
Framework for continuous improvement
Abstract:
This chapter considers occasions that lead to the continuous improvement of manufacturing processes and programs in the life sciences industry, and to the revision of SOPs. Several groups of stakeholders in the sphere of FDA-regulated industry are identified. Each of these groups tends to be associated with particular kinds of observations, observations of various deviations from the anticipated manufacturing process and product. These observations initiate an investigation and revising process that varies in emphasis but that has an underlying logic. An observation is typically escalated, triaged, and can become the basis of an investigation and RCA. At the conclusion of the investigation, programmatic remediation can be proposed in the form of corrective actions and preventive actions, and may ultimately lead to the revision of a procedure. A diligent approach to revision promotes the continuous improvement of the manufacturing process.
1.1 Introduction
There are events and situations that – when observed and acted upon – initiate the revision of processes and procedures addressed by good manufacturing practices (GMPs) in a regulated industry such as that covered by the US Food and Drug Administration (FDA) or other global regulatory agencies. These events and situations can lead to two kinds of revision of GMP processes. They can be either reactive interventions such as corrective actions, or proactive interventions such as preventive actions. An example of a proactive intervention would be the organization’s response to tracking and trending data that suggest critical action points will soon be exceeded. Another example of proactive intervention would be an organization’s response to a “close call,” where a deviation did not actually occur.
In either case, reactive or proactive intervention, systematic pursuit of revision leads to continuous improvements.
Events and situations do not generate the intervention by themselves. The key terms here are “when observed and acted upon,” focusing attention on the major groups of stakeholders that can initiate the chain of events leading to the change. The persons occupying each of these stakeholder roles must first observe, then act, to occasion the intervention.
There are five major groups of these stakeholders. These five groups make up a map of the overall sphere of the regulated life sciences industry. Each represents a different facet of regulated industry, including the following:
In brief, operational staff manufacture the regulated product, be it a drug, medical device, nutritional supplement, etc. The quality auditors function independently to monitor the activities of operational staff as well as the quality attributes – the Safety, Identity, Strength, Purity, and Quality (SISPQ) of the product. Agency investigators in turn regulate the activities of both operational and quality staff. Health care providers prescribe the product, and patients utilize the product.
Any member of these groups can make an observation that may bring about an investigation and RCA. Consider several illustrative observations:
![]() A forklift operator raised the forks too high and damaged a fire sprinkler head. The water was under high pressure and it not only flooded the area but cascaded down to the floor below, threatening to inundate production areas. After an engineer shut off the water supply, operational staff escalated this event to management.
A forklift operator raised the forks too high and damaged a fire sprinkler head. The water was under high pressure and it not only flooded the area but cascaded down to the floor below, threatening to inundate production areas. After an engineer shut off the water supply, operational staff escalated this event to management.
![]() The quality unit of a FDA regulated blood center reported an increase in the number of BacT Positive samples, which might be indicative of contamination with tuberculosis bacteria. The samples turned positive over the weekend. An investigation was initiated.
The quality unit of a FDA regulated blood center reported an increase in the number of BacT Positive samples, which might be indicative of contamination with tuberculosis bacteria. The samples turned positive over the weekend. An investigation was initiated.
![]() FDA 483 Observation to Genzyme dated 13 November 2009, for example, pointed out that Genzyme’s:
FDA 483 Observation to Genzyme dated 13 November 2009, for example, pointed out that Genzyme’s:
…Investigation AIR 1517 dated 21 June 2007 was opened because HEPA filters in the filling suite failed routine recertification. The investigation found metal particles embedded in several of the HEPA filters. However, no route [sic] cause was determined for the source of the metal contamination found in these filters.1
![]() FDA MAUDE AE Report on Genzyme Biosurgery’s Synvisc Injection dated 4 August 2008: “The [health care provider] assessed the relationship of the events – swelling both knees and right knee effusion – to synvisc as probable. He assessed the relationship of the event – allergic reaction to synvisc – as likely.”2
FDA MAUDE AE Report on Genzyme Biosurgery’s Synvisc Injection dated 4 August 2008: “The [health care provider] assessed the relationship of the events – swelling both knees and right knee effusion – to synvisc as probable. He assessed the relationship of the event – allergic reaction to synvisc – as likely.”2
![]() McNeil Consumer Healthcare recalled bottles of Tylenol eight-hour caplets after some consumers complained of a musty or moldy odor in the product.3
McNeil Consumer Healthcare recalled bottles of Tylenol eight-hour caplets after some consumers complained of a musty or moldy odor in the product.3
The outcome of an investigation can be followed by the development and execution of corrective actions and preventive actions (CAPA). The logic of this investigative and revising process may be more or less explicit in the various regulated industries, but the underlying logic is the same.
Briefly put, a CAPA can identify several kinds of remediation for a given observation. Some remediation implicates the revision of a standard operation procedure (SOP); other remediation does not. This chapter focuses on the range of intervention and remediation, giving special attention to those that lead to the revision of procedures.
