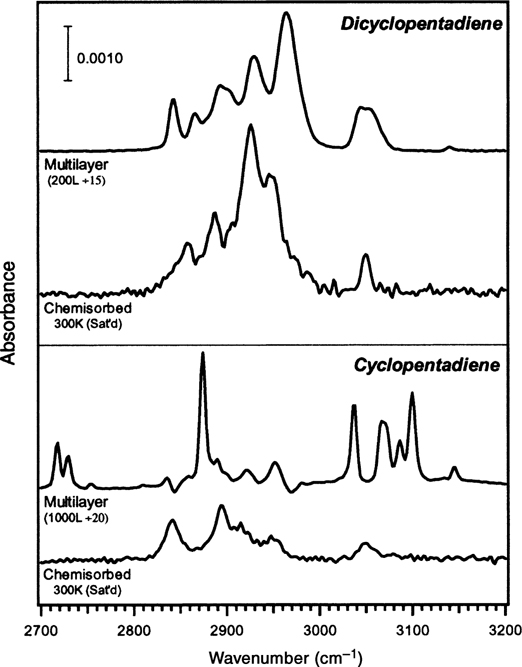
FIGURE 4.12 IR spectra of multilayers (unpolarized) and chemisorbed (p-polarized) dicyclopentadiene and cyclopentadiene on Si(100)-(2×1). The saturated chemisorbed spectra were collected after doses of 1500 and 500 L of dicyclopentadiene and cyclopentadiene, respectively. Reprinted with permission from Ref. 152. Copyright 1999 American Chemical Society.
4.3.4 [4+2]-Like Cycloaddition on Si(111)-(7×7)
Tao et al. [108] show using HREELS, XPS, and UPS that 1,3-cyclohexadiene binds to the Si(111)-(7×7) surface to form a cyclohexene-like adduct containing only a single C= C π bond. DFT calculations indicate that a [4+2]-like product is over 24 kcal/mol more stable than a [2+2]-like product [108]. Further experimental studies are necessary to differentiate between the [4+2]-like and [2+2]-like configurations of 1,3-cyclohexadiene adsorbed on Si(111)-(7×7).
Reaction pathways for 1,3-butadiene reaction with an adatom–rest atom pair on Si (111)-(7×7) were investigated using DFT calculations by Lu et al. [111]. For the more stable s-trans conformation of 1,3-butadiene, reaction pathways leading to both [4+2]-like and [2+2]-like products were investigated. The authors found a barrierless pathway to [4+2]-like cycloaddition passing through a diradical intermediate. A barrier of approximately 5 kcal/mol was found for the [2+2]-like reaction pathway, and the binding energy for the [2+2]-like product is about 19 kcal/mol less than that of the [4+2]-like product. A similar barrierless reaction pathway for [4+2]-like cycloaddition of s-cis-1,3-butadiene was calculated. The authors conclude that formation of the [4+2]-like product is both kinetically and thermodynamically favored. An experimental study of 1,3-butadiene adsorption on Si(111)-(7×7) found that reaction at adatom–rest atom pairs is the dominant pathway for adsorption, but STM images such as those in Fig. 4.13 provide evidence for 1,3-butadiene reaction at symmetric adatom–adatom pairs as well (adsorption sites labeled B and C in Fig. 4.13) [153]. The authors suggest that 1,3-butadiene may react at these sites via a concerted [4+2] cycloaddition mechanism due to the symmetry of the adatom pair (similar to the symmetry of alkenes or C(100)-(2×1) surface dimers). Further experimental work may help ascertain the bonding configuration at symmetric adatom pairs, and calculations are necessary to determine whether reaction by a concerted mechanism is, in fact, feasible at symmetric sites on the Si(111)-(7×7) surface.

FIGURE 4.13 STM images of a Si(111)-(7×7) surface exposed to 0.1 L of 1,3-butadiene at room temperature. A sample bias of 0.5 and −1.0 V was used to collect images (a) and (b), respectively, in which the unit cell is outlined by dashed lines, and adsorbed 1,3-butadiene molecules are indicated by circles. Images (c)–(h) show empty and filled state images of three adsorption sites collected at 1.0 and −2.0 V sample bias, respectively. Schematics of the proposed adsorption configurations are also shown. Reprinted with permission from Ref. 153. Copyright 2006 American Chemical Society.
4.4 CYCLOADDITION OF UNSATURATED ORGANIC MOLECULES CONTAINING ONE OR MORE HETEROATOM
Cycloaddition at semiconductor surfaces is not limited to organic molecules containing only carbon and hydrogen. For example, Ellison et al. [154] showed that azo-tert-butane primarily forms a [2+2] cycloaddition product on Si(100)-(2×1) by reacting across the N=N bond in a similar manner to the alkenes discussed earlier. The presence of one or more heteroatoms in a functional group can create uneven charge distribution, facilitating low-symmetry reaction pathways, and lone pairs on heteroatoms such as N, O, or S enable reactions to proceed through a dative-bonded precursor state. Dative bonding, which is discussed in more detail in a later chapter, is generally not possible for the simple alkenes, alkynes, and dienes discussed thus far in this chapter. In this section, reactivity of the Si(100)-(2×1) and Ge(100)-(2×1) surfaces toward heteroatom-containing organic molecules to form cycloaddition products is discussed, including mechanistic implications of dative bonding.
4.4.1 C=O-Containing Molecules
Armstrong et al. [155,156] investigated adsorption of acetaldehyde, acetone, and biacetyl on Si(100)-(2×1) using HREELS, XPS, and TPD. The authors concluded that all three molecules form di-σ products at low temperature in which the C=O and surface dimer π bonds are broken and new Si–C and Si–O bonds are formed, as in [2+2] cycloaddition. In the case of the bifunctional molecule, biacetyl, the authors conclude that only one of the ketone moieties interacts with the surface due to geometric constraints; however, Barriocanal and Doren [157] later reinterpreted the HREELS data using calculated vibrational frequencies to conclude that the thermodynamically favored [4+2] product is instead the major product. In all three cases, decomposition products are also observed even at the lowest dosing temperatures, and annealing leads to further decomposition of the adsorbates to produce both gas-phase and surface products [155,156]. Theoretical calculations have indicated that the formation of the [2+2] product across a C=O bond passes through a dative-bonded precursor state in which the oxygen atom donates a lone pair of electrons to the electrophilic “down” dimer atom at the surface [157,158]. For the reaction of glyoxal, a dialdehyde, with Si(100)-(2×1), Barriocanal and Doren [157] calculated mechanisms for both [2+2] and [4+2] cycloaddition that pass through a precursor state in which the oxygen lone pair is directed toward the dangling bond of the Si surface, and the nodal plane of the nearest π orbital is aligned along the Si–O axis, as shown in Fig. 4.14a and b. In contrast to alkenes and dienes in which only the π orbitals are available to interact with the surface, these results indicate the possibility of a reaction pathway mediated by dative bonding through an oxygen lone pair. In addition, they show optimization steps that pass through this dative-bonded state and continue without any activation barrier to both the [2+2] and [4+2] cycloaddition products. No such barrierless pathway could be found for [2+2] cycloaddition of acetone on Si(100)-(2×1) in calculations performed by Wang et al. [158]. It is hypothesized that the small (5 kcal/mol) barrier to from the dative-bonded state of acetone may arise from steric hindrance of the two methyl groups, that the conjugation of glyoxal may weaken the C=O bond allowing for a barrierless transition, or that a similar barrierless pathway exist for acetone but was not found by the calculations. Regardless of the uncertainty as to the presence or origin of an activation barrier, both studies identify the presence of dative-bonded precursor states and propose mechanisms for cycloaddition that pass through this state—a key theme in adsorption of heteroatom-containing molecules.
