
Chapter 17 Durability: Oxidation, corrosion, degradation
- 17.1 Introduction and synopsis 386
- 17.2 Oxidation, flammability and photo-degradation 387
- 17.3 Oxidation mechanisms 389
- 17.4 Resistance to oxidation, burning and photo-degradation 393
- 17.5 Corrosion: acids, alkalis, water and organic solvents 395
- 17.6 Drilling down: mechanisms of corrosion 395
- 17.7 Fighting corrosion 403
- 17.8 Summary and conclusions 416
- 17.9 Further reading and software 416
- 17.10 Exercises 417
- 17.11 Exploring design with CES 418
- 17.12 Exploring the science with CES Elements 419
The Gospel according to St Matthew reminds us that we live in a world in which ‘moth and rust doth corrupt’—a world of corrosion, degradation and decay (cover picture). Not a happy thought with which to start a chapter. But by understanding them we can, to a degree, control them. This chapter describes ways in which materials degrade or corrode, the underlying mechanisms and what can be done to slow them down.

17.1 Introduction and synopsis
The Gospel according to St Matthew reminds us that we live in a world in which ‘moth and rust doth corrupt’—a world of corrosion, degradation and decay (cover picture). Not a happy thought with which to start a chapter. But by understanding them we can, to a degree, control them. This chapter describes ways in which materials degrade or corrode, the underlying mechanisms and what can be done to slow them down.
Durability is a key material attribute, one central to the safety and economy of products. It is one of the more difficult attributes to characterise, quantify and use for selection for the following reasons.
- It is a function not just of the material but of the environment in which it operates.
- There are many mechanisms, some general, some peculiar to particular materials and environments.
- Material combinations (as in galvanic corrosion) and configuration (as in crevice corrosion) play a role.
Figure 17.1 shows some of the considerations involved. The central players are Materials and Environments. But there are other considerations in choosing materials for durability, some of them listed in the figure.
- There is the way they are used, labeled Application, because of the added constraints that this involves. Some, like aerospace, require lightweight materials, some require nonflammable materials, some biocompatibility. Some (like those of civil engineering) use materials on a vast scale, which therefore have to be cheap; others (like those of consumer electronics) on a very small scale, allowing expensive materials.
- There are the steps that can be taken to provide Protection, some generally applicable (like painting), some less so (such as the use of inhibitors).
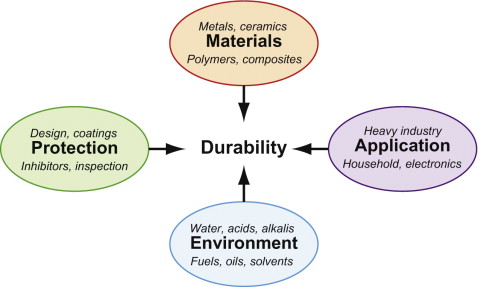
Because of these, there are preferred material choices for use in a given environment—those that, through experience, best meet both the primary constraint of resisting attack and the secondary constraints of stiffness, strength, low density, flammability, cost and so forth. We return to these in Section 17.7.
Much of our current understanding of degradation is empirical, enlightened by an understanding of the underlying mechanisms. We start by examining these. There are many. There are the chemical reactions triggered by radiation and heat, changing the properties in irreversible ways. There are electro-chemical mechanisms that appear in attack by aqueous solutions, acids and alkalis. Organic solvents attack polymers by a number of mechanisms. The mechanisms are well understood but this alone is not sufficient to guide material choice without also drawing on experience. For this reason this chapter has an unusually large number of unusually large tables: they store the empirical knowledge of corrosion resistance, preferred material and preferred corrosion-protection method. Don’t try to ‘read’ the tables; simply inform yourself of their structure and use them as look-up tables when making a material selection. The Exercises at the end of the chapter provide practice.
Because many different properties are involved here, the exploration of the science is split into two: one delving into oxidation, flammability and photo-degradation; the other into corrosion by liquids. Each section ends with a description of ways in which degradation is inhibited.
17.2 Oxidation, flammability and photo-degradation
The most stable state of most elements is as an oxide. For this reason the Earth’s crust is almost entirely made of simple or complex oxides: silicates like granite, aluminates like basalt, carbonates like limestone. Techniques of thermo-chemistry, electro-chemistry and synthesis allow these to be refined into the materials we use in engineering, but these are not, in general, oxides. From the moment materials are made they start to re-oxidise, some extremely slowly, others more quickly; and the hotter they are, the faster it happens. For safe high-temperature design we need to understand rates of oxidation.
Definition and measurement
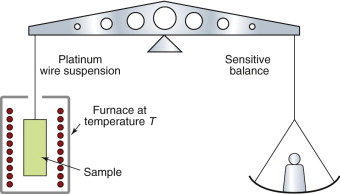
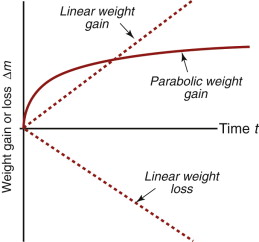
Oxidation rates of metals are measured in the way sketched in Figure 17.2: a thin sheet of the material (to give a lot of surface) is held at temperature T for an increasing time t, and the gain or loss in weight, Δm, is measured. If the oxide adheres to the material the sample gains weight in a way that is either linear (Δm ∝ t) or parabolic (Δ ∝ t1/2) in time t; if instead the oxide is volatile, the sample loses weight linearly with time (Δm ∝ −t), as in Figure 17.3.
Polymers, too, oxidise, but in a more spectacular way. That brings us to flammability.
Flammability
Ceramics and glasses do not burn. Metals, if dispersed as a fine powder, are potentially combustible, but in bulk flammability is not normally an issue, even for magnesium. Most polymers, on the other hand, are inherently flammable, although to differing degrees: some burn spontaneously if ignited; others are self-extinguishing, burning only when directly exposed to flame.
There are several ways to characterise flammability. The most logical is the Limiting oxygen index (LOI): it is the oxygen concentration, in %, required to maintain steady burning. Fresh air has about 21% oxygen in it. A polymer with an oxygen index lower than 21% will burn freely in air; one with an oxygen index that is larger will extinguish itself unless a flame is played onto it—then it burns. Thus a high oxygen index means resistance to self-sustained burning. Less logical, but much used, is the Underwriters Laboratory (UL) rating in which a strip of polymer 1.6 mm thick is held horizontally (H) or vertically (V) and ignited. Its response is recorded as a code such as HB, meaning ‘horizontal burn’. The scales are compared in Table 17.1.
Table 17.1 Flammability ratings compared
| Oxygen index (LOI) | UL 94 (1.6 mm) rating | Flammability |
|---|---|---|
| Up to 16 | Unrated | Highly flammable |
| 16–20 | HB | |
| 21–24 | HB | Slow burning |
| 25–29 | V-2 | Self-extinguishing |
| 30–43 | V-0 | |
| Over 44 | V-0 or better (e.g., 5 V) |
Photo-degradation
You don’t have to set fire to a polymer for it to oxidise. Polymers and elastomers age when exposed to light (particularly UV) and oxygen, causing loss of strength, stiffness and toughness, discoloration and loss of gloss. This is countered by additives: antioxidants, light stabilisers and fluorescent whitening agents. Some of these are so universal that the ‘standard’ grade of the polymer already contains enough of one or another of them to give it acceptable resistance; PP, ABS, PS, PET, PMMA, PC, nylons and PU all need UV protection.
17.3 Oxidation mechanisms
Mechanisms of oxidation
The rate of oxidation of most metals in dry air at room temperature is too slow to be an engineering problem; indeed, slight oxidation can be beneficial in protecting metals from corrosion of other sorts. But heat them up and the rate of oxidation increases, bringing problems. The driving force for a metal to oxidise is its free energy of oxidation—the energy released when it reacts with oxygen, but a big driving force does not necessarily mean rapid oxidation. The rate of oxidation is determined by the kinetics (rate) of the oxidation reaction, and that has to do with the nature of the oxide. When any metal (with the exception of gold, platinum and a few others that are even more expensive) is exposed to air, an ultra-thin surface film of oxide forms on it immediately, following the oxidation reaction
The film now coats the surface, separating the metal beneath from the oxygen. If the reaction is to go farther, oxygen must get through the film.
The weight-gain shown in Figure 17.3 reveals two different types of behavior. For some metals the weight gain per unit area, Δm, is linear, and this implies that the oxidation is progressing at a constant rate:
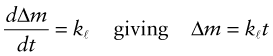
where kℓ (kg/m2.s) is the linear kinetic constant. This is because the oxide film is porous or cracks (and, when thick, spalls off) and does not protect the underlying metal. Some metals behave better than this. The film that develops on their surfaces is compact, coherent and strongly bonded to the metal. For these the weight gain per unit area of surface is parabolic, slowing up with time, and this implies an oxidation rate with the form
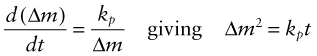
where kp (kg2/m4.s) is the parabolic kinetic constant.
Example 17.1
A metal is found, by testing, to oxidise at 500 °C with a parabolic rate constant kp = 3 × 10−10 kg2/m4.s. What is the weight gain per unit area after 1000 hours?