Following an initial section of this chapter that addresses the function of SOPs and the revision of life-cycle documents, the second section discusses the routine review of procedures. This set of routine practices should be juxtaposed to the set of exceptional practices. Thus routine practices can be taken as a baseline for the exceptional practices that are addressed in the remainder of this chapter. The third section addresses the two kinds of internally-based observations that can initiate an intervention and thereby the revision of SOPs: employee notification to supervision and quality (internal) audits. The fourth section discusses the three types of externally-based observations that may initiate an intervention and thereby the revision of a SOP: regulatory inspections, adverse event (AE) reporting, and customer quality complaints.
1.2 Procedures and change
Controlled documents such as SOPs, batch records, manufacturing orders, packaging orders, etc. provide guidance for performing tasks. For brevity’s sake, all these controlled documents are referred to herein as “procedures” or “SOPs.” The procedure identifies the tasks, the environmental and organizational setting for task performance, the resources that make up the prerequisites to each task, the sequencing of the tasks within a given process, the personnel responsible for completing the tasks, and the standards that define the satisfactory completion of the tasks. As David Peterson states:
The purpose of an SOP is straightforward: to ensure that essential job tasks are performed correctly, consistently, and in conformance with internally approved procedures. Clearly, employees’ correct, consistent performance of essential job tasks is as much a business and quality issue as it is a regulatory requirement.4
Of course, the use of SOPs in manufacturing in the life sciences industry is also a regulatory requirement. FDA has stipulated, “There shall be written procedures for production and process control designed to assure that the drug products have the SISPQ they purport or are represented to possess.” Furthermore, “such written procedures shall be followed.”5 FDA has stated similar predicate rules for written SOPs for many of the areas under its jurisdiction, as displayed in Table 1.1 FDA has even stipulated predicate rules for written SOPs that apply to itself, Good Guidance Practices.6
What are some of the consequences of the absence of written procedures?
![]() Without SOPs there is confusion about what the task is and where the task should be performed.
Without SOPs there is confusion about what the task is and where the task should be performed.
![]() Without an SOP the task performance will be poorly resourced – either under-resourced or wastefully resourced. In either case, task performance is more costly, since the under-resourced case may require rework, and the overresourced case may require cost recovery.
Without an SOP the task performance will be poorly resourced – either under-resourced or wastefully resourced. In either case, task performance is more costly, since the under-resourced case may require rework, and the overresourced case may require cost recovery.
![]() The sequencing of tasks may go awry without an SOP – a task that must precede a task may be omitted, or a task that depends upon another task may be performed too soon. Either of these could threaten the integrity of the entire regulated process.
The sequencing of tasks may go awry without an SOP – a task that must precede a task may be omitted, or a task that depends upon another task may be performed too soon. Either of these could threaten the integrity of the entire regulated process.
![]() In the absence of an SOP, personnel may be confused about who is responsible for each task. Tasks may not be successfully performed if there is no one responsible for them, or if two or more employees are responsible for them.
In the absence of an SOP, personnel may be confused about who is responsible for each task. Tasks may not be successfully performed if there is no one responsible for them, or if two or more employees are responsible for them.
![]() Without an SOP there are no standards to define the satisfactory task completion.
Without an SOP there are no standards to define the satisfactory task completion.
SOPs are not set in stone, once and forever. Business process and regulatory requirements for using SOPs incorporate the necessity of revising procedures. The revision process provides an opening for continuous improvement insofar as the changes represent improvement. FDA predicate rules call for not only “written procedures” that “shall be followed,” but the predicate rules also stipulate “written procedures, including any changes.”7
There are several aspects to this issue of “changes.” First, an SOP is a controlled document, so any change in the associated process must be documented in conformity with the organization’s change control process. Second, the FDA predicate rules stipulate that the GMPs must be “current” (i.e., cGMPs), so that changes in the associated process or practices must be captured in the procedure in a timely fashion. Third, technological change is ubiquitous in the life science industry, so there will always be the necessity of revising SOPs as an aspect of business process as well as a regulatory requirement.
Because SOPs must be and are constantly revised in a controlled fashion, it will prove useful to identify the various kinds of events and situations that may call for the revision of procedures. Neither an event nor a situation automatically calls for intervention; these must be labeled as “events or situations of interest” by an appropriate administrative procedure.
There are two fundamental kinds of occasions for revision: routine review of life-cycle documents and revision due to observations of a non-conformance, etc. In turn, there are five main kinds of observations – those that follow from escalated events, from internal audits, from regulatory (FDA) inspections, from AEs, and from customer quality complaints. These five kinds of observations correspond to the five stakeholder groups identified previously, and are displayed in Table 1.2.