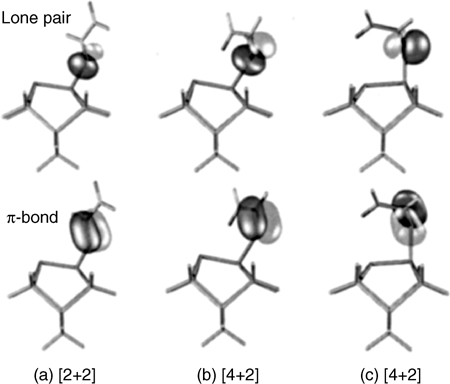
FIGURE 4.14 Calculated lone pair (top) and π (bottom) orbitals along three reaction pathways for the reaction of glyoxal with a Si9H12 cluster representing a single dimer on the Si(100)-(2×1) surface: (a) [2+2] cycloaddition of the trans isomer of glyoxal; and (b) and (c) two pathways for [4+2] cycloaddition of the cis isomer of glyoxal. The alignment of the oxygen lone pair orbital toward an Si surface atom and alignment of the π orbital nodal plane along the Si–O axis indicate a dative bond-like interaction with the surface. Reprinted with permission from Ref. 157. Copyright 2001 American Chemical Society.
In their study of acetone adsorption, Wang et al. [158] shed light on important differences between the Si(100)-(2×1) surface and the Ge(100)-(2×1) surface. Based on MIR-FTIR data, they find that acetone transfers an α-hydrogen to the Ge surface to produce an enol-like adduct at room temperature. This resembles an “ene” reaction, which falls under the category of group transfer reaction—another type of pericyclic reaction. Figure 4.15 shows a reaction coordinate diagram for acetone reaction on Ge(100)-(2×1) from DFT calculations performed by Wang et al. [158]. Based on a combination of theoretical and experimental results, they reach the conclusion that reaction of acetone on the Ge surface is under thermodynamic control. Calculations show that although both the [2+2] cycloaddition and ene reaction pathways traverse the same oxygen dative-bonded precursor state (Fig. 4.15c), the transition states lie 2 kcal/mol below the vacuum level for [2+2] cycloaddition and 2 kcal/mol above the vacuum level for ene reaction at the Ge(100)-(2×1) surface (Fig. 4.15d and b, respectively). Wang et al. conclude that acetone molecules may undergo [2+2] cycloaddition at the Ge surface, as it is kinetically favored, but because the binding energy of this product is only 12.5 kcal/mol (Fig. 4.15e), reversible desorption will quickly occur at room temperature. Eventually, the thermodynamically favored product of the ene reaction (with a 22.8 kcal/mol binding energy) is formed instead. This conclusion is substantiated by the relatively large doses required to observe the ene product at room temperature, which is consistent with the presence of an activation barrier lying slightly above the vacuum level. In an attempt to skew the product distribution toward the dative-bonded precursor state or the [2+2] cycloaddition product, the authors investigated acetone adsorption at low temperature. The IR spectrum of acetone exposed to Ge(100)-(2×1) at 115K closely resembles the multilayer spectrum of physisorbed acetone with the exception of a red-shifted C=O stretching mode, which indicates that acetone primarily adsorbs by dative bonding at this temperature.
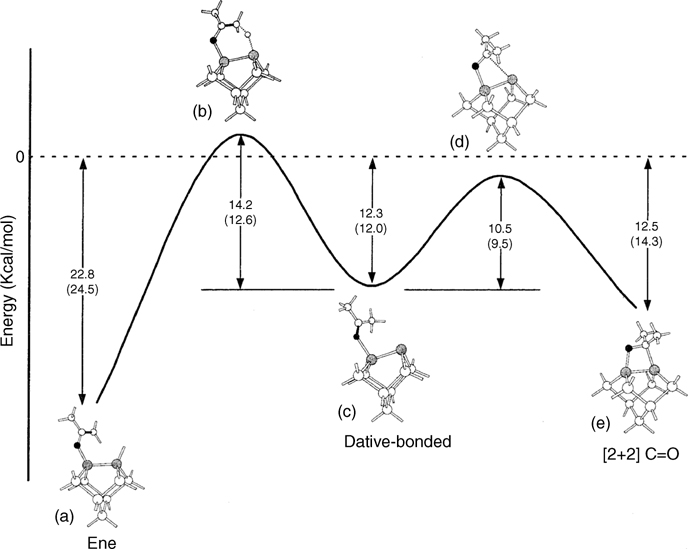
FIGURE 4.15 Calculated critical point energies on the ene and [2+2] reaction pathways of acetone on Ge(100)-(2×1). The dative-bonded precursor state shown in the center (c) can proceed through transition state (b) to the ene product (a) or through transition state (d) to the [2+2] product (e). Energies shown are in kcal/mol and were calculated using the 6-311++G(d,p) basis set (no parentheses) and 6-311+G(2df,pd) basis set (in parentheses). Reprinted with permission from Ref. 158. Copyright 2001 American Chemical Society.
DFT calculations for the [2+2] cycloaddition and ene reaction pathways for acetone on Si(100)-(2×1) were also carried out [158]. Similar reaction barriers (within the error of the calculations) were calculated for both pathways, and, like on the Ge surface, the ene product is thermodynamically favored by about 9 kcal/mol on Si(100)-(2×1). Experimental results of Armstrong et al. [156] show the formation of a [2+2] product and a product involving dissociation of the C=O bond, but no evidence for an ene reaction product was found in the HREELS spectrum at room temperature. However, STM images obtained by Hamai et al. [159] later showed the presence of adsorbates appearing as either a symmetric protrusion centered over a Si dimer or a protrusion and depression on a Si dimer. The authors attributed these to [2+2] cycloaddition and ene reaction products, respectively, and found that the ratio between the products can be changed to favor the ene product upon annealing to 420K. According to the calculations by Wang et al. [158], this result implies that the reaction is under kinetic control. Unlike on Ge(100)-(2×1), both the [2+2] and ene products have high enough binding energies on Si(100)-(2×1) (35.5 and 44.7 kcal/mol, respectively) to be observed on the experimental time scale. Thus, the presence of both products on the surface in significant quantity implies that the kinetic barriers determine the product distribution. Wang et al. [158] suggest that a barrierless pathway similar to that found for glyoxal [2+2] cycloaddition on Si(100)-(2×1) [157] may not have been located in their calculations for the reaction of acetone. If such a pathway exists, formation of the [2+2] product would be kinetically favored, thus explaining the larger percentage of [2+2] product at room temperature observed by Hamai et al. [159]. Moreover, the finding that annealing skews the product distribution toward ene product is suggested as evidence for thermodynamic control at high temperature [159], although the change in product distribution may also be rationalized in terms of the kinetics.
Wang et al. [158] note that the dative-bonded precursor states are of very similar energy (within 2 kcal/mol) on the Si(100)-(2×1) and Ge(100)-(2×1) surfaces. Compilation of data from a number of studies has shown that dative bonds are consistently only slightly stronger (~1–7 kcal/mol) on Si(100)-(2×1) than on Ge(100)-(2×1) [160]. Given this similarity in dative bond strength and the weaker bonds being formed in the reaction at the Ge surface, it is expected that higher activation barriers will generally be seen for reaction on the Ge surface. Most importantly, the studies discussed above provide clear experimental and theoretical evidence for one of the key differences between adsorption on Si(100)-(2×1) versus Ge(100)-(2×1): the strong bonds formed with Si favor kinetic control of product distribution, whereas the weaker bonds formed with Ge can lead to thermodynamic control for some reactions. This difference between the reactivity of Si and Ge surfaces may allow for much higher selectivity and the ability to control product distributions by thermal annealing on Ge(100)-(2×1).
