
7
Conclusions
Here, we will provide references and short descriptions of optimization problems from the bio-informatics field that not have been dealt with in detail so far. In particular, these include DNA sequencing problems and founder sequence reconstruction.
7.1. DNA sequencing
DNA sequencing is the process of determining the precise order of the nucleotides in a DNA molecule. The knowledge of the precise composition of DNA sequences has become very important in numerous application fields such as medical diagnosis, biotechnology, forensic biology, virology and biological systematics. Computational methods such as metaheuristics have played an important role in DNA sequencing technology. The first basic DNA sequencing technologies were developed in the 1970s (see [JAY 74]). An example of a basic DNA sequencing technology is shotgun sequencing [STA 79]. Small genomes (4,000 to 7,000 base pairs) had already been sequenced by means of shotgun sequencing in 1997 [STA 79]. The first of the next-generation sequencing technologies, known as massively parallel signature sequencing, was developed in the 1990s [BRE 00]. Another example is DNA sequencing by hybridization [BAI 88, LYS 87, DRM 89]. In the following section, we will provide several examples in which metaheuristics play an important role.
7.1.1. DNA fragment assembly
The so-called DNA fragment assembly problem is a computational problem that is a major component of DNA sequencing, often on the basis of shotgun sequencing. In shotgun sequencing, first, many copies of the DNA strands that are to be sequenced – i.e. the target sequence – are produced. These DNA strands are then cut into pieces at random sites in order to obtain fragments that are short enough (up to approx. 1,000 base pairs) to sequence directly. The fragments then need to be assembled into the most plausible DNA sequence. Moreover, in order to have a good chance assembling a DNA sequence close to the target sequence, a coverage of at least 10 is required, which means that each base from a specific position of the target sequence should appear in at least 10 DNA fragments. A variety of approaches have been applied for the assembly of the DNA fragments. One of the most popular is the overlap-layout-consensus (OLC) approach. Large eukaryotic genomes1 have been successfully sequenced with the OLC approach (see [ADA 00]).
The first step of the OLC approach consists of calculating the overlaps between the suffix and the prefix of any two fragments. In order to do so, note that both orientations of each DNA fragment must be considered. This is because it is not clear from which of the two DNA strands forming the double helix (see Chapter 1), the fragment was obtained. The overlap scores are, generally, obtained by applying a semi-global alignment algorithm such as the Smith-Waterman algorithm [SMI 81] (see also section 6.2).
The most challenging part of the OLC approach is the layout phase in which one or more contigs (sequences of DNA fragments whose respective overlaps are greater than a pre-defined threshold) are produced. This is where metaheuristics often come into play. Even without considering noise such as reading errors, etc., this problem has been shown to be NP-hard in [PEV 00]. Finally, the last phase of the OLC approach is the consensus phase in which the most likely contig is determined based on the layout phase. For an overview of the existing heuristic and metaheuristic approaches for the layout phase see Table 7.1.
Table 7.1. Heuristic and metaheuristic approaches for the layout phase in DNA fragment assembly
Type of algorithm |
Publication |
|
Genetic algorithm |
Parsons and Johnson [PAR 95a], 1995 |
|
Genetic algorithm |
Parsons et al. [PAR 95b], 1995 |
|
Genetic algorithm |
Fang et al. [FAN 05], 2005 |
|
Genetic algorithm |
Kikuchi and Chakraborty [KIK 06], 2006 |
|
Ant colony optimization |
Zhao et al. [ZHA 08], 2008 |
|
Genetic algorithm |
Nebro et al. [NEB 08], 2008 |
|
Genetic algorithm |
Hughes et al. [HUG 14], 2014 |
|
Local search |
Ben Ali et al. [BEN 15], 2015 |
|
Hybrid particle swarm optimization |
Huang et al. [HUA 15], 2015 |
|
Hybrid gravitation search |
Huang et al. [HUA 16], 2016 |
7.1.2. DNA sequencing by hybridization
DNA sequencing by hybridization works roughly as follows. The first phase of the method consists of a chemical experiment which requires a so-called DNA array. A DNA array is a two-dimensional grid whose cells typically contain all possible DNA strands – called probes – of a certain length l. For example, consider a DNA array of all possible probes of length l = 3 (see [IDU 95]):

The chemical experiment is started after the generation of the DNA array. In order to do so, the DNA array is brought together with many copies of the DNA target sequence that is to be sequenced. The target sequence possibly reacts to a probe on the DNA array, if and only if the probe is a subsequence of the target sequence. This reaction is also called hybridization. After the experiment, the DNA array allows us to identify the probes that reacted with target sequences. The set(s) of probes involved in some reaction is called the spectrum. However, it has to be taken into account that two types of errors may occur during the hybridization experiment:
- 1) negative errors: some probes do not form part of the spectrum even though they are part of the target sequence. Note that a particular type of negative error is caused by the multiple existence of a probe in the target sequence. Such a probe will appear at most once in the spectrum;
- 2) positive errors: a probe of the spectrum that does not appear in the target sequence is called a positive error.