In these equations Δm is the weight gain per unit area—that means the weight of the oxygen that has been incorporated into the oxide. The mass of the oxide itself is greater than this because it contains metal atoms (M) as well as oxygen (O). If the formula of the oxide is MO, the mass per unit area of the oxide is
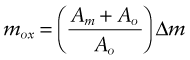
where Am is the atomic weight of the metal and Ao is that of oxygen. The thickness of the oxide is simply x = mox/ρox where ρox is the oxide density. Using equation (17.3) we find
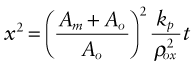
The oxide film, once formed, separates the metal from the oxygen. To react farther either oxygen atoms must diffuse inward through the film to reach the metal or metal atoms must diffuse outward through the film to reach the oxygen. The driving force is the free energy of oxidation, but the rate of oxidation is limited by the rate of diffusion, and the thicker the film, the longer this takes.
Example 17.2
The oxide of the last example is MgO. The atomic weight of magnesium is 24.3 kg/kmol and that of oxygen is 16 kg/kmol and the density of MgO is 3550 kg/m3. How thick will the oxide be after 1000 hours at 500 °C?
Figure 17.4 shows a growing oxide film. The reaction creating the oxide MO
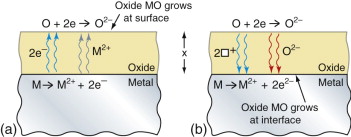
Figure 17.4 Oxidation mechanisms (a) growth by metal diffusion and electron conduction, (b) growth by diffusion of oxygen and holes.
goes in two steps. The metal first forms an ion, M2+ say, releasing electrons:
The electrons are then absorbed by oxygen to give an oxygen ion:
The problem is that the first of these reactions occurs at the metal side of the oxide film whereas the oxygen reaction is on the other side. Either the metal ions and the electrons must diffuse out to meet the oxygen, or the oxygen and electron holes (described in Chapter 14) must diffuse in to find the metal. If the film is an electrical insulator, as many oxides are, electrons cannot move through it, so it is the oxygen that must diffuse in. The concentration gradient of oxygen is that in the gas Co divided by the film thickness, x. The rate of growth of the film is proportional to the flux of atoms diffusing through the film, giving
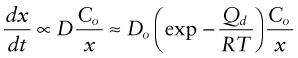
where D is the diffusion coefficient, Do and Qd are the pre-exponential and the activation energy for oxygen diffusion. Integrating gives
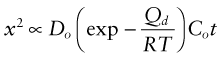
This has the same form as equation (17.4) with
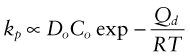
This explains why oxidation rates rise steeply with rising temperature, and why the growth is parabolic. The most protective films are those with low diffusion coefficients, and this means that they have to have high melting points. This is why the Al2O3 oxide film on aluminum, the Cr2O3 film on stainless steel and chrome plate and the SiO2 film on high silicon cast iron are so protective.
Not all oxides grow with parabolic kinetics, as we have seen. Those that show linear weight gain do so because the oxide that forms is not compact, but has cracks or spalls off because of excessive volume change, leaving the fresh surface continually exposed to oxygen. Linear weight loss, the other behaviour, occurs when the oxide is volatile, and simply evaporates as it forms.
Example 17.3
The activation energy for oxygen diffusion in MgO is 370 kJ/mol. If the test in Example 17.2 had been carried out at the temperature T1 = 450 °C rather than at T1 = 500 °C how thick would the oxide be after 1000 hours? The gas constant R = 8.31 × 10−3 kJ/mol.K.
Answer. The thickness scales with temperature as ![]() , so the thickness x1 after 1000 hours at 450 °C is less than that, xo, at 500 °C by the factor
, so the thickness x1 after 1000 hours at 450 °C is less than that, xo, at 500 °C by the factor
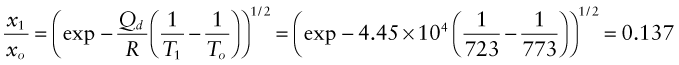
17.4 Resistance to oxidation, burning and photo-degradation
Elements for heaters, furnace components, power generation and chemical engineering plants all require materials that can be used in air at high temperatures. If it is a pure metal that you want it has to be platinum (and indeed some special furnaces have platinum windings) because the oxide of platinum is less stable than the metal itself. If you want something more affordable, it has to be an alloy.
Oxides, of course, are stable at high temperature—they are already oxidised. One way to provide high-temperature protection is to coat metals like cast irons, steels or nickel alloys with an oxide coating. Stoves are protected by enameling (a glass coating, largely SiO2); turbine blades are coated with plasma-sprayed TBCs based on the oxide zirconia (ZrO2) (Chapter 13, Section 13.6). But coatings of this sort are expensive, and, if damaged, they cease to protect.
There is another way to give oxidation resistance to metals, and it is one that repairs itself if damaged. The oxides of chromium (Cr2O3), aluminum (Al2O3), titanium (TiO2) and silicon (SiO2) have very high melting points, so the diffusion of either the metal or oxygen through them is very slow. They are also electrical insulators so electrons cannot move through them either. The mechanism of oxidation requires both diffusion and conduction, so the rate constant kp is very small: the oxide stops growing when it is a few molecules thick. These oxides adhere well and are very protective; it is they that make these otherwise-reactive metals so passive. The films can be artificially thickened by anodising—an electro-chemical process for increasing their protective power. Anodised films accept a wide range of coloured dyes, giving them a decorative as well as a protective function.
If enough chromium, aluminum or silicon can be dissolved in a metal like iron or nickel—‘enough’ means 18–20%—a similar protective oxide grows on the alloy. And if the oxide is damaged, more chromium, aluminum or silicon immediately oxidises, repairing the damage. Stainless steels (typical composition Fe–18% Cr–8% Ni), widely used high temperature equipment, and nichromes (nickel with 10–30% chromium), used for heating elements, derive their oxidation resistance in this way; so, too, do the aluminum bronzes (copper with 10% aluminum) and high silicon cast irons (iron with 16–18% silicon). To see just how important this way of suppressing oxidation has become, look back for a moment at Figure 13.8. It is the table guiding material choice at low and high temperature. All the metals above the 400 °C mark rely on alloy methods for their protection.
Flammability: how do polymers burn and how do you stop them?
The combustion of a polymer is an exothermic reaction in which hydrocarbons are oxidised to CO2 and H2O. That sounds simple, but it isn’t. Combustion is a gas phase reaction; the polymer or its decomposition-products must become gaseous for a fire to begin. When you light a candle you are melting the wax and raising it to the temperature at which it pyrolyses (400–800 °C) forming gaseous hydrocarbon decomposition products. These gasses react in the flame to produce heat, which melts and pyrolyses more wax, keeping the reaction going.
A fully developed fire results when an ignition source like a spark or cigarette ignites combustible material such as paper. Its heat radiates out causing other combustible materials (particularly polymers and fabrics) to decompose into a flammable gas mix. Flashover (Figure 17.5) occurs when these gasses ignite, instantly spreading the fire over the entire area and producing temperatures of over 1000 °C.
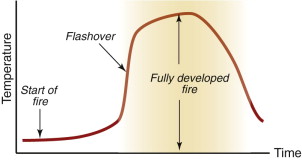
Combustion involves the reaction of free-radicals. At high temperatures hydrogen, together with one electron from its covalent bond (symbol H·) is freed from a carbon atom of the polymer molecule, leaving a highly reactive free radical R· in the gas. The hydrogen radical reacts with oxygen and the hydrocarbon radical to give CO2 and heat, releasing the H· again to propagate the reaction further.
Flame retardants work in one of two ways. Some scavenge the free radicals, tying them up harmlessly and so retarding or snuffing out the combustion reaction. These are usually compounds containing chlorine or bromine; they, too, pyrolyse to give free radicals Cl· and Br· that attach themselves to the hydrocarbon radical, removing it from the reaction. Others work by creating a protective layer of water vapor between the solid polymer and the gaseous decomposition products, limiting heat transfer, cooling it and reducing pyrolisation. Typical among these is the addition of Mg(OH)2 that decomposes at about 300 °C, releasing H2O and leaving inert MgO. Polymers containing flame retardants are identified by adding ‘FR’ to their names.
What causes photo-degradation?
As we have seen, commercial polymers are long chain, high molecular weight hydrocarbons. When exposed to radiation chemical reactions are triggered that change their chemical composition and molecular weight. These reactions, called photo-oxidation or photo-degradation, also create free radicals. They trigger a change in the physical and optical properties of the polymer, making them brittle and, if transparent, turning them white or gray. Once this starts it sets off a chain reaction that accelerates degradation unless stabilisers are used to interrupt the oxidation cycle. Heat, too, can trigger degradation in an oxygen-containing atmosphere.
Ultraviolet absorbers such as benzophenone or the benzotriazole work by preferentially absorbing UV radiation, following the grandly named Beer–Lambert law, which states the obvious: that the amount of UV radiation absorbed increases with increases in the sample thickness and stabiliser concentration. In practice, high concentrations of absorbers and sufficient thickness of the polymer are required before enough absorption takes place to effectively retard photo-degradation. Hindered amine stabilisers (HALS) are a more efficient way to stabilise against light-induced degradation of most polymers. They do not absorb UV radiation, but act to inhibit degradation of the polymer; their efficiency and longevity are due to a cyclic process wherein the HALS are regenerated rather than consumed. Low concentrations are enough to give good stabilisation.
17.5 Corrosion: acids, alkalis, water and organic solvents
Table 17.2 lists some of the more common liquids with which products have contact, with an indication of where they are found. The same environments appear in the tables that follow.