To further investigate this difference in reactivity between the Si and Ge surfaces, a series of unsaturated ketones were studied [161]. Ethylvinylketone can potentially form a number of products at the surface due to its conjugated C=C and C=O bonds. Dative bonding through the oxygen lone pair, [2+2] cycloaddition across either the C=C or C=O bonds, [4+2] hetero-Diels–Alder cycloaddition (with ethylvinylketone in the s-cis conformation), [4+2] “trans cycloaddition” (with ethylvinylketone in the s-trans conformation) and the ene reaction are, in principle, all possible for the reaction of ethylvinylketone at Si(100)-(2×1) and Ge(100)-(2×1), as shown in Fig. 4.16. Using MIR-FTIR, it was determined that ethylvinylketone primarily forms a [4+2] cycloaddition product on Ge(100)-(2×1) at room temperature [161]. Although the two possible [4+2] products—;hetero-Diels–Alder and trans—cannot be distinguished on the basis of IR spectra, calculations indicate that the hetero-Diels–Alder product should be kinetically favored at room temperature; trans cycloaddition products bridging two dimers may be present initially but may convert to the hetero-Diels–Alder product at the surface. Further study by STM may be able to definitively differentiate between these products but no such study has been reported. On Si (100)-(2×1), the IR spectrum provides evidence for the presence of a significant amount of ene product as well as [4+2] cycloaddition products. On both surfaces [2+2] cycloaddition across the C=O bond cannot be ruled out on the basis of IR spectroscopy, but the DFT calculations discussed below suggest it is unlikely to be present in significant quantity on Ge.
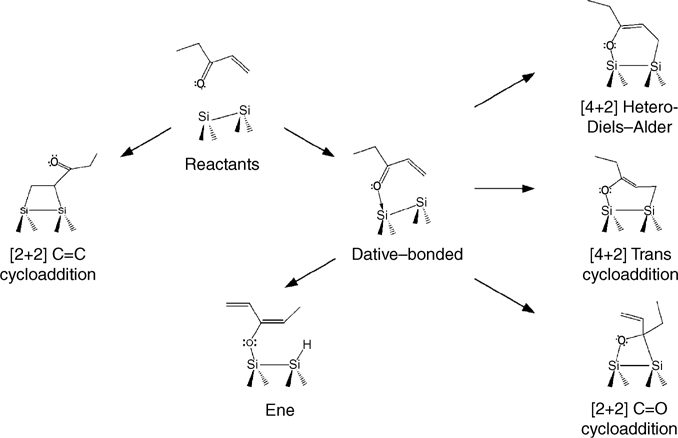
FIGURE 4.16 Schematic of possible intradimer reaction pathways for ethylvinylketone on Si(100)-(2×1). Analogous reactions pathways are also possible on Ge(100)-(2×1). Reprinted with permission from Ref. 161. Copyright 2002 American Chemical Society.
With the help of critical point energies calculated by DFT for the various reaction pathways, the difference in selectivity observed for reaction of ethylvinylketone with these two surfaces is once again explained in terms of kinetics and thermodynamics [161]. For all of the reaction pathways passing through a dative-bonded precursor state except hetero-Diels–Alder reaction, which was found to go through a barrierless pathway, higher barriers and smaller product binding energies are observed on Ge (100)-(2×1), similar to previously discussed systems. hetero-Diels–Alder cycloaddition is clearly favored kinetically consistent with the experimental observation that it is the primary product on both surfaces. However, whereas the larger activation barriers for other pathways lead to high selectivity on Ge(100)-(2×1), the comparatively small activation barriers on Si(100)-(2×1) allow other pathways to compete, as seen experimentally.
The adsorption of 2-cyclohexen-1-one was also investigated as its ring structure constrains the molecule to the s-trans conformation, thus eliminating the possibility of hetero-Diels–Alder reaction [161]. IR spectra show similar results as ethylvinylketone for adsorption at room temperature: [4+2] cycloaddition on Ge(100)-(2×1) (although presumably by trans cycloaddition rather than hetero-Diels–Alder cycloaddition) versus primarily [4+2] cycloaddition with significant quantity of ene side product on Si (100)-(2×1). Using the calculated dative-bonded and transition state energies for ethylvinylketone, which are expected to be very similar to those for 2-cyclohexen-1-one, the trans cycloaddition pathway is kinetically favored by a similar amount on both surfaces when comparing to the second most favored pathway (ene reaction on Si and C=O [2+2] cycloaddition on Ge). In Fig. 4.17, this is apparent by comparing the transition state energies of trans cycloaddition (solid lines) to the next lowest transition state energy for each surface. In both cases there is a similar (~5–6 kcal/mol) difference in transition state energies between the two most kinetically favorable pathways. Thus, kinetic arguments alone cannot be used to rationalize the difference in selectivity observed experimentally for 2-cyclohexen-1-one on Si(100)-(2×1) and Ge (100)-(2×1). However, if one also considers thermodynamics, Fig. 4.17 shows that the C=O [2+2] cycloaddition product of 2-cyclohexen- 1-one has a low binding energy on Ge (11.9 kcal/mol) and, thus, is not expected to be stable at room temperature. Similar to the case of acetone adsorption on Ge(100)-(2×1), the C=O [2+2] product may form temporarily but is expected to desorb on the time scale of the experiment at room temperature. The next most kinetically favored pathway on Ge(100)-(2×1) is the ene reaction, which leads to a more thermodynamically stable product (23.1 kcal/mol binding energy) that should be observable at room temperature. However, Fig. 4.17 shows that the transition state for ene reaction (the dashed line in Fig. 4.17) is significantly higher on Ge(100)-(2×1) than Si(100)-(2×1), leading to selective formation of trans cycloaddition product on Ge and a combination of trans cycloaddition and ene products on Si. Thus, Wang et al. conclude that both kinetic and thermodynamic factors play a role in determining product distribution on Ge(100)-(2×1), whereas kinetics are the dominant factor on Si(100)-(2×1) owing to the greater thermodynamic stability of all products on Si.
Adsorption of ethylvinylketone on Si(111)-(7×7) was later studied by Tang et al. [162]. HREELS and XPS evidence suggest that the conjugated C=C and C=O bonds of ethylvinylketone are both involved in the adsorption process with high selectivity, and a new peak in the HREELS spectrum at 1660 cm−1 is assigned to an internal C=C stretching mode. DFT calculations show that the [4+2]-like product has higher binding energy than the ene or [2+2]-like products, although binding energies for all products are high enough that they could be observed on the experimental time scale. By analogy to similar reactions on Si(100)-(2×1), the authors suggest that ethylvinylketone likely forms a dative-bonded precursor state by donation of a lone pair of electrons to an electrophilic Si adatom, and a barrierless or low activation pathway from the dative-bonded state to [4+2]-like cycloaddition may exist. Thus, they conclude that both thermodynamic and kinetic favorability of [4+2]-like cycloaddition lead to selective formation of this product, as observed experimentally. Given the complicated adsorption behavior of ethylvinylketone on Si(100)-(2×1) studied by Wang et al. [161], which appears to be dominated by kinetic factors, calculations of pathways (including transition states) for the various adsorption products may provide a more thorough understanding of why high selectivity is observed on the Si(111)-(7×7) surface.
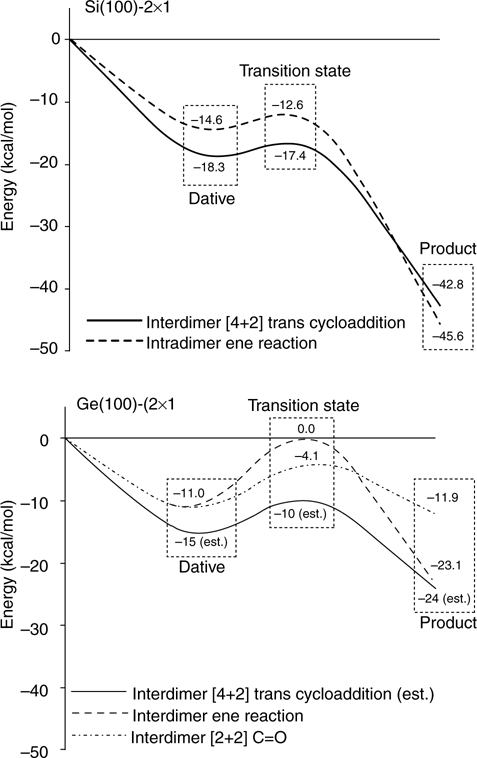
FIGURE 4.17 Calculated critical point energies on the potential energy surface for relevant reactions of 2-cyclohexen-1-one on Si(100)-(2×1) (top) and Ge(100)-(2×1) (bottom) [161]. Note that energies for trans cycloaddition on Ge(100)-(2×1) are estimated based on intradimer calculations, and energies on all pathways for dative-bonded and transition states are for ethylvinylketone, which are expected to be similar to the values for 2-cyclohexen-1-one.