After the chemical experiment, the second phase of DNA sequencing by hybridization consists of the reconstruction of the target sequence from the spectrum by means of computational optimization methods. If we assume that the obtained spectrum is free of errors, the original sequence can be reconstructed in polynomial time with an algorithm proposed by Pevzner in [PEV 89]. However, as this is not usually the case, a perfect reconstruction is not always possible. Therefore, DNA sequencing by hybridization is, generally, expressed by means of optimization problems with optimal solutions that can be shown to have a high probability of resembling the target sequence. In the following section, we present the one proposed by Baewicz et al. in [BŁA 99], which was shown to be NP-hard.
Henceforth, let the target sequence be denoted by st. The length – i.e. the number of nucleotide bases – of st is denoted by n, i.e. st ∈ {A,C,G,T}n. Furthermore, the spectrum – as obtained by the hybridization experiment – is denoted by S = {1, … , m}. More specifically, each i ∈ S is an oligonucleotide (a short DNA strand) of length l, that is, i ∈ {A,C,G,T}l. Let G = (V, A) be the completely connected directed graph defined by V = S (see [IDU 95]). To each link aij ∈ A is assigned a weight oij, which is defined as the length of the longest DNA strand, that is a suffix of i and a prefix of j (i.e. the overlap). Each directed, node-disjoint path p = (i1, … , ik) in G is considered a valid solution to the problem. The number of vertices (i.e. oligonucleotides) on a path p is called the length of the path, denoted by len(p). In the following, we denote by p[r] the rth vertex on a given path p (starting from position 1). In contrast to the length, the cost of a path p is defined as follows:
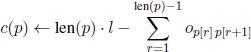
The first term sums up the length of the oligonucleotides on the path and the second term (which is subtracted from the first one) sums up the overlaps between the neighboring oligonucleotides on p. In fact, c(p) is equivalent to the length of the DNA sequence that is obtained by the sequence of oligonucleotides in p. The problem of DNA sequencing by hybridization consists of finding a solution (or path), p∗, in G with len(p∗) ≥ len(p), for all possible solutions p that fulfill c(p) ≤ n.
Consider, for example, the target sequence st = ACTGACTC. Assuming l = 3, the ideal spectrum is {ACT, CTG, TGA, GAC, ACT, CTC}. However, let us assume that the hybridization experiment results in the following faulty spectrum S = {ACT, TGA, GAC, CTC, TAA}. This spectrum has two negative errors, because ACT should appear twice, but can – due to the characteristics of the hybridization experiment – only appear once, and CTG does not appear at all in S. Moreover, S has one positive error, because it includes oligonucleotide TAA, which does not appear in the target sequence. An optimal solution in this example is p∗ = (ACT, TGA, GAC, CTC) with len(p∗) = 4 and c(p∗) = 8. The DNA sequence that is retrieved from this path is equal to the target sequence (see Figure 7.1).
7.1.2.1. Existing heuristics and metaheuristics
The first approach to solve the SBH problem was a branch and bound method proposed in [BŁA 99]. However, this approach becomes unpractical when problem size gets bigger. The algorithm was capable of solving only one out of 40 problem instances with target sequences of length 209 within one hour to optimality. Another argument against this branch and bound algorithm is the fact that an optimal solution to the SBH problem does not necessarily provide a DNA sequence that is equal to the target sequence. This implies that the importance of finding optimal solutions is not the same as for other optimization problems. Therefore, the research community has focused on heuristics and metaheuristics for tackling the SBH problem. Most of the existing approaches are either constructive heuristics or metaheuristics such as evolutionary algorithms, Tabu search, GRASP and ant colony optimization. For an overview on the existing approaches to the SBH problem, see Table 7.2.
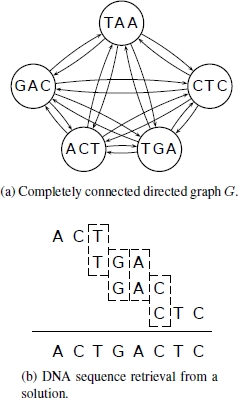
Figure 7.1. a) The completely connected directed graph G with the spectrum S = {ACT, TGA, GAC, CTC, TAA} as vertex set. The edge weights (i.e. overlaps) are not indicated for readability reasons. For example, the weight on the edge from TGA to GAC is 2, because GA is the longest DNA strand that is a suffix of TGA and a prefix of GAC. An optimal solution is p∗ = (ACT, TGA, GAC, CTC). b) How to retrieve the DNA sequence that is encoded by p∗. Note that c(p∗) = 8, which is equal to the length of the encoded DNA sequence
7.2. Founder sequence reconstruction
Given a sample of sequences from a population of individuals, a possible goal of valuable research is to study the evolutionary history of the population. For example, an important application is the discovery of the genetic basis of multi-factorial diseases. If the population under study has evolved from a relatively small number of founder ancestors, the evolutionary history can be studied by trying to reconstruct the sample sequences as fragments from the set of founder sequences. The genetic material of the population’s individuals is the result of recombination and mutation of their founders. Many findings from biological studies support the validity of this model, such as, for example, the fact that the “Ferroplasma type II genome seems to be a composite from three ancestral strains that have undergone homologous recombination to form a large population of mosaic genomes” [THY 04].
