
CHAPTER 40
Single Nucleotide Polymorphism (SNP) Mining Tools
Mir Asif Iquebal, Sarika and D Kumar
CABiN, ICAR‐IASRI, New Delhi, India
40.1 INTRODUCTION
There exist genetic variations among individuals of all organisms, and these genetic variations make individuals look phenotypically different. Single nucleotide polymorphisms (SNPs) are considered the simplest and most abundant type of genetic variations in the genome of organisms. SNPs are the markers of choice in most species for genome‐wide association studies (GWAS), phylogenetic analysis, marker‐assisted selection and genomic selection (Liu et al. 2013). They are the genetic markers of choice due to their high density and stability, and the highly automated techniques which are available for detection of SNPs (Kerstens et al., 2009).
Numerous tools are available online for mining SNPs computationally. SNP mining in NGS data has been well documented using two online open source tools: Stacks (Catchen et al., 2011; Ogden et al., 2013) and GATK (DePristo et al., 2011).
40.2 OBJECTIVE
To learn about SNP mining using Stacks, the Burrows–Wheeler algorithm (BWA) aligner, the Genome analysis toolkit (GATK) and Samtools.
40.3 PROCEDURE
We will learn to install and run the tools STACKS, BWA, GATK, Samtools, and so on, to mine SNPs in given nucleotide sequences.
40.3.1 Stacks
This is a program to study population genetics, and it is designed to work with any restriction‐enzyme‐based data, such as GBS (Genotyping by Sequencing), CRoPS, RAD‐Seq, and ddRAD‐Seq. Stacks can identify SNPs within or among populations. It has different modules to generate summary statistics and also compute parameters of population genetics, such as Fis and π, within populations, and Fst between populations.
The output of Stacks can be exported in VCF (Variant Call Format) and many other standard formats, which we can use in different programs like STRUCTURE and GenePop for downstream analysis. The SNPs predicted by Stacks can also be exported in Phylip format to predict phylogenetic trees by any standard phylogenetic software/tool. Stacks can be used to predict SNPs de novo, as well as by a reference‐based method. It is a Linux‐based program and is available for free download from the Stacks website (http://creskolab.uoregon.edu/stacks/) (Figure 40.1).

FIGURE 40.1 Screenshot of Stacks software: http://creskolab.uoregon.edu/stacks/
40.3.1.1 Stacks installation
- Untar the compressed file. tar xfvz stacks_x.xx.tar.gz
- Change directory cd stacks_x.xx
- Configure the software. ./configure
- Build the programs from the source code using “make”. make
- Become root user. make install
40.3.1.2 Install the BWA aligner
- Download the BWA software (Li and Durbin, 2010).
- Untar the compressed file tar –jxvf bwa_x.xx.tar.bz2
- Change directory cd bwa_x.xx
- Build the programs from the source code using “make”. make
Now run ./bwa from the command line to check if BWA is installed correctly. Additionally add the binaries of the softwares to the bashrc.
40.3.1.3 De novo‐based SNP mining using Stacks
The denovo_map.pl Perl wrapper script (which includes three components – ustacks, cstacks and sstacks) is used for de novo SNP mining in Stacks. The following codes have been obtained from http://catchenlab.life.illinois.edu/stacks/comp/denovo_map.php. The codes and its explanations are verbatim as available on the website.
Please also consult “Stacks”, using the following link for downloading Stacks: http://creskolab.uoregon.edu/stacks/
40.3.1.4 Steps for de novo SNP mining from example RAD‐Seq dataset
Use the denovo_map.pl script to call SNP from Mango example RAD‐Seq dataset (Figure 40.2):

FIGURE 40.2 Image of denovo_map.pl script of Stacks to call SNPs de novo from RADSeq data.