4.4.2 Nitriles
A number of studies have investigated the adsorption of nitriles and other C≡N-containing compounds on Si(100)-(2×1) [163–173] and Ge(100)-(2×1) [171,174]. In particular, acetonitrile has been the subject of a number of studies as it is the simplest organic nitrile. Several possible reactions of acetonitrile with Si(100)-(2×1) or Ge(100)-(2×1) are shown in Fig. 4.18. Tao et al. [173] used XPS and HREELS to conclude that acetonitrile binds to Si(100)-(2×1) to form a [2+2] cycloaddition product at low temperature. Their assignment is substantiated by the presence of a peak near 1610 cm−1 and absence of a peak near 2220 cm−1 in the HREELS spectra before the growth of physisorbed multilayers, indicating the presence and absence of C=Nand C≡N, respectively, although later studies have suggested that some C≡N groups may be present on the surface, as discussed below. Large shifts in C 1s and N 1s binding energies are also consistent with the proposed binding configuration based on comparison to results for acetonitrile adsorption on metal substrates. The tetra-σ (dual-[2+2]) product was also ruled out on the basis of HREELS results. TPD showed molecular desorption peaks at 400 and 467K attributable to two different chemisorbed products. The authors suggest that these may result from different on-top or bridging products within a row, similar to the configurations possible for ethylene and acetylene, and a product bridging neighboring rows; however, later DFT calculations showed that this explanation cannot account for the experimental results, as the binding energy of a product bridging rows is too low to account for either of the observed TPD peaks [175]. DFT calculations performed by Lu et al. [170] demonstrated that [2+2] cycloaddition of acetonitrile on top of a single Si dimer may occur and showed that the reaction can proceed through a dative-bonded precursor state. A study by Bournel et al. [168] using PES and NEXAFS provided evidence for the existence of multiple products on the surface at room temperature, some containing a C≡N bond tilted with respect to the substrate.
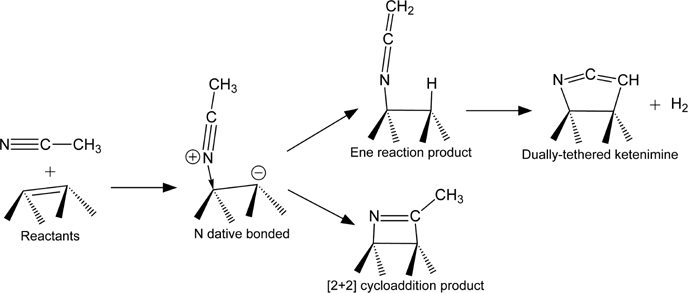
FIGURE 4.18 Schematic of possible intradimer reaction pathways for acetonitrile on Si(100)-(2×1) or Ge(100)-(2×1).
While the presence of the [2+2] product is generally agreed upon, the configurations of additional products formed upon adsorption of acetonitrile on Si(100)-(2×1) at room temperature has been the topic of some debate. A stable N dative-bonded state was predicted by Miotto et al. [176], while Mui et al. [171] suggest based on DFT calculations that α-hydrogen elimination (ene reaction) would also occur at room temperature leading to a cumulated C=C=N structure (ketenimine) bound to the surface through the nitrogen (structure labeled “Ene reaction product” in Fig. 4.18). Polarization-dependent NEXAFS studies [166] again showed the presence of multiple products but did not support the theoretical prediction of Mui et al. [171]. To reconcile the discrepancy, Schwartz and Hamers [167] more recently proposed that a C=C=N species bound to the surface at both ends can form by loss of two hydrogens (structure labeled “Dually-tethered ketenimine” in Fig. 4.18). Formation of this species is consistent with the NEXAFS polarization-dependence, and further evidence for its presence is provided by polarized-FTIR experiments showing a C=C=N mode at about 1950 cm−1 polarized parallel to the surface dimers and in the plane of the surface. Additional DFT calculations by Carniato et al. [177] agree with the Si–C=C=N–Si structure proposed by Schwartz and Hamers (although Carniato et al. note that the structure must be twisted from its unstrained geometry due to the directionality of the bonds with the surface) and suggest the presence of additional products containing C=N and C≡N bonds. Their calculations show that modes associated with both of these groups (especially the C≡N group) have significantly lower cross sections than the C=C=N mode, which may explain why the C≡N stretching mode was not observed in the HREELS spectra by Tao et al. [173] while Bournel et al. [168] found evidence for the presence of such a C≡N bond by NEXAFS. Although the most recent works by Schwartz and Hamers [167] and Carniato et al. [177] reconcile apparent discrepancies among IR, NEXAFS, and HREELS results, neither specifically address whether the proposed Si–C=C=N–Si structure is consistent with previous TPD results.
In contrast to the results on Si(100)-(2×1), Filler et al. [174] showed experimentally that acetonitrile does not react with the Ge(100)-(2×1) surface at room temperature. These results agree with DFT calculations, which predict that the dative-bonded precursor, ene reaction product and [2+2] cycloaddition product will all desorb readily at room temperature [171]. It has been suggested that the nitrile functional group could serve as a protecting group for functionalization of Ge(100)-(2×1). Upon adsorption through a second functional group, the nitrile may remain unreacted on the surface. Hydrogenation of the nitrile to an amine could then be used to convert it to a more reactive moiety for further functionalization. Filler et al. [174] also investigated adsorption on Ge(100)-(2×1) of the bifunctional nitriles 2-propenenitrile, 3-butenenitrile, and 4-pentenenitrile. Not surprisingly, the second functionality enables these molecules to adsorb on the Ge surface at room temperature. IR spectra show that the conjugated molecule 2-propenenitrile forms primarily the hetero-Diels–Alder [4+2] product and a small amount of [2+2] cycloaddition product reacted across the C=C bond. [4+2] cycloaddition of 2-propenenitrile creates a ring at the surface that includes a C=C=N ketenimine group, similar to that seen for the ene reaction product of acetonitrile. This again leads to absorption near 1950 cm−1 in the IR spectrum. Surprisingly, this mode is also observed when the nonconjugated molecules 3-butenenitrile and 4-pentenenitrile are exposed to the Ge(100)-(2×1) surface at room temperature, but Filler et al. tentatively attribute this to isomerization of these molecules to their conjugated forms before dosing them on the surface. Like acetonitrile, none of the bifunctional nitriles reacted with the Ge(100)-(2×1) surface directly through the C≡N bond at room temperature.
HREELS, XPS, and UPS results suggest that acetonitrile binds to Si(111)-(7×7) by [2+2]-like cycloaddition [178]. STM images were also used to show that acetonitrile binds at adatom–rest atom pairs, and to compare the reactivity of various sites in the unit cell. However, a later study by Bournel et al. [179] using synchrotron-based XPS and NEXAFS found evidence for two bonding configurations including one with a free C≡N group. By comparison of experimental results to DFT calculations, the authors assign one product to [2+2]-like cycloaddition bridging an adatom-rest atom pair in agreement with the previous study. The free C≡N group observed by NEXAFS is attributed to a second product involving bonding to the surface through the terminal carbon via α-hydrogen dissociation. Although this is not equivalent to the ene reaction proposed for acetonitrile adsorption on Si(100)-(2×1), and no evidence for dissociation of a second hydrogen (to form a C=C=N structure) has been reported, acetonitrile adsorption on Si(111)-(7×7) bears some similarity to that on Si(100)-(2×1): on both surfaces [2+2] cycloaddition appears to be the dominant pathway and secondary products are formed by dissociation of relatively acidic α-hydrogens.
