
The role of critical review in the revision of procedures
Abstract:
This chapter addresses changes to processes and the revision of standard operating procedures (SOPs). Emphasis is put on the necessity of SOP revisions. An illustrative problem is presented – an example of a laboratory weighing procedure and the development of a corrective action and preventive action (CAPA) project. Procedural changes in the example are evaluated and reviewed for regulatory compliance. The management of change depending upon risk assessment is considered.
3.1 Introduction
Given the ubiquitous changes in technology, procedures must be appropriately revised. The key word here is “appropriate.” Revisions – the “versioning up” – of a standard operating procedure (SOP) that adds value to a procedure can contribute to best practices. Revisions that do not add value are wasteful, and from a regulatory standpoint suggest that a process is not in control. In order to ensure that the revision is appropriate, the SOP should be subjected to critical review. Newly written procedures can also be subjected to critical review.
Approaches to the critical review of procedures vary in terms of increasing credibility of the review findings. It is a prerogative of management to weigh the benefits of increasing credibility against the costs of increasing rigor of the approach. This cost/benefit analysis must be informed by a determination of the degree of change that is involved in the revision, as well as a risk assessment of the change.
After examining the critical review of SOPs, this chapter will consider how the management of change depends upon risk assessment. It has three components: risk identification, risk analysis, and risk evaluation. The criticality and complexity of the process tends to increase the level of risk. And the appropriate level of critical review and effort supporting implementation of change is directly related to the criticality and complexity of the process.
Following the discussion of risk, an illustrative problem is presented. Over time, the variability of the potency of an active pharmaceutical ingredient (API) was seen to increase. This is a complex problem, implicating the weighing facility and instruments for the analytical standard, as well as the associated weighing procedure and calibration procedure. It is also clearly a critical problem, as the potency of the API impacts the quality attributes of the product.
A CAPA plan is developed and implemented to remediate the facility and instruments; this leads to revisions to the relevant SOPs. It is necessary to review critically the adequacy of these revised procedures. The CAPAs are tested, and the results lead to informed decision making about the changes as well as mitigation of the original problem.
This chapter concludes by considering the place that the critical review of procedures holds in a value-adding approach to program design and management.
3.2 Overview of critical review of SOPs
An SOP is a “process control;” it controls the execution of a process. The SOP can address several kinds of process – a person-to-machine process, a person-to-paper process, a person-to-person process, or a combination of the three processes.1
The SOP is a controlled document, meaning it is subject to change control. Any proposed changes to this document (and the real-world process it reflects) must be processed and approved according to the applicable change control process, as stated in the organization’s change control procedure.2 The proposed change request must indicate any impact the changes will have on process, material, product, regulatory filing, other good manufacturing practice (GMP) sites, etc. The request must identify activities, responsible parties, time frames and due dates, and deliverables comprising the proposed changes.
Given the constancy of technological change – as well as the frequency of non-conformances (unplanned deviations), associated investigations, and CAPAs – procedures must be revised. Revisions should be value-adding activities, but often are not. When the revision does add value, it can contribute to best practices in development or manufacturing. When it does not add value, it is sometimes called “procedure churn;” other times it is called “word-smithing.” From a business standpoint, procedure churn is wasteful, hence uneconomical. From a regulatory standpoint it also suggests that a process is not in control. In order to ensure that the revision adds value (i.e., adds content that is needed and no more), the SOP should be critically reviewed.
The critical review of an SOP ensures that the process addressed in the procedure, as written and executed, will attain the outcome the organization wants. The critical review of a revised procedure ensures that the proposed changes will add value to the process. Addressing “validation of both the process and process controls,” FDA defines validation as follows: “Process validation is establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality characteristics.”3 Various approaches to validation can then be ranked in terms of the “degree of assurance,” or credibility of the resulting evidence. The same is the case for the critical review of procedures.
Take a drafted SOP, whether a newly written procedure or a revision of a current one. Critical review of the SOP consists of one or more of the following approaches, listed in terms of increasing credibility of the resulting evidence:
![]() management review during the SOP approval process;
management review during the SOP approval process;
![]() expert review by subject matter experts (SMEs) or others;
expert review by subject matter experts (SMEs) or others;
Management review is the vetting of the procedure as it goes through the several iterations of the document change process. The management of each department that will be impacted by the revisions has the opportunity to review the draft, suggest changes, and sign off on the document. Everything in management review will routinely be captured in the document change process.
Management review provides the lowest degree of assurance of the resulting evidence among the several approaches. This is in good part due to the organizational role of management review in the document change process. The manager of each department, or the designee, looks for the potential impact of the revision on that department. The manager also looks for departmental responsibilities and responsible persons. Beyond those issues, there is little interest in the critical review of the procedure.