Table 17.2 Commonly encountered liquid environments*
| Environment | Where encountered |
|---|---|
| Water and aqueous solutions | |
| Water (fresh) | Fresh water is ubiquitous: any object: exposure to high humidity, rain or washing acquires a film of water containing (unless distilled) dissolved oxygen and, usually, other impurities. |
| Water (salt) | Materials in marine environments are exposed to salt water and wind-carried spray. Seawater varies in composition depending on the location. It is typically 3.5%. |
| Soils | Soils differ greatly in composition, moisture content and pH. The single most important property of a soil that determines its corrosive behaviour is its electrical resistivity—a low resistivity means that the water in the soil has high concentration of dissolved ions. A resistivity below 109 µohm.cm is very corrosive; one with a resistivity above 2 × 1010 µohm.cm is only slightly corrosive. The choice of material for use in soil depends on this and on the pH. Acidic (peaty) soils have a low pH, alkali soils (those containing clay or chalk) have a high one. |
| Body fluids | Body fluids include blood, urine, saliva, sweat and gastric fluids. All are water-based with high ion-content, some acidic, stimulating electro-chemical and acid attack. |
| Acids and alkalis | |
| Acetic acid, CH3COOH | Acetic acid is an organic acid made by the oxidation of ethanol. It is used in the production of plastics, dyes, insecticides and other chemicals. Dilute acetic acid (vinegar) is used in cooking. |
| Hydrochloric acid, HCl | Hydrochloric acid is used as a chemical intermediate, for ore reduction, for pickling steel, in acidising oil wells and in other industrial processes. In dilute form it is a component of household cleaners. |
| Hydrofluoric acid, HF | Hydrofluoric acid is used for the etching of glass, synthesis of fluorocarbon polymers, aluminum refining, as an etchant for silicon-based semiconductors and to make UF6 for uranium isotope separation. |
| Nitric acid HNO3 | Nitric acid is used in the production of fertilizers, dyes, drugs and explosives. |
| Sulfuric acid, H2SO4 | Sulfuric acid, of central importance in chemical engineering, is used in making fertilizers, chemicals, paints and in petrol refining. It is a component of acid rain. |
| Sodium hydroxide, NaOH | Sodium hydroxide of this concentration is found in some household cleaners, the making of soap and in the cleaning in food processing. |
| Fuels, oils and solvents | |
| Benzene, C6H6 | Benzene is used as an industrial solvent, as well as in the synthesis of plastics, rubbers, dyes and certain drugs. It is also found in tobacco smoke. |
| Carbon tetrachloride, CCl4 | Carbon tetrachloride is used principally to manufacture refrigerants, as a dry-cleaning solvent and as a pesticide (now banned in the United States). |
| Crude oil | Refined petroleum is not corrosive to metals, but crude oil usually contains saline water, sulfur compounds and other impurities, some acidic. |
| Diesel oil | Diesel oil is a specific fractional distillate of crude oil. It is the primary fuel for truck, shipping and nonelectric and diesel-electric rail transport. Its use for car propulsion is increasing. Diesel oil acts both as fuel and as lubricant in the engine. |
| Kerosene (paraffin oil) | Paraffin is used as aviation fuel, as well as being commonly used for heating and lighting on a domestic level. It is used to store highly reactive metals to isolate them from oxygen. |
| Lubricating oil | Oil is used as a lubricant in most metal systems with moving parts. Typically, these are mineral oils, and frequently contain sulfur. Low sulfur synthetic oils can also be produced. Lubricating oil is less corrosive than petroleum and diesel oil because it is based on hydrocarbons with higher molecular weight. |
| Petroleum (gasoline) | Petroleum is a volatile distillate of crude oil. It is used mainly to power engines, for cars, light aircraft and agricultural equipment. It often contains additives such as lead, ethanol or dyes. |
| Silicone fluid, ((CH3)2SiO)n | Silicone oils are synthetic silicon-based polymers. They are exceptionally stable and inert. They are used as brake and hydraulic fluids, vacuum pump oils, and as lubricants for metals and for textile threads during sewing and weaving of fabrics. |
| Vegetable oil | Vegetable oils are derived from olive, peanut, maize, sunflower, rape and other seed and nut crops. They are widely used in the preparation of foods. They are the basis of bio-fuels. |
| Alcohols, aldehydes, ketones | |
| Acetone, CH3COCH3 | Acetone is the simplest of ketones. It is used widely as a degreasing agent and a solvent, and as a thinning agent for polyester resins and other synthetic paints (commonly nail varnish), a cleaning agent and as an additive in automobile fuels. It is used in the manufacture of plastics, drugs, fibres and other chemicals. |
| Ethyl alcohol, C2H5OH | Ethanol is made by fermentation, and thus in alcoholic beverages. It is used medically as a solvent for disinfectants and for cleaning wounds before dressing them. It is used industrially as a solvent, a dehydrating agent and as a ‘green’ fuel for cars. |
| Methyl alcohol, CH3OH | Methanol is used in glass cleaners, stains, dyes, inks, antifreeze, solvents, fuel additives and as an extractant for oil. It is also used as a high-energy fuel for cars, aircraft and rockets, and is a possible fuel for fuel cells. |
| Formaldehyde, CH2O | Formaldehyde is used as a disinfectant in medical applications. It is used industrially to make many resins (including melamine resin and phenol formaldehyde resin) and glues, including those used in plywood. It is found in car exhausts and tobacco smoke. It is the basis of embalming fluids. |
* The CES software contains information of this sort for a larger number of environments.
Aqueous solutions, acids and alkalis
Even when pure, water causes corrosion if oxygen is available. The rusting of steel in fresh water, alone, presents enormous economic and reliability problems. Add dissolved ions (like NaCl) and the corrosion rate increases dramatically.
Strong acids like H2SO4 (sulfuric acid), HNO3 (nitric acid) and HCl (hydrochloric acid) are widely used in the chemical industry. So too are strong alkalis like NaOH (caustic soda). Household products contain some of these in dilute form, but in the home it is organic acids like vinegar (acetic acid) and milder alkalis like washing soda (sodium carbonate) that are more commonly found.
Dunk a metal into an acid and you can expect trouble. The sulfates, nitrates, chlorides and acetates of most metals are more stable than the metal itself. The question is not whether the metal will be attacked, but how fast. As with oxidation, this depends on the nature of the corrosion products and on any surface coating the metal may have. The oxide film on aluminum, titanium and chromium protects them from some acids provided the oxide itself is not damaged. Alkalis, too, attack some metals. Zinc, tin, lead and aluminum react with NaOH, for instance, to give zincates, stannates, plumbates and aluminates (as anyone who has heated washing soda in an aluminum pan will have discovered).
Ways of combating corrosion by water, acids and alkalis are described in Section 17.7. Corrosion, however, can sometimes be beautiful: the turquoise patina of copper on roofs and the rich browns of bronze statues are created by corrosion.
Oils and organic solvents
Organic liquids are ubiquitous. We depend on them as fuels and lubricants, for cooking, for removing stains, as face-cream, as nail varnish and much more—any material in service will encounter them. Metals, ceramics and glasses are largely immune to them, and some polymers can tolerate some organic liquids without problems. But not all. The identification of solvents that are incompatible with a given polymer is based largely on experience, trial and error and intuition guided by such rules as ‘like dissolves like’, meaning that nonpolar solvents like benzene (C6H6) or carbon tetrachloride (CCl4) tend to attack nonpolar polymers like polypropylene or polyethylene, while polar solvents such as acetone (CH3COCH3) tend to attack polar polymers like PMMA and nylon.
17.6 Drilling down: mechanisms of corrosion
Ions in solution and pH
Pure water, H2O, dissociates a little to give a hydrogen ion, H+, and a hydroxyl ion, OH−:
The product of the concentrations of the two ions is constant: increase one, and the other falls. This is known as Law of Mass Action:
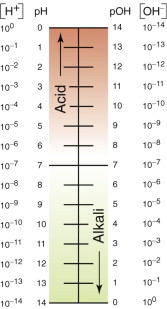
where the square brackets mean ‘molar concentration’. In pure water there are equal numbers of the two types of ion, [H+] = [OH−], and the value of the constant, when measured, is 10−14. Thus the molar concentration of both ion-types is 10−7; one H2O molecule in 107 is ionised. The pH of the ionised water is defined as the negative of the log of the hydrogen ion concentration:
so, for pure water, pH = 7.
Acids dissociate in water to give H+ ions. Sulfuric acid, for instance, dissociates
This pushes up the concentration [H+] and, because of equation (17.9), it pulls down the concentration [OH−]; weak acids have a pH of 4–6; strong ones a pH up to 0. Alkalis do the opposite. Sodium hydroxide, for example, dissociates when dissolved in water, to give OH− ions:
Weak alkalis have a pH of 8–10, strong ones a pH up to 13 (Figure 17.6).
Corrosion by acids and alkalis is an electro-chemical reaction. One-half of this is the dissociation reaction of a metal M into a metal ion, Mz+, releasing electrons e−
where z, an integer of 1, 2 or 3, is the valence of the metal. Acidic environments, with high [H+] (and thus low pH) stimulate this reaction; thus a metal such as copper, in sulfuric acid solution, reacts rapidly:
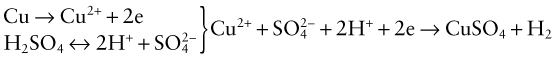
Some metals are resistant to attack by some acids because the reaction product, here CuSO4, forms a protective surface layer; thus lead-lined containers are used to process sulfuric acid because lead sulfate is protective.