Table 1.2
Correspondence between roles and observations
| Major Roles | Kinds of Observations |
| Operational staff and supervisors | Escalated events |
| Quality unit auditors | Internal audits |
| Regulatory investigators | Form 483 observations |
| Healthcare providers | Adverse events |
| Patients | Customer quality complaints |
A complex decision process lies between observation and intervention. Any one of the five kinds of observations may bring about an investigation. An observation is typically escalated, triaged, and may or may not become a record of interest in the organization’s quality management system (QMS). It is the QMS record that may or may not become the basis of an investigation and RCA. The record can also become part of a set of similar records that can be tracked and trended. If necessary, they can be investigated further. The conclusion of an investigation can be followed by the development and execution of CAPA. These elements are displayed in Figure 1.1.
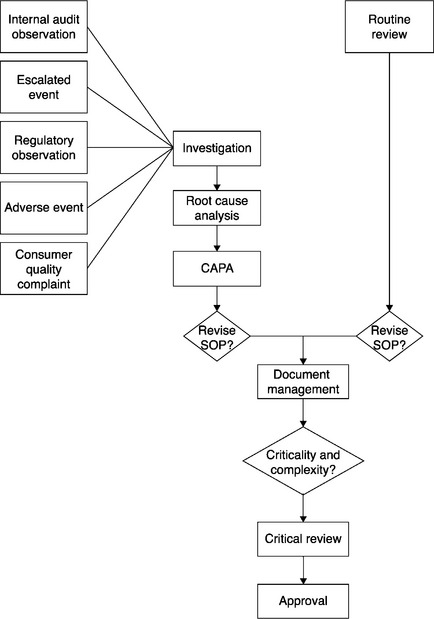
The logic of this investigative and remediation process may be more or less explicit; it is quite explicit in the case of GMPs. It is less explicit in other regulated areas. The underlying logic remains.8
Even if an observation does, on occasion, lead to the re-engineering of the GMP process, it does not necessarily lead to the revision of a procedure. A CAPA can identify several kinds of remediation for a given observation (e.g., business process redesign, risk mitigation, organizational development programs, leadership development initiatives, training intervention, etc.). Some intervention implicates the revision of an operational SOP; other intervention does not.
This section has discussed the function of SOPs and the impact of change on life-cycle documents. It makes the point that change opens the way for continuous improvement of GMP processes in the life sciences industry. The next section addresses the routine review of life-cycle documents.
1.3 Routine review of life-cycle documents
The routine review of life-cycle documents takes place on a regular schedule, typically every two years (biennial review) or every three years (triennial review). Some controlled documents, for example manufacturing orders, are continuously reviewed. At the beginning of the life-cycle, the specific version of the SOP is approved by management and quality assurance (QA) and is then implemented or “reissued.” Once implemented, like any controlled document, the SOP remains subject to change control. Either it will be unchanged during the life-cycle of that version, or it will be changed according to the organization’s change control procedure. If the SOP is unchanged, it will be diligently executed during the remainder of its life-cycle. If problems of execution occur, it will still be diligently executed until it is appropriately changed.
Should problems of execution become apparent during the life-cycle of the current version, there are two options. If the problems are not critical (i.e., presenting neither business risk nor quality risk), they can be documented by the business owner (or management in any impacted area) and filed for the next routine review. An example of a non-critical problem might be a change of name of an organizational unit that is referenced in the SOP. If the problems are critical (i.e., posing a business risk or a threat to the SISPQ of the product), a planned deviation protocol (PDP) can be prepared according to the organization’s change control procedure to revise the current version. The PDP serves as a corrective and preventive action to revise the SOP until the next routine review. As soon as the PDP is implemented, the SOP as revised will be diligently executed during the remainder of its life-cycle.
The elements of the routine review of an SOP are displayed in Table 1.3.
Table 1.3
The routine review of life-cycle documents
1. Begin the routine review of life-cycle documents such as SOPs by identifying the particular document that is due for versioning up. In the case of biennial review, this involves tracking all SOPs and provisionally tagging those that were implemented two years ago.
2. Accommodate the typical time involved in the document change process. Suppose the amount of time needed to revise an SOP is six weeks. This amount of time is added to the tracking process, so the SOPs that are being tagged are those implemented 25.5 months ago (rather than 24 months ago). This ensures enough time for those SOPs to be revised within their life-cycle.
3. Identify the business owner/author of each of those tagged SOPs.
4. Query that business owner to assess the number and substance of proposed revisions to the SOP that have been gathered over the past two years. If those proposed revisions are neither numerous nor substantial, prepare for a minor revision – sometimes called an “update” – of the SOP. If the proposed revisions are numerous and/or substantial, prepare for a full revision of the SOP.
5. Review the SOP and identify all impacted organizational units. In the case of the proposed update, contact the impacted units, indicate the assessment, and invite inputs to the update. In the case of the full revision, contact the impacted units, indicate the assessment, and propose a cross-functional meeting.
6. Prepare the draft document per the appropriate administrative procedure. If it is only an update, submit for approval (see item # 10 below).
7. List the draft document in the document management schedule.
8. If necessary, facilitate the cross-functional meeting and review the draft document to ensure all impacted areas make their inputs.