In the case of expert review, the SMEs will review the draft for both positive and negative elements. On the positive side, they will look for best practices, value-adding steps, flexibility in light of changing demands, scalability in light of changing output targets, etc. On the negative side, they will look for bottlenecks in the process, duplication of effort, unnecessary tasks, non-value-adding steps, role ambiguities (i.e., several positions responsible for a task, or no one responsible for a task), etc. It is important to document all the points raised by the experts in their review.
Expert review provides a higher degree of assurance than management review, because it is a compilation of expert opinion, and it is focused on the technical content of the procedure. The International Organization for Standardization (ISO) has stipulated that validation evidence should be “objective.”4 The same is the case for the critical review of procedures. The opinions of SMEs, while clearly not the simple prejudices of lay persons, are also not clearly “objective.”
The real-world challenge tests the procedure’s applicability by challenging it step-by-step on the floor or lab bench. This involves selecting one (or more) seasoned employee(s) within the scope of the draft procedure – not necessarily a SME – and comparing the steps as drafted in the procedure with the employee’s activities. It is important to ascertain if they align. It is equally important to consider evidence of resistance, repetition, and human factor problems like task difficulty. Once again, it is critical to document everything in the challenge.
This challenge provides a greater degree of assurance about the resulting evidence because it is an objective test of the procedure’s applicability. However, it does not control a number of internal and external threats to validity. Internal threats to validity, such as history effects or maturation effects, may provide plausible alternative explanations of the resulting evidence.5
The revised procedure may “look” better than the current procedure, but it may appear so because this particular operator, at this time and place, performs better because she or he got the prized place in the company parking lot that morning. External threats to validity (and the associated threats to “transferability”), such as expectancy effects, also called Rosenthal effects, may limit the generalizability of the resulting evidence. The operator may perform better because the vice-president of technical operations has inadvertently communicated her expectations for the real-world challenge.6
Nonetheless, it provides more credible findings for the decision either to proceed with versioning up the SOP, or to introduce further revisions in the procedure.
Suppose that management cannot make a decision about the applicable standard, say the concentration of the sanitizing solution, by means of management review, peer review, or the real-world challenge. Conducting a study based on an experimental design is an option that management can consider. Such a study will control for internal and external threats to validity, thereby providing credible findings for the requisite decision.
The proposed study is a randomized design examining the efficacy of several levels of concentration of the solution as applied to randomly selected sites in the facility7 .
The model for this randomized block design is:
β1 is the effect of the primary factor, the levels of concentration of the sanitizing solution (i = level 1; level 2,…level mean;
β2 is the effect of other program factors;
The observations will be the environmental monitoring data from the sampled sites. The primary factor, the levels of concentration of the sanitizing solution, will range from the level 1 to the level n concentration.
Immediately following sanitizing activities by the members of the sanitizing and cleaning unit, EM data will be collected and appropriately recorded.
The study hypothesis addresses the effect on bioburden of the levels of concentration of the sanitizing solution. The higher the level of concentration, the greater the reduction of the bioburden. Given the analysis of variance (ANOVA) and the F-distribution, if F0 > Fa we may conclude that the factor values are different for a given significance level a and the null hypothesis is to be rejected. Such findings will provide very credible inputs for the requisite management decision about the revision of the procedure.
These approaches to critical review are ranked in terms of increasing credibility of the results. When selecting between them, management must weigh costs against benefits, comparing the costs of increasing rigor to the benefits of increasing credibility of the findings.8 Typically, the approach to critical review of procedures that is selected will be part of a more general CAPA project.
This section has discussed how revisions to SOPs should be critically reviewed in the same manner as changes to processes such as manufacturing processes, cleaning processes, analytical methods, etc. As mentioned previously, risk assessment should be included in the weighing of costs and benefits.9 An SOP that is associated with a process or component of greater complexity and criticality will have more stringent requirements, and will require a more critical approach to the review of the procedure.