Alkalis, too, cause corrosion via an electro-chemical reaction. Zinc, for instance, is attacked by caustic soda via the steps
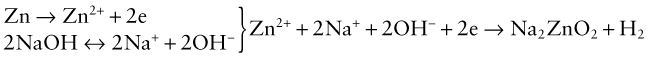
Fresh and impure water
Corrosion, as we have just seen, is the degradation of a metal by an electro-chemical reaction with its environment. Figure 17.7 illustrates in more detail the idea of an electro-chemical reaction. If a metal is placed in a conducting solution like salt water, it dissociates into ions, releasing electrons, as the iron is shown doing in the figure, via the anodic reaction
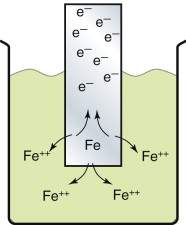
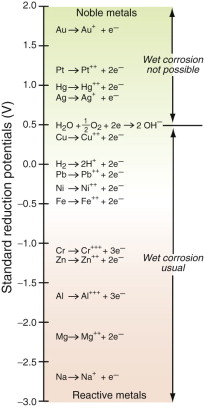
The electrons accumulate on the iron giving it a negative charge that grows until the electrostatic attraction starts to pull the Fe++ ions back onto the metal surface, stifling further dissociation. At this point the iron has a potential (relative to a standard, the hydrogen standard) of −0.44 volts. Each metal has its own characteristic potential (called the standard reduction potential), as plotted in Figure 17.8. The extra electrons enter the band structure of the metal just above the Fermi level, so the energy associated with the Fermi level determines how strongly they are held.
If two metals are connected together in a cell, like the iron and copper samples in Figure 17.9, a potential difference equal to their separation on Figure 17.8 appears between them. The corrosion potential of iron, −0.44, differs from that of copper, +0.34, by 0.78 volts, so if no current flows in the connection the voltmeter will register this difference. If a current is now allowed, electrons flow from the iron (the anode) to the copper (the cathode); the iron ionises (that is, it corrodes), following the anodic reaction of equation (17.13) and—if the solution were one containing copper sulfate—copper ions, Cu++, plate out onto the copper following the cathodic reaction
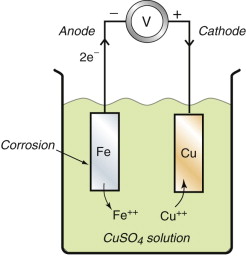
Figure 17.9 A bi-metal corrosion cell. The corrosion potential is the potential to which the metal falls relative to a hydrogen standard.
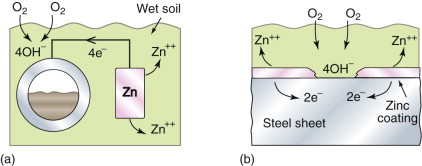
Suppose now that the liquid is not a copper sulfate solution, but just water (Figure 17.10). Water dissolves oxygen, so unless it is specially de-gassed and protected from air, there is oxygen in solution. The iron and the copper still dissociate until their corrosion potential-difference is established but now, if the current is allowed to flow, there is no reservoir of copper ions to plate out. The iron still corrodes but the cathodic reaction has changed; it is now the hydrolysis reaction
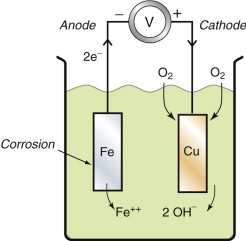
While oxygen can reach the copper, the corrosion reaction continues, creating Fe++ ions at the anode and OH− ions at the cathode. They react to form insoluble Fe(OH)2, which ultimately oxidises further to Fe2O3.H2O, rust.
Thus connecting dissimilar metals in either pure water or water with dissolved salts is a bad thing to do: corrosion cells appear that eat up the metal with the lower (more negative) corrosion potential. Worse news is to come: it is not necessary to have two metals: both anodic and cathodic reactions can take place on the same surface. Figure 17.11 shows how this happens. Here an iron sample is immersed in water with access to air. The part of the sample nearest the water surface has an easy supply of oxygen; that farther away does not. On the remoter part the ionisation reaction of equation (17.13) takes place, corroding the iron and releasing electrons that flow up the sample to the near-surface part where oxygen is plentiful, where they enable the hydrolysis reaction of equation (17.15). The hydrolysis reaction has a corrosion potential of +0.81 volts—it is shown in Figure 17.8—and the difference between this and that of iron, −0.44 volts, drives the corrosion. If the sample could be cut in two along the broken line in Figure 17.11 and a tiny voltmeter inserted, it would register the difference: 1.23 volts.
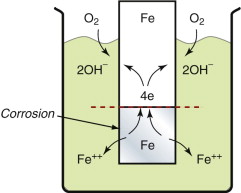
This differential oxidation corrosion is one of the most usual and most difficult to prevent: where there is water and a region with access to oxygen and one that is starved of it, a cell is set up. Only metals above the hydrolysis reaction potential of +0.81 volts in Figure 17.8 are immune.
Selective corrosion
Often, wet corrosion, instead of being uniform, occurs selectively, and when it does, it can lead to failure more rapidly than the uniform rate would suggest. Stress and corrosion acting together (‘stress-corrosion’ and ‘corrosion-fatigue’) frequently lead to localised attack, as do local changes in microstructure such as those at a welded joint. Briefly, the localised mechanisms are these.
Intergranular corrosion occurs because grain boundaries have chemical properties that differ from those of the grain, because the disregistry there gives these atoms higher energy. Pitting corrosion is a preferential attack that can occur at breaks in the natural oxide film on metals, or at precipitated compounds in certain alloys. Galvanic attack at the microstructural level appears in alloys with a two-phase microstructure, made up of regions alternately of one microstructure and of another. The two regions will, in general, lie at slightly different points on the reduction potential scale. If immersed in a conducting solution, thousands of tiny corrosion cells appear, causing the phase that lies lower in reduction potential to be eaten away.
Stress corrosion cracking is accelerated corrosion, localised at cracks in loaded components. The stress breaks the protective oxide film and the elastic energy at and near the crack tip stimulates attack there. The result is that cracks grow under a stress intensity Kscc the is far below K1c. Examples are brass in ammonia, mild steel in caustic soda and some aluminum and titanium alloys in salt water. Corrosion fatigue refers to the accelerated rate at which fatigue cracks grow in a corrosive environment. The endurance limit of some steels in salt water is reduced by a factor as large as 4. The rate of fatigue crack growth, too, increases to a level that is larger than the sum of the rates of corrosion and of fatigue acting together.
17.7 Fighting corrosion
In fighting corrosion, there are four broad strategies (Figure 17.12):
- Judicious design, meaning informed material choice and choice of geometry and configuration.
- Protective coatings, passive (meaning that the coating simply excludes the corrosive medium); and active (meaning that the coating protects even when incomplete).
- Corrosion inhibitors, chemicals added to the corrosive medium that retard the rate of the corrosion reaction.
- Monitoring, with protective maintenance or regular replacement.
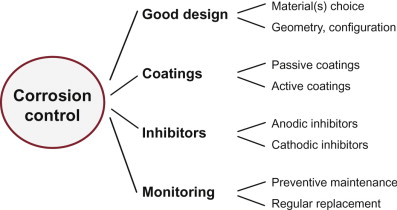
We start with the first: design.
Design: material choice
Materials differ greatly in their vulnerability to attack by corrosive media, some surviving unscathed whereas others, in the same environment, are severely attacked. There are no simple rules or indices for predicting susceptibility. The best that can be done is a ranking, based on experience. Table 17.3 lists the vulnerability of common engineering materials to 23 commonly encountered fluids, on a four-point scale from A (excellent resistance to attack) to D (unacceptably rapid attack). Tables like these (or their computer-based equivalent1) allow screening to eliminate materials that might present severe problems. But, as explained in the introduction to this chapter, there is more to it than that. The best choice of material depends not just on the environment in which it must operate, but—for economic reasons—on the application: it may be cheaper to live with some corrosion (providing regular replacement) than to use expensive materials with better intrinsic resistance. And there is the potential for protection of vulnerable materials by coatings, by inhibitors or by electro-chemical means. Experience in balancing these influences has led to a set of ‘preferred’ materials for use in any given environment. Table 17.4 presents these for the 23 environments that appear in the tables of this chapter.