9. Revise the draft document to address the comments raised in the cross-functional meeting.
10. Submit the document as revised to the management review process.
Once these steps have been completed, the SOP can be subjected to a critical review, in light of the complexity and criticality of the associated process. As the next chapter explains, this critical review can take the form of a management review, an SME review, or a step-by-step real-world challenge. Finally, the SOP is approved and implemented.
1.3.1 A different organizational approach
Some organizations take a different approach to the routine review of operational SOPs. Consider the following: an implemented, operational SOP that was due for routine biennial review would be identified. By one administrative procedure (call it “SOP Review Cycle”), an SME was assigned to review the operational SOP. The SOP Review Cycle procedure required the SME to “compare the operational SOP to current practices.” If the operational SOP was found not to deviate from “current practices,” the SME would prepare a work request for an “updating” of the operational SOP, whereupon its version number would be raised and the next due date for its revision would be stipulated. This “updating” of the operational SOP is not considered to be a versioning up. If the operational SOP was found to deviate from the “current practices,” the SOP Review Cycle required the SME to prepare a work request for a “revision” of the operational SOP. Then, by a second administrative procedure (call it “Creation and Revision of Operational Documents”), the operational SOP would be revised and its content brought into accord with the current practices. What is wrong with this picture?
Clearly this organization was not following cGMPs or, presumably, its own written operational procedures. In terms of cGMPs, FDA has stipulated that “written procedures shall be followed.” FDA did not suggest that written procedures be followed sometimes, and current practices be followed other times.
In other words, the operational practices are to conform to the written procedures, not vice versa, and moreover “any deviation from the written procedures shall be recorded and justified.”9 This recording and justification process must be timely, certainly not on a two-year cycle. There is the possibility of FDA inspectional observations regarding the failure to follow written procedures. On top of that is the possibility of FDA inspectional observations regarding inadequacy of the organization’s investigations and CAPAs.
Moreover, in the example, this approach to the routine review of life-cycle documents contradicted the organization’s change control procedure. That procedure stipulated that for proposed changes to be implemented, an operational SOP had to be appropriately requested, processed, and approved. A proposed change request had to indicate any impact the changes would have on process, material, product, regulatory filings, etc. The request also had to identify activities, responsible parties, deliverables, and time frames and due dates, comprising the proposed changes. Pending the approval of the change request, the current version of an operational SOP was to be executed diligently. There is no easy way to harmonize such contradictory SOPs as the SOP Revision Cycle procedure and the change control procedure.
This section has addressed the requirements for the routine review and controlled change of life-cycle documents such as an operational SOP. An alternate approach to routine review is discussed; however, it was found that the alternate approach posed the threat of FDA inspectional observations. The next section of this chapter examines the occasions for remediation that are due to an internally-based observation. Some of these implicate the revision of an SOP; others do not.
1.4 Intervention due to internal observation
There are two major occasions for remediation because of an internally-based observation that may require the revision of an operational SOP. The first is an observation made by an employee that requires notification to management of a business risk or quality risk. The second is an observation made by an auditor during an internal quality audit.
1.4.1 Management notification
The first kind of observation is made by an employee who is not necessarily (or even usually) an auditor. This is an internal observation of an event or situation that may constitute a business risk or quality risk. This can be an observation of either an exceptional event or a routine activity.
Any organization whose processes are “in control” will have an administrative SOP, call it “Management Notification,” that requires an employee to inform supervision of any event or situation that may impact the SISPQ of the product or constitute a business risk. FDA stipulates that it is the “person responsible for supervising” who must provide assurance of the SISPQ of the regulated product.10 And supervisory personnel and management can only provide that assurance if they are notified of threats to the SISPQ of the product in a timely fashion. Management must be informed within a specified time frame so that appropriate action can be taken (Table 1.4).
Table 1.4
Fields in typical Notice of Event form
1. Title of this event
2. Name of employee who observed this
3. Associated Quality Management System (QMS) tracking number(s)
4. Department where event was observed
5. Shift when event was observed
6. Date of discovery
7. Date of event occurrence (if known)
8. Employee involved in event
9. Other personnel involved
10. Symptom (problem type)
11. Attached files
The employee’s observation will prompt one or more of the following responses from the manager. First, the manager may triage and dismiss the event as insignificant. Second, the manager may call for immediate action to address the event that has been observed. Third, depending on the specifics of the Management Notification procedure, the manager may acknowledge receipt of the employee’s observation. The employee may be required to submit the notification to management in writing; the manager may also be required to acknowledge receipt of the notification in writing. Fourth, the manager may escalate the notification further up the line. Fifth, management may organize an investigative team for the event, a team that is charged with discovering the root cause.11
The investigative team’s report may lead to appointing a person responsible for preparing a CAPA plan (Table 1.5). In that case, management must also stipulate a due date for the CAPA. Insofar as the CAPA requires the revision of an operational SOP, the employee’s observation initiates the chain of events that lead to the revision.