3.3 Risk assessment and critical review
The critical review of a new or revised procedure should be guided by the following three fundamental questions, all elements of risk assessment:
1. What might go wrong with the associated process? Answering this question involves risk identification.
2. What is the likelihood that this will go wrong? A risk analysis addresses this second question.
3. What are the consequences? How severe are they if this goes wrong? Risk evaluation provides an answer to this question.10
The first of these three questions raises the issue of the complexity of the associated process. Definitions of complexity include the following:
![]() Interconnectedness: the organization and interaction of system components;
Interconnectedness: the organization and interaction of system components;
![]() Time-variance: the repeatability of the system’s response to control stimuli;
Time-variance: the repeatability of the system’s response to control stimuli;
![]() Information content: the amount of information needed to deal with the system from a particular perspective such as creation, use, or maintenance.11
Information content: the amount of information needed to deal with the system from a particular perspective such as creation, use, or maintenance.11
Test categorization, which measures the complexity of laboratory tests covered by the Clinical Laboratory Improvement Amendment (CLIA), provides one illustration of the measurement of complexity.12 Using the seven criteria listed in Table 3.1, a laboratory test is graded for level of complexity by assigning scores of 1, 2, or 3 within each of the criteria.
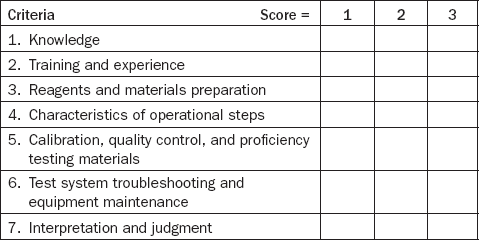
A score of “1” indicates the lowest level of complexity, and a score of “3” indicates the highest level. These scores are then totaled. Laboratory tests receiving scores of 12 or less are categorized as moderate complexity, while those receiving scores above 12 are categorized as high complexity. An analogous approach could be used to measure the complexity of another process.
The higher the complexity, the more the likelihood that something will go wrong in the process. Risk identification becomes more difficult as the process becomes more complex. This is because increasing complexity increases uncertainty about a process, “uncertainty due to combination of incomplete knowledge about a process and its expected or unexpected variability.” Since risk identification “provides the basis for further steps in the quality risk management process,”13 increased complexity of a process, as well as the associated detectability of the various hazards, makes the risk analysis – the second of the three questions, involving an estimation of risk associated with an identified hazard – more difficult.
The third question raises the issue of the criticality of the process. The Criticality Task Team of the ISPE Product Quality Life-cycle Implementation (PQLI) initiative has provided the following comments on the concept of criticality and its measurement.14 A component of a system is categorized as potentially critical, in contrast to some other component that is categorized as non-critical, in terms of the severity and probability of risk that component poses to the safety, efficacy, and quality of the product, and harm to the patient. The relative level or degree of risk a component poses is assessed relative to the probability of occurrence, detectability, and potential harm to the product or the patient.15
The more critical the process or component, the more severe the consequences should something go wrong. In brief, a procedure for supplier quality control (QC) is more complex than an SOP for signature cards; a procedure that provides guidance to a process that “touches the product” is more critical than an SOP for cartonizing a secondary package. As the International Conference on Harmonisation (ICH) has expressed it: “the level of effort, formality and documentation of the quality risk management process should be commensurate with the level of risk.”16 Thus, the review must take into account the dimensions of complexity and criticality of the associated process.
Table 3.2 displays the dimensions of complexity and criticality.

When the Complexity × Criticality is Low/Low (Scenario A in Table 3.2), the first approach (the management review of the new or revised procedure) may be appropriate. A procedure for weight checks of cartons might be an illustration of Scenario A. When the Complexity × Criticality is Med/Med (Scenario B in Table 3.2), the second approach to the critical review of the new or revised procedure, expert inputs, may be appropriate. The acquisition of a new model of a lab balance, and the issue of the revisions to the procedure for weighing this purchase requires, might be an illustration requiring more than management review. Finally, when the Complexity × Criticality is High/High (Scenario C in Table 3.2), the critical review of the revised procedure may necessitate a step-by-step challenge. The planning of a new central weigh facility, and the procedures that will be necessary in that facility, might be an illustration requiring more than SME inputs.
This section has discussed how risk assessment is an important component of the management of change. The level of critical review and appropriate work supporting implementation of the change is directly related to the criticality and complexity of the process. The next section presents an illustrative problem that requires an investigation, leading to the development of a CAPA plan that calls for changes in process as well as revision of analytical laboratory procedures.
3.4 Investigating a complex × critical problem
FDA-regulated industry must have a written procedure that defines “events of concern,” which include actual departures from established, approved SOPs, material specifications or manufacturing orders, etc. as well as potential departures (e.g., in the form of trends observed in a product-monitoring system). Regulated industry must also have a procedure for the conduct of investigations of such departures, insofar as they actually or potentially impact the quality attributes of the product.17
These procedures were followed regarding the illustrative problem presented in the following example.