Table 17.3 Corrosion-resistance ranking of materials in common environments
| Environment | Aqueous environments | Acids and alkalis | Fuels, oils and solvents | Alcohols and aldehydes | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Material | Water (fresh) | Water (salt) | Soils, acidic (peat) | Soils, alkaline (clay) | Acetic acid (10%) | Hydrochloric acid (10%) | Hydrofluoric acid (40%) | Nitric acid (10%) | Sulfuric acid (10%) | Sodium hydroxide (10%) | Sodium hydroxide (60%) | Benzene | Carbon tetrachloride | Diesel oil | Lubricating oil | Paraffin oil (kerosene) | Petroleum (gasoline) | Silicone fluids | Vegetable oils | Acetone | Ethyl alcohol (ethanol) | Formaldehyde (40%) | Methyl alcohol (methanol) | ||
| Metals | Ferrous | Cast iron | B | C | B | B | C | D | D | D | B | A | B | A | A | A | A | A | A | A | A | A | B | A | B |
| High carbon steel | B | C | B | B | C | D | D | D | D | B | B | A | A | A | A | A | A | A | A | A | B | A | B | ||
| Medium carbon steel | B | C | B | B | C | D | D | D | D | B | B | A | A | A | A | A | A | A | A | A | B | A | B | ||
| Low carbon steel | B | C | B | B | C | D | D | D | D | B | B | A | A | A | A | A | A | A | A | A | B | A | B | ||
| Low alloy steel | B | C | B | B | C | D | D | D | D | B | B | A | A | A | A | A | A | A | A | A | B | A | B | ||
| Stainless steel | A | A | A | A | A | A | C | A | B | A | A | B | A | A | A | A | A | B | A | A | A | A | A | ||
| Non-ferrous | Aluminum alloys | A | B | D | A | C | C | D | C | D | D | D | A | A | A | A | A | A | A | A | A | A | A | C | |
| Copper alloys | A | A | A | A | C | C | D | B | C | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Lead alloys | A | A | A | A | B | C | D | B | B | B | D | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Magnesium alloys | A | C | C | C | C | D | D | D | D | B | B | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Nickel alloys | A | A | A | A | A | B | D | B | B | B | B | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Titanium alloys | A | A | A | A | A | A | A | A | B | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Zinc alloys | A | B | B | A | C | D | D | D | C | C | C | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Ceramics and glasses | Glasses | Borosilicate glass | A | A | A | A | A | A | D | A | A | A | B | A | A | A | A | A | A | A | A | A | A | A | A |
| Glass ceramic | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Silica glass | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Porous | Soda-lime glass | A | A | A | A | A | A | D | A | A | A | B | A | A | A | A | A | A | A | A | A | A | A | A | |
| Brick | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Concrete | A | A | A | A | B | B | D | B | B | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Stone | A | A | A | A | C | C | D | C | C | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Technical | Alumina | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | |
| Aluminum nitride | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Boron carbide | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Silicon | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Silicon carbide | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Silicon nitride | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Tungsten carbides | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Zirconia | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Composites | Al/SiC composite | A | B | D | A | C | B | D | C | D | D | D | A | A | A | A | A | A | A | A | A | A | A | A | |
| CFRP | A | A | C | C | C | A | D | C | A | C | A | B | A | A | A | A | A | A | A | D | C | A | D | ||
| GFRP | A | A | C | C | C | A | D | C | A | C | A | B | A | A | A | A | A | A | A | D | C | A | D | ||
| Natural materials | Woods and paper | Bamboo | B | B | B | C | B | B | D | B | B | D | D | B | B | B | B | B | B | B | B | B | B | B | B |
| Cork | A | B | C | C | B | B | D | B | B | D | D | B | B | B | B | B | B | B | B | B | B | B | B | ||
| Leather | B | C | D | D | C | C | D | C | C | D | D | C | C | C | C | C | B | B | B | C | B | B | B | ||
| Paper and cardboard | D | D | D | D | D | D | D | D | D | D | D | B | B | B | B | B | B | B | B | B | B | B | B | ||
| Wood | B | B | B | C | B | B | D | B | B | D | D | B | B | B | B | B | B | B | B | B | B | B | B | ||
| Polymers | Elastomers | Butyl rubber | B | B | B | C | B | C | C | C | B | C | C | D | D | D | D | D | C | B | D | B | B | C | B |
| EVA | B | B | D | A | D | D | D | D | D | A | C | D | D | B | A | B | C | A | D | D | D | B | D | ||
| Isoprene rubber (IR) | A | A | A | A | A | A | A | A | A | A | A | D | D | D | D | D | D | A | C | A | A | A | A | ||
| Natural rubber (NR) | A | A | A | A | A | C | C | C | A | A | C | D | D | D | D | D | D | A | D | A | A | A | A | ||
| Neoprene (CR) | A | A | A | A | A | A | A | C | A | A | A | D | D | C | A | B | D | A | A | A | A | C | A | ||
| Polyurethane (elPU) | A | A | D | C | D | C | D | C | C | C | D | D | D | C | C | A | B | A | A | D | D | D | D | ||
| Silicone elastomers (SI) | A | A | A | A | A | C | D | C | C | A | A | D | D | D | D | D | D | C | C | C | A | C | A | ||
| Thermo-plastics | ABS | A | A | A | A | A | A | C | A | A | A | A | D | D | A | A | A | A | A | A | D | D | A | D | |
| Cellulose polymers (CA) | A | A | D | D | D | A | D | D | D | D | D | A | A | A | A | A | A | A | B | D | C | D | D | ||
| Ionomer (I) | A | A | A | A | A | A | A | A | A | A | A | C | D | A | C | B | B | B | B | A | C | C | C | ||
| Nylons (PA) | B | B | B | C | B | D | D | D | D | C | C | A | A | B | B | A | A | B | B | A | A | A | A | ||
| Polycarbonate (PC) | A | A | A | A | A | A | A | A | A | A | C | D | A | C | A | A | A | A | A | D | A | A | A | ||
| Polyetheretherketone (PEEK) | A | A | A | A | A | A | D | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Polyethylene (PE) | A | A | A | A | A | A | A | A | A | A | A | B | B | A | A | A | A | B | A | B | A | A | A | ||
| PET | A | A | B | C | B | A | C | A | A | C | D | A | A | A | A | A | A | B | A | C | A | A | A | ||
| Acrylic (PMMA) | A | A | A | A | A | A | D | A | D | A | A | D | C | A | A | B | A | C | A | D | C | A | D | ||
| Acetal (POM) | A | A | A | A | A | C | D | C | D | A | A | A | A | A | A | A | A | C | A | C | A | A | A | ||
| Polypropylene (PP) | A | A | A | A | A | A | A | A | A | A | A | C | C | A | A | A | A | A | B | A | A | A | A | ||
| Polystyrene (PS) | A | A | A | A | A | A | D | B | A | A | A | D | A | C | C | A | C | A | C | D | A | C | B | ||
| Teflon (PTFE) | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||
| Polyurethane (tpPUR) | A | B | C | A | C | C | D | D | B | A | D | D | D | C | C | B | B | C | A | D | D | D | D | ||
| Polyvinylchloride (tpPVC) | A | A | A | A | A | A | B | D | A | A | A | C | A | B | A | A | A | B | B | C | A | A | A | ||
| Thermo-sets | Epoxies | A | A | C | C | C | A | D | C | A | C | A | B | A | A | A | A | A | A | A | D | C | A | D | |
| Phenolics | A | A | A | D | A | A | D | A | A | D | D | A | A | A | A | A | A | B | A | A | A | A | A | ||
| Polyester | A | A | C | D | C | A | D | A | A | D | D | D | A | A | A | A | A | A | B | D | D | A | A | ||
Table 17.4 Preferred materials and coatings for given environments
| Environment | Metals | Polymers and composites | Ceramics and glasses |
|---|---|---|---|
| Water and aqueous solutions | |||
| Water (fresh) | Aluminum alloys, stainless steels, galvanised steel, copper alloys | PET, HDPE, GFRP. All polymers are corrosion free in fresh water, though some absorb up to 5%, causing swelling. | Glass, concrete, brick, porcelain |
| Water (salt) | Copper, bronze, stainless steels, galvanised steels, lead, platinum, gold, silver | PET, HDPE, GFRP. All polymers are corrosion-free in salt water, though some absorb up to 5%, causing swelling. | Glass, concrete, brick |
| Soils | Steel, bare in high-resistivity soil, coated or with galvanic protection in those with low resistivity. | HDPE, PP, PVC, most polymers (except PHB, PLA and those that are bio-degradable) corrode only slowly in soil. | Brick, pottery, glass, concrete |
| Body fluids | Cobalt-chromium alloys, nickel-titanium alloys (nitinols), nickel-chromium alloys, precious metals, implants, silver amalgam, stainless steel, titanium, gold, platinum | Acrylic, silicone, ultra high mol. wt. polyethylene (UHMWPE) | Hydroxyapatite, alumina bio-ceramic, bioglass ceramic, calcium phosphate bio-ceramic, glass-ionomer, vitreous carbon, zirconia bio-ceramic |
| Acids and alkalis | |||
| Acetic acid, CH3COOH (10%) | Aluminum, stainless steel, nickel and nickel alloys, titanium, monel | HDPE, PTFE | Glass, porcelain, graphite |
| Hydrochloric acid, HCl (10%) | Copper, nickel and nickel alloys, titanium, monel, molybdenum, tantalum, zirconium, platinum, gold, silver | HDPE, PP, GFRP, rubber | Glass |
| Hydrofluoric acid, HF (40%) | Lead, copper, stainless steel, carbon steels, monel, hastelloy C, platinum, gold, silver | PTFE, fluorocarbon polymers, rubber | Graphite |
| Nitric acid HNO3 (10%) | Stainless steel, titanium, 14.5% silicon cast iron, alloy 20 (40Fe, 35Ni, 20Cr, 4Cu) | PTFE, PVC, phenolics, PE-CTFE | Glass, graphite |
| Sulfuric acid, H2SO4 (10%) | Stainless steel, copper, nickel-based alloys, lead, tungsten, 14.5% silicon, cast iron, zirconium, tantalum, alloy 20 (40Fe, 35Ni, 20Cr, 4Cu), platinum, gold, silver | PET, PTFE, PE-CTFE, phenolics | Glass, graphite |
| Sodium hydroxide, NaOH (10%) | Nickel and its alloys, stainless steels, magnesium | PVC, LDPE, HDPE, PTFE, PE-CTFE | Glass, graphite |
| Fuels, oils and solvents | |||
| Benzene, C6H6 | Carbon steel, stainless steel, aluminum, brass | PTFE | Glass, graphite |
| Carbon tetrachloride, CCl4 | Carbon steel, stainless steel, aluminum, hastelloy C, monel, platinum, gold, silver | POM, PTFE, rubber | Glass, graphite |
| Crude oil | Carbon steel, aluminum, stainless steels | HDPE, PTFE, epoxies, polyamides | Glass, porcelain, enamelled metal |
| Diesel oil | Carbon steel, stainless steel, brass, copper, aluminum, monel | HDPE, PP, PTFE, buna (nitrile) rubber, viton, GFRP | Glass |
| Kerosene (paraffin oil) | Carbon steel, stainless steel, aluminum, monel | HDPE, PP, PTFE, GFRP | Glass |
| Lubricating oil | Stainless steel, carbon steel, aluminum | HDPE, PP, PTFE, rubber | Glass |
| Petroleum (gasoline) | Carbon steel, stainless steel, brass, aluminum, hastelloy C, alloy 20 (40Fe, 35Ni, 20Cr, 4Cu) | PTFE, HDPE, PP, buna (nitrile) rubber, viton, GFRP | Glass |
| Silicone fluid, ((CH3)2SiO)n | Carbon steel, aluminum | HDPE, PP, PET | Glass |
| Vegetable oil | Aluminum, carbon steel, stainless steel | HDPE, PP, PET | Glass |
| Alcohols, aldehydes, ketones | |||
| Acetone, CH3COCH3 | Aluminum, carbon steel, stainless steel | PTFE, HDPE, PP copolymer (PPCO) | Glass, graphite |
| Ethyl alcohol, C2H5OH | Steel, stainless steel, copper | PTFE, HDPE, PP, GFRP | Glass |
| Methyl alcohol, CH3OH | Steel, stainless steel, lead, monel | PTFE, HDPE, PP | Glass |
| Formaldehyde, CH2O | Stainless steel, monel, hastelloy C | PTFE, HDPE, PET, POM, nitrile or butyl rubber | Glass, GFRP |
Example 17.4
Use Tables 17.3 and 17.4 to identify materials that you might use for pipe-work to handle 10% sodium hydroxide. Consider material combinations as well as single materials.