4.4.3 Isocyanates and Isothiocyanates
The cumulative double bond of isocyanate and isothiocyanate functional groups provides for interesting chemistry at semiconductor surfaces, as they are able to undergo a number of reactions. In principal, two different dative-bonded states, two different [2+2] cycloaddition products and a 1,3-dipolar cycloaddition product [180] are possible for both functional groups, as shown in Fig. 4.19. Ellison and Hamers [181] reported the selective formation of a [2+2] product across the C=N bond for phenyl isothiocyanate exposed to Si(100)-(2×1) at room temperature based on XPS, IR spectroscopy, and STM. No evidence for interaction through the phenyl ring was observed. Phenyl isocyanate, on the other hand, was reported to decompose upon bonding to Si(100)-(2×1), although detailed studies of this system have not been published [11]. It is suggested that the difference between isothiocyanate and isocyanate reactivity may result from oxygen's propensity to insert into the Si–Si bonds due to its smaller size than sulfur and the larger difference in electronegativity between Si and O than between Si and S.
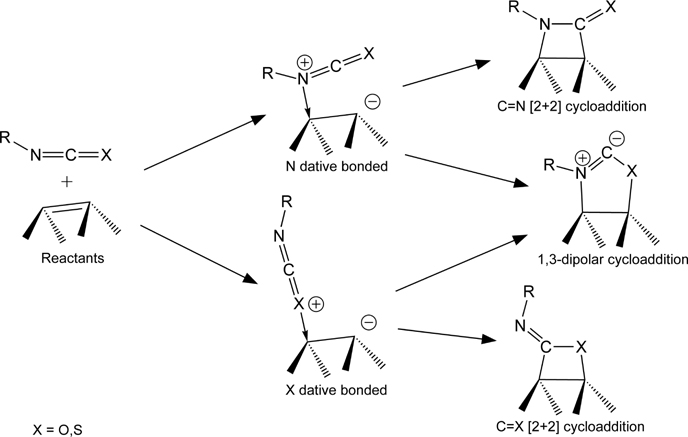
FIGURE 4.19 Schematic of possible intradimer reaction pathways for an isocyanate (X=O) or isothiocyanate (X=S) functional group on Si(100)-(2×1) or Ge(100)-(2×1).
MIR-FTIR spectra for 1,4-phenylene diisocyanate exposed to Ge(100)-(2×1) show that it adsorbs primarily by [2+2] cycloaddition across the C=N bond and at least some of the isocyanate functional groups remain unreacted [132]. No evidence for significant decomposition on this surface was reported. As this molecule was being investigated for use as a molecular layer deposition precursor, adsorption of 1,4-phenylene diisocyanate was followed by exposure to ethylenediamine. IR spectra after this exposure indicated some desorption of 1,4-phenylene diisocyanate from the surface in addition to the intended reaction between the isocyanate and amine, highlighting the relatively weak bonding between the surface and isocyanate. Unlike many adsorbates on Ge(100)-(2×1) that exhibit fairly high selectivity, phenyl isocyanate and phenyl isothiocyanate are found to form multiple adsorption products on the Ge surface and exhibit interesting coverage or time dependences [182]. Further studies are currently underway to fully understand the adsorption behavior of these molecules on Ge(100)-(2×1), but this example demonstrates that although thermodynamic factors may play a role in determining adsorption products on Ge, high selectivity does not always result.
4.5 SUMMARY
Cycloaddition reactions, including [2+2] and [4+2] Diels–Alder reactions, comprise an important means for functionalization of semiconductor surfaces and understanding the fundamental chemistry at these surfaces. Many examples of these reactions at Si(100)-(2×1), Ge(100)-(2×1), C(100)-(2×1), and Si(111)-(7×7) surfaces have been studied for alkenes, alkynes, dienes and a number of unsaturated heteroatom-containing functional groups. The fundamental reactive unit on the Group IV (100)-(2×1) surfaces, the dimer, can be described as having a partial π bond and, thus, alkene character. This enables analogy between reactions at the surface and in solution organic chemistry. Such analogy allows the extensive knowledge of organic chemistry to be used in guiding research in the field of organic functionalization of semiconductors. However, there are important differences between classic organic chemistry and chemistry at the surface. Geometric and electronic effects of the surface enable, for example, low-symmetry reaction pathways that are not observed for cycloadditions in solution or gas phase. The facile [2+2] reaction of many alkenes at semiconductor surfaces exemplifies the possibility of such reaction pathways at the surface. The heterogeneity of surface atoms on the Si(111)-(7×7) surface has led to observation of cycloaddition-like reactions believed to occur by similar mechanisms on this surface as well. Charge separation on the tilted dimers of the Si(100)-(2×1) and Ge(100)-(2×1) surfaces also facilitates dative bonding with adsorbate lone pairs, thus creating an intermediate or precursor state that is important for cycloaddition reactions of many heteroatom-containing adsorbates.
Comparing adsorption of various molecules on the three Group IV (100)-(2×1) surfaces leads to several conclusions. Adsorption on Si(100)-(2×1), the subject of the majority of studies, very often produces multiple products, the distribution of which is largely controlled by kinetic factors. These products are typically strongly bound to the surface and, upon heating, decompose before desorbing. The dimers of C(100)-(2×1) are the most direct analogue of an alkene. Although cycloadditions also occur on this surface, lower sticking coefficients are typically observed, especially in the case of [2+2] cycloaddition. This is believed to result from the symmetry of the dimers on the diamond surface and highlights the influence of the surface geometry on cycloaddition reactivity. Adsorbates tend to form weaker bonds with the Ge(100)-(2×1) surface resulting in lower binding energies and higher activation barriers. The lower binding energies have been found in many cases to favor thermodynamic control over kinetic control on the Ge surface, and higher selectivity is often achieved.
Much recent work has focused on adsorption of heteroatom-containing compounds, since the ability to dative bond to the Si(100)-(2×1) and Ge(100)-(2×1) surfaces facilitates cycloaddition. Adsorption behavior of multifunctional compounds is also of growing interest, because multiple functionalities may allow subsequent reaction; however, use of multifunctional compounds is accompanied by the challenge of achieving controlled, selective attachment to the surface. While many useful attributes have been identified in separate systems, such as higher selectivity on Ge(100)-(2×1) for some adsorbates and well-ordered adsorption of cyclopentene on Si(100)-(2×1), future work may focus on bringing together such features to functionalize semiconductor surfaces in a highly controlled fashion. As many unsaturated organic compounds readily undergo cycloaddition reactions at these surfaces, understanding the nature of cycloadditions is necessary to achieve such control.
ACKNOWLEDGMENT
This work was supported by the National Science Foundation (Grants CHE 0615087 and CHE 0910717).
REFERENCES
1. Woodward, R. B.; Hoffmann, R. J. Am. Chem. Soc. 1965, 87, 395.
2. Fleming, I. Pericyclic Reactions. Oxford University Press, Oxford, 1999.
3. Carruthers, W. Cycloaddition Reactions in Organic Synthesis. Pergamon Press, New York, 1990.
4. Fallis, A. G. Acc. Chem. Res. 1999, 32, 464.
5. Woodward, R. B.; Hoffmann, R. The Conservation of Orbital Symmetry. Academic Press, New York, 1970.
6. Choi, C. H.; Gordon, M. S. J. Am. Chem. Soc. 1999, 121, 11311.
7. Liu, H.; Hamers, R. J. J. Am. Chem. Soc. 1997, 119, 7593.
8. Liu, Q.; Hoffmann, R. J. Am. Chem. Soc. 1995, 117, 4082.
9. Lopinski, G. P.; Moffatt, D. J.; Wayner, D. D. M.; Wolkow, R. A. J. Am. Chem. Soc. 2000, 122, 3548.
10. Buriak, J. M. Chem. Commun. 1999, 1051.
11. Hamers, R. J.; Coulter, S. K.; Ellison, M. D.; Hovis, J. S.; Padowitz, D. F.; Schwartz, M. P.; Greenlief, C. M.; Russell, J. N. Acc. Chem. Res. 2000, 33, 617.