3.4.1 An illustrative problem
A QC staff member reported an increasing variability in the determination of potency of the active drug of a pharmaceutical product, a commercially available FDA-approved tablet. The variability of data on product potency did not fail specifications and was not sufficient to cause risk to patients taking the drug. However, the variability was not consistent with manufacturing data from similar products and/or processes in the same facility. Tracking and trending suggested the increasing variability should be dealt with, because it potentially impacted the quality attributes of the product. Thus the criticality of the problem was recognized from the start.
A deviation investigation team was formed including responsible manufacturing, quality assurance, and laboratory management and key personnel, and a project manager was designated. This team initiated an investigation to address the increase in data variability, beginning with a comprehensive review of the product manufacturing process. Their review focused on activities specifically influencing drug potency. Activities reviewed included active drug weighing and dispensing, active ingredient charging steps in the manufacturing process, sources of process variation, possible drug loss in processing, sampling of tablets for potency testing, and analytical testing including all associated procedures. The range of areas and activities included in the investigation highlight the complexity of the problem.
A more specialized review team was formed in the analytical area. This review team comprised the analytical department manager, SMEs, laboratory analysts, and associated personnel. This team reviewed all activities associated with the drug potency assay, including standard preparation, incoming sampling control, sample preparation, high performance liquid chromatography (HPLC) instrument control and operation, calculations, and other associated activities. The accuracy of weighing the analytical standard for the API was suspected as a potential contributing factor to the increasing variation in drug potency. Variability in weighing the analytical standard would in turn cause variation in the potency determination of the tablet product.
As part of its investigation (or perhaps “sub-investigation”), this team conducted an experimental study of the accuracy and precision of the balance used to weigh the analytical standard. The balance used to measure the analytical standard was located on a laboratory bench top in a busy location of the analytical lab. The lab was located on an upper floor of the QC laboratory building. Multiple weighings of typical standard weights at the lower calibration limit of the balance were performed according to the approved weighing procedure. All weighings were documented in controlled laboratory notebooks, including witnessing and verification by trained personnel. Mean and standard deviation of weighings were calculated. Results indicated higher than expected variation, confirming the initial reports from QC.
Observation of analysts performing the weighings also suggested that the placement of the standard weight on the balance pan affected the weight data. The weighing procedure did not require that the sample to be weighed be placed in the center of the balance pan.
A CAPA was developed, based on these experimental data and associated observations, as well as the risk analysis indicating the importance of the analytical standard weighing process.
3.4.2 CAPA for the problem
As the ICH has stated, a “pharmaceutical company should have a system for implementing corrective actions and preventive actions resulting from the investigation of […] trends from process performance and product quality monitoring.”18 Such a system came into action as a result of the investigation into the illustrative problem presented in this chapter. Because the variability of data had not yet failed specifications, the CAPA plan highlighted the preventive actions.
The preventive actions focused on upgrading the weighing procedure and facility used for weighing, followed by versioning up the relevant SOPs, as well as associated changes in support of the laboratory procedure changes. Table 3.3 outlines the CAPA plan approved by management.

This section discussed the development and implementation of a CAPA plan to respond to the results of the investigation. All the changes to the weighing facility and equipment described in the plan were completed. These included relocation of the weighing of the analytical standard to a new area with less personnel traffic, placement of the balance on a stand-alone low vibration table, and installation of a protective enclosure around the balance pan to restrict air drafts. Use of the new protective shield required a revision of the weighing procedure. Training in the use of the weighing procedure was also required.
An important implication of these changes was the necessity of revising SOPs, including the calibration and the weighing procedures. The next session highlights the factors included in the revision of the calibration SOP and the critical review of this revision.
3.4.3 Critical review of the calibration SOP
A manufacturing or lab system in a regulated framework must be demonstrably in control. Any piece of equipment that is part of that system must function according to standards. Because of normal wear and tear, the equipment will tend to deviate from those standards. Equipment calibration and preventive maintenance programs are the method to maintain acceptable equipment performance.
As seen in the CAPA plan, the criticality of the weighing procedure necessitated a review of the balance calibration requirements, standards, and guidance.19 The requirements included FDA predicate rules for calibration and the documentation of calibration activities, and internationally recognized standards for calibration. The guidance included FDA recommendations for a calibration SOP, and the directions to be gleaned from FDA inspectional observations and warning letters addressing the failure to meet these requirements, standards, and procedures.
Among regulatory documents supporting calibration, FDA requires calibration of equipment and instruments for all regulated areas, including good laboratory practice (GLP), GMP, blood processing, medical devices, and tissue processing. The agency also requires written procedures for calibration, and documentation of the calibrations (Table 3.4).