Table 1.5
1. Analyze various sources of observations to identify existing and potential causes of quality problems.
2. Investigate the root cause of the quality problem.
3. Identify remediations to correct and prevent recurrence of the quality problem.
4. Verify that the CAPA is effective and does not create further problems.
5. Implement and record changes in methods and SOPs required by the CAPA.
6. Ensure that information related to the quality problems is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems.
7. Submit information on quality problems, as well as CAPAs, to management.
8. Document all activities under the CAPA.
1.4.2 Quality audit observation
Any organization whose processes are “in control” will have a QA unit that is responsible for approving or rejecting the procedures, protocols, and specifications that impact the SISPQ of the product.12 That unit must monitor the organization’s compliance with these operational SOPs, which entails responsibility for quality audits and “reaudits of deficiencies.”13
A quality auditor, at the conclusion of the audit, will document audit findings (i.e., observations) and report these to the manager of the unit that was audited. The manager will identify a responsible person to undertake or oversee whatever investigation and RCA is required. In the real world of limited resources, Lee Vanden Heuvel and Christine Robinson point out that an investigation must balance the costs of the effort against the expected benefit of identifying the root cause.14
Typically, an investigation includes the following main steps:
![]() Evaluate the information, assess risk, take immediate remedial action;
Evaluate the information, assess risk, take immediate remedial action;
![]() Investigate and assign responsibility;
Investigate and assign responsibility;
![]() Analyze and document the root cause.15
Analyze and document the root cause.15
FDA also stipulates that when an investigation is called for under 21 CFR 211.192 and is not conducted, “the written record shall include the reason that an investigation was found not to be necessary and the name of the responsible person making such a determination.”16 When the investigation is complete, the responsible person will then prepare a CAPA plan to address the observations, along the lines suggested by the FDA and displayed in Table 1.5. The manager will also stipulate a due date for the CAPA.17
The quality audit observation will constitute the occasion for the revision of an operational SOP insofar as the remediation listed in the CAPA requires that revision. An example of this kind of observation, investigation, and CAPA would be an in-process quality observation of out of specification (OOS) levels of product variation. Management would order an investigation and then call for CAPA. The CAPA might propose that more specific directions were needed in the charging procedure (e.g., a more defined rate of addition of an ingredient) to minimize variation. These directions would be added to the operational SOP, either in the next routine biennial review or, more likely, by way of a PDP.18
As noted previously, a CAPA can call for all kinds of remediation for a given audit observation – business process redesign, risk mitigation, training intervention, organizational development initiatives, etc. Some intervention involves the revision of an operational SOP; for instance, business process redesign typically must be captured in a revised procedure. Other intervention does not; for instance, training interventions and leadership development initiatives usually do not require any change in an operational SOP. All intervention should contribute to continuous improvement of the GMP process.
This section has addressed the occasions for intervention that are a result of an internal observation – an observation of a discrepancy (or an opportunity for process improvement) that an employee escalates to supervision, or an observation made during a quality audit. In either case these may lead to the revision of an SOP. The next section examines occasions for remediation that are due to an external observation.
1.5 Intervention due to external observation
There are three major occasions for intervention that may result from an external observation of a deviation and, therefore, may lead to the revision of an operational SOP. The first is an observation made by an investigator during a regulatory inspection.19 The second is an observation associated with an AE. The third is an observation that accompanies a customer quality complaint.
1.5.1 Regulatory inspection
FDA has defined “an establishment inspection [as] a careful, critical, official examination of a facility to determine its compliance with laws administered by FDA.”20 More specifically, the inspection examines the organization’s adherence to the concepts of sanitation and GMPs, seeks assurance that all reasonable precautions are being taken to ensure the SISPQ of finished products, and seeks to identify deficiencies as well as to obtain correction of those deficiencies. This inspection can be either comprehensive or directed. The comprehensive inspection covers everything in the organization subject to FDA jurisdiction to determine the organization’s compliance status. The directed inspection covers specific areas to an assigned depth (ibid.).
In the case of a manufacturing site, this inspection will typically inspect the quality system and one of the other five systems of the FDA Quality Systems Approach: facilities and equipment, laboratory controls, production, packaging and labeling, and materials.21 A regulatory inspection can include FDA personnel reviewing records of prior inspections, walking through the facility, examining current records, interviewing employees, etc.
When “deficiencies” are observed, they are reported to management before FDA personnel has left the facility. These Form 483 observations can later become part of an FDA Warning Letter to the management of the organization. At that point, if not earlier, the organization will begin to respond. The response typically includes management’s call for an investigation of each deficiency observed during the inspection. Each investigation will include RCA, and tracking and trending of similar events or situations observed elsewhere or in prior inspections. CAPA will be developed and executed, all within a specified timeline. The remediation may require the revision of an operational SOP.