12. Bent, S. F. J. Phys. Chem. B 2002, 106, 2830.
13. Bent, S. F. Surf. Sci. 2002, 500, 879.
14. Buriak, J. M. Chem. Rev. 2002, 102, 1271.
15. Lu, X.; Lin, M. C. Int. Rev. Phys. Chem. 2002, 21, 137.
16. Filler, M. A.; Bent, S. F. Prog. Surf. Sci. 2003, 73, 1.
17. Tao, F.; Xu, G. Q. Acc. Chem. Res. 2004, 37, 882.
18. Loscutoff, P. W.; Bent, S. F. Annu. Rev. Phys. Chem. 2006, 57, 467.
19. Ma, Z.; Zaera, F. Surf. Sci. Rep. 2006, 61, 229.
20. Leftwich, T. R.; Teplyakov, A. V. Surf. Sci. Rep. 2008, 63, 1.
21. Takayanagi, K.; Tanishiro, Y.; Takahashi, M.; Takahashi, S. J. Vac. Sci. Technol. A 1985, 3, 1502.
22. Hamers, R. J.; Tromp, R. M.; Demuth, J. E. Phys. Rev. Lett. 1986, 56, 1972.
23. Northrup, J. E. Phys. Rev. Lett. 1986, 57, 154.
24. Hamers, R. J.; Tromp, R. M.; Demuth, J. E. Surf. Sci. 1987, 181, 346.
25. Wolkow, R.; Avouris, P. Phys. Rev. Lett. 1988, 60, 1049.
26. Stich, I.; Payne, M. C.; King-Smith, R. D.; Lin, J. S.; Clarke, L. J. Phys. Rev. Lett. 1992, 68, 1351.
27. Cheng, C. C.; Choyke, W. J.; Yates, J. T., Jr. Surf. Sci. 1990, 231, 289.
28. Yoshinobu, J.; Tsuda, H.; Onchi, M.; Nishijima, M. J. Chem. Phys. 1987, 87, 7332.
29. Birkenheuer, U.; Gutdeutsch, U.; Rösch, N.; Fink, A.; Gokhale, S.; Menzel, D.; Trischberger, P.; Widdra, W. J. Chem. Phys. 1998, 108, 9868.
30. Konecny, R.; Doren, D. J. Surf. Sci. 1998, 417, 169.
31. Casaletto, M. P.; Zanoni, R.; Carbone, M.; Piancastelli, M. N.; Aballe, L.; Weiss, K.; Horn, K. Phys. Rev. B 2000, 62, 17128.
32. Chua, L. H.; Jackman, R. B.; Foord, J. S. Surf. Sci. 1994, 315, 69.
33. Clemen, L.; Wallace, R. M.; Taylor, P. A.; Dresser, M. J.; Choyke, W. J.; Weinberg, W. H.; Yates, J. T. Surf. Sci. 1992, 268, 205.
34. Craig, B. I. Surf. Sci. 1995, 329, 293.
35. Fisher, A. J.; Blochl, P. E.; Briggs, G. A. D. Surf. Sci. 1997, 374, 298.
36. Huang, C.; Widdra, W.; Weinberg, W. H. Surf. Sci. 1994, 315, L953.
37. Matsui, F.; Yeoim, H. W.; Imanishi, A.; Isawa, K.; Matsuda, I.; Ohta, T. Surf. Sci. 1998, 401, L413.
38. Matsui, F.; Yeom, H. W.; Matsuda, I.; Ohta, T. Phys. Rev. B 2000, 62, 5036.
39. Mayne, A. J.; Avery, A. R.; Knall, J.; Jones, T. S.; Briggs, G. A. D.; Weinberg, W. H. Surf. Sci. 1993, 284, 247.
40. Pan, W.; Zhu, T.; Yang, W. J. Chem. Phys. 1997, 107, 3981.
41. Widdra, W.; Huang, C.; Briggs, G. A. D.; Weinberg, W. H. J. Electron Spectrosc. Relat. Phenom. 1993, 64-65, 129.
42. Widdra, W.; Huang, C.; Weinberg, W. H. Surf. Sci. 1995, 329, 295.
43. Xu, S. H.; Keeffe, M.; Yang, Y.; Chen, C.; Yu, M.; Lapeyre, G. J.; Rotenberg, E.; Denlinger, J.; Yates, J. T. Phys. Rev. Lett. 2000, 84, 939.
44. Wolkow, R. A. Annu. Rev. Phys. Chem. 2003, 50, 413.
45. Shoemaker, J.; Burggraf, L. W.; Gordon, M. S. J. Chem. Phys. 2000, 112, 2994.
46. Lu, X. J. Am. Chem. Soc. 2003, 125, 6384.
47. Lu, X.; Zhu, M.; Wang, X. J. Phys. Chem. B 2004, 108, 7359.
48. Nagao, M.; Umeyama, H.; Mukai, K.; Yamashita, Y.; Yoshinobu, J.; Akagi, K.; Tsuneyuki, S. J. Am. Chem. Soc. 2004, 126, 9922.
49. Fan, X. L.; Zhang, Y. F.; Lau, W. M.; Liu, Z. F. Phys. Rev. B 2005, 72, 165305.
50. Ryan, P. M.; Teague, L. C.; Boland, J. J. J. Am. Chem. Soc. 2009, 131, 6768.
51. Mui, C.; Bent, S. F.; Musgrave, C. B. J. Phys. Chem. A 2000, 104, 2457.
52. Hennies, F.; Fohlisch, A.; Wurth, W.; Witkowski, N.; Nagasono, M.; Novella Piancastelli, M. Surf. Sci. 2003, 529, 144.
53. Seo, J. H.; Park, J. Y.; Whang, C. N.; Kim, S. S.; Choi, D. S.; Chae, K. H. Surf. Sci. 2005, 582, L129.
54. Fink, A.; Huber, R.; Widdra, W. J. Chem. Phys. 2001, 115, 2768.
55. Kim, A.; Choi, D. S.; Lee, J. Y.; Kim, S. J. Phys. Chem. B 2004, 108, 3256.
56. Lal, P.; Teplyakov, A. V.; Noah, Y.; Kong, M. J.; Wang, G. T.; Bent, S. F. J. Chem. Phys. 1999, 110, 10545.
57. Cho, J.-H.; Kleinman, L. J. Chem. Phys. 2003, 119, 2820.
58. Miotto, R.; Ferraz, A. C. Surf. Sci. 2002, 513, 422.
59. Miotto, R.; Ferraz, A. C.; Srivastava, G. P. Surf. Sci. 2002, 507-510, 12.
60. Nishijima, M.; Yoshinobu, J.; Tsuda, H.; Onchi, M. Surf. Sci. 1987, 192, 383.
61. Taylor, P. A.; Wallace, R. M.; Cheng, C. C.; Weinberg, W. H.; Dresser, M. J.; Choyke, W. J.; Yates, J. T. J. Am. Chem. Soc. 1992, 114, 6754.
62. Carmer, C. S.; Weiner, B.; Frenklach, M. J. Chem. Phys. 1993, 99, 1356.
63. Cheng, C. C.; Taylor, P. A.; Wallace, R. M.; Gutleben, H.; Clemen, L.; Colaianni, M. L.; Chen, P. J.; Weinberg, W. H.; Choyke, W. J.; Yates Jr., J. T. Thin Solid Films 1993, 225, 196.
64. Huang, C.; Widdra, W.; Wang, X. S.; Weinberg, W. H. J. Vac. Sci. Technol. A 1993, 11, 2250.
65. Imamura, Y.; Morikawa, Y.; Yamasaki, T.; Nakatsuji, H. Surf. Sci. 1995, 341, L1091.
66. Yan, C.; Zhaohui, L. Appl. Phys. Lett. 1995, 67, 2936.
67. Widdra, W.; Huang, C.; Yi, S. I.; Weinberg, W. H. J. Chem. Phys. 1996, 105, 5605.
68. Dyson, A. J.; Smith, P. V. Surf. Sci. 1997, 375, 45.
69. Li, L.; Tindall, C.; Takaoka, O.; Hasegawa, Y.; Sakurai, T. Phys. Rev. B 1997, 56, 4648.
70. Meng, B.; Maroudas, D.; Weinberg, W. H. Chem. Phys. Lett. 1997, 278, 97.
71. Sorescu, D. C.; Jordan, K. D. J. Phys. Chem. B 2000, 104, 8259.
72. Terborg, R.; Baumgärtel, P.; Lindsay, R.; Schaff, O.; Giessel, T.; Hoeft, J. T.; Polcik, M.; Toomes, R. L.; Kulkarni, S.; Bradshaw, A. M.; Woodruff, D. P. Phys. Rev. B 2000, 61, 16697.