denovo_map.pl –m < number > –M < number > –n < number > –T < number > –S –b < number > –o < path/to/output/result/folder > –s < path/to/input/file1 > –s < path/to/input/file2 > –X “populations:‐b <1 > –t < number > –vcf ”
This script will generate results and place them in the output folder specified by the –o option. There are two input files (e.g., Mango_R1.fastq and Mango_R2.fastq) that are specified by option –s. The code for analysis is set using a number of options as given below:
- –m option: uses three identical raw reads to create a stack;
- –M option: indicates permitted mismatches (here, we have three mismatches) allowed between loci when processing a single individual;
- –n option: for the number of mismatches permissible between loci when building the catalog (here, two mismatches);
- –T option: for executing the threads (here, in this example, with 15 threads);
- –b option: batch ID for the run (here, 1);
- –S option: whether the records are to be entered in the MySQL database (here, not entered).
Additionally, our code is running a “populations program” (–X option) to generate “population genetics statistics” on batch 1 (–b option) with 100 threads to run in a parallel section (–t option) and generate results in vcf format (–vcf option).
40.3.1.5 Reference‐based SNP mining using STACKS
The reference‐based SNP mining in Stacks is done using ref_map.pl Perl, which includes three components – pstacks, cstacks and sstacks. This program needs a reference‐aligned data file and can take input data that has been aligned using Bowtie or any other aligner (like BWA); output will be in SAM (Sequence alignment/Map) format.
40.3.1.6 Steps for reference‐based SNP mining from example RAD‐Seq data
The ref_map.pl perl wrapper script is used to run reference‐based SNP mining in Stacks software (Figure 40.3):
- First index the genome by bwa index: bwa index < path/to/reference/file.fa>
- Align the reads to the reference sequence using bwa mem: bwa mem < path/to/reference/file.fa > <path/to/input/sequence/
file.fastq>
<path/to/input/sequence/file2.fastq> > <path/to/output/file.sam>
- Then call the SNPs using the ref_map.pl: ref_map.pl –T < number > –b < number > –S –o < path/to/results/folder > –s < path/to/input/file.sam > –X “populations:–b < number > –t < number > –vcf”

FIGURE 40.3 Image of ref_map.pl script of STACKS to call SNPs reference based from RAD‐Seq data.
This perl script will generate results in the output folder specified by the –o option from a reference aligned input file – e.g., MangoSeq.sam, specified by the –s option with 15 threads to execute, specified by –T option and batch id 1, for the run specified by –b option. Additionally, it can run a populations program (–X option) to generate population genetics statistics on batch 1 (–b option), with 100 threads to run in parallel section (–t option), and generate results in VCF format (–vcf option).
40.3.2 Genome Analysis Toolkit (GATK)
This is an organized programming framework (DePristo et al., 2011), which is designed to develop effective and durable analysis tools for next‐generation DNA sequencing, using the functional programming theme of MapReduce (Dean and Ghemawat, 2008; DePristo et al. 2011). The GATK has a variety of tools which primarily focus on SNP discovery and genotyping, and has a strong emphasis on data quality assurance. It has a robust architecture, a powerful processing engine, and high‐performance computing features which make it suitable to be used for a project of any size (https://www.broadinstitute.org/gatk/index.php). A reference‐based SNP mining program that uses the Linux operating system, it is available for free download from the GATK website (Figure 40.4) (https://www.broadinstitute.org/gatk/index.php).
FIGURE 40.4 Screenshot of GATK software website: https://www.broadinstitute.org/gatk/index.php.
In order to use GATK, it is first necessary to install the BWA, Samtools, and Picard tools.
40.3.2.1 BWA installation
- Untar the downloaded BWA.tar file (Li and Durbin, 2010): tar ‐jxvf bwa‐x.xx.tar.bz2
- Change the directory: cd bwa‐x.xx
- Build the program from the source code, using the “make” command: make
- Execute the command, ./bwa from the command line (to check if it is properly installed).
- Additionally, add the bwa binary to the path to make it available on the command line.
40.3.2.2 Samtools installation
- Untar the downloaded Samtools.tar file (Li et al., 2009a): tar –jxvf samtools‐x.xx.tar.bz2
- Change the directory: cd samtool‐x.xx
- Build the program from the source code, using the “make” command make
- From the command line, run ./samtools (to check if it is properly installed).