Table 3.4
FDA predicate rules for calibration
| Regulation | Calibration Predicate Rule | SOP Predicate Rule |
| 21CFR58.63 | (a) Equipment used for the generation, measurement, or assessment of data shall be adequately tested, calibrated and/or standardized. | (b) written standard operating procedures […] shall set forth in sufficient detail the methods, materials, and schedules to be used. |
| 21CFR211.68 | (a) Equipment […] shall be routinely calibrated. | […] according to a written program designed to assure proper performance. |
| 21CFR211.160; § 211.194 | (b) Laboratory controls shall include: (4) The calibration of instruments, apparatus, gauges, and recording devices at suitable intervals. | […] in accordance with an established written program. |
| 21CFR606.60; § 606.160. | (a) Equipment used in the collection, processing, compatibility testing, storage, and distribution of blood and blood components shall be […] calibrated on a regularly scheduled basis. | (b) Records shall be maintained that include, but are not limited to, the following when applicable: (5) Quality control records: (i) Calibration and standardization of equipment. |
| 21CFR820.72(a) | Each manufacturer shall ensure that all inspection, measuring, and test equipment, including mechanical, automated, or electronic inspection and test equipment, is suitable for its intended purposes and is capable of producing valid results. | Each manufacturer shall establish and maintain procedures to ensure that equipment is routinely calibrated, inspected, checked, and maintained. |
| 21CFR1271.200; § 1271.180 | (c) You must routinely calibrate according to established procedures and schedules. | (a) You must establish and maintain procedures. |
The process of calibrating equipment or instruments involves measurement standards, the calibration process itself, and the device. Measurement standards include the concepts of reliability, precision, and accuracy. FDA regulations require that equipment be calibrated according to written procedures that include measurement standards for precision and accuracy.20
Consider a balance, an instrument used to measure the weight of the product. Several questions arise: Is it a precise instrument? Is it an accurate instrument?
Regarding precision, FDA has stated that it “indicates a relative degree of repeatability, i.e., how closely the values within a series of replicate measurements agree with each other.”21 In general, reliability is inversely related to precision.
The National Institute of Standards and Technology has defined accuracy and bias as follows:
Accuracy is a qualitative term referring to whether there is agreement between a measurement made on an object and its true (target or reference) value. Bias is a quantitative term describing the difference between the average of measurements made on the same object and its true value.”22
Calibration weights are classified in accordance with the recommendations of the International Organization of Legal Metrology.23 Classes of weights include E1, E2, F1, F2, M1, M2, and M3, ranging from the highest accuracy weights (E1; maximum error at 1 kg is ± 0.5 mg) to the lowest (M3; maximum error at 1 kg is ± 500 mg). The higher-class weights are far more expensive than the lower-class weights. Each weight class is “traceable” – tested against a standard of higher accuracy (i.e., the next higher weight class).24
Speaking in general of a written procedure for the calibration of equipment, FDA has suggested25 that the SOP include the sections listed in Table 3.5.
Table 3.5
Outline of SOP for the calibration of equipment
| 1. | Purpose and scope |
| 2. | Frequency of calibration |
| 3. | Equipment and standards required |
| 4. | Limits for accuracy and precision |
| 5. | Preliminary examinations and operations |
| 6. | Calibration process description |
| 7. | Remedial action for product |
| 8. | Documentation requirements |
From a compliance standpoint, the eight topics listed in Table 3.5 are important. FDA has made inspectional observations on organizations that have not adequately addressed these topics. As an example of the failure to address the frequency of calibration, and its GXP implications, see the FDA Warning Letter to International Biologicals dated 12 June 1998: “There were no procedures outlining the frequency for calibrating the scales.”26 For an instance of not addressing required standards, see FDA Warning Letter to BTI Filtration dated 7 February 2007: “…your firm does not include the specifications for the equipment requiring calibration.”27
For an example of failing to address limits for accuracy and precision, see FDA Warning Letter to ChemSource, dated 15 November 2002:
The inspection revealed that your laboratory equipment calibration program is inadequate in the following ways: […] Failure to have written procedures describing specific calibration instructions, and limits. […] Failure to conform to the USP Section < 41 > for weight and balance determination.28
As an example of the failure to incorporate remediation steps in the calibration procedure, see FDA Warning Letter to B. Braun Medical dated 15 March 2006:
… personnel knowingly utilized […] several balances that were […] out of calibration […] In fact, your written procedures do not discuss initiating an investigation to determine whether product may have been impacted, nor discuss corrective actions for equipment that does not meet acceptable tolerance limits.29
For an instance of not meeting documentation requirements, see FDA Warning Letter to Dale Dental, dated 14 October 2004, pointing out the “Failure to [ensure] that calibration records are maintained.”30
The calibration procedure involved in the illustrative study presented in this chapter was found to meet all these standards and regulations. However, prior observations that the location of the standard weight on the balance pan affected weight data suggested that the calibration procedure should be more carefully controlled. A target weight location “X” was inscribed in the balance pan to better define the placement of the standard weight for calibration. This change necessitated a revision to the calibration SOP used by calibration technicians.