Answer. From Table 17.3, stainless steels, copper alloys and titanium alloys have an A rating in NaOH. So, too, do polyethylene and polypropylene, so they could be used to line a simple steel pipe.
From Table 17.4 stainless steel, nickel alloys (ranked B) and magnesium alloys (also B) are used.
Design: geometry and configuration
- Allow for uniform attack. Nothing lasts forever. Some corrosion can be tolerated, provided it is uniform, simply by allowing in the initial design for the loss of section over life.
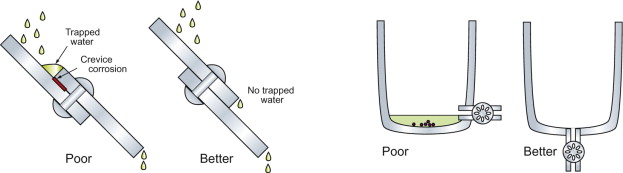
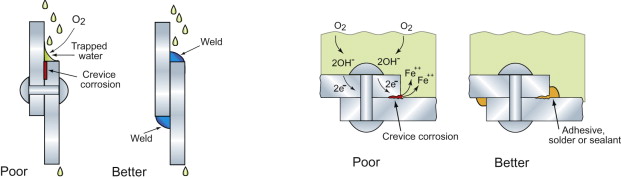
- Avoid fluid trapping (Figure 17.13). Simple design changes permit drainage and minimise the trapping of water and potentially corrosive dirt.
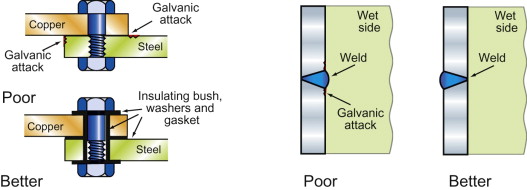
- Suppress galvanic attack (Figure 17.14). Two different metals, connected electrically and immersed in water with almost anything dissolved in it, create a corrosion cell like that of Figure 17.9 or 17.10. The metal that lies lower on the scale of reduction potential is the one that is attacked. The rate of attack can be large and localised at or near the contact. Attaching an aluminum body shell to a steel auto chassis risks the same fate, as incautious car-makers have found. Even the weld metal of a weld can differ from the parent plate in reduction potential enough to set up a cell. The answer is to avoid bi-metal couples if water is around or, when this is impossible, to insulate them electrically from each other and reduce the exposed area of the nobler metal to minimise the rate of the cathodic reaction.
- Avoid crevices (Figure 17.15). Crevices trap moisture and create the conditions for differential-aeration attack. Figure 17.15 shows riveted lap joints (the rivet, of course, made from the same metal as the plate). If water can get under the plate edge, it will. The water-air surface has free access to oxygen, but the metal between the plates does not. The result is corrosion of the joint surface, just where it is least wanted. The answer is to join by welding or soldering, or to put sealant in the joint before riveting. This is not just a joint problem—differential aeration is very hard to avoid. It is the reason that the legs of jetties and piers rust just below the water line, and why some edges of tanks are more severely rusted than others.
- Consider cathodic protection (Figure 17.16). If attaching a metal to one lower in reduction potential causes the lower one to corrode, it follows that the upper one is protected. So connecting buried steel pipe-work to zinc plates sets up a cell that eats the zinc but spares the pipes. The expensive bronze propellers of large ships carry no surface protection (the conditions are too violent for it to stay in place). Copper and bronze lie above steel in the reduction-potential table and electrical isolation is impossible. Bronze connected to steel in salt water is a recipe for disaster. The solution is to shift the disaster elsewhere by attaching zinc plates to the hull around the propeller shaft. The zinc, in both examples, acts as a sacrificial anode protecting both the bronze and the steel. The term ‘sacrificial’ is accurate—the zinc is consumed—so the protection lasts only as long as the zinc, requiring that it be replaced regularly.
- Beware of stress corrosion and corrosion-fatigue. Stresses arising from design loads can be calculated; residual stress from cold work or from thermal contraction in the heat-affected zone of welds are not so easy to predict. Their interaction with a corrosive environment can be dangerous, giving localised stress-corrosion cracking, or adding to cyclic loads to give corrosion-fatigue. Internal stresses cannot exceed the yield strength of the material (if they did the material would yield) so, as a rule of thumb, select materials that have as low a yield strength (or hardness) consistent with the other constraints of the design.
- Design for inspection and maintenance. If you can’t reach it, you can’t fix it. Design for durability ensures that parts liable to corrosion are easy to inspect, clean and, if necessary, replace.
Coatings
Metals, as we have said, are reactive. Only gold and a couple of other precious metals are stable as metals rather than as oxides. Coatings allow reactive metals—and particularly steels, the backbone of mechanical engineering—to be used in environments in which, uncoated, they would corrode rapidly. They work in more than one way.
- Passive coatings separate the material from the corrosive environment. Some, like chrome, nickel or gold plate, are inherently corrosion resistant. Polymeric coatings—paints and powder-coats—provide an electrically insulating skin that stifles electro-chemical reactions by blocking the passage of electrons. Ceramic and glass coatings rely on the extreme chemical stability of oxides, carbides and nitrides to encase the metal in a refractory shell. Passive coatings work only if they are perfect (and perfection, in this world, is unusual); if scratched, cracked or detached, corrosion starts.
- Active coatings work even when damaged. Some, like zinc, are metals that lie low on the reduction-potential scale of Figure 17.8. Zinc on steel acts as a sacrificial coat, becoming the anode of the corrosion cell, leaving the underlying steel unscathed (Figure 17.16(b)). Polymeric coatings can be made to work in this way by dispersing zinc powder in them. Others contain corrosion inhibitors (described later) that leach into the corrosive medium, weakening its potency. Cement coatings inhibit attack by providing an alkaline environment.
- Self-generated coatings rely on alloying, almost always with chromium, aluminum or silicon, in sufficient concentrations that a film of Cr2O3, Al2O3 or SiO2 forms spontaneously on the metal surface. Stainless steels, aluminum bronzes and silicon iron rely on protection of this sort. When scratched, the film immediately regrows, providing ongoing protection.
With this background, glance at Table 17.5. It lists coatings all of which you will have encountered even if you didn’t recognise them at the time. Modern engineering is totally dependent on them. They are applied in a number of ways, some illustrated in Figure 17.17.
- Metallic coatings rely on the intrinsic stability of the coat (gold); on its ability to form a self-generated, protective, oxide (chromium, aluminum, chromides and aluminides); or on its ability to provide cathodic protection (zinc, cadmium). Galvanized iron is steel sheet with a thin coating of zinc. The zinc acts as a sacrificial anode: if scratched, the exposed steel does not rust because the zinc protects it even when it does not cover it. Other platings look good but do not protect as well. A plating of copper sets up a cell that works in the opposite direction, eating the iron at a scratch rather than the copper. Chromium plating looks as if it should protect since Cr is below Fe in the table, but the chromium protects itself so well with its oxide film that it becomes passive and no cell is set up.