- Additionally, add the samtools binary to the path to make it available on the command line.
40.3.2.3 Picard tools and GATK installation
- Untar the downloaded Picard tools zip file: unzip picard‐tools‐x.xx.zip
- Change the directory: cd picard‐tools‐x.xx
- From the command line, run java –jar picard.jar –h (to check if it is properly installed). Now download GATK (latest version) from https://software.broadinstitute.org/gatk/download/, and begin the GATK installation:
- Untar the downloaded GATK tar file: tar ‐jxvf GenomeAnaysisTK‐x.xx.tar.bz2
- Change the directory: cd GenomeAnalysisTK‐x.xx
- From the command line, run java –jar GenomeAnalysisTK.jar –h (to check if it is properly installed).
40.3.3 Steps for SNP mining from chickpea data using GATK pipeline
To call SNPs using the GATK toolkit, we have to pre‐process the input bam files. Steps for pre‐processing the bam files are described below:
- Sort the bam file using the SortSam tool in Picard‐tools: java –jar < path/to/SortSam.jar > INPUT = <path/to/input/file.bam > OUTPUT = <path/to/output/file.bam > SORT_ORDER = <type > VALIDATION_STRINGENCY = <SILENT,LENIENT or STRICT>
- Mark the duplicates in the bam file using “MarkDuplicates”: java –jar < path/to/AddOrReplaceReadGroups.jar > INPUT = <path/to/Markduplicated/file.bam > OUTPUT = <path/to/output/file.bam > RGID = groupname RGLB = lib name RGPL = platformname RGPU = unit number RGSM = sample name VALIDATION_STRINGENCY = <SILENT,LENIENT or STRICT>
- Add read group information using “AddOrReplaceReadGroups”: java –jar < path/to/MarkDuplicates.jar > INPUT = <path/to/sortedfile.bam > OUTPUT = <path/to/output/file.bam > METRICS_FILE = <path/to/metrics.txt>
- Build the bam file index using “BuildBamIndex”: java –jar < path/to/BuildBamIndex.jar > INPUT = <path/to/input/file.bam VALIDATION_STRINGENCY = <SILENT,LENIENT or STRICT>
With these processed Bam files, we now can proceed further to call SNPs by GATK using the “UnifiedGenotyper” program, as described below:
java –jar < path/to/GenomeAnalysisTK.jar –T UnifiedGenotyper –R < path/to/reference/sequence.fa –I <path/to/input/file.bam –I <path/to/input/file2.bam –o <path/to/output/file.vcf VALIDATION_STRINGENCY = <SILENT,LENIENT or STRICT>
The script used in the GATK pipeline calls SNPs from two input bam files (specified by the –I option), using the reference sequence specified by the –R option, and outputs results to the output folder specified by the –o option in VCF format, using the “UnifiedGenotyper” tool. The user can select the level of stringency required, from Silent, Lenient, and Strict options.
The GATK script, running on an example dataset, is shown in Figure 40.5.

FIGURE 40.5 Image of GATK command used to mine SNPs from an example dataset.
40.4 INTERPRETATION OF RESULTS
The Variant Call Format (VCF) has its first six columns representing observed variation, and is explained as follows (see Figure 40.6):
- CHROM and POS: This gives the contig with position on which the variant occurs.
- ID: Shows the dbSNP RefSeq (rs) identifier of the SNP.
- REF and ALT: the reference base and alternative base, which vary in the samples or in the population.
- QUAL: The quality value – namely, Phred‐scaled probability that a REF/ALT polymorphism exists at this site given sequencing data.
- FILTER: The VCFs produced carry both the passing and failing filter records.