73. Cho, J.-H.; Kleinman, L.; Chan, C. T.; Kim, K. S. Phys. Rev. B 2001, 63, 073306.
74. Hofer, W. A.; Fisher, A. J.; Wolkow, R. A. Surf. Sci. 2001, 475, 83.
75. Kim, W.; Kim, H.; Lee, G.; Hong, Y.-K.; Lee, K.; Hwang, C.; Kim, D.-H.; Koo, J.-Y. Phys. Rev. B 2001, 64, 193313.
76. Mezhenny, S.; Lyubinetsky, I.; Choyke, W. J.; Wolkow, R. A.; Yates, J. T. Chem. Phys. Lett. 2001, 344, 7.
77. Morikawa, Y. Phys. Rev. B 2001, 63, 033405.
78. Silvestrelli, P. L.; Toigo, F.; Ancilotto, F. J. Chem. Phys. 2001, 114, 8539.
79. Kim, W.; Kim, H.; Lee, G.; Chung, J.; You, S.-Y.; Hong, Y.-K.; Koo, J.-Y. Surf. Sci. 2002, 514, 376.
80. Terborg, R.; Polcik, M.; Hoeft, J. T.; Kittel, M.; Sayago, D. I.; Toomes, R. L.; Woodruff, D. P. Phys. Rev. B 2002, 66, 085333.
81. Wang, F.; Sorescu, D. C.; Jordan, K. D. J. Phys. Chem. B 2002, 106, 1316.
82. Di Felice, R.; Pignedoli, C. A.; Bertoni, C. M.; Catellani, A.; Silvestrelli, P. L.; Sbraccia, C.; Ancilotto, F.; Palummo, M.; Pulci, O. Surf. Sci. 2003, 532–535, 982.
83. Silvestrelli, P. L.; Pulci, O.; Palummo, M.; Del Sole, R.; Ancilotto, F. Phys. Rev. B 2003, 68, 235306.
84. Cho, J.-H.; Kleinman, L. Phys. Rev. B 2004, 69, 075303.
85. Mineva, T.; Nathaniel, R.; Kostov, K. L.; Widdra, W. J. Chem. Phys. 2006, 125, 194712.
86. Toscano, M.; Russo, N. J. Mol. Catal. 1989, 55, 101.
87. Kim, A.; Maeng, J. Y.; Lee, J. Y.; Kim, S. J. Chem. Phys. 2002, 117, 10215.
88. Cho, J.-H.; Kim, K. S.; Morikawa, Y. J. Chem. Phys. 2006, 124, 024716.
89. Lopinski, G. P.; Moffatt, D. J.; Wayner, D. D. M.; Wolkow, R. A. Nature 1998, 392, 909.
90. Kiskinova, M.; Yates, J. T. Surf. Sci. 1995, 325, 1.
91. Madachik, M. R.; Teplyakov, A. V. J. Vac. Sci. Technol. A 2008, 26, 1241.
92. Hamers, R. J.; Hovis, J. S.; Lee, S.; Liu, H.; Shan, J. J. Phys. Chem. B 1997, 101, 1489.
93. Hovis, J. S.; Lee, S.; Liu, H.; Hamers, R. J. J. Vac. Sci. Technol. B 1997, 15, 1153.
94. Hovis, J. S.; Liu, H.; Hamers, R. J. J. Appl. Phys. A 1998, 66, S553.
95. Hovis, J. S.; Liu, H.; Hamers, R. J. Surf. Sci. 1998, 402–404, 1.
96. Liu, H.; Hamers, R. J. Surf. Sci. 1998, 416, 354.
97. Yamashita, Y.; Nagao, M.; Machida, S.; Hamaguchi, K.; Yasui, F.; Mukai, K.; Yoshinobu, J. J. Electron Spectrosc. Relat. Phenom. 2001, 114–116, 389.
98. Hovis, J. S.; Hamers, R. J. J. Phys. Chem. B 1997, 101, 9581.
99. Hovis, J. S.; Hamers, R. J. J. Phys. Chem. B 1998, 102, 687.
100. Rochet, F.; Bournel, F.; Gallet, J. J.; Dufour, G.; Lozzi, L.; Sirotti, F. J. Phys. Chem. B 2002, 106, 4967.
101. Yates, J. T., Jr. Science 1998, 279, 335.
102. Hovis, J. S.; Coulter, S. K.; Hamers, R. J.; D'Evelyn, M. P.; Russell, J. N.; Butler, J. E. J. Am. Chem. Soc. 2000, 122, 732.
103. Lee, S. W.; Hovis, J. S.; Coulter, S. K.; Hamers, R. J.; Greenlief, C. M. Surf. Sci. 2000, 462, 6.
104. Cho, J.-H.; Kleinman, L. Phys. Rev. B 2003, 67, 115314.
105. Yoshinobu, J.; Tsuda, H.; Onchi, M.; Nishijima, M. Solid State Commun. 1986, 60, 801.
106. Yoshinobu, J.; Tsuda, H.; Onchi, M.; Nishijima, M. Chem. Phys. Lett. 1986, 130, 170.
107. MacPherson, C. D.; Hu, D. Q.; Leung, K. T. Surf. Sci. 1992, 276, 156.
108. Tao, F.; Wang, Z. H.; Xu, G. Q. Surf. Sci. 2003, 530, 203.
109. Piancastelli, M. N.; Motta, N.; Sgarlata, A.; Balzarotti, A.; De Crescenzi, M. Phys. Rev. B 1993, 48, 17892.
110. Rochet, F.; Jolly, F.; Bournel, F.; Dufour, G.; Sirotti, F.; Cantin, J.-L. Phys. Rev. B 1998, 58, 11029.
111. Lu, X.; Wang, X.; Yuan, Q.; Zhang, Q. J. Am. Chem. Soc. 2003, 125, 7923.
112. Chu, S.-Y.; Anderson, A. B. Surf. Sci. 1988, 194, 55.
113. Weiner, B.; Carmer, C. S.; Frenklach, M. Phys. Rev. B 1991, 43, 1678.
114. Yoshinobu, J.; Fukushi, D.; Uda, M.; Nomura, E.; Aono, M. Phys. Rev. B 1992, 46, 9520.
115. MacPherson, C. D.; Hu, D. Q.; Doan, M.; Leung, K. T. Surf. Sci. 1994, 310, 231.
116. Rochet, F.; Dufour, G.; Prieto, P.; Sirotti, F.; Stedile, F. C. Phys. Rev. B 1998, 57, 6738.
117. De Renzi, V.; Biagi, R.; del Pennino, U. Phys. Rev. B 2001, 64, 155305.
118. Sbraccia, C.; Pignedoli, C. A.; Catellani, A.; Di Felice, R.; Silvestrelli, P. L.; Toigo, F.; Ancilotto, F.; Bertoni, C. M. Surf. Sci. 2004, 557, 80.