As the weighing procedure was reviewed, the following revisions were indicated:
Requirements to use the specified balance in the new weighing facility for weighing of analytical standards;
New steps for correct operation of the protective shield enclosure around the balance weighing pan;
Requirement to place sample to be weighed on the target “X” location on the balance pan.
The revised procedures were drafted and critically reviewed by a step-by-step real-world challenge.
This section discussed the review and revision of SOPs as part of the implementation of a more general CAPA project. It focused especially on the revision of the calibration SOP and the critical review that was part of this revision. The next section addresses the testing of the efficacy of the CAPA and the documentation of the results.
3.4.4 Testing and documenting the changes
As Gamal Amer has put it, a successful CAPA must “make necessary changes to reduce risk or eliminate it.” Moreover, it is necessary to “track and evaluate the actions taken to ensure that no additional or different risk was introduced.”31 This calls for the testing of the efficacy of the changes made, after which the completed project is fully documented, and final approval is sought.
An experimental study was conducted to evaluate the effects of the facility changes, revised calibration procedure, and revised weighing procedure. The same procedure as previously used to evaluate and characterize the balance prior to changes was used. The balance was recalibrated using the new calibration procedure. Multiple weighings of typical standard weights at the lower calibration limit of the balance were performed according to the revised weighing procedure. All weighings were again documented in controlled laboratory notebooks including witnessing and verification by trained laboratory personnel. Mean and standard deviation were calculated. Expectations were that results would be statistically equivalent or improved, relative to pre-change results. Results indicated equivalent accuracy and lower variation as reflected in lower standard deviations.
All the changes were initiated for routine use in support of commercial manufacturing. The responsible project manager developed a document describing all associated changes (Table 3.6).

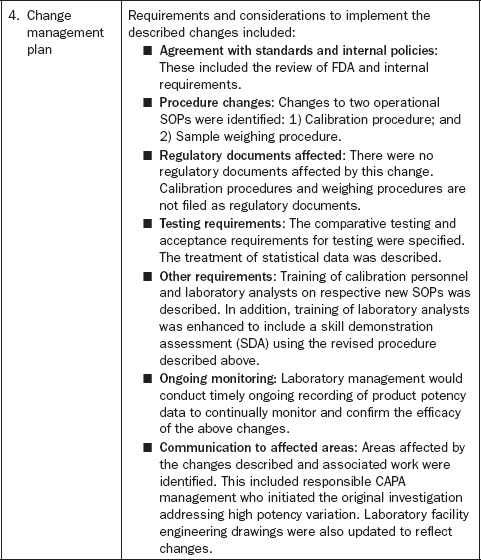
When the change management document was completed, it was affixed to the procedure change documentation as supporting evidence for the change. Implementation of the change required management approval, SME approval, and supporting experimental data.
3.5 Conclusion
This chapter addressed changes to process as well as the revision of SOPs. In order to ensure that the revision of the SOP adds value, it should be subjected to critical review. A range of approaches to the critical review of procedures, in terms of increasing credibility of the review findings, was presented. Management must select among these approaches, weighing the benefits of increasing credibility against the costs of increasing rigor of the approach appropriate for the level of risk of the procedure.
An illustrative problem involving a laboratory weighing procedure was considered, including physical facilities that might be implicated in the problem. This led to the development and implementation of a CAPA project, including changes to laboratory SOPs. FDA regulations for equipment calibration and the associated SOPs were examined. The internal calibration procedure was evaluated for consistency with FDA requirements and technical quality. It was noted that the laboratory weighing procedure must also be reviewed for technical quality. Based on all of the above, the procedural changes were implemented. Finally, the changes brought about through the CAPA by comparative experimental testing were evaluated, and the results were documented.
Critical review of procedures will prove useful in FDA regulated industry. Whatever the origin of the proposal to revise the procedure – whether it is the regularly scheduled biennial revision of a life-cycle document, or a corrective action in response to an investigation into non-conformance, etc. – revision should add as much value as possible in terms of the relative costs and benefits of the various approaches to critical review. This cost/benefit analysis must be informed by a risk assessment of the change. Adding value to procedures makes business sense. It also makes compliance sense, because it affirms that the document and its associated real-world process are both in control. Employing one of these approaches to the critical review of procedures will surely contribute to process control in the lab or manufacturing environment.