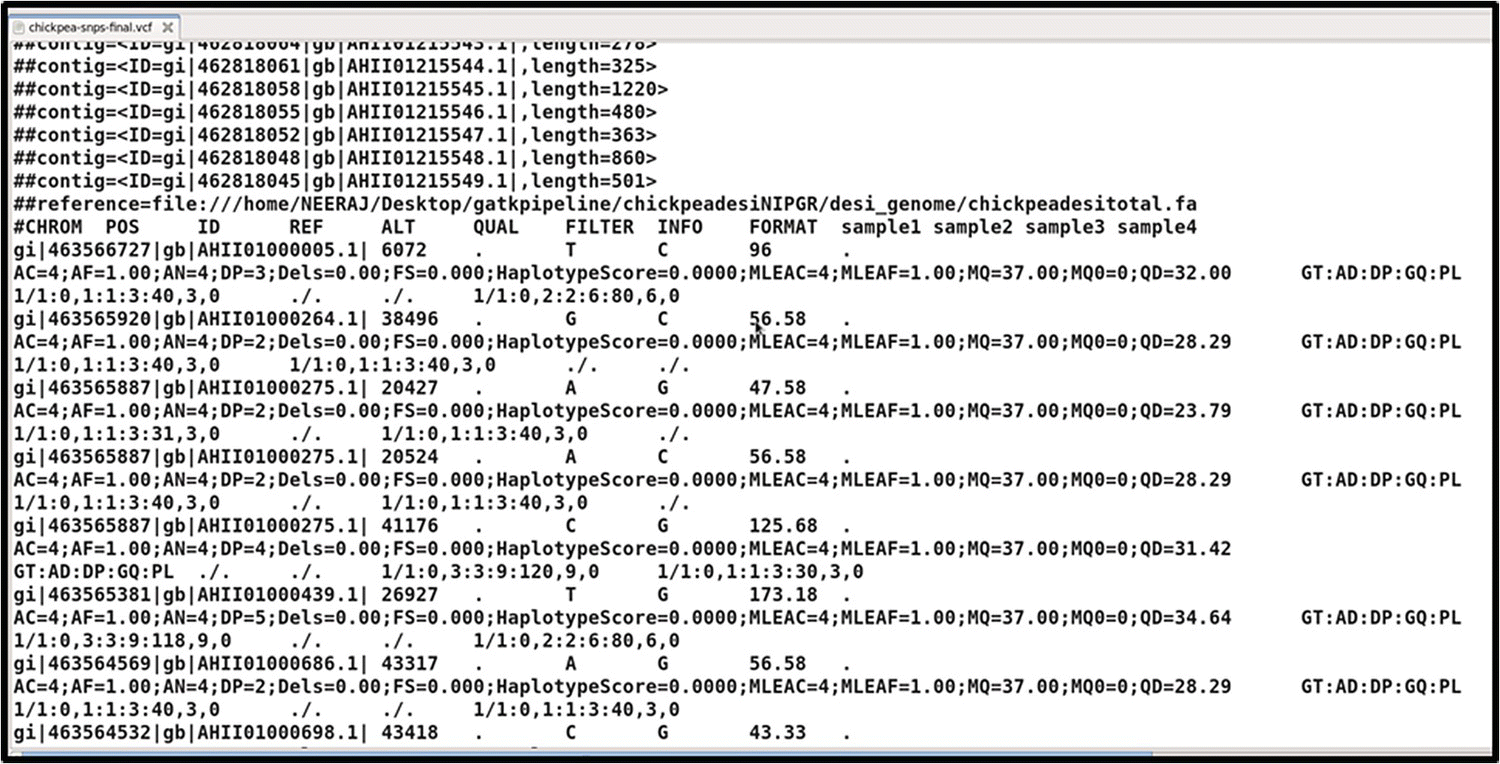
FIGURE 40.6 Result of GATK SNPs mining from an example dataset.
40.5 QUESTIONS
- 1. What are the various tools/applications for SNP mining?
- 2. Trace the sequence NC_003062.2 from the public domain database, and search for the SNPs in this sequence by the de novo approach.
- 3. How many SNP calls did you get?
- 4. How many SNPs passed for the given sequence?