119. Konecny, R.; Doren, D. J. J. Am. Chem. Soc. 1997, 119, 11098.
120. Fitzgerald, D. R.; Doren, D. J. J. Am. Chem. Soc. 2000, 122, 12334.
121. Yoshimura, T.; Tatsuura, S.; Sotoyama, W. Appl. Phys. Lett. 1991, 59, 482.
122. Yoshimura, T.; Tatsuura, S.; Sotoyama, W.; Matsuura, A.; Hayano, T. Appl. Phys. Lett. 1992, 60, 268.
123. Kubono, A.; Yuasa, N.; Shao, H.-L.; Umemoto, S.; Okui, N. Thin Solid Films 1996, 289, 107.
124. Bitzer, T.; Richardson, N. V. Appl. Phys. Lett. 1997, 71, 662.
125. Shao, H.-I.; Umemoto, S.; Kikutani, T.; Okui, N. Polymer 1997, 38, 459.
126. Bitzer, T.; Richardson, N. V. Appl. Surf. Sci. 1999, 144–145, 339.
127. Haq, S.; Richardson, N. V. J. Phys. Chem. B 1999, 103, 5256.
128. Usui, H. Thin Solid Films 2000, 365, 22.
129. Nagai, A.; Shao, H.; Umemoto, S.; Kikutani, T.; Okui, N. High Perform. Polym. 2001, 13, S169.
130. Miyamae, T.; Tsukagoshi, K.; Matsuoka, O.; Yamamoto, S.; Nozoye, H. Jpn. J. Appl. Phys. 2002, 41, 746.
131. Lee, J. S.; Lee, Y.-J.; Tae, E. L.; Park, Y. S.; Yoon, K. B. Science 2003, 301, 818.
132. Kim, A.; Filler, M. A.; Kim, S.; Bent, S. F. J. Am. Chem. Soc. 2005, 127, 6123.
133. Du, Y.; George, S. M. J. Phys. Chem. C 2007, 111, 8509.
134. Putkonen, M.; Harjuoja, J.; Sajavaara, T.; Niinisto, L. J. Mater. Chem. 2007, 17, 664.
135. Adamczyk, N. M.; Dameron, A. A.; George, S. M. Langmuir 2008 24, 2081.
136. Dameron, A. A.; Seghete, D.; Burton, B. B.; Davidson, S. D.; Cavanagh, A. S.; Bertrand, J. A.; George, S. M. Chem. Mater. 2008, 20, 3315.
137. George, S. M.; Yoon, B.; Dameron, A. A. Acc. Chem. Res. 2009, 42, 498.
138. Lee, B. H.; Im, K. K.; Lee, K. H.; Im, S.; Sung, M. M. Thin SolidFilms 2009, 517, 4056.
139. Loscutoff, P. W.; Zhou, H.; Clendenning, S. B.; Bent, S. F. ACS Nano 2009.
140. Peng, Q.; Gong, B.; VanGundy, R. M.; Parsons, G. N. Chem. Mater. 2009, 21, 820.
141. Yoon, B.; O'Patchen, J. L.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Vap. Deposition 2009, 15, 112.
142. Yoshimura, T.; Kudo, Y. Appl. Phys. Expr. 2009, 2, 015502.
143. Teplyakov, A. V.; Kong, M. J.; Bent, S. F. J. Am. Chem. Soc. 1997, 119, 11100.
144. Teplyakov, A. V.; Kong, M. J.; Bent, S. F. J. Chem. Phys. 1998, 108, 4599.
145. Hovis, J. S.; Liu, H.; Hamers, R. J. J. Phys. Chem. B 1998, 102, 6873.
146. Teplyakov, A. V.; Lal, P.; Noah, Y. A.; Bent, S. F. J. Am. Chem. Soc. 1998, 120, 7377.
147. Wang, G. T.; Bent, S. F.; Russell, J. N.; Butler, J. E.; D'Evelyn, M. P. J. Am. Chem. Soc. 2000, 122, 744.
148. Kong, M. J.; Teplyakov, A. V.; Jagmohan, J.; Lyubovitsky, J. G.; Mui, C.; Bent, S. F. J. Phys. Chem. B 2000, 104, 3000.
149. Teague, L. C.; Boland, J. J. J. Phys. Chem. B 2003, 107, 3820.
150. Teague, L. C.; Boland, J. J. Thin Solid Films 2004, 464-465, 1.
151. Teague, L. C.; Chen, D.; Boland, J. J. J. Phys. Chem. B 2004, 108, 7827.
152. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 1999, 103, 6803.
153. Baik, J.; Kim, M.; Park, C.-Y.; Kim, Y.; Ahn, J. R.; An, K.-S. J. Am. Chem. Soc. 2006, 128, 8370.
154. Ellison, M. D.; Hovis, J. S.; Liu, H.; Hamers, R. J. J. Phys. Chem. B 1998, 102, 8510.
155. Armstrong, J. L.; Pylant, E. D.; White, J. M. J. Vac. Sci. Technol. A 1998, 16, 123.
156. Armstrong, J. L.; White, J. M.; Langell, M. J. Vac. Sci. Technol. A 1997, 15, 1146.
157. Barriocanal, J. A.; Doren, D. J. J. Am. Chem. Soc. 2001, 123, 7340.
158. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 2001, 105, 12559.
159. Hamai, C.; Takagi, A.; Taniguchi, M.; Matsumoto, T.; Kawai, T. Angew. Chem. Int. Ed. 2004, 43, 1349.
160. Kachian, J. S.; Wong, K. T.; Bent, S. F. Acc. Chem. Res. 2010, 43, 346.
161. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Am. Chem. Soc. 2002, 124, 8990.
162. Tang, H. H.; Cai, Y. H.; Ning, Y. S.; Lai, Y. H.; Xu, G. Q. Surf. Sci. 2007, 601, 3293.
163. Bacalzo-Gladden, F.; Lu, X.; Lin, M. C. J. Phys. Chem. B 2001, 105, 4368.
164. Schwartz, M. P.; Hamers, R. J. Surf. Sci. 2002, 515, 75.
165. Choi, C. H.; Gordon, M. S. J. Am. Chem. Soc. 2002, 124, 6162.
166. Rangan, S.; Bournel, F.; Gallet, J. J.; Kubsky, S.; Le Guen, K.; Dufour, G.; Rochet, F.; Sirotti, F.; Carniato, S.; Ilakovac, V. Phys. Rev. B 2005, 71, 165319.
167. Schwartz, M. P.; Hamers, R. J. Surf. Sci. 2007, 601, 945.
168. Bournel, F.; Gallet, J. J.; Kubsky, S.; Dufour, G.; Rochet, F.; Simeoni, M.; Sirotti, F. Surf. Sci. 2002, 513, 37.
169. Bu, Y.; Ma, L.; Lin, M. C. J. Phys. Chem. 1993, 97, 7081.
170. Lu, X.; Xu, X.; Wu, J.; Wang, N.; Zhang, Q. New J. Chem. 2002, 26, 160.
171. Mui, C.; Filler, M. A.; Bent, S. F.; Musgrave, C. B. J. Phys. Chem. B 2003, 107, 12256.
172. Tao, F.; Sim, W. S.; Xu, G. Q.; Qiao, M. H. J. Am. Chem. Soc. 2001, 123, 9397.
173. Tao, F.; Wang, Z. H.; Qiao, M. H.; Liu, Q.; Sim, W. S.; Xu, G. Q. J. Chem. Phys. 2001, 115, 8563.
174. Filler, M. A.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Am. Chem. Soc. 2003, 125, 4928.
175. Cho, J.-H.; Kleinman, L. J. Chem. Phys. 2003, 119, 6744.
176. Miotto, R.; Oliveira, M. C.; Pinto, M. M.; de León-Pérez, F.; Ferraz, A. C. Phys. Rev. B 2004, 69, 235331.
177. Carniato, S.; Rochet, F.; Gallet, J. J.; Bournel, F.; Dufour, G.; Mathieu, C.; Rangan, S. Surf. Sci. 2007, 601, 5515.
178. Tao, F.; Chen, X. F.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2002, 106, 3890.
179. Bournel, F.; Carniato, S.; Dufour, G.; Gallet, J. J.; Ilakovac, V.; Rangan, S.; Rochet, F.; Sirotti, F. Phys. Rev. B 2006, 73, 125345.
180. Barriocanal, J. A.; Doren, D. J. J. Vac. Sci. Technol. A 2000, 18, 1959.
181. Ellison, M. D.; Hamers, R. J. J. Phys. Chem. B 1999, 103, 6243.
182. Loscutoff, P. L.; Wong, K. T.; Bent, S. F. J. Phys. Chem. C 2010 114, 14193.
1 This chapter was completed in 2009 and therefore does not capture new developments in the field occurring since 2009.
Functionalization of Semiconductor Surfaces, First Edition
Edited by Franklin (Feng) Tao and Steven L. Bernasek.
© 2012 John Wiley & Sons, Inc. Published 2012 by John Wiley & Sons, Inc.