- Polymeric coatings rely either on a solvent that, after application, evaporates (acrylic, alkyds) or on a polymerisation reaction triggered by oxygen (linseed-oil based paints) or by an added catalyst (epoxies). Painting with solvent-based paints was, until recently, the most widely used method, but the evaporating solvent is generally toxic; it is a VOC (a volatile organic compound) and, for health reasons, their use is discouraged. Water-based paints overcome the problem but do not yet produce quite as good a paint film. Polymer powder coating works by flame-spraying the polymer or dipping the component when hot into a fluidised bed of polymer powder causing a thick film of polymer to melt onto the surface. While intact, paint films and polymer powder coats work well, but as soon as the coating is damaged the protection ceases unless the underlying metal has a subcoat of something (like zinc) that protects it.
- Ceramic and glass (enamel) coatings have to be applied hot, sufficiently so to sinter the particles of ceramic or to melt the glass onto the surface of the metal. Only cement coatings are applied cold.
Table 17.5 Coatings to enhance durability
| Type and process | Materials | Typical application |
|---|---|---|
| Metallic coatings | ||
| Electro-plating | Copper, nickel, chromium, silver, gold | Passive protection of steel |
| Electroless deposition | Nickel | Passive protection of steel |
| Hot dip | Zinc | Galvanised steel sheet |
| Metal spraying | Aluminum, zinc | Protection of steel components |
| Physical vapor deposition | Aluminum | Decorative protection |
| Chemical vapor deposition | Chromides, aluminides | High-temperature protection |
| Roll-cladding | Aluminum, nickel, copper | Cladding, coinage |
| Polymer coatings | ||
| Painting and spraying | Linseed oil-based paints | Protection of wood and steel |
| Acrylics, epoxies | Automobile, aircraft bodies | |
| Alkyds | Cheap metallic components | |
| Cellulose acetate/butyrate | Cheap metallic components | |
| Hot dip and thermal spray | Nylons | Household goods |
| PTFE, PE, PP | External cladding, nonstick coats | |
| Neoprene, vinyls | Tank linings | |
| Ceramic coatings | ||
| Chemical reaction | Anodising (Al2O3), chromating (Cr2O3) | Protection of aluminum components |
| Plasma spray | Alumina (Al2O3), zirconia (ZrO2) | Protection from oxidation—turbine and chemical engineering plant |
| Vapor deposit | Titanium nitride (TiN), zirconium nitride (ZrN) | Wear and corrosion resistance |
| Enamelling | Glass | Household goods |
| Cement coat | Portland cement, potassium silicate, calcium aluminate | Corrosion protection of steel, particularly from acid attack |
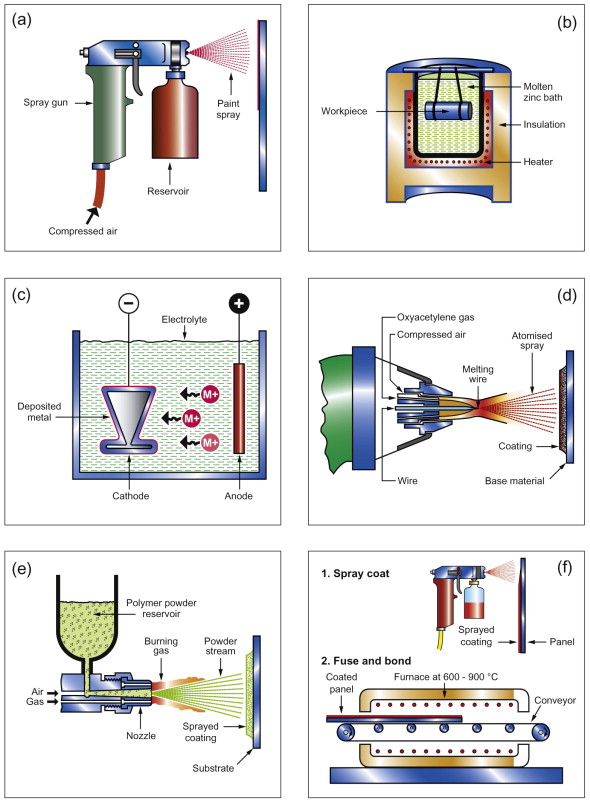
Figure 17.17 Ways of applying coatings. (a) Paint spraying. (b) Hot dip galvanising. (c) Electroplating. (d) Metal flame spraying. (e) Polymer powder spraying. (f) Enamelling.
Example 17.5
A steel component is nickel-plated. Would you expect the nickel to act as a passive or an active barrier to aqueous corrosion? Why?
Answer. Nickel lies above steel in the reduction-potential chart of Figure 17.8. It is more noble than steel, and will become the cathode in the corrosion cell. That means it acts only as a passive barrier—the steel will corrode at holes or cracks in the nickel coating.
Corrosion inhibitors
Corrosion inhibitors are chemicals that, when dissolved or dispersed in a corrosive medium, reduce (‘inhibit’) the rate of attack. Some work (like indigestion tablets) by lowering the pH or by coating the part with a gooey film. Inhibitors suppress either the anodic or the cathodic step of the corrosion reaction (see Section 17.6). The choice of inhibitor depends both on the environment and on the material to be protected. Table 17.6 lists the more common ones and the metals and environments for which they work best. Many have alarmingly polysyllabic names that only specialised corrosion consultants try to remember. The things that are useful to know are itemised here.
- PTFE ((CF2 − CF2)n) is used as a wide-spectrum inhibitor in hydrocarbon fuels, solvents and lubricants, most commonly in engine oil. It works by forming a protective layer over component surfaces, shielding them from full contact with the medium. Corrosion rates are reduced by up to 90%. Inadequate additions of PTFE lead to incomplete coverage and pitting corrosion, worsening the situation and leading to premature failure.
- Calcium bicarbonate (Ca(HCO3)2) is used in central heating, tap water and water cooling systems, where it lowers the pH and thus inhibits acidic attack. It makes the water hard, causing calcium deposits (lime scale) when water boils.
- Thiourea (C(NH2)2S) is an organic compound with a composition like that of urea but with the oxygen atom replaced by sulfur. It is the basic component of a range of organic anodic inhibitors that work by raising the activation energy of the corrosion reaction, thus decreasing the corrosion rates. Small additions can reduce the corrosion rate in hydrochloric acid by as much as 97%. Thiourea, however, is potentially carcinogenic, limiting its use to systems that do not involve human contact.
- Arsenic (III) oxide (As2O3) inhibits acidic attack of metals and, because it is an anodic inhibitor, it works well in a wide variety of acids. Even at low concentrations (0.5%) it can provide inhibition. It is, of course, toxic.
- Polyphosphates is the name of a family of polymers containing phosphate (—PO4) groups. They are found in living organisms where they perform biological functions (DNA includes polyphosphates). They inhibit corrosion of most metals in water. Polyphosphates are not toxic, allowing their use in drinking water.
- Chromates are used mostly to inhibit corrosion of metals in nonacidic environments (a few, the alkali chromates, can be used to inhibit corrosion in acids; they are used to protect aluminum). They work by forming an adsorbed layer of chromate ions, passivating the surface. Chromates, like so many of these scary compounds, can be highly toxic, limiting their use.
Table 17.6 Corrosion inhibitors and the materials for which they work
| Environment | Inhibitors (materials) |
|---|---|
| Water and aqueous solutions | |
| Water (fresh) | Calcium bicarbonate (steel, cast iron), polyphosphate (Cu, Zn, Al, Fe), calcium hydroxide (Cu, Zn, Fe), sodium silicate (Cu, Zn, Fe), sodium chromate (Cu, Zn, Pb, Fe), potassium dichromate (Mg), sodium nitrite (monel), benzoic acid (Fe), calcium and zinc metaphosphates (Zn) |
| Water (salt) | Sodium nitrite (Fe), sodium silicate (Zn), calcium bicarbonate (all metals), amyl stearate (Al), methyl-substituted dithiocarbamates (Fe) |
| Soils | Calcium nitrate is added to concrete to inhibit corrosion of steel reinforcement in soils. |
| Acids and alkalis | |
| Acetic acid, CH3COOH (10%) | Thiourea, arsenic oxide, sodium arsenate (all for Fe) |
| Hydrochloric acid, HCl (10%) | Ethylaniline, mercaptobenzotriazole, pyridine and phenylhydrazine, ethylene oxide (all used for Fe), phenylacridine (Al), napthoquinone (Al), thiourea (Al), chromic acid (Ti), copper sulfate (Ti) |
| Hydrofluoric acid, HF (40%) | Thiourea, arsenic oxide, sodium arsenate (all for Fe) |
| Nitric acid, HNO3 (10%) | Thiourea, arsenic oxide, sodium arsenate (all for Fe), hexamethylene tetramine (Al), alkali chromate (Al) |
| Sulfuric acid, H2SO4 (10%) | Phenylacridine (Fe), sodium chromate (Al), benzyl thiocyanate (Cu and brass), hydrated calcium sulfate (Fe), aromatic amines (Fe), chromic acid (Ti), copper sulfate (Ti) |
| Sodium hydroxide, NaOH (10%) | Alkali silicates, potassium permanganate, glucose (all for Al) |
| Fuels, oils and solvents | |
| Benzene, C6H6 | Anthraquinone (Cu and brass) |
| Carbon tetrachloride, CCl4 | Formamide (Al), aniline (Fe, Sn, brass, monel and Pb), mesityl oxide (Sn) |
| Diesel oil | PTFE suspension (all metals), chlorinated hydrocarbons (all metals), poly-hydroxybenzophenone (Cu) |
| Kerosene (paraffin oil) | PTFE suspension (all metals), chlorinated hydrocarbons (all metals), poly-hydroxybenzophenone (Cu) |
| Lubricating oil | PTFE suspension (all metals), chlorinated hydrocarbons organozinc compound selected such as zinc dithiophosphate and zinc dithiocarbamate (all metals), poly-hydroxybenzophenone (Cu) |
| Petroleum (gasoline) | PTFE suspension (all metals), chlorinated hydrocarbons (all metals) |
| Vegetable oil | Anthraquinone (Cu and brass) |
| Alcohols, aldehydes, ketones | |
| Ethyl alcohol, C2H5OH | Potassium dichromate, alkali carbonates or lactates (Al), benzoic acid (Cu and brass), alkaline metal sulfides (Mg), ethylamine (Fe), ammonium carbonate with ammonium hydroxide (Fe) |
| Methyl alcohol, CH3OH | Sodium chlorate with sodium nitrate (Al), alkaline metal sulfides (Mg), neutralised stearic acid (Mg), polyvinylimidazole (Cu) |
Monitoring, maintenance and replacement
Regular inspection allows early indications of corrosion to be detected. Maintenance—painting, recoating or repair—can then be carried out, minimising down-time and risk of failure. There is a design issue here: the system must allow inspection of all potentially vulnerable surfaces, and permit access for restorative work. For safety-critical components a different strategy is adopted: that of replacement at prescribed, regular intervals, too short for corrosion to have caused any serious damage.