1.5.2 Adverse event
A second kind of observation is associated with an adverse event (AE). With respect to drugs, FDA has defined an AE as “any adverse event associated with the use of a drug in humans, whether or not considered drug related.”22
Again with respect to drugs, a serious adverse event (SAE) is further defined by the FDA as:
…any adverse drug experience occurring at any dose that results in any of the following outcomes: death, a life-threatening adverse drug experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/ incapacity, or a congenital anomaly/birth defect. Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered a serious adverse drug experience when, based upon appropriate medical judgment, they may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition. (ibid)
Finally, an unexpected adverse event (UAE) is defined by the FDA as:
…any adverse drug experience that is not listed in the current labeling for the regulated product. This includes events that may be symptomatically and patho-physiologically related to an event listed in the labeling, but differ from the event because of greater severity or specificity.
The FDA explains further that:
‘Unexpected,’ as used in this definition, refers to an adverse drug experience that has not been previously observed (i.e., included in the labeling) rather than from the perspective of such experience not being anticipated from the pharmacological properties of the pharmaceutical product. (ibid)
FDA has provided analogous definitions of an AE for other regulated areas. Moreover, FDA requires “written procedures for the surveillance, receipt, evaluation, and reporting of post-marketing AEs.”23 FDA requires the investigation and reporting of AEs for many of the areas under its jurisdiction, as shown in Table 1.6. FDA encourages health care providers to report any AEs that the providers judge to be clinically significant.
Table 1.6
FDA predicate rules for adverse events
| Regulated Area | Regulation |
| Animal drugs | 21 CFR 514.80 |
| Biologies | 21 CFR 600.14, §600.80 |
| Blood processing | 21 CFR 606.171 |
| GCP | 21 CFR 310.305 |
| GMP | 21 CFR 211.198 |
| GTP | 21 CFR 1271.350(k) |
| Medical Devices | 21 CFR 803.10 |
In a typical case, an AE report is initially recorded in the organization’s drug safety information system within a specified period of time after receipt of the report (say one day). That record should include the name and title of the reporter (i.e., typically the health care provider), contact information for the reporter, the product in question including label information, and the patient’s identifier.24 A medical reviewer gives a preliminary judgment of the etiology, the “expectedness,” and the severity of the AE, and provides a medical code for the event based on the Medical Dictionary for Regulatory Activities (MedDRA) or on an internal corporate code used to describe these events. The medical reviewer also determines whether the language describing the event is correct.
This is the point of initial triage.
If the event is judged as clearly not drug related, then the report is completed at this point. However, if it is not clear or is a SAE, then the event is escalated to the next level. Depending upon the preliminary assessment of severity, the case can be expedited. If so, information on this case is provided to FDA for “each adverse drug experience that is both serious and unexpected as soon as possible.”25
Subsequent investigation of an SAE includes the following two aspects:
1. Case specific – reviewing the demographics of the patient, dosage levels and length of exposure, other medications, and other medical conditions;
Then two other medical reviewers must independently review the case narratives and lab reports including the diagnosis, clinical course, therapeutic steps, and outcome of the event. They must also independently check the medical coding and confirm the case assessment. If the blinded assessments differ, they must be reconciled. Once reconciled, the case report is submitted to the company’s Regulatory Affairs officer.
Remediation may be called for if the tracking and trending of similar events show a pattern and the trend reaches a predetermined threshold, or if the individual event is serious enough. In fact, the two factors that drive the decision to make a response are frequency and severity. If the event is a minor complaint (such as a rash) and also a frequent complaint, this may warrant a labeling change.26 Remediation will also be considered for a more serious event, such as acute renal failure, that occurs at a low frequency. Insofar as remediation requires the revision of an operational SOP, the observation and reporting of an AE initiates the revision, similar to the case of the quality audit discussed previously.
1.5.3 Customer quality complaint
A third kind of observation takes the form of a complaint made by a customer routed to the organization’s quality complaint unit (CQU). FDA has defined a customer quality complaint as “any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a [product] after it is released for distribution.”27 Processing of complaints and maintaining complaint files is an FDA requirement for many of the areas under its jurisdiction, as displayed in Table 1.7.
Table 1.7
FDA predicate rules for complaint files
| Regulated Area | Regulation |
| GCP | 21 CFR 310 |
| GMP | 21 CFR 211.198 |
| GTP | 21 CFR 864.3250; §1271.320 |
| HAACP | 21 CFR 123.8 |
| Mammography | 21 CFR 900.4 |
| Medical Devices | 21 CFR 814.9; §820.198 |
| Medicated Feeds | 21 CFR 225.115 |
Upon receiving a complaint, CQU conducts a triage whereby the complaint is classified according to type: routine complaint or urgent complaint. If the complaint is classified as routine, a response is prepared, routed via the QMS through the approval process and finalized, and the file is closed. No operational SOPs would be revised as a result of a routine complaint. If the complaint is classified as urgent, a record is opened in the QMS, the complaint is classified in terms of product quality complaint (PQC) criteria, and the complaint and supporting material are routed to the manufacturing site.