3.7 References
Amer, G. Corrective action preventive action (CAPA): a risk mitigating quality system. Pharma. Eng.. 2008; 28(3):67–72.
Claycamp, H.G. ICH Q9: Quality risk management. Presentation to the CDER Advisory Committee for Pharmaceutical Science (ACPS), Rockville, MD, CDER, 5. (October 2006):2006.
FDA. Guideline on General Principles of Process Validation. Rockville, MD, CDER, May. 1987; 1987:4.
FDA. Managing Food Safety. College Park, MD: Center for Food Safety and Applied Nutrition, April. 2006; 2006:58.
Ferrier, M. Risk-based validation of computer systems. Presentation to the NJ Chapter, ISPE, 15. (June):2006.
Fetterolf, D. Developing a sound process validation strategy. BioPharm. Internat.. 2007; 20(12):44–45.
Garcia, T., et al. PQLI Key topics – criticality, design space, and control strategy. J. Pharma. Innov.. 2008; 3(2):60–68.
International Conference on Harmonisation (ICH), Quality Risk Management Q9 Geneva: ICH Secretariat, 9 November 2005,, 2005:2–4.
International Conference on Harmonisation (ICH), Pharmaceutical Quality System – Q10 Geneva: ICH Secretariat, 4 June 2008, 2008:9.
International Organization of Legal Metrology, Weights of classes E1, E2, F1, F2, M1, M1–2, M2, M2–3 and M3, Part 1: Metrological and technical requirements (OIML R 111-1 Edition 2004), Paris: International Organization of Legal Metrology.
International Organization for Standardization (ISO). ISO 9000, Quality Management Systems: Fundamentals and Vocabulary, Geneva: ISO, December 2000. Sect.. 2000; 3(8):5.
Kermode, G.R., Sivaloganathan, S., Complexity resolution for design excellence New York: WileySivaloganathan Sangarappillai, Andrews P.T.J., eds. Design for Excellence, 2000:387–388.
Lowery, A., et al. Equipment and calibration, in Medical Device Quality Systems Manual, Chapter 7. HHS Publication FDA. 97–4179, 1996.
McMillan, J.H. Randomized field trials and internal validity. Pract. Assess., Res. Eval. 12(15). (December):2007.
National Institute of Standards and Technology (NIST), Handbook of Statistical Methods, 2003. [Chapters 2 and 3].
Nosal, R., Schultz, T. PQLI definition of criticality. J. Pharma. Innov.. 2008; 3(2):69–78.
O’Donnell, K., Greene, A. A risk management solution designed to facilitate risk-based qualification. validation, and change control activities within GMP and pharmaceutical regulatory compliance environments in the EU. J. GXP Compl. 10(4). (July):2006.
Radaev, N.N. Setting reliability requirements for products with allowance for economic factors. Meas. Tech.. 2004; 47(9):884–887.
Rivers, P. A review and analysis of the clinical laboratory improvement amendment of 1988: Compliance plans and enforcement policy. Health Care Man. Rev.. 2005; 30(2):93–102.
Rosenthal, R. Experimenter Effects in Behavioral Research. New York: Appleton-Century-Crofts; 1966.
Rosenthal, R. Covert communication in classrooms, clinics, and courtroom. Eye on Psi Chi.. 1998; 3(1):18–22.
Shadish, W.R., Cook, T., Campbell, D. Experimental and Quasi-experimental Designs for Generalized Causal Inference. Boston: Houghton Mifflin; 2002.
1According to the FDA, “Standard Operating Procedure (SOP) means a written method of controlling a practice in accordance with predetermined specifications to obtain a desired outcome.” See, for instance, FDA (2006).
2These changes usually do not include the correction of typographical errors, the addition of clarifying statements, the updating of organizational names, etc. to currently implemented procedures. Since an SOP is a life-cycle document, such “cosmetic” changes could well wait until the next regularly scheduled procedure review.
3See FDA (1987).
4The ISO standard 9000:2000 defines “validation” as “confirmation through the provision of objective evidence that the requirements for a specific intended use or application have been fulfilled …” See ISO 9000 (2000).
5See William R. Shadish, Thomas Cook, and Donald Campbell (2002); see also James H. McMillan (2007).
6On the Rosenthal effect, see Robert Rosenthal (1966); also see Rosenthal (1998).
7See William R. Shadish, Thomas Cook, and Donald Campbell (2002).
8See, for instance, N.N. Radaev (2004).