Table 7.2. Heuristic and metaheuristic approaches to the SBH problem
|
Type of algorithm |
Publication |
|
Constructive heuristic |
Baewicz et al. [BŁA 99], 1999 |
|
Constructive heuristic |
Baewicz et al. [BŁA 02a], 2002 |
|
Evolutionary algorithm |
Baewicz et al. [BŁA 02b, BŁA 05], 2002 |
|
Evolutionary algorithm |
Endo [END 04], 2004 |
|
Evolutionary algorithm |
Brizuela et al. [BRI 04a], 2004 |
|
Evolutionary algorithm |
Bui and Youssef [BUI 04], 2004 |
Tabu search |
Baewicz et al. [BŁA 00], 2000 |
|
Tabu search / scatter search hybrid |
Baewicz et al. [BŁA 04, BŁA 05], 2004 |
|
GRASP-like multi-start technique |
Fernandes and Ribeiro [FER 05], 2005 |
|
Ant colony optimization |
Blum et al. [BLU 08], 2008 |
|
Constructive heuristic |
Chen and Hu [CHE 11a], 2011 |
|
Hyper-heuristics |
Baewicz et al. [BŁA 11], 2013 |
The main problem is that the number of founder sequences as well as the founder sequences themselves are generally unknown. Nevertheless, given a specific evolutionary model, it is possible to phrase a combinatorial optimization problem whose solutions are biologically plausible founder sequences. This problem is usually called the founder sequence reconstruction problem (FSRP). From an optimization perspective, the FSRP problem is a hard combinatorial optimization problem and it is of interest per se as it constitutes a challenge for current optimization techniques.
7.2.1. The FSRP problem
The FSRP problem can technically be defined as follows: take a set of n recombinants C = {C1, … , Cn}. Each recombinant Ci is a string of length m over a given alphabet Σ, i.e. Ci = ci1ci2 … cim with cij ∈ Σ. Most of the literature consider typical biological applications where the recombinants are haplotyped sequences. Hence, Σ = {0, 1}. The symbols 0 and 1 encode the two most common alleles of each haplotype site.
A candidate solution consists of a set of kf founders ![]() = {F1, … , Fkf }. Each founder Fi is a string of length m over the alphabet Σ: Fi = fi1fi2 … fim with fij ∈ Σ ∀ j. A candidate solution
= {F1, … , Fkf }. Each founder Fi is a string of length m over the alphabet Σ: Fi = fi1fi2 … fim with fij ∈ Σ ∀ j. A candidate solution ![]() is valid if it is possible to reconstruct the set of recombinants
is valid if it is possible to reconstruct the set of recombinants ![]() from
from ![]() . This is the case when each Ci ∈
. This is the case when each Ci ∈ ![]() can be decomposed into a sequence of pi ≤ m fragments (that is, strings) Fri1Fri2 …
can be decomposed into a sequence of pi ≤ m fragments (that is, strings) Fri1Fri2 … ![]() , such that each fragment Frij appears at the same position in at least one of the founders. Therefore, a decomposition with respect to a valid solution is said to be reduced if two consecutive fragments do not appear in the same founder. Moreover, a decomposition is said to be minimal if
, such that each fragment Frij appears at the same position in at least one of the founders. Therefore, a decomposition with respect to a valid solution is said to be reduced if two consecutive fragments do not appear in the same founder. Moreover, a decomposition is said to be minimal if ![]() is minimal. In [WU 08] it was shown that for each valid solution
is minimal. In [WU 08] it was shown that for each valid solution ![]() it is possible to derive a minimal decomposition in O(n m kf) time. Henceforth, this number is called the objective function value of
it is possible to derive a minimal decomposition in O(n m kf) time. Henceforth, this number is called the objective function value of ![]() and is denoted by f(
and is denoted by f(![]() ). In biological terms, f(
). In biological terms, f(![]() ) is called the number of breakpoints of
) is called the number of breakpoints of ![]() with respect to
with respect to ![]() .
.