Management decides whether an investigation is required; if so, a responsible person is assigned the task of preparing an investigation plan, and a time schedule is developed. Preliminary tests are conducted, such as X-ray, visual inspection, and functional tests. If further investigation is warranted, the complaint moves to the quality lab or other location where more tests are conducted and evaluations made. Finally, the findings and recommendations of the investigation are reported, routed through the approval process and finalized, and the file is closed. Insofar as the remediation following from the investigation includes the revision of an operational SOP, the customer quality complaint initiates the chain of events that lead to the revision.
1.6 Conclusion
This chapter has considered the occasions that lead to re-engineering of GMP processes and, under certain conditions, the revision of SOPs. Five groups of stakeholders in the sphere of FDA-regulated industry are identified. Particular kinds of observations that can occasion intervention tend to be associated with each of these stakeholders. While there is some overlap between them, escalated events tend to be associated with employees, internal audits with auditors, Form 483 observations with regulatory investigators, AEs with health care providers, and customer quality complaints with patients.
The observations initiate an investigation and remediation process that varies in emphasis but that has an underlying logic. An observation is typically escalated, triaged, and can become a record of interest in the organization’s QMS. That record can become the basis of an investigation and RCA. It can also become part of a set of records of similar events that are tracked and trended, and investigated further. When the investigation is concluded, remediation can be proposed in the form of CAPA. The remediation is approved and enacted, and may include the revision of an SOP. Thus the initial observation can constitute an occasion for the revision of the procedure. Moreover, a diligent approach to intervention can promote the continuous improvement of the GMP process.
From an organizational standpoint, it is clearly preferable to receive internal observations rather than external observations; it is preferable to conduct investigations, and to develop and enact remediation in response to observations by internal stakeholders, rather than waiting until the organization must respond to external stakeholders. Hence it is good strategy to encourage employees not only to bring suggestions for process improvement, but also to bring observations of events and situations that may constitute a business or quality risk, to the attention of supervision. It is also good strategy to have in place an administrative procedure that provides guidance in the timely, systematic, and appropriate management response to such observations.
1.8 References
Barsky, E., Grunbaum, L. The “business” of investigations. J. GXP Comp.. 2008; 12(5):90–96.
Bartholomew, D. CAPA and root cause analysis. Pharma. Manu.. 5(4), 2006.
Campbell, K., FDA Risk-based Inspection Model. presented at Validation Week, 2008. [Philadelphia, PA, 22 October 2008].
Eckert, C. Eliminating defects in the vertical supply chain. Qual. Dig.. 2008; 28(12):24–26.
FDA. Guidance for Industry: Quality Systems Approach to Pharmaceutical cGMP Regulations, Rockville. MD: CDER/CBER. 2006. [September 2006].
FDA. Investigations Operations Manual. Rockville, MD: ORA; 2008.
Fields, T. Auditing as a component of a pharmaceutical quality system. J. GXP Comp.. 2008; 12(5):61–68.
Heuvel, L.V., Robinson, C. How many causes should you pursue? J. Qual. Particip.. 2005; 28(2):22–23.
International Conference on Harmonisation (ICH). Pharmaceutical Quality System – Q10, 2008:9. [Geneva: ICH Secretariat, 4 June 2008].
Nelson, R., et al. Good pharma-covigilance practices. Drug Safety.. 2002; 25(6):407–414.
Peterson, D.C. Assuring the effective use of standard operating procedures (SOPs) in today’s workforce. BioPharm Internat.. 2006; 19(2):42–46.
Queffelec, D., Peterson, D. Global quality assurance and regulatory compliance. BioPharm Internat.. 2008; 21(11):64.
Sandler, J. CAPA and root cause analysis based on risk management and regulatory fundamentals, presented at Validation Week. Philadelphia: PA, October; 2008. [2008].
Singer, N. More Trouble with Tylenol. New York Times, 18. (October):2010.
Spear, J. Incident investigation: a problem solving process. Prof. Safety.. 2002; 4:25–26.
Weil, S. 21 CFR Part 11: a detailed overview. ISSA J.. (April 2004):2004.
1For FDA 483 Observation to Genzyme. Available from: www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofGlobalRegulatoryOperationsandPolicy/ORA/ ORAElectronicReadingRoom/UCM191991.pdf
2For FDA MAUDE AE Report on Genzyme Biosurgery’s Synvisc injection. Available from: www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoi__id = 1099535
4See David C. Peterson (2006).
5For the requirement of “written procedures,” see 21 CFR 211.100. For the requirement that they “shall be followed,” see 21 CFR 211.22, ʃ 211.80, etc. On the regulatory side, see Denise Queffelec and David Peterson (2008):
Failure to follow established SOPs is one of the most frequently cited violations in FDA 483 s and warning letters. The frequency of SOP-related violations points to the need for all regulated companies to review their SOPs, their methods for distributing compliant SOP training curricula, their methods of validating receipt and testing for comprehension of the materials, and their documentation of SOP training activities.