9As H. Gregg Claycamp (2006) has put it in his presentation to the CDER Advisory Committee for Pharmaceutical Science (ACPS), there are a number of kinds of risk for a company – strategic risks, operational risks, financial risks, compliance risks, competitor advantage, company viability, shareholder harm, patient harm, etc. This chapter focuses on quality risks.
10See International Conference on Harmonisation (ICH) (2005).
11See G.R. Kermode and S. Sivaloganathan (2000).
12See 42 CFR 493.17. For an overview of CLIA, see Patrick Rivers et al. (2005).
13See ICH (2005) op cit.
14See Roger Nosal and Tom Schultz (2008), writing on behalf of the Product Quality Life-cycle Implementation (PQLI) initiative of the International Society for Pharmaceutical Engineering (ISPE); also Thomas Garcia et al. (2008). As Matthew Ferrier (2006) has defined it, a critical component of a process is “a component within a system where the operation, contact, data, control, alarm, or failure will have a direct impact on the quality of the product,” while a non-critical component is “a component within a system where the operation, contact, data, control, alarm, or failure will have an indirect impact, or no impact on the quality of the product.” We can interpret “quality” in this context to mean the SISPQ of the product.
15See also Roger Nosal and Tom Schultz (2008), op. cit. David Fetterolf (2007) has provided an illustration of the measurement of criticality, where each parameter of a system is assessed for its potential to affect the applicable process controls or quality attributes. Each parameter is given a numerical rating based on the likelihood and potential magnitude of impact. The parameters that have the highest likelihood and potential to affect the process are entered into range finding studies and the outcome of the studies is the relationship between the parameter’s normal operating range (ro, control space) and its proven acceptable range (ra, design space). The normal operating range is the range at which the parameter is typically controlled during routine operations, usually the range found in manufacturing instructions. It takes into account minimum and maximum values tested during initial development and a review of process history, which shows the capability of operators, facility, equipment, and utilities. The proven acceptable range is defined by minimum and maximum values for each parameter found during the range-finding studies. Range-finding studies are often designed such that the ranges studied are two or three times the normal operating range. If a parameter’s operating range is less than two times its acceptable range, i.e. ro < 2 × ra, this indicates that a deviation to the normal operating range would likely result in a failure to meet an in-process control, in-process specification, or failure of the batch.
16ICH (2005) op. cit. As Kevin O’Donnell and Anne Greene (2006) have expressed it:
risk events can have multiple causes, with multiple associated risks, some less important that [sic] others. This can result in formal risk management activities becoming costly and quite labor-intensive exercises, and should, therefore, be targeted at the most complex or critical issues” (italics added).
1721 CFR 211.192, “Production Record Review.” See also Gamal Amer (2008) on the “two types of quality events associated with risk.”
18See ICH (2008).
19A similar critical review of the weighing SOP itself was required, but not included here. In addition, that review took into account the guidance provided by US Pharmacopeia < 1251 > .
20For instance, 21 CFR 820.72(b). See Andrew Lowery et al. (1996). According to National Institute of Standards and Technology (NIST) (2003):
Calibration is a measurement process that assigns values to the property of an artifact or to the response of an instrument relative to reference standards or to a designated measurement process. The purpose of calibration is to eliminate or reduce bias in the user’s measurement system relative to the reference base.
Available from: http://www.itl.nist.gov/div898/handbook
21Lowery et al. (1996) ibid.
22See NIST (2003) op. cit. The FDA has defined accuracy as “the measure of an instrument’s capability to approach a true or absolute value. Accuracy is a function of precision and bias.” Bias, in turn, is defined as “a measure of how closely the mean value in a series of replicate measurements approaches the true value;” see Lowery et al. (1996) op cit.
23See International Organization of Legal Metrology (2004).
24As FDA has stated, “Calibration standards used for inspection, measuring, and test equipment shall be traceable to national or international standards;” see 21 CFR 820.72, “Inspection, measuring, and test equipment.”
25Lowery et al. (1996), op cit.
26Available from: www.fda.gov/foi/warning_letters/archive/1857b.pdf See also FDA Warning Letter to American Blending and Filling dated 27 September 2001, pointing out the “Failure to establish written procedures for the calibration of compounding and laboratory equipment. Instruments utilized in the manufacturing and testing of finished product are not calibrated on a routine basis.” Available from: www.fda.gov/foi/warning_letters/archive/g1833d.pdf
27Available from: www.fda.gov/foi/warning_letters/archive/b6289d.pdf
28Available from: www.fda.gov/foi/warning_letters/archive/g3695d.pdf
29Available from: www.fda.gov/foi/warning_letters/archive/g5817d.pdf
30Available from: www.fda.gov/foi/warning_letters/archive/g5014d.pdf
31See Gamal Amer (2008).