17.8 Summary and conclusions
Corrosion is like cancer: it will kill you unless something else (like wear) kills you first. Almost as much money must have been invested in corrosion research as in research on cancer, such is its damaging effect on the economy. This research has revealed what goes on at an atomic scale when materials react with oxygen, acids, alkalis, aqueous solutions, aerated water and organic solvents.
All involve electro-chemical reactions or reactions involving free radicals. In oxidation of metals, the metal ionises, releasing electrons; the electrons combine with oxygen molecules to give oxygen ions, that in turn combine with metal ions to form the oxide. As the oxide film grows, ions and electrons must diffuse through it to enable further growth; its resistance to this diffusion, and its ability to adhere to the metal surface determine how protective it is. Polymers oxidise through the action of free radicals. Radiation, including UV light, can generate the free radicals, as does heat. Polymers are protected by doping them with UV filters and with additions that absorb free radicals.
In corrosion, the electro-chemical reactions occur in the corrosive fluid rather than in the solid. A metal ionises when placed in water. The extent of ionisation determines its reduction potential. Those with the smallest (most negative) reduction potential are the most vulnerable to attack, and will corrode spontaneously even in pure water if oxygen is present. If a metal with a low reduction potential is in electrical contact with one of higher potential, the first corrodes and the second is protected.
In designing materials to resist corrosion we rely on the ability of a few of them—chromium, aluminum, silicon and titanium—to form a protective oxide film that adheres strongly to the surface. By using these as alloying elements, other metals can be given the same protection. Alternatively, metals can be protected by coating their surfaces with a corrosion-resistant film of polymer or ceramic.
17.9 Further reading and software
Bradford S.A. Corrosion Control 1993 Van Nostrand Reinhold New York, NY, USA ISBN 0-442-01088-5. (A readable text emphasising practical ways of preventing or dealing with corrosion.)
DeZurik Materials Selection Guide DeZurik Materials Selection Guide 〈 http://www.dezurik.com/ 〉. (Dezurik MSG allows the selection of materials to operate in a given environment.)
Fontanta M.G. Corrosion Engineering 3rd ed. 1986 McGraw Hill New York, USA ISBN 0-07-021463-8. (A text focussing on the practicalities of corrosion engineering rather than the science, with numerous examples and data.)
Ford F.P., Andresen P.L. Design for corrosion resistance ASM Handbook 1997 ASM International Metals Park, Ohio, USA 545-572 ISBN 0-97170-386-6. (A recent guide to design for corrosion resistance, emphasising fundamentals, by two experts in the field.)
, 2008 ProFlow Dynamics chemical resistance selector http://www.proflowdynamics.com/viewcorrosion.aspx 2008 (A free online database for finding a material suitable for use in specified environments.)
Schweitzer P.A. 4th ed. Corrosion Resistance Tables Vols. 1–3 1995 Marcel Dekker New York, USA (The ultimate compilation of corrosion data in numerous environments. Not bed-time reading.)
Schweitzer P.A. Encyclopedia of Corrosion Technology 1998 Marcel Dekker New York, USA ISBN 0-8247-0137-2. (A curious compilation, organised alphabetically, that mixes definitions and terminology with tables of data.)
Tretheway K.R., Chamberlain J. Corrosion for Science and Engineering 2nd ed. 1995 Longman Scientific and Technical Harlow, UK ISBN 0-582-238692. (An unusually readable introduction to corrosion science, filled with little bench-top experiments to illustrate principles.)
Waterman N.A., Ashby M.F. Elsevier Materials Selector 1991 Elsevier Oxford, UK ISBN 1-85-166-605-2 and, in the CRC edition, ISBN 0-8493-7790-0. (A three volume compilation of materials data for design, with extensive tables and guidelines for the oxidation and corrosion characteristics of metals and polymers.)
17.10 Exercises
- Exercise E17.1 By what mechanisms do metals oxidise? What determines the rate of oxidation?
- Exercise E17.2 The oxidation kinetics of titanium to TiO2 is limited by oxygen diffusion, with an activation energy Qd of 275 kJ/mol. If the oxide film grows to a thickness of 0.08 microns after 1 hour at 800 °C, how thick a film would you expect if it had been grown at 1000 °C for 30 minutes?
- Exercise E17.3 What is meant by the standard reduction potential? A copper and a platinum electrode are immersed in a bath of dilute copper sulfate. What potential difference would you expect to measure between them? If they are connected so that a current can flow, which one will corrode?
- Exercise E17.4 A polymer coating is required to protect components of a microchip processing unit from attack by hydrogen fluoride (HF). Use Tables 17.3 and 17.4 to identify possible choices.
- Exercise E17.5 Metal pipe work on an oil rig must carry hydrochloric acid solution to acidify the well. Use Tables 17.3, 17.4 and 17.6 to explore ways of providing and protecting the pipe.
- Exercise E17.6 A food processing plant uses dilute acetic acid for pickling onions. The acid is piped to and from holding tanks. Select a suitable material for the pipes and tanks, given that, to have sufficient strength and toughness to tolerate external abuse they must be made of a metal.
- Exercise E17.7 An automaker is concerned about the consequences of the introduction of bio-methanol, CH3OH or bio-ethanol C2H5OH into auto fuels. The particular concern is the corrosion of aluminum components, particularly the engine block, by methanol or ethanol. What steps could be taken to avoid this?
- Exercise E17.8 The automaker of Exercise E17.7 is also concerned that spillage of methanol or ethanol bio-fuel might damage the GFRP bodywork of some models. Is the concern justified?
- Exercise E17.9 The waste stream of a fertilizer plant includes dilute sulfuric acid. The dilute acid is stored in surface tanks some distance from the plant. It is suggested that the ducting to carry the acid to the tanks could, most economically, be made of wood. Is this a totally crazy suggestion?
17.11 Exploring design with CES (use Level 2 unless otherwise stated)
- Exercise E17.10 Find, by Browsing or Searching, the record for the nickel-chromium alloys called Nichromes. What are their main applications?
- Exercise E17.11 Find, by Browsing or Searching, the record for Polymer powder coating (remember to search in the Process universe, not the Materials universe). What are the three ways of applying a polymer powder coating?
- Exercise E17.12 Use a Limit stage, applied to the Level 2 Surface Treatment data table, to find surface treatment processes that impart resistance to gaseous corrosion.
- Exercise E17.13 Use a Limit stage, applied to the Level 2 Surface Treatment data table, to find surface treatment processes that impart resistance to aqueous corrosion.
- Exercise E17.14 Use the CES Search facility to find materials for food processing equipment.
- Exercise E17.15 Plastic cases for electrical plugs and switch-gear should not be made of flammable materials. Use the Search facility in CES to find polymers that are non-flammable or self-extinguishing.
- Exercise E17.16 A vat is required to hold hot caustic soda, NaOH, a strong alkali. Use the Search facility in CES to find metals that resist strong alkalis very well.
- Exercise E17.17 Pipework is required for a gherkin-pickling plant to carry vinegar (a dilute acid) at 100 °C from one vat to another. The liquid is under pressure, requiring a material with a strength of at least 100 MPa, and for ease of installation it must be able to be bent, requiring a ductility of at least 10%. Find the four cheapest materials that meet the constraints, summarised in the following table.
| Function | •Pipework for hot acetic acid |
| Constraints | |
| Objective | •Minimise cost |
| Free variable | •Choice of material |
17.12 Exploring the science with CES Elements
- Exercise E17.18 The Work function (Chapter 14) is the energy required to pluck an electron from the top of the Fermi level of a crystal and drag it away until it is isolated in vacuum. The Standard reduction potential of Figure 17.7 involves electrons dropping into energy levels just above the Fermi level. You might suspect that the two were, in some way, related. Make a graph with Standard reduction potential on the x-axis and Work function on the y-axis to find out if they are. What is your conclusion?
1 The CES Edu software, for example, has rankings for 55 environments; other such sources are listed under Further reading.