621 CFR 10.115. As Steven Weil (2004) has put it:
Table 1.1
FDA predicate rules for written SOPs
| Regulated Area | Regulation | Predicate Rule |
| Biologics | 21 CFR 600.80 | Post-market AEs |
| Food | 21 CFR 179.25 | Food irradiation |
| GCP | 21 CFR 56.101 | IRBs |
| GCP | 21 CFR 310.305; §314.80 | Post-market AEs |
| GLP | 21 CFR 58.35 | QA unit |
| GMP | 21 CFR 211.186 | Control records |
| GTP | 21 CFR 1270.31; §1271.180 | Good tissue practices |
| HAACP | 21 CFR 120.11 | Calibration |
| Medical Devices | 21 CFR 812.25 | Investigational plan |
A predicate rule is an FDA regulation such as Good Laboratory Practice (GLP) or Current Good Manufacturing Practice (cGMP). Predicate rules mandate and define: What records must be maintained; The content of the records; Whether signatures are required; How long records must be maintained.
721 CFR 211.100. Of course the changes may be improvements, or the opposite (i.e., procedure “churn”).
8This has been recognized by the International Conferenceon Harmonization (ICH) (2008) in its call for a system for implementing CAPAs resulting from the investigation of a wide range of observations, including “complaints, product rejections, non-conformances, recalls, deviations, audits, regulatory inspections and findings, and trends from process performance and product quality monitoring […] throughout the product life-cycle.”
For the predicate rules regarding GMP processes, see 21 CFR 211.192, Production record review:
Any unexplained discrepancy or the failure of a batch or any of its components to meet any of its specifications shall be thoroughly investigated. The investigation shall extend to other batches of the same drug product and other drug products that may have been associated with the specific failure or discrepancy. A written record of the investigation shall be made and shall include the conclusions and follow-up.
We take “unexplained discrepancy” to imply an observation, “other batches” to imply tracking and trending, and “conclusions and follow-up” to imply remediation.
921 CFR 211.100.
1021 CFR 211.25(b).
11See also 21 CFR 211.192 “Production record review.”
12This is explicit in 21 CFR 211.22; it is more implicit in 21 CFR 1271.160. Again, all these controlled documents are referred to herein as “procedures” or “SOPs.”
1321 CFR 1271.160 (b) (3). See also Tim Fields (2008).
14See Lee Vanden Heuvel and Christine Robinson (2005). Their Figure 1 provides a very useful overview of the tradeoff between level of effort versus return in a RCA. The ICH (2009) has stated that “The level of effort, formality, and documentation of the investigation should be commensurate with the level of risk.” Jerome Spear (2002) discusses setting priorities in such a real world.
15See also James Sandler (2008); Emma Barsky and Len Grunbaum (2008); and Doug Bartholomew (2006). For a discussion of the problems of “silo thinking,” “finger pointing,” and “jumping to conclusions” that bedevil real-world investigations, see Chris Eckert (2008).
16See 21 CFR 211.198(b) (3), ʃ 106.100(k)(2), ʃ 820.198(b), ʃ 1271.370(b), etc.
17See, for instance, 21 CFR 820.100 “Corrective and preventive action,” also ʃ 806.10 “Reports of Corrections.”
18As the ICH (2008) has pointed out, “CAPA methodology should result in product and process improvements and enhanced product and process understanding.” This highlights the role of remediation in continuous improvement.
19A regulatory inspection could be conducted by the EPA (focusing on emissions and waste), the DEA (controlled substances), etc. Chapter 6 discusses the regulatory overlap between these various agencies. This present chapter limits the discussion to the FDA.
20See the FDA Investigations Operations Manual (2008) Sect. 5.1.2, “Inspectional Approach,” and Sect. 5.51 “Drug Inspections.”
21See the FDA guidance Quality Systems Approach to Pharmaceutical cGMP Regulations (2006); also Karyn Campbell (2008).
2221 CFR 310.305(b) “Records and Reports.”
2321 CFR 310.305(a) “Records and Reports.”
24See also Robert Nelson, et al. (2002). Note that the reporter could be anyone, including the patient. Most organizations require that an employee report within one business day if a person complains about some physical problem or symptom and mentions they are taking one of the organization’s products.
2521 CFR 310.305(c) (1), “Post-marketing 15-day ‘Alert reports’.” Many organizations provide these reports in seven days. In addition, AEs relating to a regulated product are reviewed every six months for each regulated product and an updated report is filed with the FDA.
26Remediation may involve a “Dear Investigator Letter” (DIL) if it is observed during a clinical trial. A DIL is prepared if an AE occurs during a clinical trial and is confirmed or reasonably certain to be related to the regulated product. Incidentally, the term CAPA is rarely used in clinical studies. If this is a post-marketing trial or observation, then changes to the labeling may be considered.
2721 CFR 820.3(b), “Definitions.”