The optimization goal considered by the FSRP problem is the following one. Given a fixed kf, that is, a fixed number of founders, find a valid solution ![]() ∗ that minimizes the number of breakpoints. As an example, see Figure 7.2. The FSRP was first proposed by Ukkonen [UKK 02] and the problem is proved to be
∗ that minimizes the number of breakpoints. As an example, see Figure 7.2. The FSRP was first proposed by Ukkonen [UKK 02] and the problem is proved to be ![]() -complete [RAS 07] for kf > 2.
-complete [RAS 07] for kf > 2.
7.2.2. Existing heuristics and metaheuristics
This section provides an overview of existing algorithmic approaches for the FSRP. One of the first algorithms proposed for the FSRP was a dynamic programming algorithm [UKK 02]. However, this method is not efficient when the number of founders and the number or length of recombinants is high. Another dynamic programming algorithm was introduced in [RAS 07]. In [LYN 05] a branch and bound algorithm has been proposed which works independently of the lower bound method used. Although promising, this method has been evaluated only on a limited test set composed of rather small instances.

Figure 7.2. a) A set of six recombinants in matrix form. Assuming that the number of founders (kf ) is fixed to three, b) shows a valid solution, that is, a matrix of three founders. Denoting the first founder by a, the second founder by b, and the third one by c, c) shows a decomposition of the recombinant matrix into fragments taken from the founders. Note that breakpoints are marked by vertical lines. This is a decomposition with 8 breakpoints, which is the minimum value for this instance
Wu and Gusfield proposed a constructive tree-search solver, called RECBLOCK, which uses bounding information [WU 08]. RECBLOCK can be run as an exact or an approximate heuristic-solver. Although the results of RECBLOCK are very good for instances with a rather small number of founders, the computation time of RECBLOCK scales exponentially with the number of founders and makes its application infeasible for large instances. This was the motivation for developing metaheuristic approaches [BLU 03], the first of which was a Tabu search method, equipped with an effective constructive procedure [ROL 09]. An IG algorithm that achieves state-of-the-art results for large instances or in the case of many founders was presented in [BEN 10]. Furthermore, a parthenogenetic algorithm was proposed in [WU 13]. Finally, the current state-of-the-art method – which is a large neighborhood search technique based on RECBLOCK – was introduced in [ROL 12].
The FSRP is widely studied and is related to a number of different problems. In [EL 04] the authors focus on a much simpler and more tractable reconstruction problem: given a founder set and a recombinant, they want to find a minimal decomposition for the recombinant.
A similar problem, called Haplotype Coloring, has been introduced in [SCH 02] by Schwartz et al. Given a recombinant matrix, a partition of contiguous sites, called blocks, is identified according to a particular rule and each recombinant is subdivided accordingly into haplotype substrings. The objective is to assign, for each block, a different color to the distinct haplotype substrings in a block, such that the total number of color switches between two contiguous haplotype substrings in the same recombinant is minimal. This problem can be encoded into an instance of the weighted bipartite matching problem. The haplotype coloring can be solved to optimality by a O(m n2 log n) algorithm.
The FSRP formulation presented so far is based on the genetic assumption that, during the genetic evolution of the initial population of founders, mutation events are unlikely to occur. Rastas and Ukkonen generalized the FSRP by introducing a different objective function that takes into account point mutation [RAS 07], namely k + c · k′ where c > 0 is a constant. This new objective function is the sum of two contributions: k denotes the sum of the number of breakpoints across all recombinant sequences (i.e. the objective function we introduced in section 7.2.1) while k′ is the total number of point mutations. Note that if c is large, the resulting problem is equivalent to the FSRP definition as presented in section 7.2.1 because an optimal solution will not contain any mutation.
Zhang et al. [ZHA 09] present a problem that is closely related to the FSRP, called the minimum mosaic problem. This problem aims at finding a minimum mosaic, i.e. a block partitioning of a recombinant set such that each block is compatible according to the four gamete test [HUD 85] and the number of blocks is minimized. In a different way to the FSRP, this problem does not rely on the existence of a founder set. The obtained mosaic structure provides a good estimation of the minimum number of recombination events (and their location) required to generate the existing haplotypes in the population, that is, a lower bound for the FSRP. The authors provide an algorithm capable of handling large datasets.
7.3. Final remarks
In conclusion, we believe that heuristics and metaheuristics play – and will continue to play – a role of increasing importance in solving string problems from the field of bioinformatics. Just as in the case of other combinatorial optimization problems that are not related to bioinformatics, it is possible to identify a clear trend towards the application of hybrid metaheuristics, i.e. algorithmic approaches that result from combinations of metaheuristics with other techniques for optimization such as, for example, mathematical programming techniques. The current state-of-the-art algorithms for the most strings with few bad columns problem (see Chapter 4) and for longest common subsequence problems (see Chapter 3), just to name two examples, confirm this trend.